

Abstract
Among a number of laboratory strains of Listeria monocytogenes used in experimental infection, strain LO28 is highly capable of inducing robust beta interferon (IFN-β) production in infected macrophages. In this study, we investigated the molecular mechanism of the IFN-β-inducing ability of LO28 by comparing it with that of strain EGD, a low-IFN-β-inducing strain. It was found that LO28 secretes a large amount of IFN-β-inducing factor, which turned out to be cyclic di-AMP. The secretion of cyclic di-AMP was dependent on MdrT, a multidrug resistance transporter, and LO28 exhibited a very high level of mdrT expression. The introduction of a null mutation into mdrT abolished the ability of LO28 to induce IFN-β production. Examination of genes responsible for the regulation of mdrT expression revealed a spontaneous 188-bp deletion in tetR of LO28. By constructing recombinant strains of LO28 and EGD in which tetR from each strain was replaced, it was confirmed that the distinct ability of LO28 is attributable mostly to tetR mutation. We concluded that the strong IFN-β-inducing ability of LO28 is due to a genetic defect in tetR resulting in the overexpression of mdrT and a concomitant increase in the secretion of cyclic di-AMP through MdrT.
INTRODUCTION
Listeria monocytogenes is a Gram-positive, facultative intracellular parasitic bacterium which sometimes causes a severe infection in newborns, elderly people, and immunocompromised patients, with a high mortality rate (3, 8, 41, 43). L. monocytogenes invades various types of host cells, including macrophages, and is capable of replicating in the cells by means of the production of listeriolysin O (LLO), a cholesterol-dependent cytolysin. LLO disrupts the phagosomal membrane and enables L. monocytogenes to escape from the phagosome into the cytoplasm (9, 29, 30, 40).
Upon L. monocytogenes infection, macrophages produce a variety of proinflammatory cytokines that coordinately activate the host defense. It has been shown that Toll-like receptors (TLRs) participate in the production of cytokines from macrophages infected with L. monocytogenes. In general, TLRs target various extracellular pathogens and their secretory components because TLRs are localized primarily on the surfaces of cytoplasmic and endosomal membranes (1, 38). In addition, accumulating evidence indicates that various intracellular pattern recognition receptors, including nucleotide-binding oligomerization domain (NOD)-like receptors and absent in melanoma 2 (AIM2), exist in the cytosol and play an essential role in the innate immune response to L. monocytogenes. These cytosolic receptors also contribute to the production of various cytokines via recognition of molecules derived from the cytosolic L. monocytogenes (19, 21, 36, 51, 52). Therefore, these pattern recognition receptors are coincidentally activated upon L. monocytogenes infection, and the synchronous response determines the overall magnitude of innate immunity to L. monocytogenes.
Among the cytokines produced against L. monocytogenes infection, production of interleukin-6 (IL-6) and tumor necrosis factor alpha (TNF-α) is induced depending mainly on the TLR2 signal pathway, because these cytokines are produced in cells infected with heat-killed L. monocytogenes and a mutant L. monocytogenes deficient for LLO, both of which are unable to gain access to the cytoplasm, and the cytokine production by TLR2-deficient macrophages was significantly diminished compared to that by wild type macrophages (10, 14, 50). On the other hand, the production of beta interferon (IFN-β) is highly dependent on the entry of L. monocytogenes into the cytoplasm and is independent of TLRs (26, 47). L. monocytogenes that invades in the cytoplasm induces the phosphorylation of IFN regulatory factor 3 (IRF3), followed by the dimerization and translocation of IRF3 to the nucleus to induce the expression of IFN-β in cooperation with NF-κB and ATF-2/c-Jun. Subsequently, IFN-β synthesized and secreted from L. monocytogenes-infected cells binds to its receptor expressed on neighboring cells and upregulates the transcription of IRF7 via the activation of the JAK1/Tyk2-STAT1/2 signaling pathway, resulting in the enhancement of secondary IFN-β and IFN-α production (7, 45). Thus, the initial IFN-β secretion is a key step for the entire production of type I IFN. In addition to the intracellular events in host macrophages, recent reports showed that type I IFN production in infected macrophages is triggered by bacterial DNA (44) and cyclic bis-(3′→5′)-diadenosine monophosphate (cyclic di-AMP), which is secreted via MdrM, a multidrug resistance (MDR) transporter of the major facilitator superfamily (MFS) (53). Furthermore, Reutterer et al. reported that among various L. monocytogenes strains, LO28 exhibits the greatest ability to induce IFN-β production from macrophages (32). Their finding implies that LO28 possesses a strong ability to secrete immunostimulatory nucleic acids compared to that of other L. monocytogenes strains. However, the exact mechanism has not yet been elucidated.
In the present study, we investigated the mechanism by which LO28 induces a large amount of IFN-β production in infected macrophages. Here we show that the tetR (lmo2589) gene of LO28 is functionally impaired owing to a spontaneous deletion of a 188-bp gene fragment. TetR is a negative regulator of mdrT (lmo2588), a gene encoding an MDR transporter that participates in the secretion of cyclic di-AMP, which is a potent IFN-β-inducing agent. By using allelic exchange of tetR between strains LO28 and EGD, it was confirmed that the genetic defect in tetR of LO28 accounts for the upregulation of mdrT gene expression, resulting in enhanced secretion of cyclic di-AMP.
MATERIALS AND METHODS
Mice.
Female C57BL/6 mice were purchased from Japan SLC (Shizuoka, Japan). Alpha interferon receptor 1 (IFNAR1) knockout (KO) mice were kindly provided by Michel Aguet (Ecole Polytechnique Federale de Lausanne, Lausanne, Switzerland). Mice were maintained under specific-pathogen-free conditions and used at 7 to 9 weeks of age. All of the experimental procedures performed on mice were approved by the Animal Ethics and Research Committee of Kyoto University Graduate School of Medicine, Kyoto, Japan.
L. monocytogenes strains.
L. monocytogenes strain EGD has been maintained in our laboratory. L. monocytogenes strain LO28 was obtained from Yoshihiro Asano (Ehime University, Ehime, Japan). Bacteria were grown overnight in brain heart infusion (BHI) broth (Eiken Chemical, Tokyo, Japan) at 37°C with shaking. One volume of the bacterial culture was added to 100 volumes of fresh BHI broth, and bacteria were cultured for 3 h. Bacterial cells were centrifuged at 9,300 × g for 5 min, washed twice with PBS, and suspended in PBS supplemented with 10% glycerol. The aliquots were stored at −80°C until use. The CFU of bacteria were determined by plating serially 10-fold-diluted bacterial suspensions on tryptic soy agar (TSA) plates (Eiken Chemical), followed by counting of colonies after cultivation for 24 h.
Construction of L. monocytogenes mutants deficient for the hly, mdrT, or mdrM gene.
The hly gene deletion mutant was generated as described previously (13). Deletion of the gene was confirmed by PCR and DNA sequencing. The mutants deficient for mdrT (lmo2588) or mdrM (lmo1617) were constructed by the homologous recombination method. Chromosomal DNA was isolated from strains LO28 and EGD. A 1,304-bp 5′ flanking region and a 1,307-bp 3′ flanking region of the mdrT gene and a 918-bp 5′ flanking region and a 1,339-bp 3′ flanking region of the mdrM gene were amplified using PCR primer sets 1 to 4 (Table 1), respectively. The PCR fragments were treated with appropriate restriction enzymes (Table 1) and inserted into plasmid pHS-MCS, which carries a thermosensitive replication origin (13). The recombinant plasmids were introduced into LO28 and EGD by electroporation. The transformants were grown on BHI agar (Eiken Chemical) plates containing erythromycin (5 μg ml−1; Nacalai Tesque, Kyoto, Japan) at 30°C and then cultured in BHI broth containing erythromycin at 42°C to screen the clones in which the plasmid was integrated into the chromosome. Bacteria were subsequently cultured in BHI broth in the absence of erythromycin at 42°C, and erythromycin-sensitive revertants were selected. Deletion of the gene was confirmed by PCR and DNA sequencing. We thus established the mutant LO28 and EGD strains deficient for mdrT or mdrM (LO28 ΔmdrT, EGD ΔmdrT, LO28 ΔmdrM, and EGD ΔmdrM).
Table 1.
Primers used for construction of mutants
| Primer set and direction | Primer sequence (5′ to 3′)a | Restriction enzyme |
|---|---|---|
| 1 | ||
| Sense | AAGCTACGACGAGAAAAGTACG | KpnI |
| Antisense | TAGATATAATTCTAGAAAAAGAAACAGGGTCAAACT | XbaI |
| 2 | ||
| Sense | TAGATATAATTCTAGAAATTAATACCCCTTCCCATG | XbaI |
| Antisense | TAGATATAATGTCGACAGTAGCTCAGTTGGTAGAGC | SalI |
| 3 | ||
| Sense | TAGATATAATGGTACCAGTCGTGAGTTGCATAACCG | KpnI |
| Antisense | TAGATATAATGAGCTCTCACGAAAAAGCGCGTTCCA | SacI |
| 4 | ||
| Sense | TAGATATAATGAGCTCTCTCGTGCACCTTCCCTTTC | SacI |
| Antisense | TAGATATAATGTCGACTTCCAAAGAAGAGCTACGTG | SalI |
| 5 | ||
| Sense | TAGATATAATGGTACCCGACACCAACAATCAACATC | KpnI |
| Antisense | TAGATATAATGTCGACAGAACGTCGTGAGACAGTTC | SalI |
Sites recognized by restriction enzymes are underlined.
Gene complementation of LO28 ΔmdrT with the mdrT gene.
An LO28 ΔmdrT strain complemented with mdrT (LO28 ΔmdrT::mdrT) was also generated by the homologous recombination method. The mdrT-containing 3,994-bp region of LO28 was amplified by PCR using the sense primer of primer set 1 and the antisense primer of primer set 2. The DNA fragment was digested with SalI and KpnI and ligated into plasmid pHS-MCS. The recombinant plasmid was introduced into LO28 ΔmdrT, and transformants were selected by incubation at 30°C on BHI agar plates containing erythromycin. The erythromycin-resistant bacteria were then incubated at 42°C in BHI broth containing erythromycin, followed by incubation at 42°C in BHI broth in the absence of erythromycin. The clone in which the plasmid was integrated into the chromosome was selected. Complementation of the gene was confirmed by PCR and DNA sequencing.
Construction of LO28 tetRE and EGD tetRL.
A recombinant LO28 strain expressing TetR derived from EGD (LO28 tetRE) and a recombinant EGD strain expressing TetR derived from LO28 (EGD tetRL) were constructed by the homologous recombination method. To amplify a DNA fragment containing tetR and both flanking regions of strains LO28 and EGD, PCR was carried out using primer set 5 (Table 1). We obtained 2,414- and 2,602-bp DNA fragments that were derived from LO28 and EGD, respectively. Sequence analysis and comparison between these two DNA fragments revealed a 188-bp deletion in tetR of LO28, but the remaining sequence was exactly the same. DNA fragments were digested with EcoRI and XbaI and ligated into plasmid pHS-MCS. The EGD strain was transfected with the recombinant plasmid containing the LO28-derived DNA fragment, and the LO28 strain was transfected with the plasmid containing the EGD-derived DNA fragment. The transformants were grown on BHI agar plates containing 5 μg ml−1 erythromycin at 30°C and then cultured in BHI broth containing erythromycin at 42°C to obtain drug-resistant clones. Bacteria were subsequently cultured in BHI broth without erythromycin at 42°C to screen the erythromycin-sensitive revertants, and thus we established two clones, LO28 tetRE and EGD tetRL. The gene replacements in these mutants were confirmed by PCR and DNA sequencing.
Preparation of Listeria culture supernatants.
LO28, EGD, and recombinant L. monocytogenes strains were grown overnight in BHI broth at 37°C with shaking. One volume of the bacterial culture was added to 200 volumes of fresh BHI broth, and bacteria were cultured for 3 h. Bacterial cells were harvested by centrifugation at 2,380 × g for 10 min, suspended in 1/10 volume of RPMI 1640 medium (Nacarai Tesque), and cultured at 37°C for 3 h. The culture supernatants were collected and filtered through 0.2-μm filters (PALL, Port Washington, NY). Alternatively, LO28 was cultured in fresh BHI broth and then treated with 10 μg ml−1 reserpine for 30 min on ice. Bacteria were then cultured in RPMI 1640 medium for 3 h, and the culture supernatant was obtained as described above.
Preparation of Listeria cell lysate.
Bacterial cell lysates were prepared by using methods described previously, with some modification (44). Briefly, bacteria were grown overnight in BHI broth at 37°C with shaking. One hundred microliters of the overnight culture was added to 10 ml of fresh BHI broth, and bacteria were cultured for 3 h. Bacterial cells were harvested by centrifugation, washed twice with phosphate-buffered saline (PBS), and centrifuged. The bacterial pellet was freeze-thawed using liquid nitrogen, resuspend in 1 ml PBS containing 1 mg ml−1 lysozyme (Nacarai Tesque), and incubated at 37°C for 1 h. The bacterial suspension was sonicated for 2 min using an Astrason XL ultrasonic processor (Misonix, Farmingdale, NY) and centrifuged. The supernatant was collected and used as the cell lysate.
Cytokine production in Listeria-infected macrophages.
Murine peritoneal exudate cells (PECs) were obtained 3 days after an i.p. injection of 2.5 ml of 3% thioglycolate medium (Eiken Chemical). After washing with RPMI 1640 medium (Nacalai Tesque), PECs were suspended in RPMI 1640 medium supplemented with 10% fetal calf serum (FCS), seeded in 96-well tissue culture plates, and incubated at 37°C for 3 h. After removal of nonadherent cells, adherent cells were used as macrophages. To evaluate the molecular mechanism of strong IFN-β production induced by LO28 infection, macrophages were infected with LO28, EGD, and mutant strains at a multiplicity of infection (MOI) of 1. After 30 min of infection, gentamicin (10 μg ml−1; Wako Pure Chemical Industries, Osaka, Japan) was added to kill extracellular bacteria. The culture supernatants were collected 12 h after infection and stored at −80°C. For analysis of the effect of reserpine (Santa Cruz, Santa Cruz, CA) on the cytokine-inducing ability of L. monocytogenes, LO28 was treated with 2.5 and 10 μg ml−1 reserpine for 30 min on ice. Macrophages were infected with reserpine-treated L. monocytogenes at an MOI of 1 and then cultured for 12 h in the presence of reserpine at the same concentration as used for treatment with L. monocytogenes. The culture supernatant was collected, and IFN-β production was determined. Concomitantly, reserpine-treated L. monocytogenes was cultured for 12 h in RPMI 1640 medium, and the effect of reserpine on the production of cyclic di-AMP was monitored. Alternatively, macrophages were incubated for 12 h with Listeria culture supernatants, Listeria cell lysates, 1 μg ml−1 cyclic di-AMP (Biolog, Bremen, Germany), or 3 μg ml−1 cyclic di-GMP (Biolog, Bremen, Germany) in the presence or absence of a sublytic concentration of recombinant listeriolysin O (LLO) (5 μg ml−1), 0.005% saponin, or 1 μg ml−1 poly(dA·dT) (Sigma, St. Louis, MO) emulsified in Lipofectamine LTX (Invitrogen, Carlsbad, CA). The culture supernatants were collected and stored at −80°C. In one experiment, Listeria culture supernatants were treated with 1 U of Benzonase nuclease (Merck, Whitehouse Station, NJ) or 1 U of phosphodiesterase I (PDE) (USB, Cleveland, OH) in the presence of MgCl2 (final concentration, 14 mM) at 37°C for 1 h, and the cytokine-inducing activity was determined. The levels of IFN-β and IL-6 were determined with enzyme-linked immunosorbent assay (ELISA) kits for IFN-β (R&D Systems, Minneapolis, MN) and mouse IL-6 (eBioscience, San Diego, CA), respectively.
Bacterial growth in macrophages.
Macrophages were infected with various L. monocytogenes strains for 30 min at an MOI of 1. Gentamicin was added to kill extracellular bacteria, and the culture was continued. After 1, 6, and 12 h of infection, cells were washed with PBS and lysed in PBS containing 0.05% Triton X-100. Cell lysates were serially diluted and inoculated on TSA plates to count the number of viable bacteria inside the cells.
Western blot analysis.
Macrophages were infected with L. monocytogenes strains at an MOI of 10 for 30, 60, and 90 min and lysed in SDS sample buffer. The cell lysates were subjected to SDS-PAGE and subsequently transferred to polyvinylidene difluoride membranes by electroblotting. The membranes were immunoblotted with antibodies to phospho-IRF3, phospho-TBK1, TBK1, phospho-JNK or JNK (Cell Signaling, Boston, MA) or with anti-IRF3 antibody (Zymed, San Francisco, CA), followed by horseradish peroxidase (HRP)-conjugated second antibody. After soaking of the membrane in ECL Plus (GE Healthcare Bio-Sciences AB, Uppsala, Sweden), a chemiluminescent image was captured with an LAS-4000 mini-imager (Fuji Film, Tokyo, Japan).
Quantification of cyclic bis-(3′→5′)-dinucleotides.
To determine the concentrations of cyclic di-AMP and cyclic di-GMP in Listeria culture supernatants, liquid chromatography-mass spectrometry (LC-MS) analysis was carried out using an LCMS-2020 (Shimadzu, Kyoto, Japan) under the following conditions; column, Cosmosil 5 C18-AR-II (4.6 by 150 mm; Nacalai Tesque); solvent system, mobile phase of 1% (vol/vol) acetic acid (AcOH) in H2O to which acetonitrile was added with a linear gradient from 0% to 40% over 30 min; flow rate, 0.5 ml min−1; temperature, 40°C. The concentration of cyclic bis-(3′→5′)-dinucleotide was evaluated on the basis of the peak area of standard curve generated using a standard sample with a known concentration.
RT-PCR analysis.
L. monocytogenes strains were cultured to mid-log phase in BHI broth, and total bacterial RNA was extracted using an RNeasy Minikit (Invitrogen) and treated with RNase-free DNase (Promega, Madison, WI) to eliminate contaminating DNA before being subjected to reverse transcription using Superscript Vilo (Invitrogen). Quantitative real-time reverse transcription-PCR (RT-PCR) was performed on an ABI Prism 7000 (Applied Biosystems, Foster City, CA) using Express SYBR GreenER qPCR Supermix with premixed ROX (Invitrogen) with primers described in a previous study (6). Results were analyzed with ABI Prism 7000 SDS software. Gene-specific transcript levels were normalized to the amount of bglA mRNA.
Infection of mice with L. monocytogenes strains.
Mice were infected intravenously with 1.0 × 104 CFU of wild-type and mutant L. monocytogenes strains. Spleens were collected at 1 and 3 days after infection and homogenized in 5 ml of PBS. A serially 10-fold-diluted homogenate was inoculated on TSA plates, and the number of colonies was counted 1 day later. Additionally, the level of IFN-β in the homogenate was determined by ELISA.
Statistical analysis.
For comparisons between two groups, Student's t test was used. Statistical significance was determined as a P value of <0.05.
Nucleotide sequence accession numbers.
The LO28 DNA sequences identified in this study have been deposited in DDBJ under accession numbers AB671766 to AB671771 (tetR, mdrT, ladR, mdrL, marR, and mdrM, respectively).
RESULTS
LO28 but not EGD strongly induces IFN-β production from infected macrophages through the activation of the TBK1-IRF3 signaling pathway.
In order to confirm the previous report on the significant difference between LO28 and EGD (32), we first compared the IFN-β-inducing abilities of these two strains in murine macrophages. LO28 exhibited over a 50-fold-higher ability to induce IFN-β production than EGD (Fig. 1A). However, IL-6 production was similarly induced by both strains of L. monocytogenes (Fig. 1B). Moreover, there was no difference in the intracellular growth inside macrophages between LO28 and EGD (Fig. 1C), indicating that the difference in IFN-β-inducing ability is not due to a difference in cytosolic invasion after infection. Indeed, LLO mutants of both strains were incapable of inducing IFN-β production (data not shown). These data clearly showed that LO28 has a distinct ability for IFN-β induction compared to EGD. Recent reports have shown that IRF3 plays an essential role in the expression of IFN-β, and TBK1 and JNK contribute to the activation of IRF3 through phosphorylation of IRF3 at the C terminus and N terminus, respectively (39, 54). We thus determined whether the activation of these transcription factors is associated with the increased production of IFN-β in LO28-infected macrophages. Figure 1D shows that phosphorylated IRF3 was markedly increased at 90 min after infection with LO28 but not with EGD. The Western blot also revealed that phosphorylation of TBK1 was markedly induced by infection with LO28. On the other hand, there was no significant difference in the intensities of bands representative of phosphorylated JNK between macrophages infected with LO28 and those infected with EGD. These results suggested that the substantial ability of LO28 in the induction of IFN-β is due at least in part to the strong activation of the TBK1-IRF3 signaling pathway in LO28-infected macrophages. Therefore, it appears that LO28 stimulates some cytoplasmic sensor to induce a large amount of IFN-β production.
Fig 1.
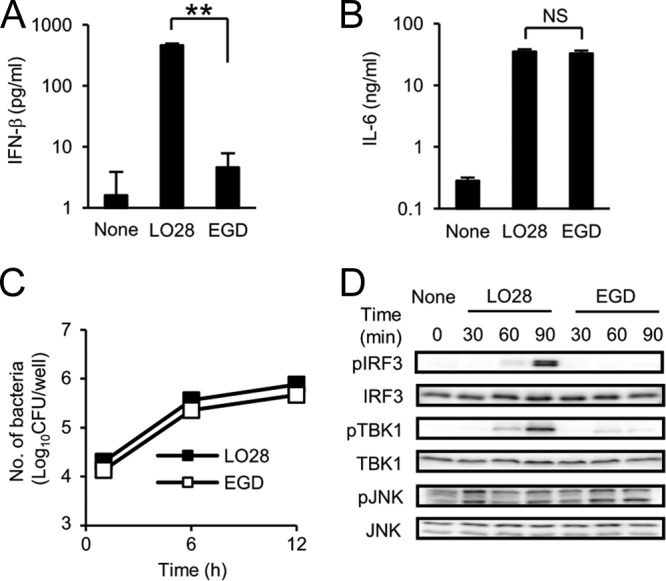
LO28 has a strong ability to induce IFN-β production through the activation of IRF3 and TBK1, whereas its ability to induce IL-6 production and its intracellular replication are comparable to those of EGD. (A and B) Macrophages were infected with LO28 or EGD at an MOI of 1 and cultured for 12 h in the presence of 10 μg ml−1 gentamicin. The culture supernatants were collected, and the production of IFN-β (Α) and IL-6 (B) was measured by ELISA. (C) Macrophages were infected with LO28 and EGD and cultured for 1, 6, and 12 h in the presence of gentamicin. Cells were lysed, and the cell lysate was inoculated on TSA plates to enumerate the intracellular bacteria. (D) Macrophages were infected with LO28 and EGD at an MOI of 10 and cultured for 30, 60, and 90 min in the presence of gentamicin. Cells were lysed, and the cell lysates were subjected to SDS-PAGE. The amounts of phospho-IRF3, phospho-TBK1, and phospho-JNK were evaluated by Western blotting. All data in panels A to C represent the means and standard deviations from triplicate assays. Similar results were obtained in two independent experiments. NS, not significant. **, P < 0.01.
Cyclic di-AMP is a factor responsible for the IFN-β-inducing ability of LO28.
We have assumed that the distinct ability of LO28 for IFN-β induction is due to the difference in some factor(s) derived from LO28, cell-associated components, or secreted bacterial products. In order to characterize the factor, we cultured LO28 and EGD in BHI broth and prepared bacterial cell lysates as well as culture supernatants. Macrophages were cultured with the lysates or the culture supernatants in the presence or absence of saponin, which introduces the bacterial components into the cytoplasm of macrophages. The culture supernatant was collected after 12 h of cultivation, and the level of IFN-β production was determined. Bacterial lysates of both strains did not induce high levels of IFN-β production even in the presence of saponin (Fig. 2A). On the other hand, the culture supernatant of LO28 exhibited a strong IFN-β-inducing activity, and the level of IFN-β production was about 10 times higher than that induced by the culture supernatant of EGD (Fig. 2B). Furthermore, the high level of IFN-β production was induced by the culture supernatant of LO28 in the absence of saponin. On the other hand, the IFN-β-inducing activity was not observed in the culture supernatant of mutant LO28 deficient for the LLO gene (LO28 Δhly), and the ability was restored by addition of LLO or saponin (Fig. 2C). The results implied that LO28 but not EGD secretes some component that promotes IFN-β production and that LLO contributes to the cytokine production as an agent to deliver the IFN-β-inducing factor to the cytoplasm by its pore-forming activity. In addition, in our preliminary study, the IFN-β-inducing activity was detected in a fraction of less than 3 kDa, and the activity was heat stable (data not shown). It has been reported that small DNA fragments and cyclic bis-(3′→5′)-dinucleotides that are derived from bacteria exert IFN-β-inducing activity (23, 44, 53). These findings raised the possibility that such Listeria-derived nucleic acids may be responsible for IFN-β induction. Therefore, we determined whether the nucleic acids play a role in the high level of IFN-β production induced by LO28 infection. The culture supernatant of LO28 was treated with phosphodiesterase I (PDE), which catalyzes hydrolysis of cyclic bis-(3′→5′)-dinucleotides, or with Benzonase, which catalyzes hydrolysis of DNA, and then the IFN-β-inducing activity was measured. As shown in Fig. 2D, treatment with Benzonase abolished the IFN-β-inducing ability of poly(dA·dT) but not that of other stimulants. In contrast, the activities of LO28 culture supernatant, cyclic di-GMP, and cyclic di-AMP were significantly diminished by treatment with PDE, suggesting that the responsible factor in the LO28 culture supernatant is a cyclic dinucleotide. To determine whether LO28 culture supernatant contains cyclic bis-(3′→5′)-dinucleotides, we performed LC-MS analysis and detected a high level of cyclic di-AMP in the culture supernatant of LO28 (Fig. 2E). Such a large amount of cyclic di-AMP was not detectable in the culture supernatant of EGD. Cyclic di-GMP was below the level of detection in the culture supernatants of LO28 and EGD. These results indicated that LO28 has the ability to secrete a large amount of cyclic di-AMP that triggers a high level of IFN-β production in infected macrophages.
Fig 2.

LO28 secretes a large amount of cyclic di-AMP that is capable of inducing IFN-β production from infected macrophages. (A to C) Bacterial cell lysates and culture supernatants of LO28, EGD, and LO28 Δhly were prepared as described in Materials and Methods. Macrophages were stimulated with the lysates (A) and the culture supernatants (B and C) for 12 h in the presence or absence of a nonlytic concentration of saponin or LLO. The culture supernatants were collected, and the IFN-β concentration was determined by ELISA. (D) Macrophages were stimulated with the culture supernatant of LO28, poly(dA·dT), cyclic di-GMP, and cyclic di-AMP, which had been treated with or without 1 unit of PDE or Benzonase. The culture supernatants were collected, and the IFN-β concentration was measured by ELISA. (E) LO28 and EGD were suspended in RPMI 1640 medium and cultured for 3 h. The culture supernatants were collected and subjected to LC-MS to measure the concentrations of cyclic di-AMP and cyclic di-GMP. All data represent the means and standard deviations from triplicate assays. Similar results were obtained in two independent experiments. n.d., not detected. NS, not significant. *, P < 0.05. **, P < 0.01.
Functional impairment of TetR in LO28 results in an enhanced expression of MdrT transporter and an increase in cyclic di-AMP secretion.
Previous reports have shown that L. monocytogenes multidrug resistance (MDR) transporters, including MdrM (Lmo1617), MdrT (Lmo2588), and MdrL (Lmo1409), are involved in the secretion of cyclic di-AMP and the induction of IFN-β production (6, 53). To determine whether these MDR transporters are involved in the enhanced secretion of cyclic di-AMP in LO28, we tested the effect of reserpine, a broad-spectrum inhibitor of MDR transporters, on cyclic di-AMP secretion from LO28. Reserpine markedly reduced both cyclic di-AMP secretion (Fig. 3A) and the IFN-β-inducing ability of LO28, whereas the treatment did not affect IL-6 production from infected macrophages (Fig. 3B), suggesting that the MDR transporters of LO28 participate in the secretion of high levels of cyclic di-AMP and in the strong IFN-β-inducing ability. To identify the major transporter that contributes to cyclic di-AMP secretion in LO28, we analyzed the expression levels of mdrM, mdrT, and mdrL in LO28 and EGD. Interestingly, the expression of mdrT was significantly upregulated in LO28 compared to EGD, whereas there was no difference in the expression of mdrM and mdrL between two strains (Fig. 3C). DNA sequence analysis revealed that both strains possess identical mdrT, mdrM, and mdrL genes, and genetic diversity was never detected in marR (lmo1618) or ladR (lmo1408), the products of which repress the expression of mdrM or both mdrM and mdrL, respectively (6). However, the tetR (lmo2589) gene of LO28, which encodes a negative regulator of mdrT, was revealed to have a 188-bp nucleotide deletion (Table 2). To analyze the influence of tetR mutation on the expression of mdrT and IFN-β-inducing ability, we constructed a mutant LO28 expressing the tetR of EGD (LO28 tetRE) and a mutant EGD expressing the tetR of LO28 (EGD tetRL). As shown in Fig. 4A, a high level of mdrT expression was detected in LO28. Strong mdrT expression was also observed in EGD tetRL, and the level was the same as that detected in LO28. In contrast, the mdrT gene expression in LO28 tetRE was as low as that detected in EGD. These results suggested that the deletion in tetR caused a loss of the regulator activity, resulting in an upregulation of mdrT expression in LO28. Moreover, L. monocytogenes strains harboring the tetR of LO28 (LO28 and EGD tetRL) secreted large amounts of cyclic di-AMP, whereas the cyclic dinucleotide was not detected in the culture supernatant of L. monocytogenes strains that express the tetR of EGD (EGD and LO28 tetRE) (Fig. 4B). In addition, EGD tetRL, as well as LO28, was capable of inducing IRF3 phosphorylation (Fig. 4C) and IFN-β secretion (Fig. 4D), but EGD or LO28 tetRE could not. The intracellular growth was not affected at all in mutants with tetR replaced (Fig. 4E). These results strongly indicated that the spontaneous deletion of the 188-bp fragment in tetR is responsible for the secretion of a large amount of cyclic di-AMP and the robust IFN-β-inducing ability of LO28.
Fig 3.

The MDR transporter of LO28 plays a critical role in cyclic di-AMP secretion and IFN-β-inducing ability. (A) LO28 was incubated with 10 μg ml−1 reserpine or dimethyl sulfoxide (DMSO) for 30 min and cultured for 3 h. The concentration of cyclic di-AMP in the culture supernatant was determined using LC-MS. (B) Macrophages were infected with LO28 that had been treated with 2.5 and 10 μg ml−1 reserpine or DMSO for 30 min at an MOI of 1 and cultured for 12 h in the presence of the same concentration of reserpine. The culture supernatants were collected, and the concentrations of IFN-β and IL-6 were measured by ELISA. (C) Total RNA was extracted from LO28 and EGD that were grown to mid-log phase in BHI broth, and the expression levels of MDR transporter genes were determined by real-time RT-PCR. The gene expression level was normalized on the basis of bglA mRNA expression. The dashed line indicates gene expression levels of MDR transporters in EGD. All data represent the means and standard deviations from triplicate assays. Similar results were obtained in two independent experiments. NS, not significant. **, P < 0.01.
Table 2.
Differences in DNA sequences of L. monocytogenes LO28 and EGD MDR transporters and their transcriptional regulators
| Gene name | Function of translated protein | Difference in DNA sequence |
|---|---|---|
| ladR (lmo1408) | Negative regulator of lmo1409 | None |
| mdrL (lmo1409) | Putative MDR transporter | None |
| mdrT (lmo2588) | Putative MDR transporter | None |
| tetR (lmo2589) | Negative regulator of lmo2588 | 188-bp deletion in lmo2589 of LO28 |
| mdrM (lmo1617) | Putative MDR transporter | None |
| marR (lmo1618) | Negative regulator of lmo1617 | None |
Fig 4.

A mutation of tetR in LO28 is the major cause of enhanced expression of mdrT and an increase in cyclic di-AMP and IFN-β production. (A) LO28, EGD, and recombinant L. monocytogenes strains with each tetR gene exchanged (LO28 tetRE and EGD tetRL) were cultured in BHI broth to the mid-log phase, and total RNA was extracted. The expression level of mdrT was determined by real-time RT-PCR. The gene expression level was normalized on the basis of bglA mRNA expression. Relative expression levels are indicated as a ratio of the mdrT expression level for the indicated strain to the mdrT expression level for EGD. (B) LO28, EGD, LO28 tetRE, and EGD tetRL were inoculated in RPMI 1640 medium and cultured for 3 h. The supernatant was collected, and the cyclic di-AMP concentration was determined using LC-MS. n.d., not detected. (C) Macrophages were infected with LO28, EGD, LO28 tetRE, and EGD tetRL at an MOI of 10 and cultured for 90 min. Cell lysates were prepared, and the level of IRF3 phosphorylation was determined by Western blotting. (D) Macrophages were infected with LO28, EGD, LO28 tetRE, and EGD tetRL at an MOI of 1 and cultured for 12 h in the presence of 10 μg ml−1 gentamicin. The culture supernatants were collected, and the IFN-β concentration was measured by ELISA. (E) Macrophages were infected with LO28, EGD, LO28 tetRE, and EGD tetRL an MOI of 1 and cultured for 1, 6, and 12 h. Cells were lysed, and the cell lysates were inoculated on TSA plates to determine the number of intracellular bacteria. All data in panels A, B, D, and E represent the means and standard deviations from triplicate assays. Similar results were obtained in two independent experiments. NS, not significant. *, P < 0.05. **, P < 0.01.
MdrT is crucial for cyclic di-AMP secretion from LO28.
To confirm that MdrT serves as an exporter of cyclic di-AMP in LO28, we constructed mdrT-deficient mutants of LO28 (LO28 ΔmdrT) and EGD (EGD ΔmdrT) and determined their abilities to secrete cyclic di-AMP. The ability of LO28 to secrete a large amount of cyclic di-AMP was completely abrogated in the mutant with mdrT deleted (Fig. 5A). Similarly, the ability to induce IFN-β production (Fig. 5B) and IRF3 phosphorylation (Fig. 5C) was markedly impaired in LO28 ΔmdrT, while the intracellular growth of this mutant was comparable to that of the wild-type strain (Fig. 5D). The inability of the mutant was restored by complementation with mdrT (Fig. 5E), supporting the idea that MdrT is critically involved. In contrast, the deficiency of mdrT did not show any influence on the ability of EGD. These data suggested that MdrT is the major exporter responsible for the secretion of a large amount of cyclic di-AMP and the resulting IFN-β-inducing ability observed in LO28.
Fig 5.

MdrT is substantially involved in cyclic di-AMP secretion and the IFN-β-inducing ability of LO28. (A) LO28 and LO28 ΔmdrT were inoculated in RPMI 1640 medium and cultured for 3 h. The culture supernatant was collected, and the cyclic di-AMP concentration was measured by LC-MS. n.d., not detected. (B) Macrophages were infected with LO28, EGD, LO28 ΔmdrT, and EGD ΔmdrT at an MOI of 1 and cultured for 12 h in the presence of 10 μg ml−1 gentamicin. The culture supernatants were collected, and the IFN-β concentration was determined by ELISA. (C) Macrophages were infected with LO28, EGD, LO28 ΔmdrT, and EGD ΔmdrT at an MOI of 10 and cultured for 90 min. Cell lysates were prepared, and the level of IRF3 phosphorylation was determined. (D) Macrophages were infected with LO28, EGD, LO28 ΔmdrT, and EGD ΔmdrT at an MOI of 1 and cultured for 6 and 12 h. Cells were lysed, and the cell lysates were inoculated on TSA plates to determine the number of intracellular bacteria. (E) Macrophages were infected with LO28, LO28 ΔmdrT and LO28 ΔmdrT::mdrT at an MOI of 1 and cultured for 12 h in the presence of 10 μg ml−1 gentamicin. The culture supernatants were collected, and the IFN-β concentration was determined by ELISA. All data in panels A, B, D, and E represent the means and standard deviations from triplicate assays. Similar results were obtained in two independent experiments. NS, not significant. **, P < 0.01.
Involvement of MdrM as a transporter of cyclic di-AMP in EGD and LO28.
Here we showed that MdrT serves as the transporter of cyclic di-AMP in LO28 and is involved in the induction of enormous IFN-β production from infected macrophages. However, MdrT was dispensable for the low level of IFN-β-inducing ability of EGD, raising the possibility that some other transporter may also be involved in the induction of IFN-β production. In this regard, it has been shown that L. monocytogenes 10403s induced IFN-β production in an MdrM-dependent manner (6). To test the possibility of additional engagement of MdrM in the beta interferon-inducing ability, we constructed mdrM-deficient mutants of LO28 and EGD (LO28 ΔmdrM and EGD ΔmdrM). We found that the deletion of mdrM in LO28 and EGD did not affect the intracellular growth in macrophages (Fig. 6A). The level of IFN-β production was partly but significantly reduced in the mdrM-defective mutant, though the level of reduction was limited compared with that in the mdrT mutant (Fig. 6B). In addition, LO28 ΔmdrM secreted less cyclic di-AMP than LO28 (Fig. 6C). In strain EGD, which induces only a marginal level of IFN-β due to the suppression of MdrT mediated by intact TetR, the IFN-β-inducing ability was reduced by deletion of mdrM (Fig. 6D). These data suggested that MdrM also contributes to the secretion of cyclic di-AMP from both LO28 and EGD. As MdrM is unlikely to be able to export a large amount of cyclic di-AMP, EGD elicits a weaker IFN-β production than LO28. In contrast, both MdrT and MdrM contribute to the secretion of cyclic di-AMP from LO28, resulting in the induction of an extremely high level of IFN-β production from infected macrophages.
Fig 6.
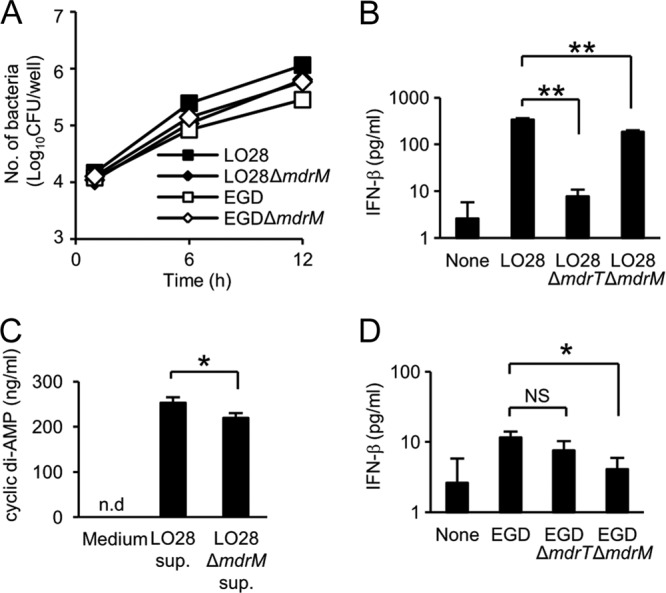
MdrM acts as a transporter of cyclic di-AMP in EGD and LO28. (A) Macrophages were infected with LO28, EGD, LO28 ΔmdrM, and EGD ΔmdrM at an MOI of 1 and cultured for 6 and 12 h. Cells were lysed, and the cell lysates were inoculated on TSA plates to determine the number of intracellular bacteria. (B) Macrophages were infected with LO28, LO28 ΔmdrT, and LO28 ΔmdrM at an MOI of 1 and cultured for 12 h in the presence of 10 μg ml−1 gentamicin. The culture supernatants were collected, and the IFN-β concentration was determined by ELISA. (C) LO28 and LO28 ΔmdrM were cultured in RPMI 1640 medium for 3 h. The culture supernatant was collected, and the cyclic di-AMP concentration was measured by LC-MS. (D) Macrophages were infected with EGD, EGD ΔmdrT, and EGD ΔmdrM at an MOI of 1 and cultured for 12 h in the presence of 10 μg ml−1 gentamicin. The culture supernatant was collected, and the IFN-β concentration was determined by ELISA. All data represent the means and standard deviations from triplicate assays. Similar results were obtained in two independent experiments. NS, not significant. *, P < 0.05. **, P < 0.01.
The strong expression of mdrT is associated with a decrease in the pathogenicity of L. monocytogenes.
Reutterer et al. showed that LO28 is more virulent than EGD due to the high level of IFN-β-inducing ability (32). In contrast, other studies have shown that L. monocytogenes expressing a high level of mdrT exhibits lower virulence than L. monocytogenes expressing a low level of mdrT (6). Therefore, we compared the virulences of LO28, EGD, LO28 ΔmdrT, and EGD ΔmdrT to clarify whether the high levels of mdrT expression and IFN-β-inducing ability serve as the virulence determinants of L. monocytogenes. We infected mice with these bacteria and then measured the IFN-β production and counted the bacterial number in spleen. Similar to the case for IFN-β production in vitro, LO28 elicited IFN-β production that was approximately twice as high as that elicited by EGD (Fig. 7A). However, the number of CFU of LO28 was approximately 10 times lower than that of EGD (Fig. 7B), indicating that LO28 is less virulent than EGD. LO28 ΔmdrT showed a marked reduction in the ability to induce IFN-β production to a level induced by EGD (Fig. 7A); however, the numbers of LO28 ΔmdrT recovered from the spleens of infected mice were significantly higher than those of LO28 (Fig. 7B). Furthermore, the deficiency of mdrT did not affect the ability of EGD to induce IFN-β production or survive in the host (Fig. 7A and B). It was suggested that the strong expression of mdrT causes not only robust IFN-β production but also attenuation of the pathogenicity in vivo. To clarify the effect of IFN-β on the host resistance to bacterial infection, we infected wild-type and IFNAR1 KO mice with LO28 and LO28 ΔmdrT and measured the bacterial number in spleen. The CFU of LO28 recovered from IFNAR1 KO mice was significantly lower than that obtained from wild-type mice (Fig. 7C). A significant reduction of the CFU was similarly observed in infection with LO28 ΔmdrT. These results suggested that IFN-β exerts a detrimental effect on the host resistance to L. monocytogenes. Interestingly, the 1-log-unit difference in the CFU between LO28 and LO28 ΔmdrT in wild-type mice was also observed in IFNAR1 KO mice. This observation may suggest that some factor other than IFN-β participates in the virulence controlled by MdrT.
Fig 7.

The expression of mdrT is inversely related to the pathogenicity of L. monocytogenes. (A) Mice were infected intravenously with 1.0 × 104 CFU of LO28, EGD, LO28 ΔmdrT, and EGD ΔmdrT. At 24 h after infection, spleens were removed and homogenized in 5 ml of PBS. The IFN-β concentration in the spleen homogenate was determined by ELISA. (B) Mice were infected intravenously with 1.0 × 104 CFU of LO28, EGD, LO28 ΔmdrT, and EGD ΔmdrT. Spleens were removed 3 days after infection and homogenized in 5 ml of PBS. The homogenate was inoculated on TSA plates to determine the number of infected bacteria. (C) Wild-type and IFNAR1 KO mice were infected intravenously with 1.0 × 104 CFU of LO28 and LO28 ΔmdrT. Spleens were obtained at 3 days after infection and homogenized in 5 ml of PBS. The homogenates were inoculated on TSA plates to determine the number of infected bacteria. All data represent the means and standard deviations for five mice. Similar results were obtained in two independent experiments. NS, not significant. *, P < 0.05. **, P < 0.01.
DISCUSSION
The innate immune system serves as the first line of pathogen surveillance and host defense via recognition of pathogen-associated molecular patterns (PAMPs). Cyclic dinucleoside monophosphates such as cyclic di-AMP and cyclic di-GMP are unique to bacteria (12, 34) and have recently been categorized as PAMPs capable of inducing type I IFN production when delivered into the cytoplasm of mammalian cells, including macrophages (23, 53). In this study, we showed that the strong IFN-β-inducing ability observed in strain LO28 is mainly due to the secretion of a large amount of cyclic di-AMP that depends on a markedly enhanced expression of MdrT transporter caused by the spontaneous genetic defect of the TetR repressor in this particular strain.
In addition to cyclic dinucleoside monophosphates, bacterial DNA is known to be a potent inducer of IFN-β expression (44). In this regard, it has been shown that the cellular response to exogenous DNA depends on a signaling pathway involving STING, TBK1, and IRF3 (17, 18, 44) but not IPS-1/MAVS, which is an adaptor molecule involved in type I IFN production in response to double-stranded and 5′-triphosphate RNAs (20, 37). It has been also shown that LO28 induces IFN-β expression in the absence of IPS-1/MAVS (25, 42, 48), while STING was required for IFN-β expression (data not shown), implying that LO28 induces IFN-β expression through the activation of a signaling pathway similar to that activated by DNA but not immunostimulatory RNA. These findings raise an alternative possibility that the amount of DNA released from LO28 may account for the strong ability of LO28 to induce IFN-β production. If this is the case, there should be a difference in the ability to induce caspase-1 activation and IL-18 production between LO28 and EGD, because L. monocytogenes promotes IL-18 production via AIM2 inflammasome-dependent caspase-1 activation and the cytokine response is mediated by DNA released from L. monocytogenes in the cytoplasm of infected cells (36, 51, 52). When we looked at the caspase-1 activation and caspase-1-dependent cytokine secretion, there was no difference in the ability to induce caspase-1 activation and IL-18 secretion between LO28 and EGD, and the levels of IL-18 induced by LO28 and EGD were decreased to the same extent by a knockdown of aim2 (data not shown). Based on these observations, we ruled out a possible contribution of bacterial DNA to the enhanced ability of LO28 in IFN-β induction. On the other hand, our study clearly indicated that LO28 has a higher ability to secrete cyclic di-AMP than EGD and that the IFN-β-inducing activity was sensitive to PDE but not to Benzonase. These findings suggested that an extremely high level of production of cyclic dinucleotide from LO28 accounts for the high IFN-β-inducing ability. In agreement with this, cyclic di-AMP has recently been shown to induce IFN-β production in a STING-dependent, IPS-1-independent manner (35, 53). Taken together, the data suggest that the strong IFN-β production induced by LO28 is dependent mainly on cyclic di-AMP released from LO28 in the cytoplasm but is not attributable to the signaling pathway activated by cytosolic DNA or RNA.
Accumulating evidence indicates that LLO is essential for the induction of IFN-β production in L. monocytogenes-infected macrophages (26, 47). It is conceivable that LLO-dependent membrane permeation and/or bacterial invasion in the cytoplasm is necessary for LO28 to trigger cyclic di-AMP-induced IFN-β production. In addition, we and others reported that LLO not only is essential for invasion of L. monocytogenes into the cytoplasm but also activates some inflammatory signal pathways, including a signaling event contributing to IFN-β production, possibly by modulating some intracellular signaling as a bacterial modulin (14, 27, 51). Thus, it appears that the amount of LLO produced in infected cells is critical for the IFN-β-inducing ability. However, we found that LO28 and EGD secreted almost the same amount of LLO in the culture supernatants (data not shown), and the nucleotide sequences of their LLO genes (hly) were identical. Therefore, the strong IFN-β-inducing ability of LO28 could not be attributed to an enhancement in the productivity of LLO or the membrane-lytic activity.
Based on the reports suggesting a central role for MDR transporters in cyclic-di-AMP secretion, we have analyzed and compared the nucleotide sequences of tetR in LO28 and EGD. This approach revealed the spontaneous deletion of a 188-bp fragment in tetR of LO28. The mutual replacement of tetR between LO28 and EGD clearly indicated that the distinct ability of LO28 for cyclic-di-AMP secretion and induction of IFN-β production is exclusively dependent on the mutation in tetR resulting in the overexpression of mdrT (Fig. 4 and 5). It seemed that the presence of intact tetR in EGD suppresses the expression of mdrT, thereby limiting the secretion of cyclic-di-AMP to below the level of detection by LC-MS.
Although type I IFNs are crucial for host resistance against infection with a variety of viruses (49), a detrimental effect of this cytokine on the host has been shown in animal models of L. monocytogenes infection. Indeed, type I IFNs have been shown to severely induce cell death of macrophages (46) and apoptosis of sensitized T lymphocytes (4) and to suppress macrophage activation induced by IFN-γ (31). In addition, it has been reported that IFNAR1 KO mice are more resistant to L. monocytogenes infection than wild-type mice (2, 24), and the susceptibility to L. monocytogenes infection was enhanced by poly(I·C)-induced type I IFNs (24). These findings raised the possibility that L. monocytogenes strains capable of inducing a high level of IFN-β, such as LO28, are more virulent than L. monocytogenes strains that have a low ability to induce IFN-β production. To test the hypothesis, we compared the pathogenicities of L. monocytogenes strains that were categorized as high (LO28) and low (EGD, LO28 ΔmdrT, and EGD ΔmdrT) IFN-β inducers. Similar to the results of previous studies by others, we found that IFN-β affects host resistance to L. monocytogenes, because IFNAR1 KO mice showed higher resistance than wild-type mice. As the higher resistance of IFNAR1 KO mice was also observed in infection with LO28 ΔmdrT, it appears that the IFNAR1-mediated detrimental effect on host resistance is elicited sufficiently by a low dose of IFN-β and that an excessive amount of this cytokine may not further contribute to the adverse outcome at all. In addition, our data clearly showed that LO28 is less virulent than LO28 ΔmdrT, suggesting that the pathogenicity of L. monocytogenes may be inversely related to the IFN-β-inducing ability. This finding may be rather controversial, because Reutterer et al. have shown that LO28 is more virulent than EGD due to the high level of IFN-β-inducing ability (32). Similar to our results, however, Crimmins et al. have reported that compared to parent L. monocytogenes, a recombinant L. monocytogenes strain expressing a high level of mdrT exhibits low pathogenicity, because significantly fewer recombinant L. monocytogenes bacteria were recovered from livers of infected mice (6). Moreover, cyclic dinucleotides have been shown to be a type of danger signal and to exert adjuvant activity (5). Therefore, it could be possible that the cyclic di-AMP may promote resistance independently of IFN-β production. Furthermore, MdrT, which serves as a transporter, may contribute to secrete some L. monocytogenes molecules other than cyclic di-AMP which are implicated in the control of L. monocytogenes replication in vivo. It is not be easy to discern the reason for the difference in the results, which were obtained by similar experimentation. Further analysis of MdrT-dependent action is necessary to determine the difference in pathogenicity between the strains.
MdrT, MdrM, and MdrL are members of the MFS, which is known as one of the largest families of transporters, and contribute to drug resistance in bacteria (11). It has been reported that MdrL is involved in the resistance of LO28 to cefotaxime and macrolides and that reserpine, an inhibitor of a wide range of efflux pumps, decreases the MIC of benzalkonium chloride (22, 33). To determine whether MdrT contributes to drug resistance of LO28, we measured the sensitivities of LO28 and the mdrT-deficient mutant to cefmenoxime, erythromycin, gentamicin, benzylpenicillin, oxacillin, tetracycline, chloramphenicol, imipenem, sulfamethoxazole/trimethoprim, and ofloxacin. There was no significant difference in drug susceptibility between these strains, suggesting that MdrT is not involved in the efflux of antibiotics in LO28 (data not shown). On the other hand, accumulating evidence indicates that MDR transporters contribute to the virulence of Gram-negative bacteria (28). For instance, Hirakata et al. have suggested that MDR efflux pumps of Pseudomonas aeruginosa export some unidentified virulence determinants, thereby increasing invasiveness into epithelial cells and lethality to mice (16). They also confirmed the role of MDR pumps in bacterial invasiveness using specific inhibitors (15). In light of the emergence of MDR organisms, MDR efflux pumps can be a novel target in the treatment of infection; however, more attention should be given to the possibility of any adverse effect of such an approach, considering the present finding that inactivation of MdrT resulted in a higher burden of bacteria as shown in our in vivo study.
L. monocytogenes is a well-characterized pathogen and has been employed as one of the most useful tools for analyzing the mechanisms of innate and acquired immunity. In addition, investigations on the strain-specific properties of bacteria often provide novel insights into pathogenesis, host-pathogen interactions, and protective immunity. In this study, we demonstrated that a deletion in the tetR gene causes a drastic change in the ability of L. monocytogenes to induce a host innate immune response, with special reference to IFN-β. The detailed mechanism for the induction of IFN-β by cyclic di-AMP remains to be determined in a future study.
ACKNOWLEDGMENTS
We thank Tomoya Takemura and Yasutaka Matsushima, Kyoto University, for assistance with LC-MS analysis and Naomasa Gotoh and Shu Minagawa, Kyoto Pharmaceutical University, for their valuable comments.
This study was supported by a grant-in-aid for scientific research on priority areas from the Ministry of Education, Science, Culture and Sports of Japan, grants-in-aid for scientific research (B and C) and a grant-in-aid for young scientists (B) from the Japan Society for the Promotion of Science, and a grant-in-aid for research on emerging and reemerging infectious diseases from the Ministry of Health, Labor and Welfare of Japan.
Footnotes
Published ahead of print 16 April 2012
REFERENCES
- 1. Akira S, Takeda K. 2004. Toll-like receptor signalling. Nat. Rev. Immunol. 4:499–511 [DOI] [PubMed] [Google Scholar]
- 2. Auerbuch V, Brockstedt DG, Meyer-Morse N, O'Riordan M, Portnoy DA. 2004. Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes. J. Exp. Med. 200:527–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bortolussi R. 2008. Listeriosis: a primer. Can. Med. Assoc. J. 179:795–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Carrero JA, Calderon B, Unanue ER. 2004. Type I interferon sensitizes lymphocytes to apoptosis and reduces resistance to Listeria infection. J. Exp. Med. 200:535–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen W, Kuolee R, Yan H. 2010. The potential of 3′, 5′-cyclic diguanylic acid (c-di-GMP) as an effective vaccine adjuvant. Vaccine 28:3080–3085 [DOI] [PubMed] [Google Scholar]
- 6. Crimmins GT, et al. 2008. Listeria monocytogenes multidrug resistance transporters activate a cytosolic surveillance pathway of innate immunity. Proc. Natl. Acad. Sci. U. S. A. 105:10191–10196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Decker T, Müller M, Stockinger S. 2005. The yin and yang of type I interferon activity in bacterial infection. Nat. Rev. Immunol. 5:675–687 [DOI] [PubMed] [Google Scholar]
- 8. Doganay M. 2003. Listeriosis: clinical presentation. FEMS Immunol. Med. Microbiol. 35:173–175 [DOI] [PubMed] [Google Scholar]
- 9. Flannagan RS, Cosío G, Grinstein S. 2009. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat. Rev. Microbiol. 7:355–366 [DOI] [PubMed] [Google Scholar]
- 10. Flo TH, et al. 2000. Human Toll-like receptor 2 mediates monocyte activation by Listeria monocytogenes, but not by group B streptococci or lipopolysaccharide. J. Immunol. 164:2064–2069 [DOI] [PubMed] [Google Scholar]
- 11. Fluman N, Bibi E. 2009. Bacterial multidrug transport through the lens of the major facilitator superfamily. Biochim. Biophys. Acta 1794:738–747 [DOI] [PubMed] [Google Scholar]
- 12. Gomelsky M. 2011. cAMP, c-di-GMP, c-di-AMP and now cGMP: bacteria use them all! Mol. Microbiol. 79:562–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hara H, et al. 2007. Cytolysin-dependent escape of the bacterium from the phagosome is required but not sufficient for induction of the Th1 immune response against Listeria monocytogenes infection: distinct role of listeriolysin O determined by cytolysin gene replacement. Infect. Immun. 75:3791–3801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hara H, et al. 2008. Dependency of caspase-1 activation induced in macrophages by Listeria monocytogenes on cytolysin, listeriolysin O, after evasion from phagosome into the cytoplasm. J. Immunol. 180:7859–7868 [DOI] [PubMed] [Google Scholar]
- 15. Hirakata Y, et al. 2009. Efflux pump inhibitors reduce the invasiveness of Pseudomonas aeruginosa. Int. J. Antimicrob. Agents 34:343–346 [DOI] [PubMed] [Google Scholar]
- 16. Hirakata Y, et al. 2002. Multidrug efflux systems play an important role in the invasiveness of Pseudomonas aeruginosa. J. Exp. Med. 196:109–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ishii KJ, et al. 2006. A Toll-like receptor-independent antiviral response induced by double-stranded B-form DNA. Nat. Immunol. 7:40–48 [DOI] [PubMed] [Google Scholar]
- 18. Ishikawa H, Barber GN. 2008. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455:674–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kanneganti TD, Lamkanfi M, Núñez G. 2007. Intracellular NOD-like receptors in host defense and disease. Immunity 27:549–559 [DOI] [PubMed] [Google Scholar]
- 20. Kawai T, et al. 2005. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 6:981–988 [DOI] [PubMed] [Google Scholar]
- 21. Kim YG, et al. 2008. The cytosolic sensors Nod1 and Nod2 are critical for bacterial recognition and host defense after exposure to Toll-like receptor ligands. Immunity 28:246–257 [DOI] [PubMed] [Google Scholar]
- 22. Mata MT, Baquero F, Pérez-Díaz JC. 2000. A multidrug efflux transporter in Listeria monocytogenes. FEMS Microbiol. Lett. 187:185–188 [DOI] [PubMed] [Google Scholar]
- 23. McWhirter SM, et al. 2009. A host type I interferon response is induced by cytosolic sensing of the bacterial second messenger cyclic-di-GMP. J. Exp. Med. 206:1899–1911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. O'Connell RM, et al. 2004. Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J. Exp. Med. 200:437–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. O'Connell RM, et al. 2005. Immune activation of type I IFNs by Listeria monocytogenes occurs independently of TLR4, TLR2, and receptor interacting protein 2 but involves TNFR-associated NF kappa B kinase-binding kinase 1. J. Immunol. 174:1602–1607 [DOI] [PubMed] [Google Scholar]
- 26. O'Riordan M, Yi CH, Gonzales R, Lee KD, Portnoy DA. 2002. Innate recognition of bacteria by a macrophage cytosolic surveillance pathway. Proc. Natl. Acad. Sci. U. S. A. 99:13861–13866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Park JM, Ng VH, Maeda S, Rest RF, Karin M. 2004. Anthrolysin O and other gram-positive cytolysins are Toll-like receptor 4 agonists. J. Exp. Med. 20:1647–1655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Piddock LJ. 2006. Multidrug-resistance efflux pumps—not just for resistance. Nat. Rev. Microbiol. 4:629–636 [DOI] [PubMed] [Google Scholar]
- 29. Portnoy DA, Auerbuch V, Glomski IJ. 2002. The cell biology of Listeria monocytogenes infection: the intersection of bacterial pathogenesis and cell-mediated immunity. J. Cell Biol. 158:409–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Portnoy DA, Jacks PS, Hinrichs DJ. 1988. Role of hemolysin for the intracellular growth of Listeria monocytogenes. J. Exp. Med. 1:1459–1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rayamajhi M, Humann J, Penheiter K, Andreasen K, Lenz LL. 2010. Induction of IFN-alphabeta enables Listeria monocytogenes to suppress macrophage activation by IFN-gamma. J. Exp. Med. 207:327–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Reutterer B, et al. 2008. Type I IFN are host modulators of strain-specific Listeria monocytogenes virulence. Cell. Microbiol. 10:1116–1129 [DOI] [PubMed] [Google Scholar]
- 33. Romanova NA, Wolffs PF, Brovko LY, Griffiths MW. 2006. Role of efflux pumps in adaptation and resistance of Listeria monocytogenes to benzalkonium chloride. Appl. Environ. Microbiol. 72:3298–3503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ryjenkov DA, Tarutina M, Moskvin OV, Gomelsky M. 2005. Cyclic diguanylate is a ubiquitous signaling molecule in bacteria: insights into biochemistry of the GGDEF protein domain. J. Bacteriol. 187:1792–1798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sauer JD, et al. 2011. The N-ethyl-N-nitrosourea-induced Goldenticket mouse mutant reveals an essential function of Sting in the in vivo interferon response to Listeria monocytogenes and cyclic dinucleotides. Infect. Immun. 79:688–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sauer JD, et al. 2010. Listeria monocytogenes triggers AIM2-mediated pyroptosis upon infrequent bacteriolysis in the macrophage cytosol. Cell Host Microbe 7:412–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schlee M, et al. 2009. Approaching the RNA ligand for RIG-I? Immunol. Rev. 227:66–74 [DOI] [PubMed] [Google Scholar]
- 38. Seki E, et al. 2002. Critical roles of myeloid differentiation factor 88-dependent proinflammatory cytokine release in early phase clearance of Listeria monocytogenes in mice. J. Immunol. 169:3863–3868 [DOI] [PubMed] [Google Scholar]
- 39. Servant MJ, et al. 2001. Identification of distinct signaling pathways leading to the phosphorylation of interferon regulatory factor 3. J. Biol. Chem. 276:355–363 [DOI] [PubMed] [Google Scholar]
- 40. Seveau S, Pizarro-Cerda J, Cossart P. 2007. Molecular mechanisms exploited by Listeria monocytogenes during host cell invasion. Microbes Infect. 9:1167–1175 [DOI] [PubMed] [Google Scholar]
- 41. Siegman-Igra Y, et al. 2002. Listeria monocytogenes infection in Israel and review of cases worldwide. Emerg. Infect. Dis. 8:305–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Soulat D, Bauch A, Stockinger S, Superti-Furga G, Decker T. 2006. Cytoplasmic Listeria monocytogenes stimulates IFN-beta synthesis without requiring the adapter protein MAVS. FEBS Lett. 580:2341–2346 [DOI] [PubMed] [Google Scholar]
- 43. Southwick FS, Purich DL. 1996. Intracellular pathogenesis of listeriosis. N. Engl. J. Med. 334:770–776 [DOI] [PubMed] [Google Scholar]
- 44. Stetson DB, Medzhitov R. 2006. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity 24:93–103 [DOI] [PubMed] [Google Scholar]
- 45. Stockinger S, Decker T. 2008. Novel functions of type I interferons revealed by infection studies with Listeria monocytogenes. Immunobiology 213:889–897 [DOI] [PubMed] [Google Scholar]
- 46. Stockinger S, et al. 2002. Production of type I IFN sensitizes macrophages to cell death induced by Listeria monocytogenes. J. Immunol. 169:6522–6529 [DOI] [PubMed] [Google Scholar]
- 47. Stockinger S, et al. 2004. IFN regulatory factor 3-dependent induction of type I IFNs by intracellular bacteria is mediated by a TLR- and Nod2-independent mechanism. J. Immunol. 173:7416–7425 [DOI] [PubMed] [Google Scholar]
- 48. Sun Q, et al. 2006. The specific and essential role of MAVS in antiviral innate immune responses. Immunity 24:633–642 [DOI] [PubMed] [Google Scholar]
- 49. Takaoka A, Yanai H. 2006. Interferon signalling network in innate defence. Cell. Microbiol. 8:907–922 [DOI] [PubMed] [Google Scholar]
- 50. Torres D, et al. 2004. Toll-like receptor 2 is required for optimal control of Listeria monocytogenes infection. Infect. Immun. 72:2131–2139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tsuchiya K, et al. 2010. Involvement of absent in melanoma 2 in inflammasome activation in macrophages infected with Listeria monocytogenes. J. Immunol. 185:1186–1195 [DOI] [PubMed] [Google Scholar]
- 52. Warren SE, et al. 2010. Cytosolic bacterial DNA activates the inflammasome via Aim2. J. Immunol. 185:818–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Woodward JJ, Iavarone AT, Portnoy DA. 2010. c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science 328:1703–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang B, et al. 2009. The TAK1-JNK cascade is required for IRF3 function in the innate immune response. Cell Res. 19:412–428 [DOI] [PubMed] [Google Scholar]