

Abstract
Porphyromonas gingivalis, a major periodontal pathogen, may contribute to atherogenesis and other inflammatory cardiovascular diseases. However, little is known about interactions between P. gingivalis and endothelial cells. E-selectin is a membrane protein on endothelial cells that initiates recruitment of leukocytes to inflamed tissue, and it may also play a role in pathogen attachment. In the present study, we examined the role of E-selectin in P. gingivalis adherence to endothelial cells. Human umbilical vein endothelial cells (HUVECs) were stimulated with tumor necrosis factor alpha (TNF-α) to induce E-selectin expression. Adherence of P. gingivalis to HUVECs was measured by fluorescence microscopy. TNF-α increased adherence of wild-type P. gingivalis to HUVECs. Antibodies to E-selectin and sialyl Lewis X suppressed P. gingivalis adherence to stimulated HUVECs. P. gingivalis mutants lacking OmpA-like proteins Pgm6 and -7 had reduced adherence to stimulated HUVECs, but fimbria-deficient mutants were not affected. E-selectin-mediated P. gingivalis adherence activated endothelial exocytosis. These results suggest that the interaction between host E-selectin and pathogen Pgm6/7 mediates P. gingivalis adherence to endothelial cells and may trigger vascular inflammation.
INTRODUCTION
Periodontitis is a disease of the supporting structures of the teeth, causing loss of attachment to the alveolar bone and eventual exfoliation of teeth (5). Severe periodontitis affects up to 20% of the population, and mild to moderate periodontitis is observed in the majority of adults (6). Gram-negative bacteria play an important role in the pathogenesis of human periodontal diseases (15, 42), and Porphyromonas gingivalis is one of the species most strongly implicated in periodontal diseases (14, 43). Several recent studies have demonstrated that P. gingivalis is able to invade and activate different cell types in the tissue surrounding teeth (endothelial and gingival epithelial cells as well as periodontal ligament cells) (12, 26, 40). Moreover, recent studies have demonstrated a transient bacteremia with potential systemic infection after a variety of dental treatment procedures (2, 19, 20, 41). Therefore, endothelial cells can act as primary target cells during infection with P. gingivalis. However, little is known about mechanisms of infection and activation of endothelial cells by P. gingivalis.
The endothelium has several important functions, which include providing a nonadhesive, nonthrombotic barrier between the blood and the underlying tissues. In atherosclerosis or in response to injury or inflammatory cytokines such as tumor necrosis factor alpha (TNF-α), the endothelium becomes activated, and selectins and cell adhesion molecules (CAMs) are rapidly induced (36, 39). In particular, members of the immunoglobulin superfamily of CAMs, such as intercellular cell adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1), as well as the selectin family members E-selectin and P-selectin, are expressed and play crucial roles in the adhesion and migration of monocyte/macrophage infiltration into atherosclerotic lesions during the early and subsequent stages of atherosclerosis in a variety of animal models (21, 47, 49). Increased expression of E-selectin and production of proinflammatory cytokines in the endothelium play a pivotal role in the generation of leukocyte infiltrates and subsequent atherosclerotic plaque formation (16, 28). P. gingivalis infection significantly increases endothelial expression of VCAM-1, ICAM-1, and E-selectin, enhances production of interleukin-6 (IL-6), IL-8, and monocyte chemoattractant protein 1 (MCP-1), and increases adhesion of THP-1 monocytes to endothelial cells (18, 46). Therefore, P. gingivalis elicits a proatherogenic response in endothelial cells. Although E-selectin is involved in vascular inflammation and is induced with P. gingivalis, the interaction between P. gingivalis and endothelial cells is not understood. In the present study, we explored the ability of E-selectin to facilitate P. gingivalis adherence to human umbilical vein endothelial cells (HUVECs). We found that activated endothelial cells interact with P. gingivalis via E-selectin on endothelial cells and via OmpA-like proteins Pgm6 and -7 of the bacterium.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
P. gingivalis ATCC 33277 was used as a wild-type strain in this study. P. gingivalis defective mutants lacking fimA were constructed as described previously (17). A P. gingivalis Pgm6/7-deficient mutant was constructed as described previously (32). This mutant did not show any sign of a polar effect on the downstream gene (data not shown). All P. gingivalis strains were grown at 37°C under anaerobic conditions (10% CO2, 10% H2, and 80% N2) on brucella HK agar (Kyokuto Pharmaceutical Industrial Co., Ltd., Tokyo, Japan) supplemented with 5% laked rabbit blood, hemin (2.5 μg/ml), menadione (5 μg/ml), and dithiothreitol (0.1 mg/ml) and in Trypticase soy broth (BD, Franklin Lakes, NJ) supplemented with yeast extract (2.5 mg/ml), hemin (2.5 μg/ml), menadione (5 μg/ml), and dithiothreitol (0.1 mg/ml). Bacterial growth was monitored by measuring the optical density at 660 nm (OD660). For infection assays, an inoculum with an infection ratio (multiplicity of infection [MOI]) of 100 bacteria per cell was added to the cell culture medium.
Cell culture conditions.
HUVECs were cultured in endothelial cell growth medium 2 (EGM-2) (Lonza, Basel, Switzerland) supplemented with fetal bovine serum, hydrocortisone, human recombinant fibroblast growth factor, vascular endothelial growth factor, recombinant insulin growth factor 1, ascorbic acid, human recombinant epidermal growth factor, gentamicin, and amphotericin B at 37°C in a humidified atmosphere of 5% CO2.
E-selectin expression.
E-selectin cDNA was constructed as described previously (53). The E-selectin cDNA was amplified by PCR with specific primers (5′-GAC AGC TAG CAT GAT TGC TTC ACA G-3′ [includes an additional NheI site] and 5′-CGG CCT CGA GTT AAA GGA TGT AAG AAG GC-3′ [includes an additional XhoI site]) and then cloned into the pcDNA3.1 vector (Invitrogen, Carlsbad, CA). For preparation of a soluble E-selectin vector, a stop codon and a unique EcoRV site were introduced by site-directed mutagenesis (Promega, Madison, WI) into the boundary between the sixth consensus repeat and the transmembrane domain, using the following oligonucleotide, which starts at nucleotide 1776: 5′-CC AAC ATT CCC TAG ATA TCT AGA CTT TCT GCT G-3′.
Measurement of E-selectin production.
An enzyme-linked immunosorbent assay (ELISA)-based method was used for quantification of E-selectin protein expression in endothelial cells. HUVECs (3.5 × 105 cells/ml) were seeded into 6-well plates and grown overnight. The cells were then stimulated with 10 ng/ml of TNF-α (PeproTec Inc., Rocky Hill, NJ) for 1, 2, 3, 4, 8, and 24 h. After removing the medium, the cell layers were washed twice with phosphate-buffered saline (PBS). Cells were lysed in a cell lysis reagent (CelLytic P; Sigma-Aldrich, St. Louis, MO) with a protease inhibitor mixture (Nacalai Tesque, Kyoto, Japan). Concentrations of E-selectin in the cell lysates were determined using a commercial ELISA kit for E-selectin (eBioscience, San Diego, CA). The cell lysates were also mixed with 4× Laemmli sample buffer without reducing agents and were fractionated by 7.5% SDS-PAGE and immunoblotted with a monoclonal antibody to E-selectin (BBIG-E4 [5D11]; R&D Systems, Abingdon, United Kingdom).
Analysis of P. gingivalis adhesion to endothelial cells.
HUVECs (2 × 106 cells) were seeded in a Lab-Tek II chamber slide system (Nalge Nunc International, Rochester, NY) that had been coated with 50 µg/ml of rat tail collagen (BD), and the cells were incubated for 24 h before administration of P. gingivalis. HUVECs grown to near confluence in each well were stimulated with TNF-α for 3 h, and then P. gingivalis cells which had been washed with EGM-2 and resuspended in EGM-2 without antibiotic at a concentration of 108 cells/ml were added to the monolayer cells at an MOI of 1:100 under 5% CO2 at 37°C for 0.5 to 3 h. Cells were then washed three times with PBS, followed each time by gentle rinsing for 5 min at room temperature, and fixed with 4% (wt/vol) paraformaldehyde at 4°C overnight. After washing three times with PBS, the cells were permeabilized with PBS containing 0.05% Triton X-100 at room temperature for 30 min. They were washed again and then blocked with PBS containing 5% (wt/vol) bovine serum albumin (BSA) at room temperature for 30 min. Bacterial cells on chamber slides were labeled with an antiserum for P. gingivalis whole cells (1:1,000 dilution) for 60 min at room temperature and then washed five times with PBS. The bacterial cells were then incubated with Alexa Fluor 488-conjugated goat anti-rabbit IgG (1:1,000 dilution; Invitrogen Co., Carlsbad, CA). Actin filaments in HUVECs or 293 cells were stained simultaneously with Alexa Fluor 568-conjugated phalloidin (1 μg/ml; Invitrogen Co.) for 60 min at room temperature in the dark. After washing 10 times with PBS, chamber slides were mounted onto a slide containing ProLong Gold antifade reagent (Invitrogen). Adherent bacteria on the cell surface were examined by fluorescence microscopy (Keyence, Osaka, Japan). We measured the area stained with Alexa 488 (corresponding to P. gingivalis) in a visual field (corresponding to 0.06 mm2) by using the Image J program. We then calculated bacterial number by dividing the area by the size (in pixels) of a P. gingivalis cell. To determine whether E-selectin is involved in P. gingivalis adherence to endothelial cells, TNF-α-pretreated HUVECs were incubated with P. gingivalis ATCC 33277 (108 cells/ml in each well) for 30 min to 3 h in the presence of various concentrations of an antibody for E-selectin (R&D Systems, Inc., Minneapolis, MN), recombinant E-selectin, or sialyl Lewis X (Calbiochem, San Diego, CA). P. gingivalis ATCC 33277 (108 cells/ml in each well) was also incubated with HEK 293 cells transfected with a human E-selectin-inserted vector for 30 min. To explore ligands for E-selectin on P. gingivalis, P. gingivalis ATCC 33277 (wild type), a FimA-deficient mutant (ΔfimA), and a Pgm6/7-deficient mutant (Δpgm6/7) (108 cells/ml) were incubated with TNF-α-pretreated HUVECs for 3 h. TNF-α-pretreated HUVECs were incubated with P. gingivalis ATCC 33277 (108 cells/ml) for 30 min in the presence or absence of envelopes isolated from wild-type or mutant P. gingivalis. TNF-α-pretreated HUVECs were incubated with P. gingivalis ATCC 33277 (108 cells/ml) for 30 min in the presence or absence of purified FimA fimbriae and Pgm6/7.
Measurement of VWF and nitric oxide.
HUVECs (3.5 × 105 cells/ml) were seeded into 12-well plates and grown overnight. The cells were then stimulated with 10 ng/ml of TNF-α for 3 h. P. gingivalis cells were inoculated into cultures at an MOI of 100, and the cultures were incubated for 30 min and 1 h. The culture media were then collected and centrifuged at 13,000 rpm for removal of bacterial cells. Concentrations of von Willebrand factor (VWF) in the supernatants were measured by use of an ELISA kit according to the manufacturer's instructions (VWF ELISA kit; American Diagnostic Inc., Stanford, CT). The concentration of NO2−/NO3− was also measured by 2,3-diaminonaphthalene (DAN) assay (24).
Preparation of P. gingivalis envelope.
Separation of whole envelopes and the outer membrane from P. gingivalis strains was performed essentially as described previously (30). Briefly, bacterial cells were washed with PBS (pH 7.5) and then resuspended in PBS (pH 7.5) containing 0.1 mM N-α-p-tosyl-l-lysine chloromethyl ketone, 0.2 mM phenylmethylsulfonyl fluoride, and 0.1 mM leupeptin. The cells were disrupted by sonication, and remaining, undisrupted bacterial cells were removed by centrifugation at 1,000 × g for 10 min. The envelope was collected as a pellet by centrifugation at 100,000 × g for 60 min at 4°C. The pellet was washed once by resuspension in PBS and recentrifuged. The final pellet was suspended in PBS.
Purification of FimA.
The major fimbriae from P. gingivalis ATCC 33277 were purified as described previously (52). The purity was ascertained by scanning of the stained SDS-polyacrylamide gel.
Purification of Pgm6/7 complex.
The functional Pgm6/7 complex was purified by two methods. First, we purified it electrophoretically from bacterial envelopes as previously reported (32). Briefly, an envelope fraction of P. gingivalis was subjected to SDS-PAGE under nonreducing conditions. A 120-kDa protein band, corresponding to Pgm6/7 heterotrimers, was excised, and then the complex was extracted electrically from a piece of gel. We used these samples for the experiments in Fig. 3E and File S3B in the supplemental material. Second, we constructed C-terminally hexahistidine-tagged Pgm6 and purified the Pgm6/7 complex from a P. gingivalis mutant by using a nickel affinity column. Briefly, we inserted a DNA fragment consisting of the pgm7 open reading frame (ORF) associated with the DNA sequence encoding Gly-Ser-Ser-hexahistidine into the vector pT-COW (13), bearing a powerful promoter of the 350-bp upper region of ragA (31). The constructed plasmid was introduced into a pgm7 deletion mutant of P. gingivalis (32). The cell lysate was applied to a nickel affinity column, and the bound proteins were eluted. Although a hexahistidine tag was associated with Pgm7 alone, the Pgm6/7 complex was obtained. We used these samples for the experiments in Fig. 3F and G and File S3C in the supplemental material.
Fig 3.

Pgm6/7 in P. gingivalis mediates the interaction with activated endothelial cells. (A) P. gingivalis ATCC 33277 (wild type), a FimA-deficient mutant (ΔFimA), and a Pgm6/7-deficient mutant (ΔPgm6/7) (108 cells/ml in each well) were incubated with TNF-α-pretreated HUVECs for 3 h. Other procedures are described in the legend to Fig. 1A. Bars, 10 μm. (B) P. gingivalis ATCC 33277 (wild type) and a FimA-deficient mutant (ΔFimA) (108 cells/ml in each well) were incubated with TNF-α-pretreated HUVECs for 30 min. Other procedures are described in the legend to Fig. 1A. (C) P. gingivalis ATCC 33277 (wild type) and a Pgm6/7-deficient mutant (Pgm6/7) (108 cells/ml in each well) were incubated with TNF-α-pretreated HUVECs for 30 min. Other procedures are described in the legend to Fig. 1B. Data are means ± SD (n = 3). *, P < 0.01 versus no TNF-α. (D) Inhibitory effects of P. gingivalis envelopes on TNF-α-induced adhesion of P. gingivalis to HUVECs. HUVECs were incubated with TNF-α (10 ng/ml) for 3 h. Cells were then washed and incubated with P. gingivalis ATCC 33277 (108 cells/ml in each well) for 30 min in the presence or absence of envelopes isolated from wild-type or mutant P. gingivalis. Other procedures are described in the legend to Fig. 1B. Data are means ± SD (n = 3). *, P < 0.01 versus no TNF-α; †, P < 0.01 versus control. (E) Effects of extracted Pgm6/7 and FimA on TNF-α-induced adhesion of P. gingivalis to HUVECs. HUVECs were incubated with TNF-α (10 ng/ml) for 3 h. Cells were then washed and incubated with P. gingivalis ATCC 33277 (108 cells/ml in each well) for 30 min in the presence or absence of purified Pgm6/7 and FimA. Other procedures are described in the legend to Fig. 1B. Data are means ± SD (n = 3). *, P < 0.01 versus no TNF-α; †, P < 0.01 versus Pgm6/7 fraction. (F) Inhibitory effect of P. gingivalis Pgm6/7 on TNF-α (10 ng/ml)-induced adhesion of P. gingivalis to HUVECs. HUVECs were incubated with TNF-α (10 ng/ml) for 3 h. Cells were then washed and incubated with P. gingivalis ATCC 33277 (108 cells/ml in each well) for 30 min in the presence or absence of purified Pgm6/7. Other procedures are described in the legend to Fig. 1B. Data are means ± SD (n = 3). *, P < 0.01 versus no TNF-α; †, P < 0.01 versus Pgm6/7 (0 ng/ml). (G) Inhibitory effect of P. gingivalis Pgm6/7 on TNF-α-induced adhesion of P. gingivalis to HUVECs. HUVECs were incubated with TNF-α (10 ng/ml) for 3 h. Cells were then washed and incubated with P. gingivalis ATCC 33277 (108 cells/ml in each well) for 30 min in the presence or absence of purified Pgm6/7. Other procedures are described in the legend to Fig. 1A. Bars, 10 μm.
RESULTS
TNF-α augments adherence of P. gingivalis to endothelial cells by inducing expression of E-selectin.
We first examined induction of E-selectin expression by TNF-α by using ELISA and Western blotting of HUVEC cultures. TNF-α induced a time-dependent expression of E-selectin in HUVECs (see Files S1 and S2 in the supplemental material). E-selectin expression was maximal 3 h after TNF-α addition. No basal expression of E-selectin was found. To determine whether E-selectin expression in endothelial cells is involved in adhesion of P. gingivalis to the cells, we incubated HUVECs with TNF-α (10 ng/ml) for 0.5 to 3 h, and then P. gingivalis ATCC 33277 cells (108 cells/ml in each well) were added to the culture medium for 0.5 to 3 h. Cells were then washed, and attachment of P. gingivalis to the cells was observed by fluorescence microscopy. Attachment of P. gingivalis to HUVECs increased time dependently without pretreatment of TNF-α (Fig. 1A and B). Pretreatment with 10 ng/ml of TNF-α significantly enhanced the level of attachment in HUVEC cultures.
Fig 1.
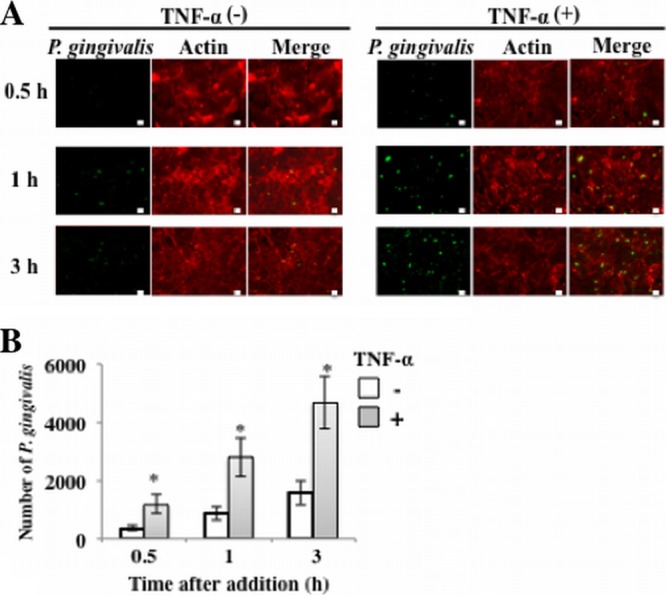
Adherence of P. gingivalis to HUVECs is enhanced by stimulation with TNF-α. (A) HUVECs were incubated with TNF-α (10 ng/ml) for 0.5 to 3 h. P. gingivalis ATCC 33277 cells (108 cells/ml in each well) were then added to the culture medium for 0.5 to 3 h. Cells were then washed, and attachment of P. gingivalis to the cells was observed by fluorescence microscopy. P. gingivalis was stained with Alexa Fluor 488 (green), and actin of endothelial cells was visualized with Alexa Fluor 568 (red). Bars, 10 μm. (B) HUVECs were incubated with TNF-α (10 ng/ml) for 0.5 to 3 h. P. gingivalis ATCC 33277 cells (108 cells/ml in each well) were then added to the culture medium for 0.5 to 3 h. Cells were then washed, and attachment of P. gingivalis to the cells was observed by fluorescence microscopy. The attachment levels are expressed as numbers of P. gingivalis cells per 60,430 mm2 (means ± standard deviations [SD] [n = 3]). *, P < 0.01 versus no TNF-α.
To clarify the role of E-selectin in P. gingivalis adherence to HUVECs, we examined the effect of anti-E-selectin antibodies on P. gingivalis adherence to HUVECs. HUVECs were pretreated with TNF-α and then incubated with P. gingivalis for 30 min in the presence of antibodies for E-selectin or control IgG. Antibodies to E-selectin inhibited P. gingivalis adherence to TNF-α-pretreated HUVECs (Fig. 2A).
Fig 2.

Adherence of P. gingivalis to TNF-α-activated endothelial cells was mediated by E-selectin. (A) Inhibitory effect of anti-E-selectin antibodies. HUVECs were incubated with TNF-α (10 ng/ml) for 3 h. Cells were then washed and incubated with P. gingivalis ATCC 33277 (108 cells/ml in each well) for 30 min in the presence of antibodies for E-selectin or control IgG. Other procedures are described in the legend to Fig. 1B. Data are means ± SD (n = 3). *, P < 0.01 versus no TNF-α; †, P < 0.01 versus no anti-E-selectin antibodies. (B) Inhibitory effect of sialyl Lewis X. HUVECs were incubated with TNF-α (10 ng/ml) for 3 h. Cells were then washed and incubated with P. gingivalis ATCC 33277 (108 cells/ml in each well) for 30 min in the presence of purified sialyl Lewis X (0 to 10 ng/ml). Other procedures are described in the legend to Fig. 1B. Data are means ± SD (n = 3). *, P < 0.01 versus no TNF-α; †, P < 0.01 versus no sialyl Lewis X. (C) Adherence of P. gingivalis was augmented in HEK293 cells transfected with an expression vector for E-selectin. P. gingivalis ATCC 33277 (108 cells/ml in each well) was incubated with 293 cells transfected with a human E-selectin-inserted vector for 0 min. Other procedures are described in the legend to Fig. 1A. Bars, 10 μm. (D) Adherence of P. gingivalis was augmented in 293 cells transfected with an expression vector for E-selectin. P. gingivalis ATCC 33277 (108 cells/ml in each well) was incubated with 293 cells transfected with a human E-selectin-inserted vector for 30 min. Other procedures are described in the legend to Fig. 1B. Data are means ± SD (n = 3). *, P < 0.01 versus control.
E-selectin mediates the rolling of leukocytes on activated endothelial cells through binding of the carbohydrate antigen sialyl Lewis X (37). Therefore, we examined the effect of sialyl Lewis X on interactions between P. gingivalis and endothelial cells. Sialyl Lewis X inhibited TNF-α-induced P. gingivalis adherence to HUVECs at a concentration of 0.1 μg/ml (Fig. 2B). To assess the effect of E-selectin overexpression on the upregulation of P. gingivalis adherence to endothelial cells, we transfected an E-selectin-inserted plasmid into HUVECs. Expression of E-selectin was confirmed by Western blotting 24 h after transfection (Fig. 2C). Adherence of P. gingivalis significantly increased in E-selectin-transfected HEK 293 cells (Fig. 2D). These results suggest that TNF-α augments P. gingivalis adherence to HUVECs by inducing expression of E-selectin.
P. gingivalis interacts with TNF-α-stimulated endothelial cells via Pgm6/7.
The initial adherence of P. gingivalis to host cells is mediated by multiple adhesins, including FimA and HagB (44, 45). To determine whether an interaction occurs between the major fimbriae and E-selectin, we examined adherence to endothelial cells of P. gingivalis defective in FimA alone (ΔFimA). TNF-α increased the adherence to endothelial cells of FimA-deficient P. gingivalis as well as wild-type P. gingivalis, and the degrees of adherence were similar (Fig. 3A and B). We next examined whether a major outer membrane protein of P. gingivalis that is homologous to the OmpA protein in Escherichia coli, namely, the Pgm6/7 complex, mediates P. gingivalis adherence to HUVECs. The Pgm6/7-deficient mutant (ΔPgm6/7) was incubated with TNF-α-pretreated HUVECs, and attachment of P. gingivalis to the cells was observed. TNF-α increased adherence of wild-type P. gingivalis to endothelial cells but failed to increase adherence of ΔPgm6/7 P. gingivalis to endothelial cells (Fig. 3C). To clarify whether Pgm6/7 mediates P. gingivalis adherence to HUVECs, we prepared envelopes from wild-type, ΔFimA, and ΔPgm6/7 P. gingivalis cells and examined the effects on the interaction between wild-type P. gingivalis and HUVECs. Envelope peptides from wild-type P. gingivalis or ΔFimA P. gingivalis suppressed adherence of P. gingivalis to TNF-α-pretreated HUVECs (Fig. 3D). However, envelope peptides from ΔPgm6/7 P. gingivalis did not affect P. gingivalis adherence. In addition, the Pgm6/7 fraction from P. gingivalis ATCC 33277 suppressed TNF-α-augmented P. gingivalis adherence, but the FimA fraction from the same strain did not (Fig. 3E). Furthermore, purified Pgm6/7 inhibited TNF-α activation of P. gingivalis adherence to HUVECs at concentrations as low as 0.25 ng/ml (Fig. 3F and G). These results suggest that the P. gingivalis peptide Pgm6/7 plays a role in the adherence of P. gingivalis to endothelial cells.
P. gingivalis interaction with endothelial cells via E-selectin induces endothelial exocytosis and NO production.
Finally, to determine whether E-selectin-mediated adherence of P. gingivalis activates endothelial cells and increases vascular inflammation, we investigated induction of VWF and nitric oxide in TNF-α-pretreated endothelial cells by stimulation with P. gingivalis. HUVECs were incubated with TNF-α (10 ng/ml) for 3 h, and then the cells were washed and incubated with P. gingivalis for 0 to 1 h. The release of VWF into the medium was measured by ELISA. P. gingivalis triggers endothelial exocytosis, as measured by endothelial release of VWF. Release of VWF by stimulation with P. gingivalis was also enhanced by pretreatment of HUVECs with TNF-α (Fig. 4). TNF-α pretreatment of HUVECs before P. gingivalis stimulation for 30 min significantly increased NO2− release into the medium (Fig. 5). Anti-E-selectin antibodies inhibited activation of NO release by P. gingivalis in TNF-α-pretreated HUVECs. These results suggest that interaction of P. gingivalis with endothelial cells via E-selectin activates the endothelial cells and enhances proinflammatory responses of the cells to this bacterium.
Fig 4.
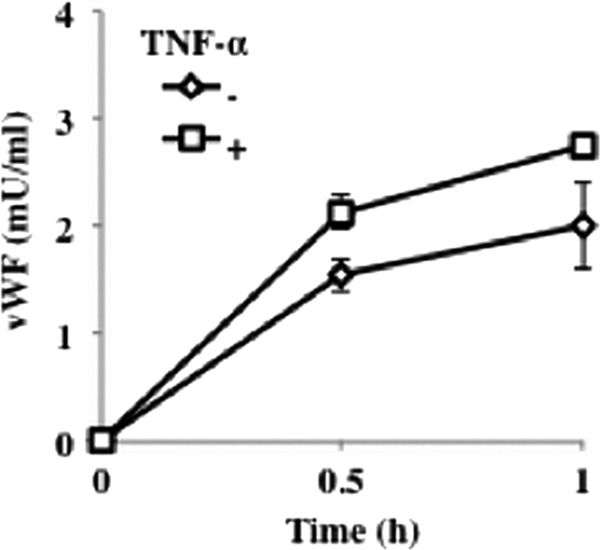
Endothelial VWF exocytosis in response to P. gingivalis is augmented by pretreatment with TNF-α. HUVECs were incubated with TNF-α (10 ng/ml) for 3 h. Cells were then washed and incubated with P. gingivalis ATCC 33277 (108 cells/ml in each well) for 0 to 1 h. The release of VWF into the medium was measured by ELISA. Data are means ± SD (n = 3).
Fig 5.
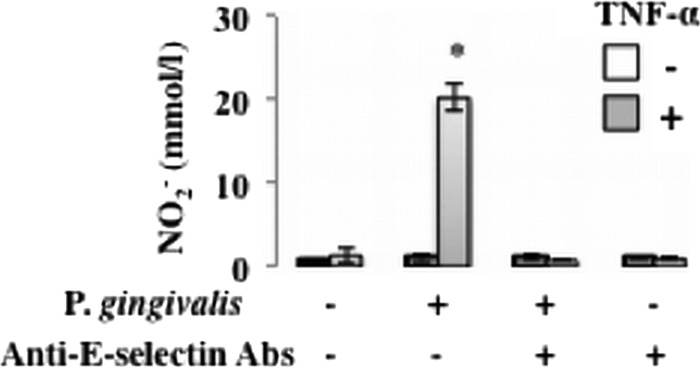
P. gingivalis-induced nitric oxide release from activated endothelial cells is mediated by E-selectin. HUVECs were incubated with TNF-α (10 ng/ml) for 3 h. Cells were then washed and incubated with P. gingivalis ATCC 33277 (108 cells/ml in each well) for 30 min in the presence or absence of an antibody for E-selectin. The release of nitric oxide into the medium was measured by DAN assay. Data are means ± SD (n = 3). *, P < 0.01 versus no TNF-α.
DISCUSSION
P. gingivalis adherence to and invasion of endothelial cells have been reported by several investigators (9, 46). However, this is the first report on the adhesion of activated endothelial cells by P. gingivalis. HUVECs activated by TNF-α increased the adherence of P. gingivalis through E-selectin expression, interacting with the OmpA-like proteins Pgm6 and -7 in P. gingivalis.
One of the initial events in atherogenesis is the activation of endothelial cells, which then express cell surface adhesion molecules such as endothelial leukocyte adhesion molecule (E-selectin), VCAM-1, and ICAM-1 (8, 10, 22). These endothelial adhesion molecules in turn facilitate the attachment of blood leukocytes to endothelial surfaces (34). In the present study, we demonstrated that one of the periodontopathogens adheres to endothelial cells via E-selectin.
P. gingivalis can invade many cell types, including human oral epithelial cells (33, 51), human gingival fibroblasts or epithelial cells (3, 26), human coronary artery smooth muscle cells, and HCAECs (11). Adhesion of P. gingivalis to host cells is multimodal (27) and involves a variety of cell surface and extracellular components, including fimbriae, proteases, hemagglutinins, and lipopolysaccharide (LPS) (8). Among the large array of virulence factors produced by P. gingivalis, the major fimbriae (FimA) as well as cysteine proteinases (gingipains) contribute to the attachment to and invasion of many types of mammalian cells, including oral epithelial cells (4) and endothelial cells. P. gingivalis strains deficient in FimA fimbriae had an attenuated capacity to adhere to and invade epithelial cells and endothelial cells (33, 46, 51). Invasive P. gingivalis strains and their purified fimbriae activate expression of cytokines and cell adhesion molecules in endothelial cells (46). However, our data showed that Pgm6/7 rather than FimA is associated with P. gingivalis adherence to TNF-α-treated endothelial cells. Although we do not know exact mechanisms, P. gingivalis cells adhere to activated endothelial cells through their Pgm6/7 complex, in a manner different from the fimbria-integrin interaction. TNF-α activates endothelial cells to express adhesion molecules as well as proinflammatory cytokine and chemokine receptors and promotes synthesis and release of a variety of inflammatory cytokines and chemokines to support recruitment of activated leukocytes to an inflammatory lesion (38). TNF-α promotes the inflammatory cascade within the arterial wall during development of atherosclerosis (1). In addition, P. gingivalis has been detected within atherosclerotic plaques from vascular tissues (25, 54). Therefore, TNF-α may also augment adherence of P. gingivalis, as well as that of leukocytes, in part through inducing E-selectin expression. Weibel-Palade bodies (WPBs) are endothelial granules that store VWF and other vascular modulators (48, 50). Endothelial cells secrete WPBs in response to vascular injury, releasing VWF, which triggers platelet rolling. Endothelial exocytosis is one of the earliest responses to vascular damage and plays a pivotal role in thrombosis and inflammation (29). In this study, we demonstrated that P. gingivalis interaction with endothelial cells via E-selectin activates endothelial cells, enhances endothelial exocytosis (Fig. 4), and may enhance atherogenesis and thrombosis (e.g., Buerger disease) (7, 23).
Pgm6/7 in P. gingivalis, which shares a low level of homology with E. coli OmpA, exists as a heterotrimer comprising Pgm6 and Pgm7 and plays a role in the outer membrane integrity of this organism. OmpA in E. coli K1 has been reported to interact with a glycoprotein (Ecgp) of human brain microvascular endothelial cells for invasion (35). Therefore, P. gingivalis invasion into endothelial cells should be investigated in the near future, especially regarding whether Pgm6/7 is involved in the invasion. How does Pgm6/7 bind to E-selectin? The adhesion activity of E-selectin is mediated primarily by the binding of sialyl Lewis X on the leukocyte to the carbohydrate-binding domain of the protein. E-selectin recognizes the carbohydrate structure of sialyl Lewis X. Pgm6/7 is also a glycoprotein, and therefore it may bind to E-selectin through its carbohydrate side chain. However, we need additional experiments to reveal the mechanism.
Collectively, in the present study, we clarified a new host-pathogen interaction, i.e., the interaction between Pgm6/7, a major outer membrane protein of P. gingivalis, and E-selectin of activated endothelial cells. This finding raises the possibility that chronic infection of the vasculature by pathogens such as P. gingivalis could exacerbate systemic vascular diseases such as coronary heart disease, stroke, and diabetes mellitus.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants-in-aid for scientific research (22390354 and 21659436 to K.M.) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Footnotes
Published ahead of print 16 April 2012
Supplemental material for this article may be found at http://iai.asm.org/.
REFERENCES
- 1. Aggarwal BB, Natarajan K. 1996. Tumor necrosis factors: developments during the last decade. Eur. Cytokine Netw. 7:93–124 [PubMed] [Google Scholar]
- 2. Amar S, et al. 2003. Periodontal disease is associated with brachial artery endothelial dysfunction and systemic inflammation. Arterioscler. Thromb. Vasc. Biol. 23:1245–1249 [DOI] [PubMed] [Google Scholar]
- 3. Amornchat C, Rassameemasmaung S, Sripairojthikoon W, Swasdison S. 2003. Invasion of Porphyromonas gingivalis into human gingival fibroblasts in vitro. J. Int. Acad. Periodontol. 5:98–105 [PubMed] [Google Scholar]
- 4. Andrian E, Grenier D, Rouabhia M. 2006. Porphyromonas gingivalis-epithelial cell interactions in periodontitis. J. Dent. Res. 85:392–403 [DOI] [PubMed] [Google Scholar]
- 5. Brown LJ, Oliver RC, Loe H. 1989. Periodontal diseases in the U.S. in 1981: prevalence, severity, extent, and role in tooth mortality. J. Periodontol. 60:363–370 [DOI] [PubMed] [Google Scholar]
- 6. Burt B. 2005. Position paper: epidemiology of periodontal diseases. J. Periodontol. 76:1406–1419 [DOI] [PubMed] [Google Scholar]
- 7. Chen Z, et al. 2007. Synergistic contribution of CD14 and HLA loci in the susceptibility to Buerger disease. Hum. Genet. 122:367–372 [DOI] [PubMed] [Google Scholar]
- 8. Cutler CW, Kalmar JR, Genco CA. 1995. Pathogenic strategies of the oral anaerobe, Porphyromonas gingivalis. Trends Microbiol. 3:45–51 [DOI] [PubMed] [Google Scholar]
- 9. Deshpande RG, Khan MB, Genco CA. 1998. Invasion of aortic and heart endothelial cells by Porphyromonas gingivalis. Infect. Immun. 66:5337–5343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dong ZM, et al. 1998. The combined role of P- and E-selectins in atherosclerosis. J. Clin. Invest. 102:145–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dorn BR, Dunn WA, Jr, Progulske-Fox A. 1999. Invasion of human coronary artery cells by periodontal pathogens. Infect. Immun. 67:5792–5798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dorn BR, Dunn WA, Jr, Progulske-Fox A. 2001. Porphyromonas gingivalis traffics to autophagosomes in human coronary artery endothelial cells. Infect. Immun. 69:5698–5708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gardner RG, Russell JB, Wilson DB, Wang GR, Shoemaker NB. 1996. Use of a modified Bacteroides-Prevotella shuttle vector to transfer a reconstructed beta-1,4-d-endoglucanase gene into Bacteroides uniformis and Prevotella ruminicola B(1)4. Appl. Environ. Microbiol. 62:196–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Griffen AL, Becker MR, Lyons SR, Moeschberger ML, Leys EJ. 1998. Prevalence of Porphyromonas gingivalis and periodontal health status. J. Clin. Microbiol. 36:3239–3242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Haffajee AD, Socransky SS. 2005. Microbiology of periodontal diseases: introduction. Periodontol. 2000 38:9–12 [DOI] [PubMed] [Google Scholar]
- 16. Hansson GK. 2005. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 352:1685–1695 [DOI] [PubMed] [Google Scholar]
- 17. Hasegawa Y, et al. 2009. Anchoring and length regulation of Porphyromonas gingivalis Mfa1 fimbriae by the downstream gene product Mfa2. Microbiology 155:3333–3347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hashizume T, Kurita-Ochiai T, Yamamoto M. 2011. Porphyromonas gingivalis stimulates monocyte adhesion to human umbilical vein endothelial cells. FEMS Immunol. Med. Microbiol. 62:57–65 [DOI] [PubMed] [Google Scholar]
- 19. Heimdahl A, et al. 1990. Detection and quantitation by lysis-filtration of bacteremia after different oral surgical procedures. J. Clin. Microbiol. 28:2205–2209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Herzberg MC, Weyer MW. 1998. Dental plaque, platelets, and cardiovascular diseases. Ann. Periodontol. 3:151–160 [DOI] [PubMed] [Google Scholar]
- 21. Hope SA, Meredith IT. 2003. Cellular adhesion molecules and cardiovascular disease. I. Their expression and role in atherogenesis. Intern. Med. J. 33:380–386 [DOI] [PubMed] [Google Scholar]
- 22. Iiyama K, et al. 1999. Patterns of vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 expression in rabbit and mouse atherosclerotic lesions and at sites predisposed to lesion formation. Circ. Res. 85:199–207 [DOI] [PubMed] [Google Scholar]
- 23. Iwai T. 2009. Periodontal bacteremia and various vascular diseases. J. Periodontal Res. 44:689–694 [DOI] [PubMed] [Google Scholar]
- 24. Kleinhenz DJ, Fan X, Rubin J, Hart CM. 2003. Detection of endothelial nitric oxide release with the 2,3-diaminonapthalene assay. Free Radic. Biol. Med. 34:856–861 [DOI] [PubMed] [Google Scholar]
- 25. Kurihara N, et al. 2004. Detection and localization of periodontopathic bacteria in abdominal aortic aneurysms. Eur. J. Vasc. Endovasc. Surg. 28:553–558 [DOI] [PubMed] [Google Scholar]
- 26. Lamont RJ, et al. 1995. Porphyromonas gingivalis invasion of gingival epithelial cells. Infect. Immun. 63:3878–3885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lamont RJ, Jenkinson HF. 1998. Life below the gum line: pathogenic mechanisms of Porphyromonas gingivalis. Microbiol. Mol. Biol. Rev. 62:1244–1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Libby P. 2002. Inflammation in atherosclerosis. Nature 420:868–874 [DOI] [PubMed] [Google Scholar]
- 29. Matsushita K, et al. 2003. Nitric oxide regulates exocytosis by S-nitrosylation of N-ethylmaleimide-sensitive factor. Cell 115:139–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Murakami Y, Imai M, Nakamura H, Yoshimura F. 2002. Separation of the outer membrane and identification of major outer membrane proteins from Porphyromonas gingivalis. Eur. J. Oral Sci. 110:157–162 [DOI] [PubMed] [Google Scholar]
- 31. Nagano K, et al. 2007. Characterization of RagA and RagB in Porphyromonas gingivalis: study using gene-deletion mutants. J. Med. Microbiol. 56:1536–1548 [DOI] [PubMed] [Google Scholar]
- 32. Nagano K, et al. 2005. Trimeric structure of major outer membrane proteins homologous to OmpA in Porphyromonas gingivalis. J. Bacteriol. 187:902–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Njoroge T, Genco RJ, Sojar HT, Hamada N, Genco CA. 1997. A role for fimbriae in Porphyromonas gingivalis invasion of oral epithelial cells. Infect. Immun. 65:1980–1984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Osterud B, Bjorklid E. 2003. Role of monocytes in atherogenesis. Physiol. Rev. 83:1069–1112 [DOI] [PubMed] [Google Scholar]
- 35. Prasadarao NV, et al. 2003. Cloning and expression of the Escherichia coli K1 outer membrane protein A receptor, a gp96 homologue. Infect. Immun. 71:1680–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Read MA, et al. 1995. The proteasome pathway is required for cytokine-induced endothelial-leukocyte adhesion molecule expression. Immunity 2:493–506 [DOI] [PubMed] [Google Scholar]
- 37. Rosen SD, Bertozzi CR. 1994. The selectins and their ligands. Curr. Opin. Cell Biol. 6:663–673 [DOI] [PubMed] [Google Scholar]
- 38. Ross R. 1999. Atherosclerosis—an inflammatory disease. N. Engl. J. Med. 340:115–126 [DOI] [PubMed] [Google Scholar]
- 39. Rothlein R, et al. 1988. Induction of intercellular adhesion molecule 1 on primary and continuous cell lines by pro-inflammatory cytokines. Regulation by pharmacologic agents and neutralizing antibodies. J. Immunol. 141:1665–1669 [PubMed] [Google Scholar]
- 40. Sandros J, Papapanou P, Dahlen G. 1993. Porphyromonas gingivalis invades oral epithelial cells in vitro. J. Periodontal Res. 28:219–226 [DOI] [PubMed] [Google Scholar]
- 41. Sconyers JR, Crawford JJ, Moriarty JD. 1973. Relationship of bacteremia to toothbrushing in patients with periodontitis. J. Am. Dent. Assoc. 87:616–622 [DOI] [PubMed] [Google Scholar]
- 42. Slots J. 1979. Subgingival microflora and periodontal disease. J. Clin. Periodontol. 6:351–382 [DOI] [PubMed] [Google Scholar]
- 43. Slots J, Ting M. 1999. Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis in human periodontal disease: occurrence and treatment. Periodontol. 2000 20:82–121 [DOI] [PubMed] [Google Scholar]
- 44. Sojar HT, Han Y, Hamada N, Sharma A, Genco RJ. 1999. Role of the amino-terminal region of Porphyromonas gingivalis fimbriae in adherence to epithelial cells. Infect. Immun. 67:6173–6176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Song H, Belanger M, Whitlock J, Kozarov E, Progulske-Fox A. 2005. Hemagglutinin B is involved in the adherence of Porphyromonas gingivalis to human coronary artery endothelial cells. Infect. Immun. 73:7267–7273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Takahashi Y, Davey M, Yumoto H, Gibson FC, III, Genco CA. 2006. Fimbria-dependent activation of pro-inflammatory molecules in Porphyromonas gingivalis infected human aortic endothelial cells. Cell. Microbiol. 8:738–757 [DOI] [PubMed] [Google Scholar]
- 47. Tedder TF, Steeber DA, Chen A, Engel P. 1995. The selectins: vascular adhesion molecules. FASEB J. 9:866–873 [PubMed] [Google Scholar]
- 48. Wagner DD, et al. 1991. Induction of specific storage organelles by von Willebrand factor propolypeptide. Cell 64:403–413 [DOI] [PubMed] [Google Scholar]
- 49. Wang G, et al. 2002. Increased monocyte adhesion to aortic endothelium in rats with hyperhomocysteinemia: role of chemokine and adhesion molecules. Arterioscler. Thromb. Vasc. Biol. 22:1777–1783 [DOI] [PubMed] [Google Scholar]
- 50. Weibel ER, Palade GE. 1964. New cytoplasmic components in arterial endothelia. J. Cell Biol. 23:101–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Weinberg A, Belton CM, Park Y, Lamont RJ. 1997. Role of fimbriae in Porphyromonas gingivalis invasion of gingival epithelial cells. Infect. Immun. 65:313–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yoshimura F, Takahashi K, Nodasaka Y, Suzuki T. 1984. Purification and characterization of a novel type of fimbriae from the oral anaerobe Bacteroides gingivalis. J. Bacteriol. 160:949–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yoshizaki K, Wakita H, Takeda K, Takahashi K. 2008. Conditional expression of microRNA against E-selectin inhibits leukocyte-endothelial adhesive interaction under inflammatory condition. Biochem. Biophys. Res. Commun. 371:747–751 [DOI] [PubMed] [Google Scholar]
- 54. Zaremba M, Gorska R, Suwalski P, Kowalski J. 2007. Evaluation of the incidence of periodontitis-associated bacteria in the atherosclerotic plaque of coronary blood vessels. J. Periodontol. 78:322–327 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.