

Abstract
The human pathogen Helicobacter pylori employs a diverse collection of outer membrane proteins to colonize, persist, and drive disease within the acidic gastric environment. In this study, we sought to elucidate the function of the host-induced gene HP0289, which encodes an uncharacterized outer membrane protein. We first generated an isogenic H. pylori mutant that lacks HP0289 and found that the mutant has a colonization defect in single-strain infections and is greatly outcompeted in mouse coinfection experiments with wild-type H. pylori. Furthermore, we used protease assays and biochemical fractionation coupled with an HP0289-targeted peptide antibody to verify that the HP0289 protein resides in the outer membrane. Our previous findings showed that the HP0289 promoter is upregulated in the mouse stomach, and here we demonstrate that HP0289 expression is induced under acidic conditions in an ArsRS-dependent manner. Finally, we have shown that the HP0289 mutant induces greater expression of the chemokine interleukin-8 (IL-8) and the cytokine tumor necrosis factor alpha (TNF-α) in gastric carcinoma cells (AGS). Similarly, transcription of the IL-8 homolog keratinocyte-derived chemokine (KC) is elevated in murine infections with the HP0289 mutant than in murine infections with wild-type H. pylori. On the basis of this phenotype, we renamed HP0289 ImaA for immunomodulatory autotransporter protein. Our work has revealed that genes induced in vivo play an important role in H. pylori pathogenesis. Specifically, the outer membrane protein ImaA modulates a component of the host inflammatory response, and thus may allow H. pylori to fine tune the host immune response based on ImaA expression.
INTRODUCTION
The human pathogen Helicobacter pylori infects half of the world's population and causes chronic infection that elevates the risk of multiple gastric diseases, including gastric adenocarcinoma (30, 54, 73). In an effort to better understand H. pylori pathogenesis, Castillo et al. identified a set of H. pylori genes that were expressed at higher levels when the bacterium was in the mouse stomach than when the bacterium was in the lab setting. Host-induced genes have been shown to be crucial for colonization and virulence in a number of bacterial species, including H. pylori (15–17, 35, 44, 49, 66, 78). This previous work used recombination-based in vivo expression technology (RIVET) to identify host-induced H. pylori genes (17). The RIVET system utilizes fusions of transcriptional promoters to a promoterless gene encoding a recombinase protein. If these promoters are transcribed, for example in the mouse stomach, the recombinase is created and mediates site-specific recombination events that convert the strain from antibiotic resistance to antibiotic sensitivity (71). The previous study analyzed ∼71% of the genome and found six promoters induced in vivo (17). Two of the promoters (Pivi10 and Pivi66) regulated genes, mobABD and cagZ, respectively, that were important for mouse stomach colonization (17). Of the four remaining unstudied promoters from the RIVET screen, one regulated a gene, HP0289, that was previously annotated as a toxin-like outer membrane protein in the complete genome sequence of H. pylori strain 26695 (67). Here we explore how HP0289 contributes to H. pylori pathogenesis.
The H. pylori genome is predicted to encode more than 30 outer membrane proteins (OMPs), or approximately 4% of the bacterium's coding potential (3, 23). This level of dedication to omp genes is not commonly seen in other bacterial species, and only a small percentage of these OMPs have actually been characterized in H. pylori (3, 7, 23, 29). The abundance of specialized OMPs in the H. pylori proteome has been proposed to allow the bacterium to persist within an environment that is demanding and often changing (39). Of the well-studied H. pylori omp genes, a number encode proteins that play a prominent role in H. pylori pathogenesis (29). The most notable of these proteins include the vacuolating cytotoxin protein VacA and the host antigen-specific adhesins BabA and SabA. VacA belongs to a family of OMPs called autotransporters, a class of proteins that appear three other times in the H. pylori proteome (26). While a number of H. pylori OMPs have been characterized, VacA is the only OMP with an autotransporter domain that has been well described; thus, a void exists in our knowledge of the remaining autotransporters.
Autotransporters are a family of Gram-negative bacterial secreted proteins that can be toxins, proteases, or adhesins (23, 28). These proteins are called autotransporters because originally they were thought to contain all of the machinery necessary for secretion to the outer membrane. Recent work, however, suggests that many interact with an additional protein, the beta-barrel assembly machinery BAM (29, 65). All autotransporters possess a conserved domain structure, which consists of the following: (i) an N-terminal signal peptide that facilitates Sec-dependent secretion across the inner membrane; (ii) a generally nonconserved central region called the passenger domain, which confers the effector function of the protein; and (iii) a C-terminal beta-barrel domain that is the hallmark of the autotransporter family and is critical for protein translocation across the outer membrane (29). Passenger domains represent the surface-exposed component of the protein and typically adopt an extended right-handed beta helix structure (11, 82). These domains are extremely diverse in both sequence and function, making it difficult to predict what a particular autotransporter does (29). Known autotransporter functions include the following: (i) binding to host proteins to mediate adhesion, invasion, immunoglobulin binding, or intracellular movement; (ii) interacting with other bacterial molecules to mediate agglutination and biofilm formation; (iii) acting as intracellular toxins; and (iv) behaving as proteases that target such proteins as host immunoglobulin. The autotransporter studied here, HP0289, was originally annotated as a toxin-like outer membrane protein; however, there is no experimental evidence for such a function (76).
H. pylori colonization causes chronic inflammation, a host response that is considered one of the primary risk factors for adenocarcinoma (30). H. pylori strains are highly variable and are well documented to contain various combinations of genes that enhance inflammation, including genes of the cag pathogenicity island (cagPAI), oipA, babA, and sabA (25, 33, 51, 86). Recent reports suggest that HP0289 might also vary between strains. Specifically, Kawai et al. analyzed 20 H. pylori genome sequences and found that the highly carcinogenic East Asian (hspEAsia lineage) strains had several changes, including a large deletion in HP0289 that removed 83% of the protein (38). Indeed, Lee et al. previously demonstrated that East Asian clinical isolates induce epithelial cells to produce significantly higher proinflammatory cytokine levels than do Western strains; however, the strains in this specific study were not analyzed for the presence of HP0289 (43). These findings thus suggest that loss of HP0289 may create H. pylori strains that are more proinflammatory.
In this study, we characterize the H. pylori autotransporter HP0289 and show that it contributes to murine stomach colonization and is under the control of the acid-sensing ArsRS two-component system. Additionally, we demonstrate that the protein decreases expression of inflammatory chemokines and cytokines in both cultured epithelial cells and infected stomachs.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
All bacterial strains are described in Table 1. H. pylori strain LSH100, a mouse-adapted descendant of the clinical isolate G27 (19, 50), was used for proteinase K digestions, all of the mouse colonization, gene expression, and AGS cell experiments. Strain 26695 was used for additional AGS cell inflammation experiments (1), and H. pylori strain SS1 (42) was used for murine infection and ImaA subcellular localization experiments. H. pylori strains were maintained on Columbia blood agar base (Difco, Detroit, MI) supplemented with 5% defibrinated horse blood (Hemostat Laboratories, Dixon, CA), 0.2% (wt/vol) β-cyclodextrin (Sigma) plus 5 μg/ml trimethoprim, 8 μg/ml amphotericin B, 50 μg/ml cycloheximide, 10 μg/ml vancomycin, 5 μg/ml cefsulodin, and 2.5 U/ml polymyxin B (CHBA) to inhibit the growth of unwanted microbes under 10% CO2, 7 to 10% O2 and balance N2, at 37°C. Liquid H. pylori cultures were grown in 1× Ham's F-12 medium (Gibco, Grand Island, NY) containing 10% heat-inactivated fetal bovine serum (FBS) (Gibco) or brucella broth supplemented with 10% fetal bovine serum (BB10). The antibiotic chloramphenicol (Cm) was used for selection at a concentration of 13 μg/ml. H. pylori strains were stored at −80°C in brain heart infusion medium supplemented with 10% fetal bovine serum, 1% (wt/vol) β-cyclodextrin, 25% glycerol, and 5% dimethyl sulfoxide.
Table 1.
H. pylori strains used in this study
| Strain | Description or relevant genotype | Reference |
|---|---|---|
| G27 | Wild type (NSH57 parent strain) | 19 |
| NSH57 | Mouse-adapted isolate of G27 | 8 |
| LSH100 | NSH57 with repaired fliM allele | 50 |
| SS1 | Wild type | 42 |
| 26695 | Wild type | 1 |
| KO954 | SS1 ΔimaA::cat | This study |
| KO1370 | LSH100 ΔimaA::cat | This study |
| KO1371 | LSH100 ΔarsS::cat | This study |
| KO1163 | SS1 cagE::kan | This study (strain provided by David McGee) |
| KO1372 | LSH100 cagE::kan | This study |
| KO1373 | LSH100 cagE::kan ΔimaA::cat | This study |
| KO1374 | 26695 ΔimaA::cat | This study |
Acid exposure.
H. pylori bacteria cultured for ∼36 h on CHBA were resuspended in sterile BB10, and their concentrations were determined by optical density (optical density at 600 nm [OD600]). For the 2-h acid treatment to examine imaA and ureA transcript levels, the cell suspensions were diluted to an OD600 of 1.75 in 1 ml of BB10 and then centrifuged at 2,500 × g for 8 min. The resulting pellet was resuspended in 2 ml of BB10 at a pH of either 5 or 7 and then incubated at 37°C under H. pylori culture conditions for 2 h. For the time course experiments measuring ImaA protein levels, cells were prepared the same way, except that the cultures were diluted to an OD600 of 0.220 at the initiation of the incubation period. The cell density of each sample taken at every time point was then normalized with the OD600 to ensure that equal amounts of protein were being examined for each respective culture.
Mammalian cell culture.
AGS (ATCC CRL 1739) human gastric epithelial cells were obtained directly from the American Type Culture Collection (ATCC) and maintained in Dulbecco's modified Eagle's medium (DMEM) (Lonza, Walkersville, MD) containing 10% FBS at 37°C with 10% CO2. To assay interleukin-8 (IL-8) production, AGS cells were seeded at 1 × 105 cells/ml in 24-well tissue culture dishes and incubated for 24 h. After this period, H. pylori, cultured for ∼36 h on CHBA, were scraped from a plate and resuspended in sterile DMEM plus FBS to a concentration of 1 × 107 to create a multiplicity of infection (MOI) of 100. H. pylori concentrations were determined by OD600, assuming 3 × 108 bacteria/ml/OD600 unit. AGS cells were infected for 2 h under 10% CO2. After 2-h incubation, culture supernatant was removed, and AGS monolayers were washed twice in 1× phosphate-buffered saline (PBS), and then the cells were resuspended in TRIzol for RNA isolation.
Construction of H. pylori mutants.
H. pylori SS1 ΔimaA::cat mutant was created using splicing by overlap extension (SOE) PCR with the primers D1 (5′-GCCCTTAGTTCAGGTGTGGCAGTTTAAGG), D2 (5′-CAAGGAGGATCCCGGCCGCGGCTACCTTCTCATTTCCTAGATAGTAGCC), D3 (5′-ATCCACTTTTCAATCTATATCACGGTTGCCGGGAATGTGGGCATGCGAGTGGCG), and D4 (5′-GTTTTAGCGTCAATGTTGGGGTTGATTCTAATGG) that amplified the imaA chromosomal region and primers catF (F stands for forward) (17) and catR2 (R stands for reverse) (17) that amplified the cat gene. This gene deletion extends from 7 bp upstream of the imaA start codon to 31 bp upstream of the imaA stop codon and places a terminatorless cat gene in the same transcriptional orientation as imaA. To generate the deletion in H. pylori LSH100, genomic DNA from H. pylori SS1 ΔimaA::cat mutant was used to naturally transform wild-type strain LSH100 to create strain KO1370 (KO stands for knockout). The deletion in 26695 was produced by natural transformation of wild-type 26695 with genomic DNA from strain KO1370, to generate strain KO1374. Selection was done on CHBA containing Cm, and proper integration was confirmed with PCR using primers D4 and catR2. To generate the LSH100 ΔarsS::cat mutant (KO1371), the chloramphenicol resistance cassette (cat) was inserted into the arsS gene (HP0165) by SOE PCR. In brief, primers were generated that reside approximately 300 bp upstream of arsS, ArsS1.1 (5′-AACCCTATGATCCTAAGGAATTA) and ArsS3.1 (5′-ATCCACTTTTCAATCTATATCAACGCAAAACCCCTTAACTCC), and downstream of arsS, ArsS2.2 (5′-GGCTTCCTGTAGCGTCCTTATG) and ArsS4.1 (5′-CCCAGTTTGTCGCACTGATAAGAGAACATGTTCAAACGATTGA). The two arsS PCR products were spliced to a third PCR product that contained the nonpolar cat allele generated from the primers catR2 and catF (17). The PCR product composed of the cat gene flanked by upstream and downstream regions of the arsS gene was then cloned into the TOPO-TA vector (Invitrogen) to generate plasmid pcat-arsS. This plasmid was then used to naturally transform strain LSH100 to Cm resistance. Proper integration was confirmed by PCR using primers ArsS1.1 and ArsS2.2 and by sequencing of that PCR product. The original cagE mutant was a kind gift from David McGee and Kylie Nolan (Table 1). It consists of an insertion of the aphA3 gene at a unique BglII site in cagE (HP0544, cag23) that is ∼600 bp from the start site of the 3,000-bp gene. We used genomic DNA from this strain to naturally transform either wild-type strain LSH100 or mutant strain KO1370 to kanamycin resistance.
Mouse colonization experiments.
H. pylori strains used for colonization analyses were passaged minimally in the lab (two or three times) and then inoculated into either Ham's F-12 culture medium (75) for H. pylori LSH100 or BB10 for H. pylori SS1 for ∼18 h, as described above. After this period, cells were analyzed to determine motility and cell concentration (OD600) prior to infection. For all infections, 4- to 6-week-old male FVB/N mice (Charles River) were housed in an Association for the Assessment and Accreditation of Laboratory Animal Care-accredited facility in microisolator cages with free access to standard food and water. All animal procedures were approved by the Institutional Animal Care and Use Committee. Approximately 1 ml of H. pylori cells containing 9 × 107 to 1 × 108 CFU/ml were used to orally gavage the mice. For coinfections, wild-type and mutant cells were grown separately and then combined in equal concentrations. To determine the true CFU/ml of each culture, all cultures were serially diluted and plated on CHBA. Infections were allowed to persist for 2 to 3 weeks, after which time mouse stomachs were excised as described before (57), homogenized using the Bullet Blender (Next Advance, Averill Park, NY), and then plated on CHBA with or without Cm (described above) supplemented with 10 μg of nalidixic acid/ml and 200 μg of bacitracin/ml. For the coinfection, the mouse stomachs were plated on both nonselective CHBA and CHBA supplemented with Cm as described previously (74). The cell counts obtained from the input and output data allowed us to calculate the competitive index, as follows: (CFU/g of mutant strain output/CFU/g of wild-type strain output)/(CFU/g of mutant strain input/CFU/g of wild-type strain input).
For keratinocyte-derived chemokine (KC) (mouse IL-8 homolog) detection in mouse tissue, infections persisted for 3 weeks (2). When excising the stomach, half of the tissue was placed in BB10 for plating and the other half was placed immediately in liquid N2 and then stored at −80°C within an hour of extraction. To isolate RNA, tissue samples were suspended in TRIzol and then homogenized using the Polytron (Kinematica, Switzerland) automated tissue homogenizer.
RNA preparation.
Total RNA was isolated from H. pylori strains LSH100 and its isogenic mutants, ΔimaA::cat and ΔarsS::cat mutant strains, using TRIzol reagent (Invitrogen, Carlsbad, CA) combined with RNeasy columns (Qiagen, Valencia, CA). Bacterial cells were pelleted and resuspended in 1 ml of TRIzol at room temperature for 5 min before 200 μl of chloroform was added. Samples were then centrifuged (12,000 × g, 15 min, 4°C), and the aqueous layer was removed and placed into new tubes. RNA was precipitated by combining 500 μl of isopropanol with the aqueous layer and incubating at room temperature for 10 min, followed by a centrifugation as described above. The RNA pellet was washed with 75% ethanol, dried, and resuspended in RNase-free water. To remove contaminating genomic DNA from purified RNA, samples were treated with 4 U of RNase-free DNase I (Ambion) for 3 h at 37°C, followed by further purification using the Qiagen RNeasy spin columns as specified in the manufacturer's instructions. RNA was ultimately eluted in RNase-free water, RNA concentrations were quantified on a Nanodrop spectrophotometer (Nanodrop, Wilmington, DE), and the absence of contaminating genomic DNA was confirmed BY PCR. RNA was immediately transcribed into cDNA (see below), and the remaining sample was stored at −80°C.
RNA was isolated from AGS cells in a similar manner. Briefly, 1 ml TRIzol reagent was added directly to cells in the culture dish per 10 cm2 of culture dish surface. The cells, including infecting H. pylori cells, were lysed directly in the culture dish by pipetting the cells up and down several times. Homogenized samples then underwent the same preparation as described above, except the DNase I treatment and secondary purification with the Qiagen RNeasy kit were omitted.
cDNA synthesis and quantitative real-time PCR.
Total RNA served as a template for cDNA synthesis using the Tetro cDNA synthesis kit (Bioline, London, United Kingdom). cDNA synthesis was carried out following the manufacturer's protocol, starting with 0.5 to 1 μg total RNA, 50 ng random hexamers, and 10 mM deoxynucleoside triphosphates (dNTPs) per 20-μl reaction mixture. The mixture was incubated at 65°C for 10 min before being combined with 10 μl of master mix, which includes the reverse transcriptase enzyme (200 U/μl). The reaction proceeded for 1 h at 37°C until the reverse transcriptase enzyme was inactivated at 70°C for 15 min. Quantitative real-time PCR was performed using the Opticon 2 real-time cycler (Bio-Rad, Hercules, CA) and SYBR green supermix reagents (Bioline, London, United Kingdom). For relative expression of imaA and ureA, transcript levels were normalized to the levels of groEL (69) in each sample. Transcripts were amplified with HP0289 For1.1 (For stands for forward) (5′-TAACGATCCAAAACGCTTCC) and HP0289 Rev1.1 (Rev stands for reverse) (5′-TCCCTTGAGGCGAGAGTGATT), UreA F1 and UreA R1 (36), and groEL F (JVO-529) and groEL R (JVO-5298) (69). Il8 (55) and TNF-α (84) expression levels from AGS cells were normalized to 18S (55) rRNA, and KC levels from mouse tissues were normalized to GAPDH (24). All reactions were performed in triplicate, and a melting curve analysis was used to ensure that a single product was amplified with each primer set. To validate RNA purity, no reverse transcriptase control reactions were also performed. imaA gene expression at low pH was determined from 4 independent experiments, and statistical differences were evaluated with the Mann-Whitney U-test. Relative expression of Il8 was determined from 5 independent experiments, and statistical differences were evaluated by Student's t test. All differences in gene expression were calculated by the ΔΔCT method (47).
ELISA.
Enzyme-linked immunosorbent assays (ELISAs) for human IL-8 were performed using the Human IL-8 EASIA kit (Invitrogen). AGS cells were infected by either wild-type H. pylori or its isogenic mutant, ΔimaA::cat mutant strain, at concentrations of 1 × 107, 2 × 106, and 1 × 105 cells/ml, and culture supernatant was preserved for ELISA at 4, 6, 12, and 24 h postinfection.
Proteinase K treatment of H. pylori cells.
Digestion of H. pylori outer membrane proteins with the extracellular protease, proteinase K, was conducted as described previously by Sabarth et al. (63). H. pylori cells grown for 48 h on CHBA plates were collected with an inoculation loop and suspended in 1 ml of phosphate-buffered saline. Cells were centrifuged at 5,000 × g for 10 min and then resuspended in PBS at a concentration of 3 × 108 cells/ml, based on the OD600. Cells were treated with either 40 or 400 μg/ml proteinase K for 30 min at room temperature in 1× PBS. The reaction was halted with the addition of 5 mM phenylmethylsulfonyl fluoride (PMSF); the cells were then washed twice in PBS. After a final centrifugation at 5,000 × g for 5 min, the cells were resuspended in PBS and then diluted into NuPAGE 4× sample buffer (Invitrogen, Carlsbad, CA) for subsequent Western blot analysis.
Sarcosine preparation of H. pylori outer membrane.
The sarcosine-insoluble outer membrane fraction was prepared as described previously (7) with slight modifications. H. pylori wild-type strain SS1 and its isogenic mutant, ΔimaA::cat mutant strain, were grown on CHBA for 48 h. To optimize the outer membrane yield, each respective strain was grown to confluent growth on two full CHBA plates, with all of the cells utilized for fractionation. Cells were collected using a sterile inoculation loop and suspended in 1 ml of 20 mM Tris-HCl (pH 7.5) and collected by centrifugation (8,000 × g, 10 min, 4°C). The pellet was then resuspended in 1 ml of 20 mM Tris-HCl containing a protease inhibitor (1 mM PMSF) and a cell wall hydrolase (0.25 mg/ml lysozyme). The resuspended pellet was sonicated 9 times for 15 s each time (Fisher sonicator, 80% amplitude), and unbroken bacteria were removed by centrifugation (6,000 × g, 10 min, 4°C). Total membranes were isolated by centrifugation for 45 min at 50,000 × g and 4°C. The supernatant containing the soluble fraction was removed, and the total membrane pellet was washed once in PBS, resuspended in 1 ml sonication buffer containing 2.0% (wt/vol) sodium lauryl sarcosine, and incubated at room temperature for 30 min. The inner membrane fraction was separated by centrifugation (50,000 × g, 45 min, 4°C), and the pellet containing the outer membrane was resuspended in sarcosine for an additional treatment to optimize outer membrane purity. The resuspended pellet was incubated at room temperature for another 30 min and then centrifuged (50,000 × g, 45 min, 4°C). The final pellet was resuspended in 1 ml of 20 mM Tris-HCl and stored at −20°C.
Western blotting and ImaA antibody creation.
The ani-ImaA-1 polyclonal antibody was prepared in rabbits using a 19-amino-acid peptide (amino acids 2065 to 2084) from the passenger domain of the ImaA protein (Open Biosystems, Huntsville, AL). The antibody specifically recognizes ImaA, as well as several unidentified nonspecific proteins that were significantly different in size.
Proteins for Western blot analysis were resuspended in 4× NuPAGE sample buffer (Invitrogen, Carlsbad, CA) with 0.025% 2-mercaptoethanol and heated at 70°C for 15 min. Samples were separated on 3 to 8% NuPAGE Tris-acetate gels for 60 min at 150 V. Following electrophoresis, the proteins were transferred to polyvinylidene difluoride membranes (Bio-Rad, Hercules, CA) with the Bio-Rad semidry transfer cell for 35 min at 16 V. The membranes were then incubated with a 1:300 dilution of anti-ImaA-1 antibody or a 1:2,000 dilution of anti-GST-TlpA22 (GST stands for glutathione S-transferase) antibody (83) for ∼18 h at 4°C. For visualization, blots were incubated with goat anti-rabbit antibody conjugated to horseradish peroxidase (Santa Cruz Biotech) at a dilution of 1:2,000 for 1 h, followed by incubation with luminol, p-coumaric acid, and hydrogen peroxide. Luminescent blots were visualized by exposure to Ultra Cruz autoradiography film (Santa Cruz Biotech).
RESULTS
H. pylori ImaA is predicted to be an autotransporter.
To examine the functional significance of the uncharacterized H. pylori genes that were upregulated in the stomach (17), we employed in silico sequence analyses of each protein to identify signature domains. The gene predicted to be regulated by promoter Pivi77, HP0289, is predicted to encode a protein with all of the typical characteristics of an autotransporter (Fig. 1). On the basis of experiments described below, we designate HP0289 as ImaA (immunomodulating autotransporter). imaA is transcribed monocistronically (69), and the protein encoded by this gene is 2,893 amino acids in length with a calculated molecular mass of 311 kilodaltons. The SignalP algorithm predicts that ImaA bears an N-terminal signal peptide with a signal peptidase cleavage site between positions 42 and 43 (VYA-NN) (10). ImaA also carries the highly conserved C-terminal beta-barrel autotransporter domain, readily identified by domain finding software such as the Simple Modular Architecture Research Tool (SMART) (67). The passenger domain of ImaA, which likely confers the effector function of the protein, is 2,581 amino acids in length. This region has little conservation with any other previously characterized protein, with the exception of three “VacA2” regions that are 59 amino acids long. In the initial sequencing of H. pylori, HP0289 was annotated as a toxin-like protein, with small regions of similarity noted between VacA, HP0289 and two other H. pylori autotransporters (HP0610 and HP0922) (76). The main block of homology is at the C-terminal autotransporter domain. There were, however, additional regions of low similarity (26 to 31%) at several spots in the central passenger domain called VacA2 regions, although these regions do not correspond to a functional portion of VacA. Based on this homology, ImaA has been annotated as a VacA paralog. Our analysis, however, suggests that ImaA is an autotransporter that is not specifically related to VacA.
Fig 1.

ImaA contains hallmarks of autotransporter proteins. The schematic diagram shows the highly conserved domains in ImaA as predicted by SignalP 3.0 and SMART and the N-terminal (N′) signal peptide (SP) and the C-terminal end (C′). The signal peptide probability was 1.00, and the autotransporter E value was 5 × 10−7. Numbers below and above the schematic diagram indicate the amino acid positions of predicted domains within ImaA.
ImaA promotes mouse stomach colonization.
RIVET studies in several organisms have identified pathogen colonization and virulence factors (15–17, 44, 49, 66, 78). Therefore, we first examined whether imaA was required for mouse stomach colonization, a model routinely utilized in H. pylori studies. We generated an imaA mutant, ΔimaA::cat, in which nearly the entire imaA open reading frame (ORF) is replaced with the chloramphenicol acetyltransferase (cat) gene, and thus is a null allele. This allele was used to replace the endogenous imaA locus in the H. pylori strain LSH100 (50). LSH100 is a mouse-adapted derivative of H. pylori G27 (19). LSH100 arose from mouse adaptation of strain G27 to create strain NSH57, followed by repair of a mutation in the fliM locus to the original wild-type sequence to yield LSH100 (50). We used the LSH100 strain, because both it and the original RIVET strain, mG27, were derived from the same parent, but LSH100 infects mice more consistently. Because of their high genetic relatedness, we felt that experiments with both mG27 and LSH100 would not have revealed significantly different conclusions. Unless noted otherwise, all subsequent in vivo and in vitro H. pylori infections, localization, and gene expression experiments were done with strain LSH100 to maintain consistency with our mouse-infecting strain. Male FVB/N mice were infected with wild-type LSH100 or its isogenic ΔimaA::cat mutant for 2 to 3 weeks. These time points have been widely used in other H. pylori murine colonization studies and have been shown to accurately reflect colonization levels at longer infection time points (8, 18, 27, 52, 58, 72, 74, 80). While the imaA mutant was able to sustain infection for these lengths of time, the output CFU/gram stomach was significantly lower than that obtained from wild-type infections (Fig. 2A). To address whether the colonization defect of the ΔimaA::cat strain would be altered by the presence of wild-type H. pylori, we carried out coinfection experiments with equal concentrations of wild-type and ΔimaA::cat strains. Two weeks postinfection, we determined the ratio of CFU/g of stomach for mutant and wild-type bacteria and calculated a competitive index. In all infections, the imaA mutant was greatly outcompeted by the wild-type bacteria (Fig. 2B). These results demonstrate that H. pylori requires ImaA to reach wild-type gastric colonization levels. We did not complement the imaA::cat mutant, because imaA is over 8,000 base pairs in length and therefore would be readily targeted by H. pylori's extensively developed restriction-modification system, which comprises over 4% of the genome (46). Despite advances in methods to circumvent the H. pylori restriction-modification system, gene complementation remains one of the most difficult endeavors in H. pylori molecular genetics (22).
Fig 2.

(A) The H. pylori LSH100 ΔimaA::cat mutant colonizes mice at levels significantly lower than those of the wild-type H. pylori LSH100 parent. Oral single-strain infection studies with the wild-type H. pylori or ΔimaA::cat mutant were carried out using male FVB/N mice. Infections persisted for 2 or 3 weeks. Single-strain infections were conducted with 7 mice for the wild-type strain and with 10 mice for the ΔimaA::cat mutant. Each circle represents the value for one infected mouse, derived from independent 2-week (open circles) or 3-week (filled circles) infections, and the solid or broken line represents the mean for the group of mice. The values between the two groups of mice in panel A were significantly different (P = 0.01 by Student's t test) and are indicated by the bracket and asterisk. (B) The H. pylori LSH100 ΔimaA::cat mutant is outcompeted by the wild-type strain in a coinfection colonization assay. Each point represents the competitive index for one mouse stomach for eight mice in two independent infections. The competitive index is a ratio and is calculated as follows: (mutant output/wild-type output)/(mutant input/wild-type input). The values were significantly different (P < 0.001 by Student's t test) compared to a hypothetical strain with no defect (competitive index [CI] of 1). (C) The H. pylori SS1 ΔimaA::cat mutant colonizes mice at levels that are comparable to those of wild-type H. pylori SS1. These infections persisted for 2 weeks, with four mice for the wild-type strain and six mice for the ΔimaA::cat mutant strain. (D) The H. pylori SS1 ΔimaA::cat mutant is not outcompeted for mouse colonization in a coinfection with wild-type H. pylori in two independent infections of eight mice.
ImaA has been shown to be important in other H. pylori strains. Specifically, an imaA (HP0289) transposon mutant in H. pylori strain G1.1 was outcompeted by the wild-type H. pylori strain for gerbil colonization (37). To expand this analysis, we checked whether a third strain, SS1, would similarly need imaA for stomach colonization. We found, surprisingly, that H. pylori SS1 ΔimaA::cat colonized as well as the wild type did in both single-strain and competition infections (Fig. 2C and D). This strain difference is not surprising, given that others have observed that there is extensive variability in whether particular H. pylori proteins are essential for mouse colonization (8). Thus, these results suggest that ImaA is needed by some strains and that ImaA's importance is possibly dependent on each strain's unique interactions with the host.
ImaA is secreted to the outer membrane of the cell.
We next wanted to confirm the in silico prediction that ImaA is exported to the outer membrane of the cell. We first generated a peptide antibody directed at the passenger domain of the protein that accurately detects mature ImaA protein from whole-cell lysates (Fig. 3). We then used proteinase K digestion to assess whether ImaA was surface localized in H. pylori LSH100 and mG27. Proteinase K does not diffuse across the outer membrane of Gram-negative bacteria and thus cleaves only proteins residing on the bacterial surface. This approach has been widely used to assess autotransporter surface localization (20, 45, 77). As predicted for a surface-localized protein, ImaA is digested by the protease and is thus surface exposed in strain LSH100 (Fig. 3A) as well as in strain mG27 (not shown). To demonstrate that proteinase K treatments were not breaching the membrane and degrading internal proteins, we determined that the TlpABC chemoreceptors localized on the inner membrane were not digested (Fig. 3A). To further validate these findings and to assess whether ImaA localization to the outer membrane is conserved in strain SS1, we performed subcellular fractionation experiments with the detergent sarcosine, which selectively solubilizes the inner membrane and thus enables separation from the outer membrane. Western blot analysis demonstrated that ImaA is in the outer membrane fraction, as well as somewhat in the inner membrane (Fig. 3B). A control blot, using antibody that detects the inner membrane protein, showed that the fractions were fairly pure (Fig. 3B). Inner membrane-localized ImaA may represent protein that is transiting to the outer membrane or an indication of incomplete membrane separation. Other studies have detected autotransporter proteins in both inner and outer membrane fractions (5). Of note, these analyses were performed in three different H. pylori strain backgrounds, providing strong evidence that ImaA is translocated to the outer membrane. In addition, we did not detect any ImaA in concentrated supernatant from H. pylori cultures (data not shown), suggesting that ImaA stays associated with the outer membrane.
Fig 3.
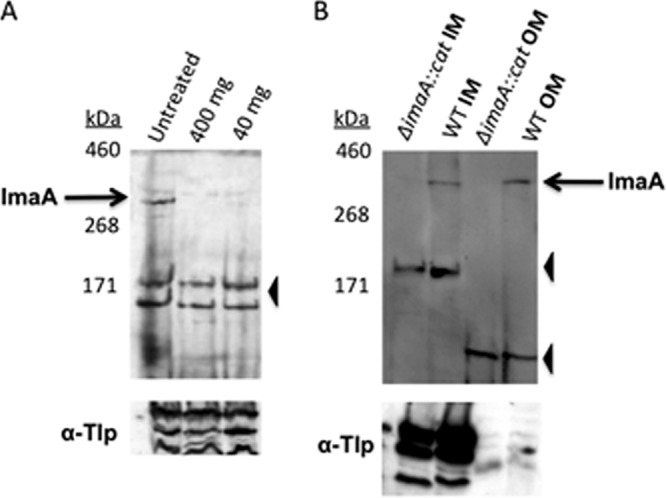
ImaA localizes to the outer membrane. (A) Whole cells of H. pylori strain LSH100 were treated with different concentrations of proteinase K (40 or 400 mg ml−1) or with no proteinase K as a control. The top panels show blots probed with anti-ImaA-1, while the bottom panels are probed with anti-GST-TlpA22 antibody (α-Tlp), which recognizes inner membrane chemoreceptors (83). Similar results were obtained with strain mG27 (not shown). (B) Sarcosine-insoluble outer membrane (OM) fractions and sarcosine-soluble inner membrane (IM) fractions were obtained from wild-type (WT) and H. pylori SS1 ΔimaA::cat mutant cells and then probed with anti-ImaA-1. In both panels, the positions of full-length ImaA are indicated by black arrows labeled ImaA, and the positions of nonspecific proteins recognized by the anti-ImaA serum are indicated by black arrowheads.
ImaA is a member of the acid-responsive ArsRS regulon.
The RIVET studies showed that imaA transcription is upregulated within the host environment (17). Therefore, we wanted to identify the signal responsible for inducing imaA in vivo. In a recently published H. pylori whole transcriptome paper, Sharma et al. demonstrated that imaA/hp0289 is induced 10-fold at low pH (69). Acidic pH is the key environmental signal for activating the H. pylori two-component regulatory system ArsRS (acid-responsive signaling) (60, 62). We thus examined whether imaA is a member of the ArsRS regulon by creating a null mutant for the histidine kinase ArsS and observing imaA gene expression through quantitative real-time PCR (qRT-PCR) under neutral and acidic conditions. The response regulator, ArsR, is an essential gene so the ArsS mutant serves as the ArsRS representative (9). We employed the housekeeping gene, groEL, for normalization, as used in previous work (69). After 2 h of acidic pH exposure, imaA expression increased ∼10-fold in the wild-type background but not in the arsS mutant (Fig. 4A). Furthermore, imaA expression is depressed in the arsS deletion strain, even at neutral pH (Fig. 4A). These results suggest that imaA is a member of the ArsRS regulon. We additionally compared the expression of imaA to that of a known ArsRS-regulated acid-induced gene, ureA (61). We found that ureA gene expression in wild-type H. pylori was induced ∼14-fold in acid over expression at neutral pH in a partially arsS-dependent manner (Fig. 4B), similar to the findings of Pflock et al. (59). At neutral pH, ureA required ArsS for expression more so than imaA did. These results thus show that the experimental conditions affect ArsS regulon members as expected. Furthermore, our data support that imaA is a member of the ArsRS regulon, due to the ArsS-dependent increased expression in acid, but that it is not regulated identically to ureA.
Fig 4.

The ArsRS two-component regulatory system influences imaA transcription. (A) Quantitative real-time PCR (qRT-PCR) was performed using cDNA generated from the H. pylori LSH100 strain or its isogenic mutant, ΔarsS::cat mutant, that were exposed to either neutral or acidic BB10 for 2 h. The levels of expression of the genes in the wild type (WT) at pH 5 and in the ΔarsS::cat mutant at pH 5 are shown relative to the values for the WT at pH 7 from four independent biological replicates, each performed in triplicate, and normalized to the housekeeping gene groEL. Values for imaA expression that are significantly higher or lower (P < 0.05 by Wilcoxon rank sum test) are indicated by an L-shaped bracket and asterisk. (B) The ureA gene responds to acid and depends on the ArsRS regulatory system for expression. qRT-PCR was performed with the same cDNA that was used in the imaA transcription analysis, four independent biological replicates, each performed in triplicate. Values for ureA expression that are significantly higher or lower (P < 0.05 by Wilcoxon rank sum test) are indicated by an L-shaped bracket and asterisk. (C) Western blots with the anti-ImaA-1 antibody showing ImaA expression at multiple time points under both neutral and acidic conditions in strain LSH100 or in the ΔarsS::cat mutant strain. These data are representative of the results of three independent time course experiments.
We next examined whether ImaA protein levels were affected by pH. For these experiments, we grew H. pylori cultures at pH 5 and then sampled them after 2, 5, or 8 h. Despite observing a 10-fold increase in imaA mRNA (Fig. 4A), we did not detect any elevation in ImaA protein levels at low pH (Fig. 4C). We did observe, however, that ImaA protein expression was arsS dependent at low pH (Fig. 4D). This observation suggested that H. pylori relies on ArsRS to maintain ImaA expression under acidic conditions. All together, these results thus show that acid induces imaA transcription and that ArsRS is needed to maintain both imaA transcript and ImaA protein levels at acidic pH.
Loss of imaA creates H. pylori strains that induce elevated Il8 transcription.
We next examined whether loss of imaA influenced levels of inflammatory mediators in mouse and in vitro cell culture models. Colonization of H. pylori in the stomach results in the release of the chemoattractant IL-8 in humans or its analog, KC, in mice. IL-8 stimulates the infiltration of neutrophils into the gastric mucosa, leading to chronic inflammation (31, 56); therefore, Il8 transcription levels are often used as a readout for a proinflammatory response (41). To examine ImaA's influence on Il8/KC levels, we performed KC or Il8 qRT-PCR on mouse tissue or AGS gastric epithelial cells infected with H. pylori. In mice infected with H. pylori LSH100 (wild-type) or ΔimaA::cat mutant strains for 3 weeks, a time point used by others for similar analyses (2), we found very low levels of KC overall. There was, however, elevated KC in mice infected with H. pylori ΔimaA::cat compared to those infected with the wild-type strain (Fig. 5A). While the difference in KC levels between uninfected mice and mice infected with the ΔimaA::cat mutant was significant, there was minimal difference in KC levels between uninfected mice infected and mice infected with wild-type H. pylori (Fig. 5A). These experiments thus suggest that ImaA's normal function is to decrease KC levels. To confirm this finding, we employed the well-established AGS human gastric cell model to investigate Il8 levels. AGS cells infected with wild-type H. pylori for 2 h revealed an ∼75-fold induction in Il8 transcription in AGS cells compared to the uninfected cells, while the ΔimaA::cat mutant strain generated a significantly greater ∼189-fold induction in Il8 levels (Fig. 5B). To confirm that Il8 transcript levels seen in ΔimaA::cat mutant infections translated to increased levels of the protein product, we next measured secreted IL-8 levels with an ELISA. Il8 transcript levels were measured from AGS cells that were infected with H. pylori at an multiplicity of infection (MOI) of 100; however, when we measured IL-8 protein levels at this MOI, we saw no difference in cytokine production between ΔimaA::cat mutant and wild-type H. pylori infections (data not shown). We reasoned that the amount of H. pylori might be saturating the IL-8 protein production, so we lowered the MOI. When we used an MOI of 1, we witnessed elevated cytokine levels in the ΔimaA::cat mutant compared to wild-type H. pylori infections at 4, 6, and 12 h postinfection, with hour 12 providing a significant difference in IL-8 between the wild-type bacterial and ΔimaA::cat mutant infections (Fig. 5C). These data suggest that ImaA serves to modulate the amount of IL-8 that is generated during infection.
Fig 5.

KC and Il8 levels are significantly elevated in Δima::cat mouse and AGS cell infections, respectively. (A) Male FVB/N mice were infected with either the wild-type LSH100 strain or its isogenic mutant, the ΔimaA::cat mutant, for 3 weeks. Quantitative RT-PCR (qRT-PCR) was performed on whole gastric tissue to analyze the expression of KC using primers by the method of Yamaoka et al. (84). Mouse samples are the same as in Fig. 2A and include 5 mice infected with the wild-type LSH100 strain, 8 mice infected with the ΔimaA::cat mutant, and 6 uninfected mice. There was a significant difference (P < 0.05 by Student's t test) in KC between uninfected mice and mice infected with the ΔimaA::cat mutant as indicated by the L-shaped bracket and asterisk. (B) Il8 transcript levels are elevated in AGS cells infected with the ΔimaA::cat mutant. For transcript analysis, AGS cells were infected with either wild-type LSH100 or its isogenic single mutants, the ΔimaA::cat or cagE::kan mutant, or the ΔimaA::cat cagE::kan double mutant at an MOI of 100. qRT-PCR was performed to analyze the expression of interleukin-8 (Il8) using primers by the method of Nazarenko et al. (55) after 2 h of infection. These data represent 5 independent infections (biological replicates) with reactions done in triplicate. All differences in expression were calculated by the ΔΔCT method (47) and statistically significant differences (P < 0.01 by Student's t test) for AGS cell infections are indicated by an L-shaped bracket and asterisk. (C) ELISAs for IL-8 levels were conducted on culture media taken from AGS cells infected with either wild-type H. pylori or with the ΔimaA::cat mutant (MOI of 1) at the following time points: 4, 6, and 12 h postinfection. Data show two biological replicates, each done with two technical replicates. Values that were statistically significantly different (P < 0.03 by Wilcoxon rank sum test) are indicated by an asterisk. (D) Il8 expression levels in AGS cells infected with either wild-type strain 26695 or its isogenic mutant, the ΔimaA::cat mutant strain. Data represent 5 independent infections with reactions done in triplicate. The differences in expression were analyzed by the ΔΔCT method (47). Values that were statistically significantly different (P = 0.01 by Student's t test) are indicated by an L-shaped bracket and asterisk. (E) Analysis of TNF-α transcript levels in AGS cells infected with wild-type LSH100 or its isogenic mutant, the ΔimaA::cat mutant strain. Data represent 5 independent infections with reactions done in triplicate. The differences in expression were analyzed by the ΔΔCT method (47). Values that were statistically significantly different (P < 0.05 by Student's t test) are indicated by an L-shaped bracket and asterisk.
We next examined whether the inflammation phenotype associated with the loss of imaA in H. pylori LSH100 was common to other CagA-positive H. pylori strains, so we created an ΔimaA::cat mutant in the widely used CagA-positive strain, 26695, and performed AGS cell infections. Similar to the response we witnessed with H. pylori LSH100, the H. pylori 26695 ΔimaA::cat mutant infections induced higher levels of Il8 transcription overall than the wild-type H. pylori infections did (Fig. 5D). Wild-type H. pylori 26695 induced an ∼106-fold increase in Il8 transcription compared to uninfected AGS cells, while the 26695 ΔimaA::cat mutant generated a significantly greater ∼274-fold increase in Il8 transcription.
In addition to IL-8, H. pylori infection promotes the production of numerous proinflammatory cytokines. To test whether the ΔimaA::cat mutant induces elevated concentrations of other immune mediators, we measured transcript levels of the proinflammatory cytokine tumor necrosis factor alpha (TNF-α). TNF-α is associated with an increased severity and distribution of gastritis in infected individuals (70). We found that AGS cells infected with wild-type H. pylori produced an ∼7-fold increase in TNF-α transcript levels compared to uninfected cells, while AGS cells infected with the ΔimaA::cat mutant displayed a significantly greater 19-fold increase in TNF-α levels (Fig. 5E). Taken together, these results suggest that in the absence of ImaA, there is a stronger induction of the mammalian proinflammatory pathway. When mouse tissue was examined for TNF-α, there was no difference in the levels between the uninfected mice and mice infected with either the wild-type or ΔimaA::cat mutant strain (data not shown). This outcome is not entirely unexpected, as H. pylori-induced TNF-α levels appear to be much smaller than H. pylori-induced IL-8 levels (85).
The cagPAI type IV secretion system underlies the enhanced IL-8 production seen in imaA mutant infections.
H. pylori is known to control IL-8 levels by action of the type IV secretion system (T4SS) encoded by the cagPAI. To establish whether the increase in inflammation we witnessed in the ΔimaA::cat infections was dependent on the activity of the cagPAI type IV secretion system (cag-T4SS), we created the ΔimaA::cat ΔcagE::kan double mutant and examined Il8 levels using the same in vitro infection model. cagE encodes a putative ATPase and is required for IL-8 induction (21). cagE mutants had dramatically decreased levels of Il8 transcription (Fig. 5B), as predicted from other studies with strains related to H. pylori G27 (6). The cagE effect was dominant over the imaA Il8 upregulation, as both a ΔcagE single mutant and the ΔcagE ΔimaA::cat double mutant induced Il8 levels that were not significantly above that of the uninfected AGS cells (Fig. 5B). This outcome suggests that the immunomodulatory activity of ImaA requires cagPAI function.
DISCUSSION
H. pylori relies on multiple outer membrane proteins to chronically persist within the gastric environment (32, 51, 68). In this study, we demonstrate that a previously uncharacterized H. pylori autotransporter, HP0289 or ImaA, is important for host colonization and dampens the inflammatory response. Furthermore, we show that imaA is under the control of the acid-responsive ArsRS two-component regulatory system. Our findings thus support the hypothesis that the in vivo-induced imaA gene contributes to H. pylori pathogenesis and that the protein product normally decreases the inflammatory response brought about by the action of the cagPAI.
H. pylori must adapt to the changing landscape of the stomach during the course of a chronic infection (12). One way the bacterium can accomplish this adaptation is through tailoring the expression of virulence genes to particular conditions. We found that imaA transcription is under the control of the ArsRS regulon. Whole-genome transcriptional profiling of H. pylori strains cultured at low pH identified more than 100 genes that were differentially expressed in an ars-dependent manner, although imaA was not one of them (62). Similarly, others have found that there is some variability in H. pylori gene expression at low pH. For example, α-carbonic anhydrase (HP1186) expression has been shown to be repressed in some cases and upregulated in others at low pH (53, 81). We found that imaA expression is induced under acidic conditions, similar to the findings of Sharma and colleagues (69), and furthermore, we found that transcriptional control of imaA is mediated to a significant degree by the ArsRS two-component system. While imaA mRNA was greatly increased at low pH, we did not detect a corresponding increase in ImaA protein levels at low pH. This paradox of increased transcript levels not directly translating to increases in protein levels is not unprecedented in studies of the H. pylori ArsRS system. Loh et al. recently examined the proteomes of wild-type H. pylori and an isogenic arsS mutant under neutral and acidic conditions and compared them to the previously established transcriptional profiles for each strain under these conditions (48). They found very few acid-responsive protein changes in either strain, as only 15 proteins were differentially expressed in total. imaA likely belongs to the group of more than 100 genes that show altered transcriptional profiles at low pH but do not exhibit clear changes in protein levels. Loh et al. attributed the discrepancy between transcript and protein levels to posttranscriptional regulatory processes, which may dilute alterations in acid-induced protein expression (48). Despite discrepancies in protein levels, it is clear that ArsS is important for expression of imaA at low pH, demonstrating that imaA is under the control of the ArsRS regulon.
We show here that ImaA is important for mouse colonization. ImaA was previously found to be crucial for colonization in a different animal model, the gerbil, as part of a global transposon mutagenesis screen done in strain G1.1 that was evaluated in a competition model (37). Thus, these two studies demonstrate that ImaA's presence is necessary to achieve wild-type gastric colonization in multiple animal models. Conversely, a third analyzed strain, SS1, tolerates the loss of imaA in murine infections. Of note, strain SS1 does express ImaA (Fig. 3). There are many differences between strain SS1 and strain LSH100/G27, the most notable of which is that SS1 has an inactive cagPAI T4SS, while that of G27 and its mouse-selected variants is active (8). Strain G1.1, like SS1, does not secrete CagA (25, 34). Thus, ImaA appears to be important for colonization in both Cag-positive and Cag-negative strains, and furthermore, ImaA may have roles in the host that are not limited to affecting the cagPAI, although those remain to be determined.
A rodent colonization defect is unusual with H. pylori outer membrane proteins. The H. pylori adhesin proteins BabAB or SabA do not display any colonization defects, and AlpAB exhibits a defect that is statistically insignificant (4, 68). In fact, the only characterized H. pylori outer membrane protein to display a rodent colonization defect is the autotransporter VacA (64). VacA, like ImaA, possesses immunomodulatory activity albeit through suppression of T-cell activation (13). However, unlike VacA, ImaA appears to act while cell associated, as we were unable to identify an ImaA secreted peptide in cell culture supernatant. Interestingly, it was recently shown that mutants deficient for the laminin binding proteins AlpA and AlpB caused greater levels of inflammation in gerbil infections (68). This outcome, however, was not attributed to any inherent AlpAB immunomodulatory properties but rather the mutant's inability to intimately adhere to gastric epithelial cells and express other immunosuppressive proteins. We were unable to detect any in vitro adherence ability associated with ImaA (data not shown), so we believe it operates in a different manner from AlpAB.
A central component of H. pylori-induced inflammation is delivery of proinflammatory molecules into host cells via the cagPAI T4SS. We have demonstrated that the ΔimaA::cat mutant evokes a significant increase in expression of the proinflammatory cytokines IL-8, and TNF-α compared to AGS cell infections with wild-type H. pylori. While the bulk of the inflammation experiments were done with the H. pylori G27 derivative, LSH100, we also found that ImaA had a similar effect in strain 26695, suggesting that ImaA function is conserved. Furthermore, we found that the imaA mutant inflammation phenotype requires a functional cagPAI T4SS. This outcome suggests that ImaA acts to diminish the normal cagPAI-mediated induction of proinflammatory cytokines. The cagPAI T4SS aids the delivery of two effectors capable of inducing IL-8 expression in epithelial cells, CagA and peptidoglycan (14, 79). While we do not know how ImaA interacts with the cagPAI, it is not unprecedented for H. pylori outer membrane proteins to influence cagPAI T4SS activity. The ABO/Lewis b (Leb) blood group antigen binding protein, BabA, facilitates interactions between the cagPAI T4SS machinery and the host cell. Strains null for babA induce reduced levels of IL-8 in infected host cells, opposite to what we see in imaA mutant infections (33). Interestingly, the protein homology/analogy recognition Engine (PHYRE) predicts that ImaA has homology to the bacterial integrin binding protein, invasin (E = 1.8 × 10−4). Components of the cagPAI T4SS pilus bind directly to the α5β1 integrin receptor to facilitate secretion of CagA and peptidoglycan into the host cell cytoplasm (40). Thus, it is possible that ImaA and the cagPAI T4SS machinery compete for integrin binding and that in the absence of ImaA, there is increased T4SS binding and therefore, enhanced effector molecule secretion into host cells.
In conclusion, we have determined that the H. pylori host-induced HP0289 gene encodes a surface-localized autotransporter protein that we designate ImaA. ImaA promotes colonization of the host stomach and diminishes the inflammatory response. Specifically, ImaA decreases the amount of Il8 transcript generated by the H. pylori cagPAI. imaA expression is furthermore controlled by the acid-sensing two-component regulatory system ArsRS in response to acid. These findings support the notion that genes induced in vivo play a central role in H. pylori pathogenesis. Furthermore, they suggest that H. pylori has sophisticated mechanisms to modulate the host inflammatory response by controlling expression of a protein that decreases bacterially triggered inflammatory gene expression.
ACKNOWLEDGMENTS
We thank David McGee and Kylie Nolan for providing the H. pylori SS1 cagE::kan mutant. We are also grateful to Fitnat Yildiz and Annah Rolig for critical reading of the manuscript.
This work was supported by a Research Scholar Grant (RSG-05-249-01-MBC) from the American Cancer Society (to K.M.O.) and by grant AI050000 (to K.M.O.) from the National Institutes of Allergy and Infectious Disease (NIAID) at the National Institutes of Health.
The contents of this article are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Footnotes
Published ahead of print 7 May 2012
REFERENCES
- 1. Akopyants NS, Eaton KA, Berg DE. 1995. Adaptive mutation and cocolonization during Helicobacter pylori infection of gnotobiotic piglets. Infect. Immun. 63:116–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Algood HM, Gallo-Romero J, Wilson KT, Peek RM, Cover TL. 2007. Host response to Helicobacter pylori infection before initiation of the adaptive immune response. FEMS Immunol. Med. Microbiol. 51:577–586 [DOI] [PubMed] [Google Scholar]
- 3. Alm RA, et al. 2000. Comparative genomics of Helicobacter pylori: analysis of the outer membrane protein families. Infect. Immun. 68:4155–4168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Amieva MR, El-Omar EM. 2008. Host-bacterial interactions in Helicobacter pylori infection. Gastroenterology 134:306–323 [DOI] [PubMed] [Google Scholar]
- 5. Ashgar SS, et al. 2007. CapA, an autotransporter protein of Campylobacter jejuni, mediates association with human epithelial cells and colonization of the chicken gut. J. Bacteriol. 189:1856–1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bach S, Makristathis A, Rotter M, Hirschl AM. 2002. Gene expression profiling in AGS cells stimulated with Helicobacter pylori isogenic strains (cagA positive or cagA negative). Infect. Immun. 70:988–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Baik SC, et al. 2004. Proteomic analysis of the sarcosine-insoluble outer membrane fraction of Helicobacter pylori strain 26695. J. Bacteriol. 186:949–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Baldwin DN, et al. 2007. Identification of Helicobacter pylori genes that contribute to stomach colonization. Infect. Immun. 75:1005–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Beier D, Frank R. 2000. Molecular characterization of two-component systems of Helicobacter pylori. J. Bacteriol. 182:2068–2076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bendtsen JD, Nielsen H, von Heijne G, Brunak S. 2004. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 340:783–795 [DOI] [PubMed] [Google Scholar]
- 11. Benz I, Schmidt MA. 2011. Structures and functions of autotransporter proteins in microbial pathogens. Int. J. Med. Microbiol. 301:461–468 [DOI] [PubMed] [Google Scholar]
- 12. Blaser MJ. 1997. The versatility of Helicobacter pylori in the adaptation to the human stomach. J. Physiol. Pharmacol. 48:307–314 [PubMed] [Google Scholar]
- 13. Boncristiano M, et al. 2003. The Helicobacter pylori vacuolating toxin inhibits T cell activation by two independent mechanisms. J. Exp. Med. 198:1887–1897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brandt S, Kwok T, Hartig R, König W, Backert S. 2005. NF-kappaB activation and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proc. Natl. Acad. Sci. U. S. A. 102:9300–9305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bron PA, Grangette C, Mercenier A, de Vos WM, Kleerebezem M. 2004. Identification of Lactobacillus plantarum genes that are induced in the gastrointestinal tract of mice. J. Bacteriol. 186:5721–5729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Camilli A, Mekalanos JJ. 1995. Use of recombinase gene fusions to identify Vibrio cholerae genes induced during infection. Mol. Microbiol. 18:671–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Castillo AR, Woodruff AJ, Connolly LE, Sause WE, Ottemann KM. 2008. Recombination-based in vivo expression technology identifies Helicobacter pylori genes important for host colonization. Infect. Immun. 76:5632–5644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chevalier C, Thiberge JM, Ferrero RL, Labigne A. 1999. Essential role of Helicobacter pylori gamma-glutamyltranspeptidase for the colonization of the gastric mucosa of mice. Mol. Microbiol. 31:1359–1372 [DOI] [PubMed] [Google Scholar]
- 19. Covacci A, et al. 1993. Molecular characterization of the 128-kDa immunodominant antigen of Helicobacter pylori associated with cytotoxicity and duodenal ulcer. Proc. Natl. Acad. Sci. U. S. A. 90:5791–5795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dautin N, Barnard T, Anderson D, Bernstein H. 2007. Cleavage of a bacterial autotransporter by an evolutionarily convergent autocatalytic mechanism. EMBO J. 26:1942–1952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Day AS, et al. 2000. cagE is a virulence factor associated with Helicobacter pylori-induced duodenal ulceration in children. J. Infect. Dis. 181:1370–1375 [DOI] [PubMed] [Google Scholar]
- 22. Donahue JP, Israel DA, Peek RM, Blaser MJ, Miller GG. 2000. Overcoming the restriction barrier to plasmid transformation of Helicobacter pylori. Mol. Microbiol. 37:1066–1074 [DOI] [PubMed] [Google Scholar]
- 23. Dossumbekova A, et al. 2006. Helicobacter pylori outer membrane proteins and gastric inflammation. Gut 55:1360–1361 [PMC free article] [PubMed] [Google Scholar]
- 24. Eaton KA, Benson LH, Haeger J, Gray BM. 2006. Role of transcription factor T-bet expression by CD4+ cells in gastritis due to Helicobacter pylori in mice. Infect. Immun. 74:4673–4684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Eaton KA, et al. 2001. Role of Helicobacter pylori cag region genes in colonization and gastritis in two animal models. Infect. Immun. 69:2902–2908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Finn RD, et al. 2010. The Pfam protein families database. Nucleic Acids Res. 38:D211–D222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Harris AG, et al. 2003. Catalase (KatA) and KatA-associated protein (KapA) are essential to persistent colonization in the Helicobacter pylori SS1 mouse model. Microbiology 149:665–672 [DOI] [PubMed] [Google Scholar]
- 28. Henderson I, Nataro J. 2001. Virulence functions of autotransporter proteins. Infect. Immun. 69:1231–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Henderson I, Navarro-Garcia F, Desvaux M, Fernandez R, Ala'Aldeen D. 2004. Type V protein secretion pathway: the autotransporter story. Microbiol. Mol. Biol. Rev. 68:692–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Herrera V, Parsonnet J. 2009. Helicobacter pylori and gastric adenocarcinoma. Clin. Microbiol. Infect. 15:971–976 [DOI] [PubMed] [Google Scholar]
- 31. Huang J, O'Toole PW, Doig P, Trust TJ. 1995. Stimulation of interleukin-8 production in epithelial cell lines by Helicobacter pylori. Infect. Immun. 63:1732–1738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ilver D, et al. 1998. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science 279:373–377 [DOI] [PubMed] [Google Scholar]
- 33. Ishijima N, et al. 2011. BabA-mediated adherence is a potentiator of the Helicobacter pylori type IV secretion system activity. J. Biol. Chem. 286:25256–25264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Israel DA, et al. 2001. Helicobacter pylori strain-specific differences in genetic content, identified by microarray, influence host inflammatory responses. J. Clin. Invest. 107:611–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Janakiraman A, Slauch JM. 2000. The putative iron transport system SitABCD encoded on SPI1 is required for full virulence of Salmonella typhimurium. Mol. Microbiol. 35:1146–1155 [DOI] [PubMed] [Google Scholar]
- 36. Janzon A, et al. 2009. Presence of high numbers of transcriptionally active Helicobacter pylori in vomitus from Bangladeshi patients suffering from acute gastroenteritis. Helicobacter 14:237–247 [DOI] [PubMed] [Google Scholar]
- 37. Kavermann H, et al. 2003. Identification and characterization of Helicobacter pylori genes essential for gastric colonization. J. Exp. Med. 197:813–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kawai M, et al. 2011. Evolution in an oncogenic bacterial species with extreme genome plasticity: Helicobacter pylori East Asian genomes. BMC Microbiol. 11:104 doi:10.1186/1471-2180-11-104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kuipers EJ, et al. 1995. Long-term sequelae of Helicobacter pylori gastritis. Lancet 345:1525–1528 [DOI] [PubMed] [Google Scholar]
- 40. Kwok T, et al. 2007. Helicobacter exploits integrin for type IV secretion and kinase activation. Nature 449:862–866 [DOI] [PubMed] [Google Scholar]
- 41. Lamb A, et al. 2009. Helicobacter pylori CagA activates NF-kappaB by targeting TAK1 for TRAF6-mediated Lys 63 ubiquitination. EMBO Rep. 10:1242–1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lee A, et al. 1997. A standardized mouse model of Helicobacter pylori infection: introducing the Sydney strain. Gastroenterology 112:1386–1397 [DOI] [PubMed] [Google Scholar]
- 43. Lee KH, et al. 2004. Alanine-threonine polymorphism of Helicobacter pylori RpoB is correlated with differential induction of interleukin-8 in MKN45 cells. J. Clin. Microbiol. 42:3518–3524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lee SH, Butler SM, Camilli A. 2001. Selection for in vivo regulators of bacterial virulence. Proc. Natl. Acad. Sci. U. S. A. 98:6889–6894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lenz JD, et al. 2011. Expression during host infection and localization of Yersinia pestis autotransporter proteins. J. Bacteriol. 193:5936–5949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lin LF, Posfai J, Roberts RJ, Kong H. 2001. Comparative genomics of the restriction-modification systems in Helicobacter pylori. Proc. Natl. Acad. Sci. U. S. A. 98:2740–2745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25:402–408 [DOI] [PubMed] [Google Scholar]
- 48. Loh JT, Gupta SS, Friedman DB, Krezel AM, Cover TL. 2010. Analysis of protein expression regulated by the Helicobacter pylori ArsRS two-component signal transduction system. J. Bacteriol. 192:2034–2043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lowe AM, Beattie DT, Deresiewicz RL. 1998. Identification of novel staphylococcal virulence genes by in vivo expression technology. Mol. Microbiol. 27:967–976 [DOI] [PubMed] [Google Scholar]
- 50. Lowenthal AC, et al. 2009. Functional analysis of the Helicobacter pylori flagellar switch proteins. J. Bacteriol. 191:7147–7156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mahdavi J, et al. 2002. Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science 297:573–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Marchetti M, Rappuoli R. 2002. Isogenic mutants of the cag pathogenicity island of Helicobacter pylori in the mouse model of infection: effects on colonization efficiency. Microbiology 148:1447–1456 [DOI] [PubMed] [Google Scholar]
- 53. Merrell DS, Goodrich ML, Otto G, Tompkins LS, Falkow S. 2003. pH-regulated gene expression of the gastric pathogen Helicobacter pylori. Infect. Immun. 71:3529–3539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Montecucco C, Rappuoli R. 2001. Living dangerously: how Helicobacter pylori survives in the human stomach. Nat. Rev. Mol. Cell Biol. 2:457–466 [DOI] [PubMed] [Google Scholar]
- 55. Nazarenko I, et al. 2002. Multiplex quantitative PCR using self-quenched primers labeled with a single fluorophore. Nucleic Acids Res. 30:e37 doi:10.1093/nar/30.9.e37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Obonyo M, Guiney DG, Harwood J, Fierer J, Cole SP. 2002. Role of gamma interferon in Helicobacter pylori induction of inflammatory mediators during murine infection. Infect. Immun. 70:3295–3299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ottemann KM, Lowenthal AC. 2002. Helicobacter pylori uses motility for initial colonization and to attain robust infection. Infect. Immun. 70:1984–1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pappo J, et al. 1999. Helicobacter pylori infection in immunized mice lacking major histocompatibility complex class I and class II functions. Infect. Immun. 67:337–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pflock M, Dietz P, Schär J, Beier D. 2004. Genetic evidence for histidine kinase HP165 being an acid sensor of Helicobacter pylori. FEMS Microbiol. Lett. 234:51–61 [DOI] [PubMed] [Google Scholar]
- 60. Pflock M, et al. 2006. Characterization of the ArsRS regulon of Helicobacter pylori, involved in acid adaptation. J. Bacteriol. 188:3449–3462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pflock M, Kennard S, Delany I, Scarlato V, Beier D. 2005. Acid-induced activation of the urease promoters is mediated directly by the ArsRS two-component system of Helicobacter pylori. Infect. Immun. 73:6437–6445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Pflock M, Kennard S, Finsterer N, Beier D. 2006. Acid-responsive gene regulation in the human pathogen Helicobacter pylori. J. Biotechnol. 126:52–60 [DOI] [PubMed] [Google Scholar]
- 63. Sabarth N, et al. 2005. Identification of Helicobacter pylori surface proteins by selective proteinase K digestion and antibody phage display. J. Microbiol. Methods 62:345–349 [DOI] [PubMed] [Google Scholar]
- 64. Salama NR, Otto G, Tompkins L, Falkow S. 2001. Vacuolating cytotoxin of Helicobacter pylori plays a role during colonization in a mouse model of infection. Infect. Immun. 69:730–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sauri A, et al. 2009. The Bam (Omp85) complex is involved in secretion of the autotransporter haemoglobin protease. Microbiology 155:3982–3991 [DOI] [PubMed] [Google Scholar]
- 66. Saviola B, Woolwine SC, Bishai WR. 2003. Isolation of acid-inducible genes of Mycobacterium tuberculosis with the use of recombinase-based in vivo expression technology. Infect. Immun. 71:1379–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Schultz J, Milpetz F, Bork P, Ponting CP. 1998. SMART, a simple modular architecture research tool: identification of signaling domains. Proc. Natl. Acad. Sci. U. S. A. 95:5857–5864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Senkovich OA, et al. 2011. Helicobacter pylori AlpA and AlpB bind host laminin and influence gastric inflammation in gerbils. Infect. Immun. 79:3106–3116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sharma CM, et al. 2010. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 464:250–255 [DOI] [PubMed] [Google Scholar]
- 70. Shibata J, et al. 1999. Regulation of tumour necrosis factor (TNF) induced apoptosis by soluble TNF receptors in Helicobacter pylori infection. Gut 45:24–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Slauch JM, Camilli A. 2000. IVET and RIVET: use of gene fusions to identify bacterial virulence factors specifically induced in host tissues. Methods Enzymol. 326:73–96 [DOI] [PubMed] [Google Scholar]
- 72. Styer CM, et al. 2010. Expression of the BabA adhesin during experimental infection with Helicobacter pylori. Infect. Immun. 78:1593–1600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Suerbaum S, Michetti P. 2002. Helicobacter pylori infection. N. Engl. J. Med. 347:1175–1186 [DOI] [PubMed] [Google Scholar]
- 74. Terry K, Williams SM, Connolly L, Ottemann KM. 2005. Chemotaxis plays multiple roles during Helicobacter pylori animal infection. Infect. Immun. 73:803–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Testerman TL, McGee DJ, Mobley HL. 2001. Helicobacter pylori growth and urease detection in the chemically defined medium Ham's F-12 nutrient mixture. J. Clin. Microbiol. 39:3842–3850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Tomb JF, et al. 1997. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388:539–547 [DOI] [PubMed] [Google Scholar]
- 77. Van Gerven N, Sleutel M, Deboeck F, De Greve H, Hernalsteens JP. 2009. Surface display of the receptor-binding domain of the F17a-G fimbrial adhesin through the autotransporter AIDA-I leads to permeability of bacterial cells. Microbiology 155:468–476 [DOI] [PubMed] [Google Scholar]
- 78. Veal-Carr WL, Stibitz S. 2005. Demonstration of differential virulence gene promoter activation in vivo in Bordetella pertussis using RIVET. Mol. Microbiol. 55:788–798 [DOI] [PubMed] [Google Scholar]
- 79. Viala J, et al. 2004. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat. Immunol. 5:1166–1174 [DOI] [PubMed] [Google Scholar]
- 80. Watanabe T, et al. 2010. NOD1 contributes to mouse host defense against Helicobacter pylori via induction of type I IFN and activation of the ISGF3 signaling pathway. J. Clin. Invest. 120:1645–1662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wen Y, Feng J, Scott DR, Marcus EA, Sachs G. 2007. The HP0165-HP0166 two-component system (ArsRS) regulates acid-induced expression of HP1186 alpha-carbonic anhydrase in Helicobacter pylori by activating the pH-dependent promoter. J. Bacteriol. 189:2426–2434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Williams CL, Haines R, Cotter PA. 2008. Serendipitous discovery of an immunoglobulin-binding autotransporter in Bordetella species. Infect. Immun. 76:2966–2977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Williams SM, et al. 2007. Helicobacter pylori chemotaxis modulates inflammation and bacterium-gastric epithelium interactions in infected mice. Infect. Immun. 75:3747–3757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Yamaoka Y, et al. 2002. Helicobacter pylori infection in mice: role of outer membrane proteins in colonization and inflammation. Gastroenterology 123:1992–2004 [DOI] [PubMed] [Google Scholar]
- 85. Yamaoka Y, et al. 1997. Induction of various cytokines and development of severe mucosal inflammation by cagA gene positive Helicobacter pylori strains. Gut 41:442–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Yamaoka Y, Kwon DH, Graham DY. 2000. A M(r) 34,000 proinflammatory outer membrane protein (oipA) of Helicobacter pylori. Proc. Natl. Acad. Sci. U. S. A. 97:7533–7538 [DOI] [PMC free article] [PubMed] [Google Scholar]