

Abstract
The capsule of Neisseria meningitidis is the major virulence factor that enables this bacterium to overcome host immunity elicited by complement and phagocytes, rendering it capable of surviving in blood. As such, nonencapsulated N. meningitidis isolates are generally considered nonpathogenic. Here, we consider the inherent virulence of two nonencapsulated N. meningitidis isolates obtained from our national surveillance of infected blood cultures in Canada. Capsule deficiency of both strains was confirmed by serology and PCR for the ctrA to ctrD genes and siaA to siaC genes, as well as siaD genes specific to serogroups B, C, Y, and W135. In both strains, the capsule synthesis genes were replaced by the capsule null locus, cnl-2. In accordance with a lack of capsule, both strains were fully susceptible to killing by both human and baby rabbit complement. However, in the presence of cytidine-5′ monophospho-N-acetylneuraminic acid (CMP-NANA), allowing for lipooligosaccharide (LOS) sialylation, a significant increase of resistance to complement killing was observed. Mass spectrometry of purified LOS did not reveal any uncommon modifications that would explain their invasive phenotype. Finally, in a mouse intraperitoneal challenge model, these nonencapsulated isolates displayed enhanced virulence relative to an isogenic mutant of serogroup B strain MC58 lacking capsule (MC58ΔsiaD). Virulence of all nonencapsulated isolates tested was below that of encapsulated serogroup B strains MC58 and B16B6. However, whereas no mortality was observed with MC58ΔsiaD, 5/10 mice succumbed to infection with strain 2275 and 2/11 mice succumbed to strain 2274. Our results suggest the acquisition of a new virulence phenotype by these nonencapsulated strains.
INTRODUCTION
Neisseria meningitidis remains a common cause of invasive bacterial disease in humans of all age groups. One of the most important virulence factors of this bacterium is the polysaccharide capsule, and nearly all N. meningitidis isolates recovered from patients with invasive meningococcal disease (IMD) are encapsulated, with capsule serogroups A, B, C, Y, and W135 being the most frequent causes of disease. In contrast, meningococci obtained from healthy carriers are often nonserogroupable, due either to phase variation of capsule expression (13), inactivation of genes involved in capsule synthesis by genetic mobile elements, such as an insertion sequence (12), or absence of genes required for capsule production, leading to a capsule null phenotype (6).
The Neisseria meningitidis capsule synthesis genes are clustered together into 5 regions, denoted as A through E and arranged in the order of B-D-A-C-E-D′ or B-D′-E-C-A-D, with D′ a truncated duplicate copy of region D (35). Region A contains genes required for synthesis of the serogroup-specific polysaccharide. Region B contains genes encoding enzymes required for the synthesis of the lipid moiety of the capsule, which allows the polysaccharide to be anchored to the bacterial outer membrane. Region C contains genes involved in capsule transport and genes in region D are involved in lipooligosaccharide (LOS) synthesis, while the function of region E remains unknown. Certain N. meningitidis strains have lost genes for the transport and synthesis of the capsule polysaccharide, and the regions A and C are replaced by a stretch of about 110 bp of DNA termed the capsule null locus (cnl). Three cnl alleles encoding sequence variants have been identified in N. meningitidis, and additional alleles are found in the related but naturally nonencapsulated Neisseria lactamica and Neisseria gonorrhoeae (6).
Two independent carriage studies (6, 27) have found the prevalence of capsule null mutants to be ∼16% among nasopharyngeal meningococci in healthy children and young adults. Furthermore, most null mutants were found within a few genetic clones of sequence type 53 (ST-53), ST-1117, ST-845, ST-198, and ST-1136, as defined by multilocus sequence typing (MLST). While these are usually regarded as nonpathogenic commensal strains, there have been 3 reports of N. meningitidis strains with a cnl allele causing IMD. The first case occurred in 2001, with an infection developing in an immunocompromised 42-year-old adult suffering from a common acute lymphatic leukemia and established chronic graft-versus-host disease after peripheral blood stem cell transplantation without T-cell depletion (36). Shortly after initiation of systemic antibiotic treatment, the symptoms of sepsis resolved in this patient. The second documented case, in 2004, developed in a previously healthy 13-year-old girl, with the course of infection appearing typical of IMD in teenagers. Her initial flu-like symptoms quickly progressed to septic shock with widespread petechial rashes and significant disseminated intravascular coagulopathy that resulted in death about 5 h after initial hospitalization (14). The most recent report described 3 IMD cases with no epidemiological link but which were caused by a single strain with a cnl allele circulating among healthy carriers in Burkina Faso (8). Two of the cases happened in March and April 2003 in children aged 12 and 13 years, while the third case involved an 11-year-old girl in April 2004. All three patients had symptoms of meningitis (fever, headache, stiff neck, and turbid cerebrospinal fluid with pus cells) but without petechia or loss of consciousness, and all three patients responded to treatment with chloramphenicol.
Although IMD cases caused by cnl strains are still rarely encountered in our routine surveillance program in Canada, there have been two instances of IMD with cnl strains, in one patient who had fatal disease in British Columbia in 2004 (strain 2274) and a second patient in Ontario in 2006 (strain 2275). In this study, we have characterized these cnl strains and investigated their pathogenicity by using an in vivo mouse model of sepsis, with the objective to compare the intrinsic virulence of invasive cnl isolates relative to nonencapsulated mutants of encapsulated disease isolates.
MATERIALS AND METHODS
Bacterial strains.
Strains 2274 and 2275 were provided by the provincial Public Health Laboratories in British Columbia and Ontario, respectively, for phenotypic and genetic analysis at the National Microbiology Laboratory. N. meningitidis MC58 and B16B6 were obtained from ATCC (Manassas, VA). MC58ΔsiaD was obtained by insertion of a kanamycin cassette into the siaD gene of B16B6 and transformation into MC58 (the siaD construct was generously provided by Rolando Pajon and Anthony B. Schryvers, University of Calgary). Other meningococci used in this study were from the Public Health Agency of Canada's culture collection.
Serogrouping, serotyping, and serosubtyping.
Serogroup determination was done by standard bacterial agglutination test (22) using rabbit antisera against all 13 serogroups (A, B, C, D, H, I, K, L, W135, X, Y, Z, and 29E) (2). Detection of serogroups A, B, C, W135, X, Y, Z. and 29E by PCR was accomplished using primers and PCR conditions described by Borrow and colleagues (in 1997 and 1998), Taha (in 2000), and Bennett et al. (in 2004) (3–5, 32). The serotype and serosubtype antigens were determined by indirect whole-cell enzyme-linked immunosorbent assay (1) and using a monoclonal antibody typing kit (Rijksinstituut voor Volksgezondhei en Milieu, National Institute of Public Health, The Netherlands).
Genetic analysis and MLST.
Sequencing of the porB and porA genes, which encode the serotype and serosubtype antigens, respectively, was performed following the protocol and nomenclature of Sacchi et al. (25, 26). Detection of the capsule null locus and determination of the cnl allele by PCR amplification and sequencing were performed according to methods described by Claus et al. (6). MLST was done according to the method described by Maiden et al. (22), and sequence types (STs) were assigned according to information presented on the Neisseria MLST website (http://pubmlst.org/neisseria).
In vitro susceptibility of meningococci to complement.
Baby rabbit serum was obtained from Pel Freeze, Inc. (Brown Deer, WI) and aliquoted into small volumes sufficient for each assay to prevent repeated freeze-thawing of the complement source. Human serum was obtained by venipuncture of healthy volunteers by using a BD vacutainer and quickly processing the samples on ice after allowing them to coagulate. Coagulated blood samples were spun down at 10,000 × g for 10 min, and serum was recovered, aliquoted, and immediately frozen at −80°C. The in vitro testing of susceptibility of meningococci to complement was based on modifications of the method described by Vogel et al. (36). Briefly, bacteria were grown in the presence or absence of 20 μM CMP-N-acetylneuraminic acid (CMP-NANA), as indicated, and approximately 105 CFU were mixed with various concentrations (0%, 5%, 10%, 20%, or 40%) of baby rabbit serum or of human serum in a final volume of 50 μl of minimum essential medium (Invitrogen Corp., Burlington, ON, Canada). After incubation at 37°C for 30 min, serial dilutions were made, and 15-μl aliquots were removed from each dilution for plating on agar plates (GC agar supplemented with IsoVitaleX [Becton Dickinson]). After incubation (18 to 24 h) at 37°C with 5% CO2, the numbers of CFU on the agar plates were counted and converted to represent the number of bacteria present in the serum-bacteria mixture.
Mass spectrometric analysis of LOS structures.
N. meningitidis strains were grown overnight on GC agar plates, and the lawn of growth was resuspended in 1 ml of phosphate-buffered saline (PBS). The samples were heat inactivated for 30 min at 65°C, washed thrice with PBS, pelleted at 10,000 × g in Eppendorf tubes, and lypophilized.
Lyophilysates were dissolved in 180 μl of H2O and 20 μl of proteinase K (0.25 mg/ml) and incubated for 4 h at 37°C; the samples were heated to 65°C for 10 min and lyophilized. The samples were then dissolved in 180 μl of buffer (20 mM ammonium acetate, pH 7.4) and 10 μl each of RNase (0.1 mg/ml of 20 mM ammonium acetate) and DNase (0.2 mg/ml of 20 mM ammonium acetate), incubated for 4 h at 37°C, and then lyophilized. Samples were then dissolved in 200 μl of anhydrous hydrazine and incubated for 1.5 h at 37°C under N2. Excess hydrazine was destroyed by treating the ice-chilled reaction mixtures with 5 volumes of “dry ice-cold” acetone, i.e., the acetone was cooled in a dry ice bath prior to addition to the hydrazine reaction mixture and pelleting the O-deacylated lipopolysaccharide (LPS-OH) at 8,000 × g for 15 min. The LPS-OH was washed twice with acetone and dissolved in H2O and lyophilized. Capillary electrophoresis-electrospray-mass spectrometry (CE-ES-MS) experiments were performed as described previously (30).
Mouse infection studies.
Animal experiment procedures were approved by the Animal Ethics Review Committee of the University of Toronto. Six-week-old male C57BL/6 mice were purchased from Jackson Laboratories. Mouse intraperitoneal infection studies were performed as described by Gorringe et al. (9). Briefly, the N. meningitidis strains were grown overnight at 37°C with 5% CO2 on chocolate agar plates, the lawn of growth was harvested and transferred to Mueller-Hinton broth supplemented with 60 μg/ml of the iron chelator deferoxamine mesylate and agitated for 4 h at 37°C to induce expression of virulence factors. The neat suspension was adjusted with Mueller-Hinton broth to the target density, and 1 volume of suspension was admixed with 1 volume of 40 mg/ml of human holo-transferrin (Sigma) as a source of iron for the meningococci. A total of 107 or 108 viable CFU in a final volume of 500 μl per animal was injected intraperitoneally. Viable CFU counts in the inoculum were monitored by plating serial dilutions onto GC agar plates and counting colonies grown after overnight incubation at 37°C, 5% CO2. Mice were weighed and monitored for signs of sickness every 6 h to 12 h from 36 h prior to infection until 48 h after infection. The following scheme was applied to assign clinical scores (with higher scores reflecting aggravated symptoms): loss of body weight (5 to 10%, score of 1; 10 to 15%, 2; 15 to 20%, 3); grooming (0 to 2); posture (0 to 2); appearance of eyes and nose (0 to 2); breathing rate (0 to 2); unprovoked behavior (0 to 2); provoked behavior (0 to 2); dehydration (0 to 2); diarrhea (0 to 2).
Protein expression analysis.
N. meningitidis strains were grown overnight on GC agar supplemented with IsoVitaleX (Becton Dickinson), and the lawn of growth was swabbed and resuspended in phosphate-buffered saline supplemented with 1 mM MgCl2. The suspension was adjusted to 2 × 1010 CFU/ml, and 50 μl of suspension was mixed with 50 μl of 2× SDS loading buffer and boiled for 10 min. Next, 12% SDS-polyacrylamide gels were loaded with 20 μl of sample per lane for silver staining of whole protein or for Western blot analysis of Opa proteins, or with 5 μl of sample per lane for Western blot analysis of pilin. After electrophoresis, either the silver staining procedure was performed using the SilverSnap kit II (Thermo Scientific) or protein was blotted onto a nitrocellulose membrane and probed for Opa proteins using MAb 4B12/C11 or, for pilin protein, MAb 5H10.1.1.
opa gene typing.
For fingerprinting of opa genes, the method described by Khaki et al. was used (18). Briefly, PCR was performed with primers that bind to all the different Opa alleles (forward primer, ATGTGCAGGCGGATTTAGCT; reverse primer, AGACTTCGTGGGTTTTG) and with genomic DNA of the N. meningitidis strains as template. The reaction mixture contained the following: 3 μl 10× ThermoPol buffer, 2 mM each deoxynucleoside triphosphate, 200 μM forward and reverse primer, 2 U ThermoPol Taq DNA polymerase (all reagents from New England BioLabs) in a total volume of 30 μl. The cycler settings were initial denaturing at 94°C for 10 min and then 30 cycles of 1 min denaturing at 94°C, 1 min annealing at 64°C, and 1 min elongation at 72°C. The PCR products (∼670 bp) were sodium acetate precipitated, and pellets were resuspended in distilled water and subsequently subjected to restriction enzyme digestion by using 30 U/μg DNA of HpaI (New England BioLabs). Approximately 500 ng of DNA was loaded per lane on a 12% polyacrylamide gel and electrophoretically separated. DNA bands were visualized by ethidium bromide staining of the gel.
RESULTS
Phenotypic and genetic characterization.
The first cnl strain isolated from an IMD patient in Canada (strain 2274) was previously characterized and described in detail (14). Briefly, the strain was recovered in 2004 in British Columbia, Canada, from cerebrospinal fluid of an immunocompetent 13-year-old girl before she succumbed to the infection. The isolate was found to be nonencapsulated and to harbor the cnl-2 allele. Analysis of 16S rRNA as well as porA gene analysis confirmed that the isolate was N. meningitidis.
The second cnl strain (strain 2275) has not been previously described. It was isolated from blood cultures of a 13-year-old male in Ontario, Canada, in 2006. The isolate was identified by standard biochemical tests as N. meningitidis. As with strain 2274, however, this isolate failed to agglutinate the serogrouping antisera. Genogrouping by PCR also failed to identify the serogroup, and PCR to detect the ctrA gene was also negative. Instead, the cnl-2 allele could be identified in this strain.
Serotyping and serosubtyping as well as PorB and PorA VR typing results for both strains are shown in Table 1 (applying the nomenclature of Sacchi et al. [25, 26]). Strain 2274 was identified as NG:15:P1.− (with NG indicating nongroupable). Despite not being serosubtypeable, PorA VR types could be assigned for this strain. Strain 2275 was identified as NG:15:P1.6. Note that the P1.6 epitope, which is not listed under the Neisseria.org or PubMLST.org databases because it is not located in VR1 or VR2, is actually located in VR3 (PorA surface-exposed loop V). Multilocus sequence typing identified both strains as ST-198. Additionally, the fetA genes of both isolates were sequenced, and both were determined to be type F5-5.
Table 1.
Phenotypic and genetic characterization of Neisseria meningitidis capsule null locus (cnl) strains recovered from invasive meningococcal disease patients in Canada
Considering that the strains possess similar serological typing patterns and fetA sequences, it remains possible that the two cnl-2 strains may have recently diverged by simple horizontal acquisition of one of the porA alleles. To further compare the two strains, we sought to determine differences in their protein expression profiles. As shown in Fig. 1A, a one-dimensional protein gel of whole-cell lysates visualized by silver staining revealed several differences between strains 2274 and 2275, and even moreso between these two strains and MC58. Moreover, we observed what appeared to be different Opa protein variants expressed by strains 2274 and 2275, and these two strains also displayed obvious differences with respect to their pilin (Fig. 1A). Since phase-variable expression of antigens by N. meningitidis can alter protein profiles through the random on-and-off switching of about 47 different proteins during cell division (24), we assessed differences among the two nonencapsulated isolates by opa gene typing (Fig. 1B). Opa proteins are N. meningitidis adhesins that can be considered fast evolutionary clocks, since each of the four opa genes are highly variable due to frequent recombination within and between strains. Indeed, the opa fingerprint indicates sequence differences in at least one of their opa genes. Given that 2274 and 2275 have different porA genes, this indicates that at least two genes diverged between the two nonencapsulated isolates.
Fig 1.
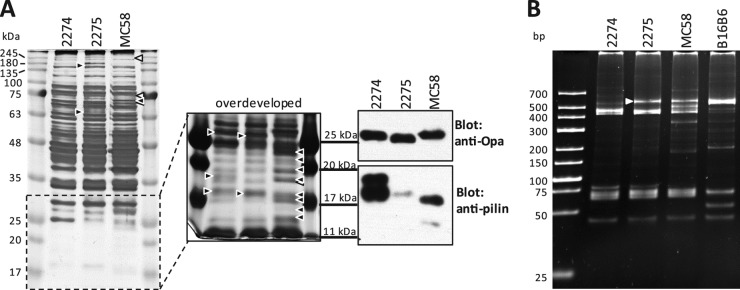
Phenotypic characterization of nonencapsulated N. meningitidis strains. (A) Whole-cell lysates of strains 2274, 2275, and MC58 were analyzed on a 12% polyacrylamide gel and visualized by silver staining (left). The indicated low-molecular-mass portion of the gel after prolonged silver stain development is shown to the right. Differences in band intensities or band presence are indicated by arrowheads. Black arrowheads indicate the presence of a protein band missing or expressed at a significantly different level or with a different molecular mass than in the other strain(s). White arrowheads indicate protein bands missing or significantly fainter in MC58 than in 2274 and 2275. Immunoblots for Opa and pilin are shown on the right (note that the molecular mass markers are aligned with those of the adjacent [overdeveloped] silver-stained gel.) (B) PCR-based opa typing of strains 2274, 2275, MC58, and B16B6. The white arrowhead indicates a band that was detected in strain 2275 but not 2274.
Mass spectrometric analysis of the LOS structure of nonencapsulated pathogenic N. meningitidis strains.
The O-deacylated lipooligosaccharide of the nonencapsulated N. meningitidis strains was analyzed by capillary electrophoresis-electrospray-mass spectrometry in order to assess any abnormalities in their carbohydrate composition or lipid A modifications that could account for the observed virulence of these strains. The results of the mass spectrometry analysis are summarized in Table 2. No obvious differences in LOS composition between the two cnl strains were observed. With an m/z of 952 as determined by MS-MS analysis, the O-deacylated lipid A portion does not harbor any modifications. The carbohydrate composition, as well, showed a stereotypical architecture of the core oligosaccharide, with one phosphonoethanolamine group positioned at HepII. Interestingly, there was no evidence for any sialic acid modifications of the LOS, an addition which is often found in pathogenic Neisseria as a means of molecular mimicry to counteract killing by the complement system (23, 37).
Table 2.
Mass spectrometric analysis of LOS from Neisseria meningitidis capsule null locus (cnl) strainsa
| Strain | Observed ions (m/z) |
Molecular mass (Da) |
Relative intensityb | Lipid A-OHc | Core OSd | Proposed composition | ||
|---|---|---|---|---|---|---|---|---|
| (M − 3H)3− | (M − 2H)2− | Observed | Calculated | |||||
| 2274 | 929.8 | 1,395.5 | 2,792.7 | 2,792.6 | 1.0 | 952 | 1,841 | 3Hex, 2HexNAc, PEtn, 2Hep, 2 Kdo, lipid A-OH |
| 2275 | 930.1 | 1,395.5 | 2,793.1 | 2,792.6 | 1.0 | 952 | 1,841 | 3Hex, 2HexNAc, PEtn, 2Hep, 2 Kdo, lipid A-OH |
Negative-ion CE-MS and CE–ES–MS-MS data and proposed compositions of O-deacylated LOS are shown. Average mass units (in Da) were used for calculation of molecular masses, based on the proposed composition, as follows: Hex, 162.15; Hep, 192.17; HexNAc, 203.19; Kdo, 220.18; PEtn, 123.05.
Relative to most intense peak in mass spectrum.
As determined by MS-MS analyses, following introduction of separated LPS-OH glycoforms into the mass spectrometer by CE.
As deduced from lipid A-OH size determinations.
Susceptibility to bactericidal complement present in human and baby rabbit serum.
Despite the absence of LOS modifications, we considered that the ability to establish systemic disease may be attributable to an intrinsic resistance to the bactericidal activity of serum complement. To test this hypothesis, both cnl strains as well as encapsulated meningococcal control strains were exposed to various concentrations (5%, 10%, 20%, and 40%) of normal rabbit baby serum or human serum and then tested for survival. Encapsulated meningococci belonging to serogroup B (strain MC58; H44/76), serogroups Y (strain 860060) and X (strain 860800), maintained viability after 30 min of exposure to medium containing up to 40% serum from either origin, human or rabbit. No bacteria were recovered when strains 2275 and 2274 were incubated with 20% human serum. In rabbit serum, strain 2275 was effectively killed by as little as 10% rabbit serum, while strain 2274 required 20% serum for complete killing (Fig. 2). Therefore, both cnl strains were susceptible to both human and rabbit complement, consistent with their lack of the serum protection that would be afforded by capsular polysaccharide. It is interesting that the capsule-deficient mutant strain MC58ΔsiaD was also susceptible to killing by both human and rabbit serum, yet it required significantly higher concentrations of both human and rabbit serum for complete killing.
Fig 2.
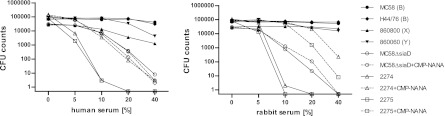
Relative susceptibility of N. meningitidis isolates to killing with human versus rabbit serum as the source of complement. A total of ∼105 CFU of each strain was exposed to dilutions of serum for 30 min at 37°C in minimal essential medium, and then serial dilutions were plated onto GC agar plates to assess viable CFU counts. Nonencapsulated strains 2274 and 2275 as well as the capsule-deficient strain MC58ΔsiaD, grown in the presence or absence of 20 μM CMP-NANA, were compared to encapsulated clinical isolates MC58 (serogroup B), H44/76 (serogroup B), 86800 (serogroup X), and 860060 (serogroup Y).
In considering that the cnl strains were isolated from invasive infections, their susceptibility to complement-mediated killing seems counterintuitive. Indeed, these findings were in contrast with a previous report that indicated that N. meningitidis cnl isolates from Burkina Faso were naturally resistant to human complement-mediated killing (8). Since some neisserial strains can sialylate their LOS when provided with an exogenous source of sialic acid, we exposed the nonencapsulated strains to CMP-NANA, a substrate for this activity. This condition allowed a substantial increase in the resistance of 2274 and 2275 to complement-mediated killing, making them comparable to that of MC58ΔsiaD, which in turn did not display any changes through growth in the presence of CMP-NANA. In rabbit serum, the cnl strains 2274 and 2275 showed an even more marked increase upon exposure to CMP-NANA, each becoming substantially more resistant than MC58ΔsiaD. When combined, these in vitro serum killing assays suggest that exogenous LOS sialylation of strains 2274 and 2275 bestows them with an increased resistance to complement-dependent killing.
Virulence of cnl strains upon intraperitoneal infection of mice.
The human-restricted tropism of N. meningitidis is an inherent obstacle to a deeper understanding of its virulence mechanisms in vivo. However, intraperitoneal infection of rodents still represents the best model available by which to assess the potential for strains to cause invasive meningococcemia (9); while these models do not consider highly specialized adaptation to life in humans, they do allow direct comparisons between the baseline virulence potentials of pathogenic and nonpathogenic meningococcal strains. With this intent, we sought to compare the relative ability of the cnl strains 2274 and 2275 versus the wild type and a nonencapsulated siaD mutant of serogroup B N. meningitidis in the mouse intraperitoneal infection model. As expected, rapid onset of severe clinical symptoms and high mortality were observed in mice infected with the encapsulated, highly virulent serogroup B control strains MC58 and B16B6 (Fig. 3A, right panels). Upon infection with 107 CFU, seven out of eight mice (87.5%) succumbed to infection with B16B6 and six out of eight mice (75%) succumbed to infection with MC58.
Fig 3.

The mouse intraperitoneal infection model of invasive meningococcal disease. (A) Mice were infected with 107 CFU of encapsulated N. meningitidis strain MC58 or B16B6 (right panels) or with 108 or 107 CFU of N. meningitidis MC58ΔsiaD (capsule-deficient mutant) or the nonencapsulated clinical isolates strain 2274 and strain 2275 (center and left). From 36 h prior to infection until 48 h after infection, mice were monitored every 12 h (and also at 18 h postinfection) for survival (upper panels), loss of body weight (middle panel), and clinical scores, assigned to reflect morbidity (lower panels). Statistic analyses were performed using GraphPad Prism 5. The Mantel-Cox log rank test was used for survival analysis (*, P < 0.05). A one-way analysis of variance applying Bonferroni's post hoc test was used for statistical analysis of body weight (*, P < 0.05, **, P < 0.01). The Kruskal-Wallis test with application of Dunn's post hoc test was used to analyze clinical scoring data (*, P < 0.05). For body weight and clinical score data, only the lowest significance levels of differences between strain 2275 and the other two groups are indicated. na, no statistics applicable because only one (B16B6) or two (MC58) animals survived. (B) Assessment of bacteremia in intraperitoneally infected mice. Tail vein blood samples were drawn at the indicated times after infection of mice and plated to assess viable bacteria in the peripheral blood. Depicted is the percentage of mice testing culture positive for N. meningitidis. Numbers on top of columns indicate the numbers of culture-positive animals among the total number of tested animals.
Upon infection with 107 CFU of the nonencapsulated meningococcal strains, the mice displayed obvious clinical symptoms, including a clear drop in body weight (∼5% by 24 h), but the mice tended to recover within 2 days of infection (Fig. 3A, center panels). In order to understand whether the nonencapsulated strains had the potential to cause lethal infection in this model, mice were inoculated with a 10-fold-higher dose of bacteria. These mice showed a pronounced loss of body weight, averaging about 10% by 24 h, and this symptom was consistently longer for mice infected with strain 2275 than strain 2274 or MC58ΔsiaD at 108 CFU. Reflecting the changes in body weight, the clinical scores, used as measures of morbidity, were significantly higher in mice infected with 108 CFU of strain 2275 relative to strain 2274 or MC58ΔsiaD (Fig. 3A, left panels). The effect of strain 2274 on the mouse clinical scores was not different than that caused by MC58ΔsiaD.
While the progression of clinical symptoms of the infection caused by the cnl locus and MC58ΔsiaD tended to follow a similar trend, particularly when comparing the capsule-deficient serogroup B mutant and strain 2274, the end points of infection clearly differed in that both cnl strains caused mortality whereas the MC58ΔsiaD strain did not. While no mortality was seen upon infection with MC58ΔsiaD, 2 of 11 mice (18%) succumbed to infection with strain 2274, while 5 out of 10 (50%) mice died after infection with strain 2275 (Fig. 3A, upper left panel). It is, however, pertinent that while the mortality caused by strain 2275 reached statistical significance when using the Mantel-Cox test to compare it to MC58ΔsiaD, the effects of strain 2274 did not.
In order to correlate disease progression with the kinetics of bacterial sepsis, blood was sampled and cultured to detect invasive meningococci during the course of an infection. As expected, most mice infected with encapsulated strain MC58 or B16B6 displayed pronounced bacteremia until the end of the experiment (or death of the mice). However, bacteremia was also apparent with all three nonencapsulated strains. Whereas most mice (3/4 tested) cleared MC58ΔsiaD within 12 h from their bloodstream, a large proportion of mice infected with strain 2274 (3/7) or strain 2275 (4/6) remained bacteremic until that time point, and several remained bacteremic at the end of the experiment (Fig. 3B).
In summary, while encapsulated N. meningitidis strains are clearly more virulent than any of the nonencapsulated strains tested, the cnl strain 2275 showed higher virulence in the mouse intraperitoneal challenge model than the capsule-deficient mutant of an otherwise-encapsulated strain. While the effects of cnl strain 2274 were not statistically different from those caused by MC58ΔsiaD, cnl 2274 did persist in the bloodstream and caused two mice to succumb. Combined, these results suggest that the cnl strains have a higher intrinsic virulence in this model.
DISCUSSION
The two IMD cases caused by cnl strains in Canada occurred in two separate parts of the country, with one case in the most western province of British Columbia and the other case in eastern Canada, in the province of Ontario. Furthermore, these two cases were separated by about 2 years (spring seasons of 2004 and 2006). While both isolates have the cnl-2 allele, are ST-198, and fetA type F5-5, their protein expression profiles display variations, they have different porA VR types, and differ in at least one opa allele. Our observation that both strains can use an exogenous source of sialic acid to modify their surface in a manner that allows them to resist the bactericidal activity of serum may help to explain their abilities to cause invasive meningococcal disease in the mouse model and in humans.
A meningococcal carriage study in Germany (6) that examined nonencapsulated isolates from healthy children and young adults found that cnl-2 and cnl-1 were the two common allele types in isolates that carried the null locus and that these occurred at almost equal frequencies (75 out of 136 [55%] nonencapsulated isolates carried the cnl-1 allele, while 60 of 136 [44%] were cnl-2). Furthermore, 26 of the cnl-2 isolates were typed as ST-198, and an additional 18 isolates belonged to STs related to ST-198 (as either single or double allelic variants). In other words, of the 60 isolates with the cnl-2 allele, 73% belonged to the ST-198 clonal complex (6). The remaining 16 cnl-2 isolates belonged to 2 other clonal complexes. Therefore, finding two Canadian cnl-2 null mutants that both belong to ST-198 may reflect the fact that this is one of the most prevalent cnl-2-harboring N. meningitidis lineages carried, rather than reflecting the emergence of a new strain in Canada.
The fact that these cnl strains can cause fatal IMD in apparently healthy individuals prompted us to investigate their virulence properties. N. meningitidis is highly adapted to life in humans, and certain virulence factors that contribute to colonization cannot be accounted for in animal models of infection, due to their exquisite specificity for human proteins (10, 11, 16, 29). However, the mouse intraperitoneal challenge model has been widely used as a means to compare the overall virulence of meningococcal isolates (7, 15, 19, 34, 38) and the efficacy of novel vaccine candidates (20, 21, 31) Herein, we used this model to understand whether the Canadian cnl-2 strains were intrinsically more virulent than a nonencapsulated mutant of an encapsulated strain. These in vivo challenge studies revealed that at least one of the nonencapsulated cnl-2 isolates, 2275, can be considered more virulent than a capsule-deficient mutant of the well-characterized strain MC58. While the other cnl-2 strain, 2274, was able to cause some mortality in the mouse model, which was not observed with MC58ΔsiaD, this effect did not reach statistical significance. This effect must be considered in context, since encapsulated strains MC58 and B16B6 caused 75 to 87.5% mortality at a 10-fold-lower dose of infection. However, the heightened virulence of the cnl-2 strain 2275 relative to the genetically engineered capsule-deficient strain suggests that it has acquired some means by which to persist and cause disease in vivo, both in the mouse model and in the patients from which it was derived. Future functional genomic studies aimed at understanding the diversity of cnl strains will hopefully reveal whether specific attributes correlate with the invasive potential of certain isolates.
It is interesting that both Canadian isolates and the German IMD-derived nonencapsulated strain all belonged to ST-198 and had the cnl-2 allele, and that they were susceptible to complement present in normal serum from both rabbit and human. This is in contrast to the capsular polysaccharide locus (cps) null strain involved in 3 IMD cases in Burkina Faso in Africa where, in 2003 and 2004, apparently healthy children had systemic infections caused by isolates that were typed as ST-192, had the cnl-3 allele, and were resistant to normal human serum (8). It should be noted that we observed that the availability of CMP-NANA, which allows exogenous sialylation of LOS, conferred increased serum resistance for both Canadian cnl-2 isolates, whereas the serum sensitivity of the cnl-3 Burkina Faso isolates was not affected by exposure to CMP-NANA. The ability of both cnl strains 2274 and 2275 to sialylate their surface may explain the increased virulence of these strains in mice and, by extension, humans. However, since the virulence of N. meningitidis can be viewed as a multigenic trait, the relative contribution of this and other new or established virulence factors remains unclear.
While an important model in which to study meningococcal virulence, mouse infection studies lack some key components to mimic human disease in detail. N. meningitidis is a human-restricted pathogen that specifically interacts with human-derived forms of host proteins, such as transferrin (29), factor H (10), carcinoembryonic antigen-like cell adhesion molecules (11), and CD46 (16), allowing important functions, such as the acquisition of iron, evasion of complement killing, and adhesion to host cells in vivo. The increased virulence of the cnl strains tested herein cannot be attributed to these well-described virulence factors, implying that more broadly effective attributes, such as endogenous sialylation of surface antigens or other factors, must explain the outcome in this model. In this context, it is pertinent to consider that not all sequence types of encapsulated meningococci are associated with invasive disease. For example, ST-136 and ST-53 are found frequently in carriers but are almost never isolated from patients with invasive disease, whereas other STs, such as ST-32 and ST-11, are considered hyperinvasive for their potential to cause epidemic outbreaks of invasive meningococcal disease (http://pubmlst.org/neisseria) (17). Comparative genomics have already proven to be an effective means to unravel the differences in whole genomes from carriage and disease strains of Neisseria meningitidis (17, 28), and genome sequencing of a broad set of cnl strains may prove informative in gaining deeper insight into their genetic arsenal.
The capsule of N. meningitidis is generally appreciated as being the most important virulence factor for protection against host defense systems in the blood; however, certain strains of Neisseria gonorrhoeae have a propensity to cause disseminated disease despite the fact that this close relative to N. meningitidis does not express any capsular polysaccharide (33). As such, a strict view that a capsule is absolutely required for the invasive potential of a pathogenic Neisseria sp. does not hold true. The IMD-causing nonencapsulated N. meningitidis strains have the potential to cause disease even in vaccinated populations, since there is no protective immunity induced against nonencapsulated strains by the currently licensed, capsular carbohydrate-based meningococcal vaccines. This prompts consideration of the value of replacing today's capsule-based vaccines with a protein subunit-based or other subunit vaccine that broadly protects against all meningococci. Perhaps more immediately, it means that meningococci must not be summarily excluded as the cause of invasive bacterial disease based solely upon the presence or absence of capsule.
ACKNOWLEDGMENT
This work was supported by the Alberta Heritage Foundation for Medical Research through the Interdisciplinary Team in Vaccine Design and Implementation.
Footnotes
Published ahead of print 16 April 2012
REFERENCES
- 1. Abdillahi H, Poolman JT. 1987. Whole-cell ELISA for typing Neisseria meningitidis with monoclonal antibodies. FEMS Microbiol. Lett. 48:367–371 [PubMed] [Google Scholar]
- 2. Ashton FE, Ryan A, Diena BB, Frasch CE. 1979. Evaluation of the antiserum agar method for the serogroup identification of Neisseria meningitidis. Can. J. Microbiol. 25:784–787 [DOI] [PubMed] [Google Scholar]
- 3. Bennett DE, Mulhall RM, Cafferkey MT. 2004. PCR-based assay for detection of Neisseria meningitidis capsular serogroups 29E, X, and Z. J. Clin. Microbiol. 42:1764–1765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Borrow R, et al. 1998. siaD PCR ELISA for confirmation and identification of serogroup Y and W135 meningococcal infections. FEMS Microbiol. Lett. 159:209–214 [DOI] [PubMed] [Google Scholar]
- 5. Borrow R, et al. 1997. Non-culture diagnosis and serogroup determination of meningococcal B and C infection by a sialyltransferase (siaD) PCR ELISA. Epidemiol. Infect. 118:111–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Claus H, Maiden MC, Maag R, Frosch M, Vogel U. 2002. Many carried meningococci lack the genes required for capsule synthesis and transport. Microbiology 148:1813–1819 [DOI] [PubMed] [Google Scholar]
- 7. Deghmane AE, et al. 2010. Emergence of new virulent Neisseria meningitidis serogroup C sequence type 11 isolates in France. J. Infect. Dis. 202:247–250 [DOI] [PubMed] [Google Scholar]
- 8. Findlow H, et al. 2007. Three cases of invasive meningococcal disease caused by a capsule null locus strain circulating among healthy carriers in Burkina Faso. J. Infect. Dis. 195:1071–1077 [DOI] [PubMed] [Google Scholar]
- 9. Gorringe AR, Reddin KM, Voet P, Poolman JT. 2001. Animal models for meningococcal disease. Methods Mol. Med. 66:241–254 [DOI] [PubMed] [Google Scholar]
- 10. Granoff DM, Welsch JA, Ram S. 2009. Binding of complement factor H (fH) to Neisseria meningitidis is specific for human fH and inhibits complement activation by rat and rabbit sera. Infect. Immun. 77:764–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gu A, Zhang Z, Zhang N, Tsark W, Shively JE. 2010. Generation of human CEACAM1 transgenic mice and binding of Neisseria Opa protein to their neutrophils. PLoS One 5:e10067 doi:10.1371/journal.pone.0010067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hammerschmidt S, et al. 1996. Modulation of cell surface sialic acid expression in Neisseria meningitidis via a transposable genetic element. EMBO J. 15:192–198 [PMC free article] [PubMed] [Google Scholar]
- 13. Hammerschmidt S, et al. 1996. Capsule phase variation in Neisseria meningitidis serogroup B by slipped-strand mispairing in the polysialyltransferase gene (siaD): correlation with bacterial invasion and the outbreak of meningococcal disease. Mol. Microbiol. 20:1211–1220 [DOI] [PubMed] [Google Scholar]
- 14. Hoang LM, et al. 2005. Rapid and fatal meningococcal disease due to a strain of Neisseria meningitidis containing the capsule null locus. Clin. Infect. Dis. 40:e38–e42 [DOI] [PubMed] [Google Scholar]
- 15. Holbein BE. 1981. Differences in virulence for mice between disease and carrier strains of Neisseria meningitidis. Can. J. Microbiol. 27:738–741 [DOI] [PubMed] [Google Scholar]
- 16. Johansson L, et al. 2003. CD46 in meningococcal disease. Science 301:373–375 [DOI] [PubMed] [Google Scholar]
- 17. Joseph B, et al. 2010. Comparative genome biology of a serogroup B carriage and disease strain supports a polygenic nature of meningococcal virulence. J. Bacteriol. 192:5363–5377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Khaki P, et al. 2009. Molecular typing of Neisseria gonorrhoeae isolates by Opa-typing and ribotyping in New Delhi, India. Int. J. Microbiol. 2009:934823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li MS, et al. 2009. A Neisseria meningitidis NMB1966 mutant is impaired for invasion of respiratory epithelial cells, survival in human blood and for virulence in vivo. Med. Microbiol. Immunol. 198:57–67 [DOI] [PubMed] [Google Scholar]
- 20. Li Y, Wooldridge KG, Javed MA, Tang CM, Ala'aldeen DA. 2009. Secreted proteins of Neisseria meningitidis protect mice against infection. Vaccine 27:2320–2325 [DOI] [PubMed] [Google Scholar]
- 21. Li Y, et al. 2006. Immunization with live Neisseria lactamica protects mice against meningococcal challenge and can elicit serum bactericidal antibodies. Infect. Immun. 74:6348–6355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Maiden MC, et al. 1998. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl. Acad. Sci. U. S. A. 95:3140–3145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mandrell RE, et al. 1991. Endogenous sialylation of the lipooligosaccharides of Neisseria meningitidis. J. Bacteriol. 173:2823–2832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Martin P, et al. 2003. Experimentally revised repertoire of putative contingency loci in Neisseria meningitidis strain MC58: evidence for a novel mechanism of phase variation. Mol. Microbiol. 50:245–257 [DOI] [PubMed] [Google Scholar]
- 25. Sacchi CT, et al. 1998. Proposed standardization of Neisseria meningitidis PorA variable-region typing nomenclature. Clin. Diagn. Lab. Immunol. 5:845–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sacchi CT, et al. 1998. Correlation between serological and sequencing analyses of the PorB outer membrane protein in the Neisseria meningitidis serotyping system. Clin. Diagn. Lab. Immunol. 5:348–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sadler F, et al. 2003. Genetic analysis of capsular status of meningococcal carrier isolates. Epidemiol. Infect. 130:59–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schoen C, et al. 2008. Whole-genome comparison of disease and carriage strains provides insights into virulence evolution in Neisseria meningitidis. Proc. Natl. Acad. Sci. U. S. A. 105:3473–3478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schryvers AB, Morris LJ. 1988. Identification and characterization of the transferrin receptor from Neisseria meningitidis. Mol. Microbiol. 2:281–288 [DOI] [PubMed] [Google Scholar]
- 30. St. Michael F, et al. 2005. Structural analysis of the lipopolysaccharide of Pasteurella multocida strain VP161: identification of both Kdo-P and Kdo-Kdo species in the lipopolysaccharide. Carbohydr. Res. 340:59–68 [DOI] [PubMed] [Google Scholar]
- 31. Sun Y, et al. 2005. Identification of novel antigens that protect against systemic meningococcal infection. Vaccine 23:4136–4141 [DOI] [PubMed] [Google Scholar]
- 32. Taha MK. 2000. Simultaneous approach for nonculture PCR-based identification and serogroup prediction of Neisseria meningitidis. J. Clin. Microbiol. 38:855–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Thiery G, Tankovic J, Brun-Buisson C, Blot F. 2001. Gonococcemia associated with fatal septic shock. Clin. Infect. Dis. 32:E92–E93 [DOI] [PubMed] [Google Scholar]
- 34. Vicente D, Esnal O, Perez-Trallero E. 2012. Fatal Neisseria meningitidis serogroup X sepsis in immunocompromised patients in Spain: virulence of clinical isolates. J. Infect. 64:184–187 [DOI] [PubMed] [Google Scholar]
- 35. Vogel U, Claus H, Frosch M. 2001. Capsular operons. Methods Mol. Med. 67:187–201 [DOI] [PubMed] [Google Scholar]
- 36. Vogel U, et al. 2004. Bacteremia in an immunocompromised patient caused by a commensal Neisseria meningitidis strain harboring the capsule null locus (cnl). J. Clin. Microbiol. 42:2898–2901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wetzler LM, Barry K, Blake MS, Gotschlich EC. 1992. Gonococcal lipooligosaccharide sialylation prevents complement-dependent killing by immune sera. Infect. Immun. 60:39–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zarantonelli ML, et al. 2008. Hyperinvasive genotypes of Neisseria meningitidis in France. Clin. Microbiol. Infect. 14:467–472 [DOI] [PubMed] [Google Scholar]