

Abstract
Enterotoxigenic Escherichia coli (ETEC) produces both heat-labile (LT) and heat-stable (ST) enterotoxins and is a major cause of diarrhea in infants in developing countries and in travelers to those regions. In addition to inducing fluid secretion, LT is a powerful mucosal adjuvant capable of promoting immune responses to coadministered antigens. In this study, we examined purified A subunit to further understand the toxicity and adjuvanticity of LT. Purified A subunit was enzymatically active but sensitive to proteolytic degradation and unable to bind gangliosides, and even in the presence of admixed B subunit, it displayed low cyclic AMP (cAMP) induction and no enterotoxicity. Thus, the AB5 structure plays a key role in protecting the A subunit from proteolytic degradation and in delivering the enzymatic signals required for secretion. In contrast, the A subunit alone was capable of activating dendritic cells and enhanced immune responses to multiple antigens following intranasal immunization; therefore, unlike toxicity, LT adjuvanticity is not dependent on the AB5 holotoxin structure or the presence of the B subunit. However, immune responses were maximal when signals were received from both subunits either in an AB5 structure or with A and B admixed. Furthermore, the quality of the immune response (i.e., IgG1/IgG2 balance and mucosal IgA and IL-17 secretion) was determined by the presence of an A subunit, revealing for the first time induction of Th17 responses with the A subunit alone. These results have important implications for understanding ETEC pathogenesis, unraveling immunologic responses induced by LT-based adjuvants, and developing new mucosal vaccines.
INTRODUCTION
Enterotoxigenic Escherichia coli (ETEC) produces both heat-labile (LT) and heat-stable (ST) enterotoxins and is a major cause of diarrhea in infants in developing countries and in travelers to those regions (28). In addition to inducing fluid secretion, LT is a powerful adjuvant, capable of promoting immune responses to coadministered antigens. For these reasons, LT has been extensively examined in attempts to elucidate the mechanisms of both enterotoxicity and adjuvanticity.
The LT holotoxin is composed of an enzymatically active A subunit noncovalently associated with a pentameric B subunit. LT has an AB5 structure and shares 80% homology with cholera toxin (CT) (reviewed in reference 24). Upon cell contact, the B subunit mediates binding and internalization of the toxin, aided by an endoplasmic reticulum (ER) retention sequence on the A subunit, eventually resulting in retrograde transport through the Golgi apparatus to the ER. Concurrent proteolytic cleavage and disulfide bond reduction in the ER separates the A subunit into its two components: the enzymatically active A1 domain and a smaller A2 peptide that links A1 to the B subunit. Transport of A1 into the cytoplasm of intestinal epithelial cells results in binding to ADP-ribosylation factors (ARF); ADP-ribosylation of numerous cellular proteins, including Gsα; irreversible activation of adenylate cyclase; cyclic AMP (cAMP) accumulation; and deregulation of ion transport mechanisms on the luminal surfaces of intestinal epithelial cells. This ultimately leads to an osmotic gradient favoring intestinal water secretion into the lumen of the bowel (22) and secretory diarrhea. The ability of LT to induce cAMP accumulation and fluid secretion can be prevented or reduced by mutations to the ganglioside-binding sites in the B subunit or to the active site or the protease-sensitive sites in the A subunit (7, 19, 27, 48).
Administration of LT by virtually any mucosal route first activates innate immune responses, including secretion of inflammatory cytokines, dendritic cell (DC) recruitment and activation, and initiation of antigen presentation (1, 21, 47, 50, 54). Subsequently, antigen-specific adaptive immune responses develop, including IgG1/IgG2a antibodies, mucosal IgA, and mixed Th1/Th2/Th17/Treg cellular responses, depending upon the intrinsic nature of the coadministered antigen, the type of attenuating mutation to the LT (if any), and the route of immunization (3, 6, 13, 24, 37, 41, 42, 53, 55). The mechanisms through which LT functions as a mucosal adjuvant are not well understood, and conflicting results have been obtained by different investigators using different preparations of LT, different antigens, and different routes of immunization. Most evidence suggests that the ability of LT to function as a mucosal adjuvant is related to its ability to induce cAMP, and molecules that are completely enzymatically inactive are less effective as mucosal adjuvants than those that retain at least a minimal level of cAMP activation (7, 36, 39). The B subunit can itself promote immune responses to coadministered antigens, although these responses are less potent and more skewed toward Th2 and regulatory phenotypes than those seen with the holotoxin or certain AB5 mutants (3, 20, 32, 52, 56). The adjuvant properties of the B subunit have been difficult to clearly establish because many studies have been performed using B subunit isolated by dissociation chromatography and contaminated with small amounts of LT holotoxin or with lipopolysaccharide (LPS). The contribution of the A subunit has been more challenging to define because purified A subunit is hydrophobic and difficult to keep in solution.
Our previous studies have shown that many of the differences in enzymatic activity, enterotoxicity, and adjuvanticity observed between LT and CT are a function of the A subunits of the two toxins (5). In this study, we examined both the A subunit and the A1 domain of LT to further understand the toxicity and adjuvanticity of the molecule. We used purified recombinant versions of both the A subunit and the A1 domain, each with a 3-histidine tag, here referred to as A and A1, respectively, and compared their sensitivity to proteolytic degradation and enzymatic activity to those of native LT and recombinant B subunit. We also examined the binding properties and uptake of A and A1 by both epithelial cells and dendritic cells, their ability to induce accumulation of intracellular cAMP, and their potential enterotoxicity. To better understand the mechanisms of adjuvanticity of LT, we evaluated the abilities of both A and A1 to activate dendritic cells and to induce serum and mucosal antibodies to two different, unrelated antigens following intranasal immunization. Finally, we compared antigen-specific IL-17 secretion by splenocytes isolated from animals immunized with antigen in combination with A, LT, or B subunit and examined the correlation between LT-induced antigen-specific secretion of interleukin 17 (IL-17) and production of antibody isotypes. The results obtained demonstrate that the AB5 structure plays a key role in protecting the A subunit from proteolytic degradation and inducing secretion; however, adjuvanticity is not dependent on the AB5 holotoxin structure, although induction of systemic antibody responses are maximized with signals received from both the A and B subunits. Furthermore, the quality of the immune response (i.e., IgG1/IgG2 balance, mucosal IgA, and IL-17 secretion) is determined by the presence of the A subunit, revealing for the first time induction of Th17 responses with only the A subunit or A subunit-containing adjuvants.
MATERIALS AND METHODS
Toxin and subunit purification.
For this study, we used purified preparations of native LT isolated from recombinant clones expressing LT from the human isolate of E. coli H10407, a nontoxic mutant of LT from H10407 designated LT(R192G/L211A) or dmLT, and B subunit (11, 12, 48, 55). All of these molecules possess ganglioside receptor binding properties and were purified by galactose affinity chromatography in our laboratory as described previously (10, 11, 55). Both the A subunit and A1 were derivatives of the H10407 LT clone. His-tagged A subunit and A1 were prepared using the LT-containing plasmid pCS96 (7) and by first introducing NcoI and XhoI restriction sites into the flanking regions of the A subunit (722-bp) or A1 (558-bp) DNA fragment using mutagenesis primers. A- or A1-encoding DNA was then excised using NcoI and XhoI and subcloned into pET28b (Novagen) following the manufacturer's instructions so that the C terminus of A or A1 ended with a 6-histadine tag. His-tagged A or A1 was excised with NheI and XhoI and transformed into the E. coli expression strain BL21(DE3) STAR (Invitrogen) following the manufacturer's instructions. Transformed cells were grown in LB-kanamycin broth at 37°C to an optical density at 600 nm (OD600) of 0.75, and then 0.8 mM isopropyl-β-d-thiogalactopyranoside (IPTG) was added for an additional 2.5 h of culture. Inclusion bodies (IBs) were then isolated using BugBuster Reagent BBR (Novagen) following the manufacturer's instructions. The IBs were solubilized with 6 M urea, pH 8.2, in 200 mM Na2HPO4/NaH2PO4 buffer before stepwise dialysis to remove the urea. Purified proteins were prepared from solubilized IBs using a His-trap HP column (GE Healthcare). The composition and purity of each protein was confirmed by SDS-PAGE and Limulus amebocyte lysate assay (Lonza Inc., Walkersville, MD). The endotoxin content of the final products was <1 endotoxin unit (EU)/mg.
Visualization of proteins by SDS-PAGE.
Toxins and subunits were electrophoresed on a NuPAGE 10% Bis-Tris Gel (Invitrogen, Carlsbad, CA) under reducing conditions with or without prior boiling (5 min; 95°C). Protein bands were visualized after staining with 0.25% Coomassie blue. For trypsinization, 10 μg of each toxin was subjected to 10, 20, or 30 min or 1 h of digestion with 10 ng of trypsin (Sigma, St. Louis, MO) at 37°C prior to boiling and electrophoresis.
Detection of enzymatic activity.
Toxins and subunits were analyzed for in vitro ADP-ribosyltransferase activity in a cell-free assay, as previously described (33, 57). Briefly, LT, A, A1, and B were incubated for 1 h at 37°C with NAD (Sigma) and recombinant ARF4 (rARF4) (Abnova, Taiwan) in a high-potassium buffer containing 2.5 mM dithiothreitol at pH 7.4. The reactions were stopped with SDS sample buffer (Invitrogen), and the products were electrophoresed on a NuPAGE 10% Bis-Tris Gel for Western blot analysis. The blots were probed with the His-tagged macrodomain rAF1521 (a generous donation from Andreas Ladurner) followed by anti-His-horseradish peroxidase (HRP) antibodies (Qiagen Inc., Valencia, CA) to detect ADP-ribosylated proteins or with anti-ARF antibodies (Abcam, Cambridge, MA).
Detection of GM1 binding by ELISA.
The quantitative GM1 binding enzyme-linked immunosorbent assay (ELISA) was performed as previously described (26, 58). Briefly, wells were coated with 10 μg of GM1 monosialoganglioside (Sigma) each prior to addition of samples or were coated directly with 0.1 μg of LT, A, A1, or B. The plates were incubated, washed, and blocked before the addition of samples at 0.1 μg/well. After washing, anti-LT A subunit (LTA) or anti-LT B subunit (LTB) polyclonal rabbit sera prepared in our laboratory were added and detected with goat anti-rabbit-HRP (Sigma). Absorbance was determined spectrophotometrically at 405 nm.
Detection of intracellular LT by Western blotting.
Detection of intracellular toxins and subunits by Western blot analysis of treated cells was performed as previously described (48). DCs were expanded from bone marrow cultures at low cell densities in the presence of recombinant granulocyte-macrophage colony-stimulating factor (rGM-CSF) (ebioscience, San Diego, CA) for 7 days in 10% RPMI (38). Nonadherent suspension cells were then collected and further purified for CD11+ DCs (>90%) using a 13% Histodenz (Sigma) density gradient, followed by negative selection with a Mouse DC Enrichment Kit (Stem Cells Technology, Vancouver, BC, Canada). INT407 cells were propagated as previously described (48). Confluent epithelial cell monolayers or 1 × 106 DCs were treated with 10 μg/ml of toxins or subunits and, after 24 h, extensively washed and lysed. Cell lysates were analyzed by Western blot probing with anti-LTA or anti-LTB antibodies and detected with HRP-tagged donkey anti-rabbit IgG (Santa Cruz Biotechnology, Santa Cruz, CA).
Evaluation of dendritic cell maturation.
DCs were expanded from bone marrow cultures as described above. Suspension cells (∼75% CD11c+) were harvested, washed, and immediately plated in a 96-well plate at 2.5 × 105 cells per well. The cells were treated with toxins or subunits, including molar concentrations of A and B equivalent to the molarity of individual A and B subunits in 1 μg/ml LT holotoxin. After 24 h, the cells were washed, resuspended in flow cytometry buffer (PBS containing 1% bovine serum albumin [BSA], 0.1% NaN3), incubated with Fc Block (BD Biosciences, San Jose, CA), and stained with anti-mouse CD80-fluorescein isothiocyanate (FITC), CD86-phycoerythrin (PE) (ebioscience), and CD11c-peridinin chlorophyll protein (PerCP)-Cy5.5 (BD Biosciences, Sparks, MD). The stained cells were run on a FACS Calibur (BD Biosciences) and analyzed for percent mature DCs (defined as CD80hi CD86hi CD11c+) using FlowJo (TreeStar, Ashland, OR).
Detection of enzymatic activity in human colonic epithelial cells by cAMP assay.
Caco-2 cells were treated as previously described (7, 48), except 3-h treatments consisted of subunit proteins, the cAMP analog N6,2′-O-dibutyryladenosine 3′,5′-cyclic monophosphate sodium salt (d-cAMP) (Sigma), or trypsin-activated toxins. The cells were washed and lysed, and intracellular cAMP levels were assessed using a cAMP assay kit (R&D Systems, Minneapolis, MN) following the manufacturer's instructions.
Patent mouse assay for in vivo enterotoxicity.
The patent (nonoccluded gut) mouse assay was performed as previously described (7). Groups of three BALB/c mice each were administered oral doses containing 25 μg or 125 μg of toxins or subunits in 0.5 ml normal saline. After 3 h of incubation, the animals were sacrificed, their intestines were excised, and individual gut/carcass (G/C) ratios were calculated. All animal studies were approved by the Tulane University Institutional Animal Care and Use Committee.
Intranasal immunizations.
Mice were immunized intranasally with 10 μl containing 10 μg tetanus toxoid (TT) (Statens Serum Institute, Denmark) or chicken egg ovalbumin (OVA) (Sigma) alone or with 5 μg of one of the test adjuvants. For TT experiments, groups of six BALB/c mice each were immunized 3 times at weekly intervals and sacrificed 1 week after the last immunization; for OVA experiments, groups of five BALB/c mice each were immunized 3 times at monthly intervals and sacrificed 2 weeks after the last immunization. Serum was collected by cardiac puncture. A bronchoalveolar lavage (BAL) was performed with 0.8 ml saline containing a protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN). Fecal pellets were collected directly from the colon at the time of sacrifice. The fecal pellets were weighed, dissolved in 1.5 ml PBS containing 0.5% Tween 20 and a protease inhibitor cocktail, and centrifuged, and the supernatants were stored at −20°C until they were assayed.
Detection of antibodies and Th17 responses.
Quantitative anti-TT and anti-OVA ELISAs were performed on individual serum, BAL fluid, and fecal samples as previously described (26, 58), with wells coated with 0.1 μg of antigen each. For Th17 analysis, an antigen restimulation assay was performed using splenocytes isolated and cultured at 1 × 105 cells with 0.1 μg/well antigen (OVA or TT) in a 96-well plate with 200 μl medium. After 3 days in culture, the plates were centrifuged and the supernatants were collected. Samples were analyzed by Bio-plex using Milliplex MAP Mouse Cytokine/Chemokine Kits (Millipore, Billerica, MA).
Data analysis and toxin diagrams.
Statistical analysis was performed using Prism (GraphPad Software, Inc., La Jolla, CA) for one-way analysis of variance (ANOVA) with Tukey's multiple-comparison posttest or Spearman's rank order correlation coefficient (r) analysis. For correlation analysis, data were log transformed for calculations and graphed with a linear best-fit line. The structures of LT, A subunit, and the A1 domain were derived from the previously published structure of partially activated E. coli heat-labile enterotoxin (Protein Data Bank identifier [ID], 1LTB) (44), using Jmol, an open-source Java viewer for chemical structures in three dimensions (3D) (http://www.jmol.org/).
RESULTS
The isolated A subunit and A1 are enzymatically active but more sensitive to proteolytic degradation than the LT holotoxin.
LT holotoxin has an AB5 toxin structure consisting of one A subunit and five B subunit monomers that form the B subunit pentamer (Fig. 1A). Trypsin cleavage and disulfide bond reduction are required for activation and further separation of the A subunit into an enzymatically active A1 domain and A2 peptide. To confirm the structure and purity of the isolated A subunit and A1 domain used in this study, we performed an SDS-PAGE analysis with or without prior boiling or trypsin digestion. As seen in Fig. 1B, on SDS-PAGE without boiling, 1 μg of native LT separates into its 28-kDa A subunit and the 56-kDa B subunit pentamer, reflecting the AB5 structure of the native molecule. Upon boiling, the A subunit remains intact and the B subunit separates into 11.5-kDa monomers. If LT is treated with trypsin prior to boiling, the A subunit further separates into the 21-kDa A1 domain and a 6-kDa A2 peptide. The recombinant A subunit and A1 domain both migrated as expected on SDS-PAGE with and without boiling and were indistinguishable from native A subunit and native A1, respectively. However, it was evident that incubation with trypsin completely degraded recombinant A and A1 to bands of <6 kDa, indicating enhanced sensitivity to enzymatic degradation compared to native A subunit in the AB5 holotoxin structure. This was verified by increasing the concentrations of LT, A, and A1 to 10 μg (i.e., 10×) prior to trypsin treatment for 10, 20, and 30 min and SDS-PAGE (Fig. 1C, arrowhead). A1 was especially sensitive to degradation, whereas the A subunit appeared to be first cleaved into an A1-size band before fragmentation. The enhanced sensitivity to trypsin degradation is most likely a function of conformational changes in A and A1 in the absence of B subunit, exposing more trypsin-sensitive residues. Despite the enhanced sensitivity to proteolytic degradation, A and A1 maintained in vitro ADP-ribosyltransferase activity. Both A and A1 retained the ability to ADP-ribosylate ARF proteins (Fig. 1D). The level of ADP-ribosylation by A1 was greater than that by A, as expected, based on LT's known requirement for trypsin activation to express full enzymatic activity (19).
Fig 1.
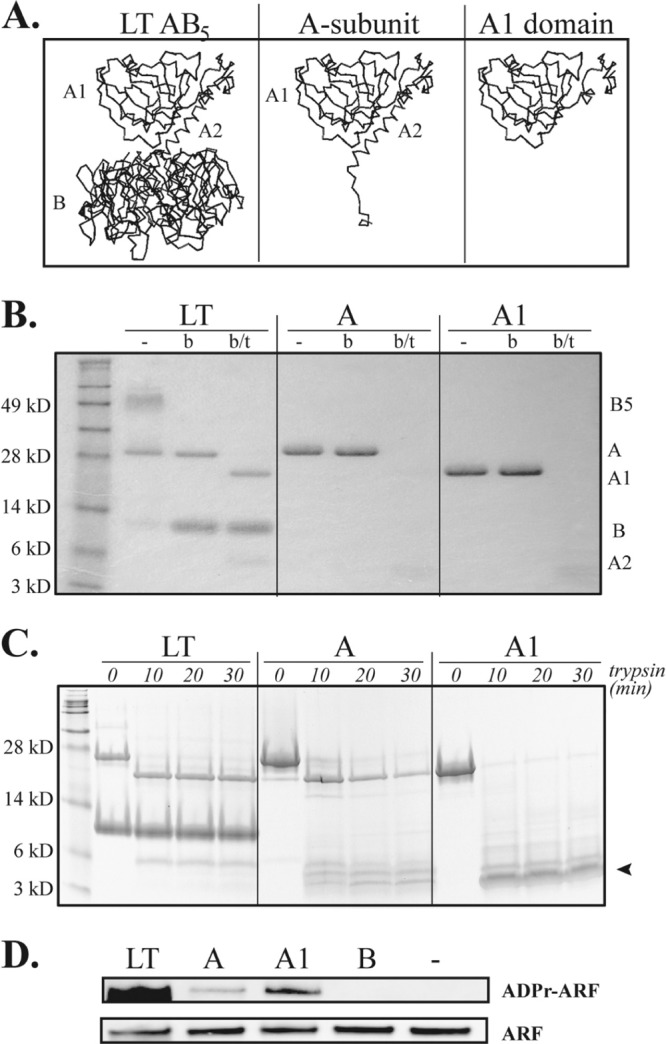
A and A1 are enzymatically active but more sensitive to proteolytic degradation than LT holotoxin. (A) Ribbon diagrams of structures of LT AB5 holotoxin, A subunit, and the A1 domain. (B) SDS-PAGE gel of 1 μg of unboiled (−), boiled (b), or boiled and trypsin-treated (b/t) LT or subunits. A and A1 exhibited the expected SDS-PAGE migration; however, A, and especially A1, displayed enhanced sensitivity to trypsin degradation. (C) SDS-PAGE gels with 10 μg (i.e., 10×) of trypsin-treated LT or subunits, confirming proteolytic degradation of A and A1. (D) Western blots identifying ADP-ribosylation of ARF receptor protein in vitro, verifying the retained enzymatic activities of LT, A, and A1. The results shown are representative of at least 3 independently conducted experiments.
The A subunit does not bind to GM1 and has altered cellular internalization and ability to induce dendritic cell maturation and cAMP accumulation compared to LT holotoxin.
LT holotoxin binds and is internalized through the interaction of its B subunit with ubiquitous cell surface receptors, particularly GM1 ganglioside (29). Indeed, this GM1-B subunit interaction is critical to both the in vivo toxicity and oral adjuvanticity of LT (27). There is no known extracellular receptor for the A subunit, and it was important to establish whether isolated A or A1 could bind to the known LT receptor GM1, be internalized into epithelial cells, activate immune cells, or induce accumulation of cAMP when added extracellularly. The carboxyl terminus of A2 extends through the hydrophobic pore in the B subunit and may make contact with the epithelial cell surface, so association with GM1 needs to be evaluated. To assess GM1 binding by the A subunit, a GM1-binding ELISA was performed using ELISA microtiter plates either coated with GM1 prior to addition of toxins or subunits or directly coated with the toxins or subunits and then probed with anti-LTA or anti-LTB. As seen in Fig. 2A, LT, A, and B were each detectable by the appropriate antibody when ELISA plates were directly coated with them; however, when toxins or subunits were applied to GM1-coated wells, only LT and the B subunit were detected. No anti-LTA binding was detected in plates coated with GM1 prior to the addition of A, demonstrating that the A subunit does not bind to GM1.
Fig 2.

The A subunit does not bind to GM1 and has altered cellular internalization and ability to induce dendritic cell maturation and cAMP accumulation compared to LT holotoxin. (A) GM1-binding ELISA with LT A and B subunits detected with rabbit anti-LTA or anti-LTB. (B) Western blots of cell lysates of epithelial cells or dendritic cells probed with anti-LTA or anti-LTB rabbit sera 24 h after treatment with A and A1. Both were internalized, but the band intensities differed from that of LT. untx, untreated. (C) Percent mature DCs after 24 h of culture with A or A1 (0.1, 1, or 10 μg) alone or admixed with B subunit (1 μg) or in equal molar (e.m.) ratios equivalent to that in LT holotoxin. Both A and A1 induced dose-dependent maturation of DCs (P < 0.05), which was greatly enhanced with admixed B subunit (B versus any combination of A plus B or A1 plus B, P < 0.05). (D) cAMP levels in epithelial cells after 3 h of incubation with 1 μg A or A1 alone or admixed with 1 μg B subunit compared to d-cAMP or trypsinized LT. (E) cAMP levels in epithelial cells treated with escalating doses (1 to 100 μg) of A1. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant compared to untreated cells as calculated by one-way ANOVA with Tukey's multiple-comparison posttest. The error bars indicate standard errors of the means.
Without a GM1-binding step, it was unclear if either A or A1 could undergo cellular internalization. To evaluate binding and internalization, intestinal epithelial cells and DCs were incubated with LT, A, A1, or B for 24 h and washed extensively, and cell lysates were analyzed for the presence of the A or B subunit by Western blotting (Fig. 2B). Despite the absence of a B subunit to facilitate cell binding, both A and A1 were detectable in cell lysates of both intestinal epithelial cells and dendritic cells in Western blots probed with anti-LTA or anti-LTB. The detection level depended on the cell type, but there was consistently less binding and internalization of A1 than A. LT is internalized and traffics intracellularly by several mechanisms (8); which binding or internalization mechanism(s) is employed by A and A1 is unknown, though it is probably distinct from that of LT. The most likely explanation for the presence of A and A1 intracellularly is normal endocytic or phagocytic uptake.
These results indicate that A and A1 can be internalized by both DCs and epithelial cells, but they do not reveal any downstream cellular effects of internalization. LT holotoxin is a potent inducer of maturation of DCs and intracellular cAMP accumulation in intestinal epithelial cells (27, 48, 50). The ability of either A or A1 to promote these in vitro responses was evaluated alone or when admixed with B subunit. Admixing with B subunit would reveal if a second B subunit-mediated signal was present or required for cellular activity. We first examined the abilities of A and A1 to activate dendritic cells (defined as increasing expression of CD80 and CD86 on CD11c+ cells). As seen in Fig. 2C, both A and A1 induced dose-dependent maturation of DCs (P < 0.05). This was more evident with A1 than with A, possibly a reflection of the fact that A1 does not require proteolytic activation. Both LT holotoxin and B subunit were able to induce maturation of DCs more effectively than isolated A or A1 (LT versus any dose of A or A1, P < 0.05; B versus any dose of A or A1, P < 0.05), indicating that DC maturation can also be triggered by GM1 binding and does not require the enzymatic component. However, it was also evident that even small amounts of A or A1 coadministered with B subunit were able to enhance DC maturation compared to B subunit alone (B versus any combination of A and B or A1 and B, P < 0.05). Furthermore, when A and B were coadministered at molar concentrations equivalent to that of native LT A and B subunits, the percentage of DC maturation was identical to that seen with native LT. This suggests that the B subunit and A subunit deliver separate maturation signals to DCs that act either additively or synergistically.
It was also not clear whether GM1-independent uptake of A or A1 by epithelial cells would result in induction of cAMP. The in vitro ADP-ribosylation studies (Fig. 1D) demonstrated that both A and A1 retain enzymatic activity in an in vitro cell-free assay but not whether they would have any effect on intact cells. Induction of cAMP by LT normally follows delivery of A1 to the cytosol from the ER after reduction and proteolytic cleavage from A2. For these studies, intact CaCo-2 cells were incubated with A or A1, washed, and lysed, and intracellular cAMP levels were assessed using a cAMP ELISA (Fig. 2D). We initially examined the same amounts of A and A1, alone or in combination with isolated B subunit, that were used for the DC activation studies (Fig. 2C). There was a small but not significant increase in intracellular cAMP under these conditions. For reference, 0.1 μg of LT induced approximately 200 pMol/ml of cAMP in this assay, equivalent to the levels achieved with 1 mM d-cAMP. In a separate series of experiments, CaCo-2 cells were incubated with increasing doses of A1 (1 to 100 μg) before lysis and analysis for intracellular accumulation of cAMP. In these studies, there was a dose-dependent increase in intracellular cAMP induced by A1 (Fig. 2E), but 100 μg of A1 induced accumulation of only 60 pMol/ml cAMP, indicating that isolated A and A1 are >1,000-fold less active at inducing cAMP than LT holotoxin when incubated with intact cells. These results show that free A subunit is internalized by epithelial cells (Fig. 2B) and subsequently induces low levels of cAMP (Fig. 2C), indicating that enzymatically active A or A1 enters the cytosol.
The A subunit has no detectable in vivo enterotoxicity, either alone or admixed with the B subunit.
A patent (nonoccluded gut) mouse assay was performed to examine potential enterotoxicity of the A subunit. Groups of 3 BALB/c mice each were orally inoculated with 25 μg or 125 μg of LT or 125 μg of A, A plus B, or dmLT. As seen in Fig. 3, mice orally inoculated with 125 μg of A alone or admixed with 125 μg of B subunit maintained normal gut/carcass mass ratios (G/C < 0.09) after 3 h, consistent with negative saline controls and equivalent to that seen with a nontoxic mutant of LT (dmLT). Despite its lack of toxicity, dmLT has adjuvant activity equivalent to that of native LT (37, 48, 55) and was used in the adjuvant studies described below. In contrast, both 25 μg and 125 μg of native LT significantly increased intestinal fluid accumulation in this model (P < 0.001). These results are consistent with the low levels of cAMP induced by A and A1 (and dmLT) and indicate the importance of the AB5 toxin structure to the secretory effects of LT, as the A subunit, even admixed with B subunit, does not induce detectable secretion.
Fig 3.

The A subunit has no detectable in vivo enterotoxicity, either alone or admixed with the B subunit. Groups of 3 BALB/c mice each were orally inoculated with 25 μg or 125 μg of LT or 125 μg of A, A plus B, or dmLT. G/C ratios were determined after 3 h. G/C ratios of >0.09 are positive. Neither A alone nor A admixed with B induced intestinal fluid secretion. ***, P < 0.001 compared to saline controls. The error bars indicate standard errors of the means.
The A subunit is a mucosal adjuvant for coadministered antigens.
In the following study, we examined both A and A1 for the ability to function as a mucosal adjuvant for an unrelated, coadministered antigen (TT) either alone or, based on the DC activation studies, when admixed with B subunit. For these studies, groups of six BALB/c mice each were immunized intranasally with TT or TT admixed with B, A, A1, A plus B, or dmLT and analyzed for serum and mucosal anti-TT antibodies. As seen in Fig. 4A and B, intranasal immunization with TT alone induced serum anti-TT IgG responses in immunized animals that were significantly increased when the immunizations included any of the test adjuvants (B, P < 0.05; A1, P < 0.01; A, dmLT, and A plus B, P < 0.001). Importantly, both A and A1 functioned as mucosal adjuvants for increasing antigen-specific serum IgG even though they lack a known receptor on epithelial and immune cells, do not have a receptor-targeting delivery system (e.g., B subunit), and induce very low levels of cAMP.
Fig 4.

The A subunit acts as a mucosal adjuvant for coadministered antigens. Groups of 6 BALB/c mice each were immunized intranasally three times at weekly intervals with TT alone or in combination with one of the test adjuvants. Serum and mucosal anti-TT antibody responses were evaluated by ELISA 1 week after the final dose. The test adjuvants included B, A, A1, dmLT, and A plus B. Both group mean OD values (A) and medians of individual antibody responses (B to F) demonstrate that A and A1 can function as adjuvants and elicit greater responses than the B subunit or no adjuvant but lower responses than dmLT or A plus B for serum IgG (B), IgG1 (C), and IgG2a (D). Mucosal anti-TT IgA responses were detected in BAL fluid (E) and fecal pellets (F) of immunized mice. Strikingly, induction levels of IgA antibodies were similar between A-, A1-, dmLT-, and A-plus-B-adjuvanted groups, suggesting that induction of mucosal IgA against coadministered antigens is A subunit dependent and that very low levels of residual enzymatic activity are both necessary and sufficient for adjuvanticity. Individual animal results are represented as individual points and group medians in the bar graphs. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant compared to no-adjuvant (TT only) controls unless otherwise indicated.
We next compared the quality of the immune responses generated by the different adjuvants by analyzing the anti-TT antibody isotypes in serum, BAL fluid, and feces. As seen in Fig. 4C, the A subunit elicited a significantly greater serum anti-TT IgG1 response than the B subunit or no adjuvant (P < 0.001) but lower serum anti-TT IgG1 responses than dmLT or A plus B (P < 0.001), indicating synergy between the two subunits for promotion of serum antigen-specific IgG1 responses. Interestingly, only dmLT (P < 0.05) and A plus B (P < 0.01) significantly increased the serum anti-TT IgG2a responses (Fig. 4D) compared to no adjuvant. Moreover, B alone suppressed the TT-specific IgG2a response, consistent with type 2 polarization. Specifically, there was no detectable serum anti-TT IgG2a in 5 of 6 animals immunized with TT and B. A1 did not significantly enhance the serum anti-TT IgG1 or IgG2a response compared to TT alone, which may be a reflection of the greater sensitivity of this molecule to proteolytic degradation compared to A or AB5.
As seen in Fig. 4E and F, the presence of anti-TT IgA in BAL fluid or fecal pellets, a measure of mucosal immune responses, followed a different pattern. In the absence of an adjuvant, mice developed no detectable BAL fluid anti-TT IgA, and mice receiving TT admixed with B developed significantly lower levels of BAL fluid anti-TT IgA antibodies than those immunized with TT admixed with A (P < 0.001) or A plus B (P < 0.001). Similar levels of BAL fluid IgA were detected in all groups receiving an A subunit adjuvant, including TT admixed with A, A1, dmLT, and A plus B (P < 0.001). A, and A plus B were not different, suggesting that the separate A and B signals that support induction of serum anti-TT IgG1 and IgG2a (Fig. 4C and D, respectively) may not be required for induction of antigen-specific BAL fluid IgA responses. Similar responses were seen in fecal samples analyzed for anti-TT IgA (Fig. 4F). These data suggest that induction of mucosal IgA against coadministered antigens is A subunit dependent and that very low levels of residual enzymatic activity are both necessary and sufficient for adjuvanticity.
The A subunit promotes Th17 responses, and IL-17 expression is correlated with serum IgG2a and BAL fluid IgA in OVA-immunized animals.
For these studies, groups of five BALB/c mice each were immunized intranasally 3 times at monthly intervals with OVA alone or combined with one of the test adjuvants (B, A, dmLT, A and B, or LT). OVA was selected as the antigen for these experiments because it is generally less immunogenic than toxoid antigens (e.g., tetanus and diphtheria) and could provide additional information about adjuvanticity. The dosing schedule was selected to approximate the WHO Expanded Program on Immunization (EPI) vaccine schedule for childhood vaccines (30-day intervals). As shown in Fig. 5A, intranasal immunization with OVA admixed with any of the test adjuvants enhanced the magnitude of the serum anti-OVA IgG response compared to animals immunized intranasally with OVA alone. This was observed not only for B, dmLT, and LT, all of which possess receptor-binding B subunits, but also for A, which lacks any known mechanism for receptor-mediated uptake. Unlike TT (Fig. 4), which induced a significant serum anti-TT IgG response when administered intranasally with no adjuvant, OVA alone failed to induce a detectable serum anti-OVA IgG response, demonstrating that A can also function as an adjuvant for weakly immunogenic antigens, such as OVA. The addition of A and A-containing adjuvants (A and B, dmLT, and LT) not only increased the serum anti-OVA IgG response, but directed the quality of anti-OVA IgG1, IgG2a, and IgA isotype expression, similar to that seen when TT was the antigen (data not shown). Taken together, the findings with TT and OVA clearly demonstrate that the A subunit can function as a mucosal adjuvant even without the B subunit or the AB5 structure.
Fig 5.

The A subunit promotes Th17 responses, and IL-17 expression is correlated with serum IgG2a and BAL fluid IgA in OVA-immunized animals. Groups of 5 BALB/c mice each were immunized intranasally three times at monthly intervals with OVA alone or in combination with one of the test adjuvants. Two weeks after the final dose, serum and mucosal anti-OVA antibody responses were evaluated by ELISA. The test adjuvants were B, A, A plus B, dmLT, and LT. (A) Intranasal immunization with OVA admixed with any of the test adjuvants enhanced the magnitude of the serum anti-OVA IgG response compared to animals immunized intranasally with OVA alone. (B) IL-17 secretion by OVA-stimulated splenocytes from BALB/c mice immunized with OVA and test adjuvants. The A-plus-B adjuvant group excludes one high-value outlier. All groups immunized with a test adjuvant that included an A subunit, whether part of an AB5 complex, alone, or admixed with B, developed antigen-specific Th17 responses. The error bars indicate standard errors of the means. (C and D) Secretion of IL-17 was significantly correlated with serum anti-OVA IgG2a (C) and BAL fluid anti-OVA IgA (D). Spearman's rank order correlation coefficient (r) and P values are indicated for log-transformed data.
Experimental evidence has shown that LT, some mutant LTs, and CT promote Th17 cell development (6, 17, 35, 37, 43, 53). To assess the contribution of the A subunit to Th17 responses in these animals, we evaluated splenocytes from the OVA-immunized mice for secretion of IL-17 in an antigen restimulation assay. Secretion of IL-17 in response to OVA was observed only in splenocytes from mice receiving adjuvants containing an A subunit, including LT, A, A plus B, and dmLT (Fig. 5B). Interestingly, the dmLT-adjuvanted groups developed the highest antigen-induced IL-17 response (P < 0.01), followed by A and A plus B, and finally LT, indicating that low levels of enzymatic activity are required for induction of Th17 responses and that higher levels of cAMP may have a suppressive effect. Similar levels of IL-17 secretion after antigen restimulation were also seen in TT-immunized mice (data not shown). Collectively, this evidence indicates that the presence of an A subunit is responsible for directing the development of Th17 responses after administration of LT-based adjuvants.
Recently, Th17 responses have been correlated with increased levels of antigen-specific IgA and IgG2a (30, 45). To determine if the IL-17 induced by the A subunit is associated with antigen-specific immunoglobulin responses, we performed correlation analyses between IL-17 secretion by splenocytes after OVA restimulation and induction of anti-OVA antibodies. As seen in Fig. 5C and D, IL-17 secretion was significantly correlated with anti-OVA serum IgG2a (r = 0.49; P = 0.006) and BAL fluid IgA (r = 0.68; P < 0.001) responses, but not serum IgG, serum IgG1, or fecal IgA responses (data not shown). The correlation of IL-17 production with BAL fluid IgA and not fecal IgA following intranasal immunization is consistent with the compartmentalization of the mucosal immune response following immunization at different mucosal sites (15, 16, 59). It is likely that IL-17 production induced by LT-based adjuvants promotes mucosal IgA production in a site-specific manner (e.g., oral administration induces local gut Th17 responses that correlate with fecal IgA). Nevertheless, these data strongly point to an immunologic mechanism whereby LT's A subunit induces production of Th17 that promotes specific antibody responses (IgG2a and IgA).
DISCUSSION
The purpose of this study was to use isolated and purified A subunit and the A1 domain from the heat-labile enterotoxin of E. coli to further elucidate the mechanisms of enterotoxicity and adjuvanticity of LT. We employed a wide range of in vitro and in vivo analyses, including protease sensitivity, ADP-ribosyltransferase activity, ganglioside binding, intracellular localization, and cAMP induction, to further evaluate the contribution of the A subunit to enterotoxicity (fluid secretion in the patent mouse assay) and adjuvanticity (DC maturation, increased serum and mucosal antibodies, and Th17 cell development in response to coadministered antigens). The findings from these analyses are summarized in Table 1.
Table 1.
Properties of the LT holotoxin, dmLT, A subunit, A1 domain, and B subunit
| Analysis | Detection levela |
|||||
|---|---|---|---|---|---|---|
| LT | dmLT | A + B | A | A1 | B | |
| In vitro | ||||||
| Protease sensitivity | + | ++ | ND | +++ | ++++ | NO |
| Enzymatic activity | +++ | +++b | ND | + | ++ | NO |
| GM1 binding | +++ | +++ | ND | NO | ND | +++ |
| Intracellular detection | +++ | ++ | ND | ++ | + | +++ |
| cAMP induction | ++++ | + | + | + | + | NO |
| DC maturation | ++++ | ++++ | ++++ | + | + | +++ |
| In vivo | ||||||
| Secretion | ++++ | NO | NO | NO | NO | NO |
| Intranasal adjuvant | Yes | Yes | Yes | Yes | Yes | Yes |
| IgG | ++++ | ++++ | ++++ | +++ | +++ | ++ |
| IgG1 | ++++ | ++++ | ++++ | +++ | ++ | ++++ |
| IgG2a | ++++ | ++++ | ++++ | +++ | + | NO |
| Mucosal IgA | ++++ | ++++ | ++++ | +++ | +++ | + |
| Th17 response | ++ | ++++ | +++ | +++ | ND | NO |
Individual properties are scaled by detection level from lowest (+) to highest (++++). ND, not determined; NO, not observed; Yes, observed.
Data not shown elsewhere in this work.
We found strong evidence that the AB5 toxin structure prevents proteolytic degradation of the A subunit, most likely keeping the molecule in a structural conformation that sequesters trypsin-sensitive residues. This would be important, not only for targeting the A subunit to the cell, but for protecting it from the protease-rich environment of the gut lumen or proteases in endocytic vesicles. The arginine at position 192 that is normally cleaved and required for proteolytic activation of LT is in a surface-exposed loop that must be cleaved for the toxin to be biologically activated. In the absence of the B subunit, the A subunit conformation is changed, and it is likely that additional trypsin-sensitive residues are exposed. The consequence is rapid and complete degradation of the A subunit, as opposed to the proteolytic cleavage and activation seen with native LT, in which A is part of the AB5 complex. These results correlate with previous studies on the enhanced proteolytic sensitivity of LT mutants with altered structural stability (36, 40, 49).
The B subunit also mediates binding and internalization of the toxin aided by the ER retention sequence on A2, eventually resulting in retrograde transport through the Golgi apparatus to the ER (9). Delivery of A1 to the cytosol, where the NAD-glycohydrolase and ADP-ribosyltransferase activities of the A subunit associated with fluid secretion occur, is clearly more efficient when the B subunit delivers A to the cell. Nonetheless, the small amounts of enzymatically active A and A1 taken up by cultured intestinal epithelial cells are sufficient to induce low levels of cAMP, but not fluid, secretion in an in vivo animal model of enterotoxicity. Thus, the AB5 structure plays a key role in protecting the A subunit from proteolytic degradation and delivering the enzymatic signals required for secretion. These findings are consistent with what has been shown about the functions and relationship of the A and B subunits in other systems.
Because of increased sensitivity to proteolytic degradation and inability to induce accumulation of high levels of cAMP, it would be reasonable to assume that neither A nor A1 can function as an adjuvant. However, the results of this study demonstrate that the A subunit is a potent adjuvant by itself. Despite the reduced enzymatic activity and lack of enterotoxicity, the A subunit is capable of enhancing systemic and mucosal antibodies to codelivered antigens following intranasal immunization. Further research may reveal that the A subunit has limited adjuvanticity when delivered by other mucosal routes, such as the highly protein-degradative gastrointestinal mucosa. Our adjuvant analyses did find differences between the adjuvant properties of A and A1. This is presumably due to differences in enzymatic activity, structural instability, and/or the lack of an ER retention signal in A1 that alters intracellular trafficking/compartmentalization. The last is suggested by our preliminary microscopic analysis, which shows sequestering of A1 intracellularly in a punctate, vesicular pattern different from that of A or LT (E. B. Norton and J. D. Clements, unpublished data). Regardless, our results show that free A subunit is internalized by a GM1-independent mechanism and that A subunit treatment of epithelial cells, DCs, and nasal mucosa results in measurable, functional outcomes (i.e., accumulation of low levels of cAMP, cell maturation, and adjuvanticity, respectively). The low levels of cAMP induced in epithelial cells by A and A1 (Fig. 2D and E) confirm that enzymatically active A subunit is able to enter the cytosol. At this point, we can only conclude that internalization of free A subunit is GM1 independent and is possible in multiple cell types, but the exact nature of cellular entry is unknown.
The results of this investigation show that by using an isolated A subunit, adjuvanticity can be separated from the toxicity of the holotoxin, a conclusion similar to what we recently reported with dmLT (48). However, in the absence of the structural stability provided by the B subunit, it is unclear how isolated A subunit functions as an adjuvant. Traditionally, the adjuvanticity of LT-based adjuvants is thought to be related to induction of intracellular cAMP, as some of the in vitro effects can be mimicked by cAMP analogs or derivatives (1, 2, 47). Moreover, molecules that are completely enzymatically inactive are less effective as mucosal adjuvants than those that retain at least a minimal level of cAMP activity (7, 36, 39). However, studies on mechanisms that control the immune response have clearly demonstrated that high cAMP induction is linked to T-cell suppression and regulation (4, 31). Similarly, administration of LT or CT (which stimulates even higher cAMP accumulation than LT) has been found to enhance T-regulatory responses (references 3 and 34 and data not shown). In the present study, molecules that induced low levels of cAMP, such as isolated A subunit and dmLT, promoted robust immune responses to codelivered antigens, at times higher than those induced by native LT. In particular, we observed a trend of detoxified mutant LT or A subunit inducing more IL-17 secretion and IgG2a than native holotoxin, as seen in previous studies (6; Norton and Clements, unpublished). Therefore, a more accurate hypothesis might be that some cAMP induction is necessary for adjuvanticity but higher cAMP induction leads to toxicity and regulated immune responses. It is also possible that cAMP-independent effects contribute in part to adjuvant responses; the stimulatory effects of CT on human B cells have been linked to non-cAMP-derived enzymatic effects (23). Low-level cAMP induction and cAMP-independent responses as key initiators of adjuvanticity of the A subunit and mutant LTs are intriguing ideas that merit further study.
A previously published study by de Haan et al. also examined recombinant His-tagged forms of the A subunit with and without ADP-ribosylation activity—LTA(His)10 and LTA-E112K(His)10, respectively—for the ability to act as adjuvants or as immunogens (18). Similar to our findings, these investigators showed that LTA(His)10 can stimulate serum and mucosal antibody responses to a coadministered antigen (influenza subunit protein). However, the LTA(His)10-adjuvanted antigen-specific antibody responses were comparable in both magnitude and isotype distribution to those observed after immunization with antigen and LT or the enzymatically inactive mutant LT(E112K), LTA-E112K(His)10, or recombinant B subunit. Based on the comparability of A subunit immune responses to those seen with LT, inactive mutants, or B subunit, the authors concluded that the A subunit by itself, independent of any enzymatic activity, is a potent adjuvant for intranasally administered antigens. This conclusion is surprising, given the repeated superiority of LT and enzymatically active mutants to active-site mutants, such as LT(E112K) and the B subunit, in enhancing immune responses to coadministered antigens (7, 39). Differences in protein preparation, including the number of histidine residues, purification techniques, and possible impurities, such as LPS, might explain the differences between their study and our study and other published data.
We observed several important contributions of each subunit to aspects of LT-mediated adjuvanticity, although the innate antigenicity of the test antigens used in this study (OVA and TT) each contributed to the immune outcomes. First, A and B admixed repeatedly induced higher levels of serum antibodies to coadministered antigens than either A or B alone, which was similar to our findings with the in vitro DC maturation; however, mucosal IgA levels were similar as long as an active A subunit was present, whether alone, admixed with B, or in an AB5 structure. This suggests that adjuvanticity is not dependent on the AB5 holotoxin structure, although induction of systemic antibody responses is maximal, with at least two signals received from both A and B subunits. Second, our data revealed that the quality of this response (i.e., IgG1/IgG2 balance and mucosal IgA and Th17 responses) is dependent on the presence of an active A subunit, with few alterations induced by admixed A and B subunits. For instance, Th17 responses have previously been observed with LT, LT mutants, and CT enterotoxins (6, 17, 35, 37); however, this study for the first time reports induction of Th17 responses with the A subunit alone or the A subunit containing adjuvants (i.e., A plus B). Th17 responses are linked to B-cell help for IgG2a class switching (45) and enhanced polymeric Ig receptor expression in the airways, leading to elevated secretory IgA levels (30). Our correlation analyses are consistent with this model and also suggest a previously unknown mechanism for how Th17 responses contribute to LT-based effects. We did find that Th17 responses and IgG2a induction were not perfectly correlated, and it is likely that other IgG2a class-switching signals (e.g., T-bet upregulation, IL-27, gamma interferon [IFN-γ], and/or CD40L) are also contributing factors (14, 25, 46, 60). Similarly, secretion of IgA in the lungs was only partially correlated with Th17 responses. Mechanisms that regulate IgG production differ from those of IgA production, with the latter including T-independent processes (51). It is possible that the induction of systemic IgG antibodies occurs via a T-dependent method, initiated by both A and B subunits, which would explain their synergy for the magnitude of the IgG response; while the induction of IgA antibodies could be through both T-dependent (Th17 and Th1 responses) and T-independent mechanisms that are initiated by the A subunit. Exactly which immunologic mechanisms contribute to mucosal IgA and IgG antibody isotype induction is unknown and merits additional study.
In summary, the results of this study reveal unique contributions of the A and B subunits to the biological activity of LT, including (i) that the AB5 structure plays a key role in protecting the A subunit from proteolytic degradation and in enterotoxicity; (ii) that adjuvanticity is not dependent on the AB5 holotoxin structure, although induction of systemic antibody responses are maximized with signals received from both the A and B subunits; (iii) that the quality of the immune response (i.e., IgG1/IgG2 balance and mucosal IgA and IL-17 secretion) is determined by the presence of the A subunit; (iv) that free A subunit is internalized by a GM1-independent mechanism and A subunit treatment of epithelial cells, DCs, and nasal mucosa results in measurable, functional outcomes; and (v) that induction of Th17 responses is dependent on the presence of the A subunit. By critically examining isolated and admixed A and B subunits from LT, we have furthered the understanding of toxin biology, enterotoxicity, and adjuvanticity. These results have important implications for understanding ETEC pathogenesis, unraveling immunologic responses induced by LT-based adjuvants, and developing new mucosal vaccines.
ACKNOWLEDGMENT
This work was supported in part by a grant from the National Institutes of Health (EB006493-04).
Footnotes
Published ahead of print 23 April 2012
REFERENCES
- 1. Anosova NG, et al. 2008. Cholera toxin, E. coli heat-labile toxin, and non-toxic derivatives induce dendritic cell migration into the follicle-associated epithelium of Peyer's patches. Mucosal. Immunol. 1:59–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bagley KC, Abdelwahab SF, Tuskan RG, Fouts TR, Lewis GK. 2002. Cholera toxin and heat-labile enterotoxin activate human monocyte-derived dendritic cells and dominantly inhibit cytokine production through a cyclic AMP-dependent pathway. Infect. Immun. 70:5533–5539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Basset C, et al. 2010. Cholera-like enterotoxins and regulatory T cells. Toxins (Basel) 2:1774–1795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bopp T, et al. 2007. Cyclic adenosine monophosphate is a key component of regulatory T cell-mediated suppression. J. Exp. Med. 204:1303–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bowman CC, Clements JD. 2001. Differential biological and adjuvant activities of cholera toxin and Escherichia coli heat-labile enterotoxin hybrids. Infect. Immun. 69:1528–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brereton CF, et al. 2011. Escherichia coli heat-labile enterotoxin promotes protective Th17 responses against infection by driving innate IL-1 and IL-23 production. J. Immunol. 186:5896–5906 [DOI] [PubMed] [Google Scholar]
- 7. Cheng E, Cardenas-Freytag L, Clements JD. 1999. The role of cAMP in mucosal adjuvanticity of Escherichia coli heat-labile enterotoxin (LT). Vaccine 18:38–49 [DOI] [PubMed] [Google Scholar]
- 8. Chinnapen DJ, Chinnapen H, Saslowsky D, Lencer WI. 2007. Rafting with cholera toxin: endocytosis and trafficking from plasma membrane to ER. FEMS Microbiol. Lett. 266:129–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cieplak W, Jr, Messer RJ, Konkel ME, Grant CC. 1995. Role of a potential endoplasmic reticulum retention sequence (RDEL) and the Golgi complex in the cytotonic activity of Escherichia coli heat-labile enterotoxin. Mol. Microbiol. 16:789–800 [DOI] [PubMed] [Google Scholar]
- 10. Clements JD, El-Morshidy S. 1984. Construction of a potential live oral bivalent vaccine for typhoid fever and cholera-Escherichia coli-related diarrheas. Infect. Immun. 46:564–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Clements JD, Finkelstein RA. 1979. Isolation and characterization of homogeneous heat-labile enterotoxins with high specific activity from Escherichia coli cultures. Infect. Immun. 24:760–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Clements JD, Flint DC, Engert RF, Klipstein FA. 1983. Cloning and molecular characterization of the B subunit of Escherichia coli heat-labile enterotoxin. Infect. Immun. 40:653–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Clements JD, Hartzog NM, Lyon FL. 1988. Adjuvant activity of Escherichia coli heat-labile enterotoxin and effect on the induction of oral tolerance in mice to unrelated protein antigens. Vaccine 6:269–277 [DOI] [PubMed] [Google Scholar]
- 14. Collins JT, Shi J, Burrell BE, Bishop DK, Dunnick WA. 2006. Induced expression of murine gamma2a by CD40 ligation independently of IFN-gamma. J. Immunol. 177:5414–5419 [DOI] [PubMed] [Google Scholar]
- 15. Czerkinsky C, Holmgren J. 2012. Mucosal delivery routes for optimal immunization: targeting immunity to the right tissues. Curr. Top. Microbiol. Immunol. 354:1–18 [DOI] [PubMed] [Google Scholar]
- 16. Czerkinsky C, Holmgren J. 2010. Topical immunization strategies. Mucosal Immunol. 3:545–555 [DOI] [PubMed] [Google Scholar]
- 17. Datta SK, et al. 2010. Mucosal adjuvant activity of cholera toxin requires Th17 cells and protects against inhalation anthrax. Proc. Natl. Acad. Sci. U. S. A. 107:10638–10643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. de Haan L, Holtrop M, Verweij WR, Agsteribbe E, Wilschut J. 1999. Mucosal immunogenicity and adjuvant activity of the recombinant A subunit of the Escherichia coli heat-labile enterotoxin. Immunology 97:706–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dickinson BL, Clements JD. 1995. Dissociation of Escherichia coli heat-labile enterotoxin adjuvanticity from ADP-ribosyltransferase activity. Infect. Immun. 63:1617–1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Donaldson DS, Tong KK, Williams NA. 2011. Mucosal administration of the B subunit of E. coli heat-labile enterotoxin promotes the development of Foxp3-expressing regulatory T cells. Mucosal Immunol. 4:227–238 [DOI] [PubMed] [Google Scholar]
- 21. Fahlen-Yrlid L, et al. 2009. CD11c(high)dendritic cells are essential for activation of CD4+ T cells and generation of specific antibodies following mucosal immunization. J. Immunol. 183:5032–5041 [DOI] [PubMed] [Google Scholar]
- 22. Field M. 2003. Intestinal ion transport and the pathophysiology of diarrhea. J. Clin. Invest. 111:931–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Francis ML, et al. 1995. cAMP-independent effects of cholera toxin on B cell activation. III. Cholera toxin A subunit-mediated ADP-ribosylation acts synergistically with ionomycin or IL-4 to induce B cell proliferation. J. Immunol. 154:4956–4964 [PubMed] [Google Scholar]
- 24. Freytag LC, Clements JD. 2005. Mucosal adjuvants. Vaccine 23:1804–1813 [DOI] [PubMed] [Google Scholar]
- 25. Gerth AJ, Lin L, Peng SL. 2003. T-bet regulates T-independent IgG2a class switching. Int. Immunol. 15:937–944 [DOI] [PubMed] [Google Scholar]
- 26. Glenn GM, et al. 2007. Safety and immunogenicity of an enterotoxigenic Escherichia coli vaccine patch containing heat-labile toxin: use of skin pretreatment to disrupt the stratum corneum. Infect. Immun. 75:2163–2170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Guidry JJ, Cardenas L, Cheng E, Clements JD. 1997. Role of receptor binding in toxicity, immunogenicity, and adjuvanticity of Escherichia coli heat-labile enterotoxin. Infect. Immun. 65:4943–4950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gupta SK, et al. 2008. Part III. Analysis of data gaps pertaining to enterotoxigenic Escherichia coli infections in low and medium human development index countries, 1984–2005. Epidemiol. Infect. 136:721–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Holmgren J. 1973. Comparison of the tissue receptors for Vibrio cholerae and Escherichia coli enterotoxins by means of gangliosides and natural cholera toxoid. Infect. Immun. 8:851–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jaffar Z, Ferrini ME, Herritt LA, Roberts K. 2009. Cutting edge: lung mucosal Th17-mediated responses induce polymeric Ig receptor expression by the airway epithelium and elevate secretory IgA levels. J. Immunol. 182:4507–4511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kambayashi T, Wallin RP, Ljunggren HG. 2001. cAMP-elevating agents suppress dendritic cell function. J. Leukoc. Biol. 70:903–910 [PubMed] [Google Scholar]
- 32. Khan S, et al. 2007. Ability of SPI2 mutant of S. typhi to effectively induce antibody responses to the mucosal antigen enterotoxigenic E. coli heat labile toxin B subunit after oral delivery to humans. Vaccine 25:4175–4182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lai CY, Xia QC, Salotra PT. 1983. Location and amino acid sequence around the ADP-ribosylation site in the cholera toxin active subunit A1. Biochem. Biophys. Res. Commun. 116:341–348 [DOI] [PubMed] [Google Scholar]
- 34. Lavelle EC, et al. 2003. Cholera toxin promotes the induction of regulatory T cells specific for bystander antigens by modulating dendritic cell activation. J. Immunol. 171:2384–2392 [DOI] [PubMed] [Google Scholar]
- 35. Lee JB, Jang JE, Song MK, Chang J. 2009. Intranasal delivery of cholera toxin induces th17-dominated T-cell response to bystander antigens. PLoS One 4:e5190 doi:10.1371/journal.pone.0005190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lobet Y, Cluff CW, Cieplak W., Jr 1991. Effect of site-directed mutagenic alterations on ADP-ribosyltransferase activity of the A subunit of Escherichia coli heat-labile enterotoxin. Infect. Immun. 59:2870–2879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lu YJ, et al. 2010. Options for inactivation, adjuvant, and route of topical administration of a killed, unencapsulated pneumococcal whole-cell vaccine. Clin. Vaccine Immunol. 17:1005–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lutz MB, et al. 1999. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J. Immunol. Methods 223:77–92 [DOI] [PubMed] [Google Scholar]
- 39. Lycke N, Tsuji T, Holmgren J. 1992. The adjuvant effect of Vibrio cholerae and Escherichia coli heat-labile enterotoxins is linked to their ADP-ribosyltransferase activity. Eur. J. Immunol. 22:2277–2281 [DOI] [PubMed] [Google Scholar]
- 40. Magagnoli C, et al. 1996. Mutations in the A subunit affect yield, stability, and protease sensitivity of nontoxic derivatives of heat-labile enterotoxin. Infect. Immun. 64:5434–5438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McNeal MM, et al. 2007. Intrarectal immunization of mice with VP6 and either LT(R192G) or CTA1-DD as adjuvant protects against fecal rotavirus shedding after EDIM challenge. Vaccine 25:6224–6231 [DOI] [PubMed] [Google Scholar]
- 42. McNeal MM, et al. 2006. Protection against rotavirus shedding after intranasal immunization of mice with a chimeric VP6 protein does not require intestinal IgA. Virology 346:338–347 [DOI] [PubMed] [Google Scholar]
- 43. McNeal MM, et al. 2007. IFN-gamma is the only anti-rotavirus cytokine found after in vitro stimulation of memory CD4+ T cells from mice immunized with a chimeric VP6 protein. Viral Immunol. 20:571–584 [DOI] [PubMed] [Google Scholar]
- 44. Merritt EA, et al. 1994. Structure of partially-activated E. coli heat-labile enterotoxin (LT) at 2.6 A resolution. FEBS Lett. 337:88–92 [DOI] [PubMed] [Google Scholar]
- 45. Mitsdoerffer M, et al. 2010. Proinflammatory T helper type 17 cells are effective B-cell helpers. Proc. Natl. Acad. Sci. U. S. A. 107:14292–14297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mohr E, et al. 2010. IFN-{gamma} produced by CD8 T cells induces T-bet-dependent and -independent class switching in B cells in responses to alum-precipitated protein vaccine. Proc. Natl. Acad. Sci. U. S. A. 107:17292–17297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Negri DR, et al. 2009. Cholera toxin and Escherichia coli heat-labile enterotoxin, but not their nontoxic counterparts, improve the antigen-presenting cell function of human B lymphocytes. Infect. Immun. 77:1924–1935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Norton EB, Lawson LB, Freytag LC, Clements JD. 2011. Characterization of a mutant Escherichia coli heat-labile toxin, LT(R192G/L211A), as a safe and effective oral adjuvant. Clin. Vaccine Immunol. 18:546–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pande AH, et al. 2007. Conformational instability of the cholera toxin A1 polypeptide. J. Mol. Biol. 374:1114–1128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Petrovska L, et al. 2003. Modulation of dendritic cell endocytosis and antigen processing pathways by Escherichia coli heat-labile enterotoxin and mutant derivatives. Vaccine 21:1445–1454 [DOI] [PubMed] [Google Scholar]
- 51. Puga I, Cols M, Cerutti A. 2010. Innate signals in mucosal immunoglobulin class switching. J. Allergy Clin. Immunol. 126:889–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schnitzler AC, Burke JM, Wetzler LM. 2007. Induction of cell signaling events by the cholera toxin B subunit in antigen-presenting cells. Infect. Immun. 75:3150–3159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Smiley KL, et al. 2007. Association of gamma interferon and interleukin-17 production in intestinal CD4+ T cells with protection against rotavirus shedding in mice intranasally immunized with VP6 and the adjuvant LT(R192G). J. Virol. 81:3740–3748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Soriani M, Bailey L, Hirst TR. 2002. Contribution of the ADP-ribosylating and receptor-binding properties of cholera-like enterotoxins in modulating cytokine secretion by human intestinal epithelial cells. Microbiology 148:667–676 [DOI] [PubMed] [Google Scholar]
- 55. Summerton NA, et al. 2010. Toward the development of a stable, freeze-dried formulation of Helicobacter pylori killed whole cell vaccine adjuvanted with a novel mutant of Escherichia coli heat-labile toxin. Vaccine 28:1404–1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tacket CO, Pasetti MF, Edelman R, Howard JA, Streatfield S. 2004. Immunogenicity of recombinant LT-B delivered orally to humans in transgenic corn. Vaccine 22:4385–4389 [DOI] [PubMed] [Google Scholar]
- 57. Trepel JB, Chuang DM, Neff NH. 1977. Transfer of ADP-ribose from NAD to choleragen: a subunit acts as catalyst and acceptor protein. Proc. Natl. Acad. Sci. U. S. A. 74:5440–5442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Uddowla S, Freytag LC, Clements JD. 2007. Effect of adjuvants and route of immunizations on the immune response to recombinant plague antigens. Vaccine 25:7984–7993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wu HY, Russell MW. 1997. Nasal lymphoid tissue, intranasal immunization, and compartmentalization of the common mucosal immune system. Immunol. Res. 16:187–201 [DOI] [PubMed] [Google Scholar]
- 60. Yoshimoto T, et al. 2004. Induction of IgG2a class switching in B cells by IL-27. J. Immunol. 173:2479–2485 [DOI] [PubMed] [Google Scholar]