

Abstract
In mammalian cells, nitric oxide (NO·) is an important signal molecule with concentration-dependent and often controversial functions of promoting cell survival and inducing cell death. An inducible nitric oxide synthase (iNOS) in various mammalian cells produces higher levels of NO· from l-arginine upon infections to eliminate pathogens. In this study, we reveal novel pathogenic roles of NO· generated by bacteria in bacterium-host cell cocultures using Moraxella catarrhalis, a respiratory tract disease-causing bacterium, as a biological producer of NO·. We recently demonstrated that M. catarrhalis cells that express the nitrite reductase (AniA protein) can produce NO· by reducing nitrite. Our study suggests that, in the presence of pathophysiological levels of nitrite, this opportunistic pathogen hijacks host cell signaling and modulates host gene expression through its ability to produce NO· from nitrite. Bacterium-generated NO· significantly increases the secretion of tumor necrosis factor alpha (TNF-α) and modulates the expression of apoptotic proteins, therefore triggering host cell programmed death partially through TNF-α signaling. Furthermore, our study reveals that bacterium-generated NO· stalls host cell division and directly results in the death of dividing cells by reducing the levels of an essential regulator of cell division. This study provides unique insight into why NO· may exert more severe cytotoxic effects on fast growing cells, providing an important molecular basis for NO·-mediated pathogenesis in infections and possible therapeutic applications of NO·-releasing molecules in tumorigenesis. This study strongly suggests that bacterium-generated NO· can play important pathogenic roles during infections.
INTRODUCTION
In mammalian cells, nitric oxide (NO·) is a highly reactive and diffusible signaling molecule that plays key roles in modulating both physiological and pathological processes, such as immune response, cell survival, and cell death (reviewed in references 8 and 36). The diverse functions of NO· are often determined by its concentration or its source of production. Low levels of endogenous NO· are produced from l-arginine by constitutively expressed nitric oxide synthase in neuronal cells (nNOS, also known as NOS1) and endothelial cells (eNOS, also known as NOS3) to mediate normal physiological processes. Higher levels of NO· can be produced by an inducible nitric oxide synthase (iNOS, also known as NOS2) in different cell types, but mainly in macrophages, upon infection to kill pathogens (reviewed in reference 9). The higher levels of NO· produced by iNOS have been implicated in various human inflammatory diseases and neurodegenerative diseases (reviewed in references 9 and 12) and in chronic inflammation-related tumorigenesis (18, 33). Recent studies have also shown that chemical-generated higher levels of exogenous NO· can induce tumor cell apoptosis in vitro and in animal studies (37; reviewed in reference 1), raising interests in therapeutic exploration of NO·-releasing reagents as antitumor prodrugs.
On the other hand, a complete bacterial denitrification pathway involves a set of denitrification enzymes to sequentially reduce nitrate (NO3−) to gaseous nitrogen (N2), with NO· as an intermediate product (reviewed in reference 69). This complete denitrification pathway has been studied mostly as an alternative means of generating energy used by anaerobes under oxygen-limited or strict anaerobic growth conditions. Recently, various truncated denitrification pathways have been identified in several human pathogens, such as Mycobacterium tuberculosis (56), Neisseria meningitidis (3), and Neisseria gonorrhoeae (48). However, the possible involvement of NO· generated by bacteria in bacterial pathogenesis was not previously elucidated. Recently, we discovered and characterized a truncated denitrification pathway (see Fig. 1A) that was highly upregulated in Moraxella catarrhalis cells grown in a biofilm (64–66).
Fig 1.

M. catarrhalis truncated denitrification genes exert nitrite-dependent adverse effects on host cells. (A) The truncated denitrification pathway in M. catarrhalis involves reduction of nitrate (NO3−) to nitrite (NO2−) by the nitrate reductase complex NarGHJI, reduction of NO2− to nitric oxide (NO) by nitrite reductase AniA, and reduction of NO· to nitrous oxide (N2O) by nitric oxide reductase NorB. (B) Nitrite dose-dependent disruption of the host HBE cell monolayer by M. catarrhalis O35E. Microscopic images of Giemsa-stained HBE cells that were cocultured with wild-type M. catarrhalis O35E cells in medium containing different levels of nitrite (0, 100, 500, 1,000 and 5,000 μM) for 24 h. (C) Identification of the required M. catarrhalis denitrification enzyme for the nitrite-dependent disruption of a host cell monolayer. Microscopic images of HBE cells that were cocultured with wild-type M. catarrhalis O35E (wt) and O35E narGH mutant, O35E aniA mutant, and O35E norB mutant in MEM* containing 5 mM nitrite at 37·C in a CO2 incubator for 24 h. (D) Confirmation of the M. catarrhalis AniA protein responsible for NO·-mediated pathogenesis in vitro. Microscopic images of HBE cells that were cocultured with the mutant M. catarrhalis O35E aniA(pWW115) or the complemented mutant O35E aniA(pWW161) in MEM* containing 5 mM nitrite for 24 h. Each image in panels B to D shows a representative image from at least three repeated experiments. All microscopic images were captured using a 20× objective. Bars, 200 μm.
Moraxella catarrhalis is a Gram-negative obligate aerobe that has been recognized as one of the top three bacterial causes of the most common childhood infectious disease, acute otitis media (AOM) (reviewed in reference 45). M. catarrhalis forms biofilms on the middle ear mucosa in children with otitis media (29). In addition, M. catarrhalis is the second most common bacterial cause of exacerbations of chronic obstructive pulmonary disease (COPD) (46). M. catarrhalis infection increases airway inflammation and the levels of proinflammatory cytokines interleukin-8 (IL-8) and tumor necrosis factor alpha (TNF-α) in sputum samples from COPD patients (49, 54). It was unknown which bacterial gene function may affect host secretion of TNF-α in exacerbations of COPD caused by M. catarrhalis. Little is known about the pathogenic mechanism used by this opportunistic pathogen due to the lack of an animal model for studying M. catarrhalis pathogenesis.
Recent studies have shown that the M. catarrhalis nitrite reductase AniA (also known as a major anaerobically induced outer membrane protein) can produce NO· from nitrite (64, 66) and is most likely expressed in vivo (52). The NO· generated by the AniA protein is further detoxified by the M. catarrhalis nitric oxide reductase NorB (64). Importantly, a combined nitrate/nitrite concentration in middle ear effusion (MEE) from patients with otitis media was as high as 0.5 to ∼1 mM (35), providing in vivo substrates for bacterial denitrification enzymes to produce NO·.
In this study, we investigated the possible pathogenic effects of bacterium-generated NO· using the human bronchial epithelial cell line 16HBE14o (hereafter abbreviated as HBE) (17) as host cells in a bacterium-host cell interaction model. We utilized wild-type M. catarrhalis O35E and its well-defined denitrification mutant strains as biological producers of NO· or as controls. Our data show that in the presence of pathophysiological levels of nitrite, the M. catarrhalis cells exert adverse effects on the host cell monolayer and reduce host cell viability by the bacterium's ability to generate NO· from nitrite. Using state-of-the-art confocal microscopy live-cell imaging technology, we reveal that bacterium-generated NO· stalls host cell division, resulting in a cascade of apoptosis of dividing cells followed by death of the adjacent cell with apoptotic-cell-like morphological changes. These observations are further mechanistically supported by the results of investigating NO·-modulated host gene expression using human protein arrays. Our data show that bacterium-generated NO· significantly induces host cell secretion of TNF-α, elevates the levels of a crucial apoptosis effector (activated caspase-3) and decreases the levels of inhibitors of apoptosis proteins (IAPs, including cellular IAP 1 [cIAP1], cIAP2, X-linked inhibitor of apoptosis [XIAP], and survivin), therefore triggering host cell programmed death partially through TNF-α signaling. Importantly, the reduced levels of survivin, an essential physiological regulator of cell division (reviewed in reference 2), may be mechanistically linked to the observation of NO· stalling host cell division and inducing dividing cell apoptosis.
To the best of our knowledge, this is the first study to provide novel insight into important pathogenic roles of bacterium-generated NO·, including modulating host gene expression and host cell cycle, stimulating the secretion of the most potent cell death signaling molecule and the proinflammatory cytokine TNF-α, and inducing host cell death.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
M. catarrhalis strains used in this study are listed in Table 1. The base medium and bacterial culture conditions were described previously (64).
Table 1.
Bacterial strains and plasmids used in this study
| Bacterial strain or plasmid | Description or genotype | Source or reference |
|---|---|---|
| Bacterial strains | ||
| ATCC 43617 | Wild-type strain | ATCC |
| O35E | Wild-type strain | 31 |
| O35E narGH | A narGH deletion mutant of strain O35E | 65 |
| O35E aniA | The DNA sequence between primer pair WW217-WW218 (Fig. 1C) was replaced with a kan cartridge | 66 |
| O35E aniA(pWW115) | aniA::kan mutant containing a cloning vector pWW115 | This study |
| O35E aniA(pWW161) | aniA::kan mutant containing a wild-type aniA gene in trans | This study |
| O35E norB | The DNA sequence between primer pair WW248-WW349 (Fig. 1C) was replaced with a kan cartridge | 64 |
| ETSU-9 | Wild-type strain | Steven Berk |
| 7169 | Wild-type strain | Anthony Campagnari |
| Plasmids | ||
| pWW115 | Specr; cloning vector for M. catarrhalis | 63 |
| pWW161 | pWW115 containing the wild-type aniA gene of M. catarrhalis ATCC 43617 | This study |
Cocultures of M. catarrhalis and human airway epithelial cells.
HBE cells were routinely cultured in tissue culture medium MEM* [minimal essential medium (MEM) (Cellgro) containing 10% (vol/vol) heat-inactivated fetal bovine serum (FBS) (HyClone) and 1× Glutamax-I (Gibco)] with supplementation of 1× penicillin-streptomycin (MP Biomedicals, LLC) in a 5% CO2 tissue culture incubator at 37·C. M. catarrhalis-HBE cell cocultures (multiplicity of infection [MOI] of 10 to 50) were incubated for up to 24 h in MEM* with or without nitrite (up to 5 mM NaNO2). Human hemoglobin (1 mg/ml) was added as an NO· scavenger, caspase inhibitors (caspase-3 fmk inhibitor Z-DEVD-fmk [benzyloxycarbonyl-Asp-Glu-Val-Asp-fluoromethyl ketone] [R&D Systems], pancaspase fmk inhibitor Z-VAD-fmk [benzyloxycarbonyl-Val-Ala-dl-Asp-fluoromethyl ketone] [R&D Systems], or caspase fmk inhibitor control Z-FA-fmk [z-Phe-Ala-fluoromethyl ketone] [R&D Systems]) were added (to a final level of 100 μM) as appropriate.
Cloning of M. catarrhalis aniA gene for complementation study.
A DNA fragment containing the wild-type aniA gene was amplified using the oligonucleotide primer pairs CTGGATCCATGTTCATCTTGACACGCCAT and TGGAGCTCAATGAAAACCAGCGGAT together with the M. catarrhalis ATCC 43617 genomic DNA as the template. (The extra nucleotides encoding restriction digestion sites BamHI and SacI are underlined.) The PCR amplicon was digested with both BamHI and SacI and then ligated into cloning vector pWW115 (63) that had been digested with the same restriction enzymes. The ligation reaction mixture was used to transform M. catarrhalis O35E aniA mutant cells (66). A plasmid (designated pWW161) was isolated from one kanamycin- and spectinomycin-resistant transformant and was confirmed to contain an ATCC 43617 wild-type aniA gene by DNA sequencing and Western blotting (data not shown). The cloning vector pWW115 was also used to transform M. catarrhalis O35E aniA mutant cells to generate O35E aniA(pWW115) as a control.
Examination of the host HBE cell morphology.
In tissue cultures, a low population of dead cells (1 to 5%) is normally observed. Therefore, for unbiased observations, we routinely monitor the morphology of HBE cell monolayer using a low-magnification (20×) objective. The cells were fixed by incubation in methanol for 5 min and then stained with a modified Giemsa stain reagent (Sigma-Aldrich). Cell images were captured using a Olympus IX51 microscope (20× objective), an Olympus DP72 camera, and the manufacturer's imaging software (DP2-BSW [Olympus]).
Confocal microscopy study together with immunofluorescent labeling was included in the investigation of possible host cell death mechanisms. For confocal microscopy live-cell image studies, M. catarrhalis-HBE cells (MOI of ∼10) were cocultured in chambered coverglass slides in Dulbecco modified Eagle medium (DMEM) (GIBCO) containing 10% FBS (HyClone) and 1× Glutamax-I (GIBCO) with or without 1 mM nitrite. The cocultures were incubated at 37·C in a CO2 incubator (XLmulti S DARK LS [PECON]) and were imaged using Axio Observer Z.1 and a Yokogawa CSU-X1 Spinning Disk confocal microscope (Zeiss) (63× objective) in 10-min or 15-min intervals for up to 15 h. The following immunofluorescent dyes were also added to the media to distinguish apoptotic cells from necrotic cells: (i) a cell-permeant DNA dye, Hoechst 33258 (Invitrogen), to stain the nuclei (pseudocolored dark blue) of all cells; (ii) a cell-impermeant DNA dye, YOYO-1 iodide (Invitrogen), to detect extracellular DNA (pseudocolored green) that was released by necrotic cells or to costain cellular DNA (pseudocolored turquoise) of late apoptotic and necrotic cells that lost membrane integrity; and (iii) a cell-impermeant annexin V Alexa Fluor 594 conjugate (Invitrogen) to detect apoptotic cells (38, 61). Because early apoptotic cells lose membrane asymmetry resulting in the phosphatidylserine translocation to the outside of cells, annexin V specifically stains phosphatidylserine red (pseudocolor) to visualize apoptotic cells.
For fixed-slide confocal microscopy analysis, M. catarrhalis-HBE cell cocultures in chambered glass slides were fixed with 4% formaldehyde at 24 h postinfection. The host cell actin cytoskeleton was stained (pseudocolor) red with phalloidin-Alexa Fluor 594 conjugate (Invitrogen). Bacterial cells were labeled with the mouse monoclonal antibody 24B5 (15) against the M. catarrhalis outer membrane protein UspA1 and then stained (pseudocolored) green with Alexa Fluor 488-conjugated goat anti-mouse IgG antibody (Invitrogen). The ProLong Gold (Invitrogen) antifade reagent was used to mount coverslips. The fixed slides were imaged using the confocal microscope (100× objective).
Measurement of host HBE cell viability.
At 0 h and 24 h postinfection, HBE cells from each coculture were collected, and the live HBE cell counts were determined using an automated cell counter (Countess [Invitrogen]) following the manufacturer's protocol. This study was performed in three repeated infection experiments, and in each experiment, cocultures were performed in triplicate for statistical analysis.
Detection of the expression of inflammatory cytokines by HBE cells.
HBE cells, with or without infection (MOI of ∼10) with wild-type M. catarrhalis O35E or O35E aniA mutant cells, were cultured in MEM* with or without the addition of 1 mM nitrite. We examined the secretion of HBE cell inflammatory cytokines using a Proteome Profiler Human Cytokine Antibody Array (R&D Systems) following the manufacturer's protocol. The human cytokine antibody array (spotted in duplicate) blots from two repeated experiments were scanned and quantified using a FluorChemQ MultiImage III system and software (Alpha Innotech). The intensities of cytokine protein arrays were quantified using the FluorChemQ software, and the resulting arbitrary densitometry units were used as measurements of cytokine levels. The positive control of HBE cells cultured alone was used to normalize cytokine protein arrays. The relative levels of cytokines secreted by HBE cells that were cocultured with wild-type O35E cells or O35E aniA mutant cells versus HBE cells alone were calculated.
In addition, the levels of secreted TNF-α and IL-1α were measured using the LEGEND MAX Human TNF-α and IL-1α enzyme-linked immunosorbent assay (ELISA) kits (BioLegend, Inc.) following the manufacturer's protocols.
Determination of host apoptotic protein levels.
To determine host apoptotic protein levels, HBE cells were cocultured with M. catarrhalis O35E (wild type) or O35E aniA mutant cells (MOI of ∼10) in MEM* supplemented with 1 mM nitrite. At 24 h postinfection, the medium was removed, and cell lysates were prepared for detecting host apoptotic protein levels using the Proteome Profiler human apoptosis array kit (R&D Systems) following the manufacturer's protocol. The human apoptosis array blots from at least two repeated experiments were scanned using a FluorChemQ MultiImage III system (Alpha Innotech). The intensities of apoptotic protein arrays were quantified using the FluorChemQ software, and the resulting arbitrary densitometry units were used as measurements of apoptotic protein levels. The positive control of HBE cells cocultured with M. catarrhalis O35E aniA cells was used to normalize apoptotic protein arrays. The relative levels of apoptotic proteins in HBE cells cocultured with wild-type O35E cells versus with O35E aniA mutant cells were calculated.
RESULTS
Establishing an in vitro M. catarrhalis-human airway epithelial cell coculture model for studying M. catarrhalis pathogenesis.
M. catarrhalis is a strict human pathogen, and a workable animal model for studying M. catarrhalis pathogenesis remains to be developed. Chinchilla nasopharyngeal colonization studies showed that M. catarrhalis colonized the nasopharynxes of chinchillas without apparent disease symptoms (4, 32, 43). To investigate whether bacterium-generated NO· could be involved in pathogenesis, we optimized an in vitro system using HBE cells as the host in bacterium-host cell cocultures. After 24 h of coculture in MEM*, the number of CFU of M. catarrhalis O35E cells that grew attached to HBE cells was shown to be approximately 2 orders of magnitude higher than that of unattached cells [(6.8 ± 2.4) × 105 CFU/well and (5.8 ± 2.1) × 103 CFU/well, respectively], indicating that the majority (∼99.9%) of M. catarrhalis cells grow attached to host cells in biofilms (see the proposed pathogenic model of M. catarrhalis-generated NO· in Discussion).
When M. catarrhalis O35E-HBE cell cocultures were incubated in MEM* containing low levels of sodium nitrite, O35E cells had little or no apparent effect on the host cell monolayer in the presence of low levels (0 to 100 μM) of nitrite (Fig. 1B). At increased nitrite levels (500 μM or higher), M. catarrhalis O35E cells disrupted the newly developed monolayer of host cells in a nitrite dose-dependent manner, progressing from the edges to the inner area of the host cell monolayer (Fig. 1B). This study showed that M. catarrhalis O35E cells are able to disrupt a monolayer of host cells in MEM* containing nitrite at approximately 500 μM, a level equivalent to pathophysiological conditions (35).
Importantly, the nitrite-dependent disruption of the newly formed host cell monolayer was observed during coculturing with all wild-type M. catarrhalis strains tested, including M. catarrhalis 7169 and ETSU-9 (data not shown).
M. catarrhalis O35E AniA is responsible for nitrite-dependent disruption of host cell monolayer.
To identify the denitrifying enzyme of M. catarrhalis responsible for the observed adverse effect on host cells, the wild-type M. catarrhalis O35E strain and its denitrification-deficient mutant derivatives, O35E narGH (65), O35E aniA (66), and O35E norB (64) strains, were used to infect HBE cells. The wild-type M. catarrhalis O35E strain and narGH and norB mutant strains all expressed detectable levels of AniA protein in cocultures by Western blot analysis (data not shown), whereas the O35E aniA mutant did not express AniA protein (66). Infection with the above-mentioned M. catarrhalis O35E strains does not have an apparent effect on host cell monolayer in MEM* (data not shown). However, in MEM* containing a low level of nitrite, only the aniA mutant cells had no apparent effect on the morphology of a host cell monolayer at 24 h postinfection (Fig. 1C), suggesting that AniA is required for the nitrite-dependent disruption of a host cell monolayer. As expected, the monolayer of HBE cells was not affected by the addition of nitrite alone (Fig. 1C, HBEC-only panel).
To confirm that the M. catarrhalis AniA protein is required for the nitrite-dependent disruption of a host cell monolayer, we performed a complementation study. As expected, the M. catarrhalis O35E aniA strain carrying the plasmid vector did not have apparent harmful effects on a monolayer of host cells [Fig. 1D, aniA (pWW115) panel]. The O35E aniA mutant containing a wild-type aniA gene in trans regained the ability to exert deleterious nitrite-dependent effects on the host cell monolayer [Fig. 1D, aniA (pWW161) panel]. To the best of our knowledge, this is the first study identifying and directly confirming that a functional M. catarrhalis AniA protein is responsible for an opportunistic pathogenic effect of M. catarrhalis on the host cell monolayer exacerbated by the presence of nitrite.
Bacterium-generated NO· is responsible for reduced host cell viability.
The above study (Fig. 1) indicated that M. catarrhalis-expressed AniA might be responsible for the nitrite-dependent adverse effects on the host cell monolayer by reducing nitrite to NO· (64, 66). We monitored the viability of HBE cells to assess cytotoxicity. As expected, at 24 h postinfection, HBE viability was not significantly affected by nitrite when cultured alone (Fig. 2A, HBEC bars with and without nitrite [P < 0.184]) or cocultured with an M. catarrhalis O35E aniA mutant (Fig. 2A, aniA bars with and without nitrite [P < 0.438]). In contrast, when HBE cells were cocultured with AniA-expressing wild-type O35E or O35E norB mutant cells, live HBE cell counts were significantly lower (P < 0.034 or P < 0.003) in MEM* containing 5 mM nitrite (Fig. 2A, wt bars with and without nitrite) than in MEM* alone (without nitrite).
Fig 2.
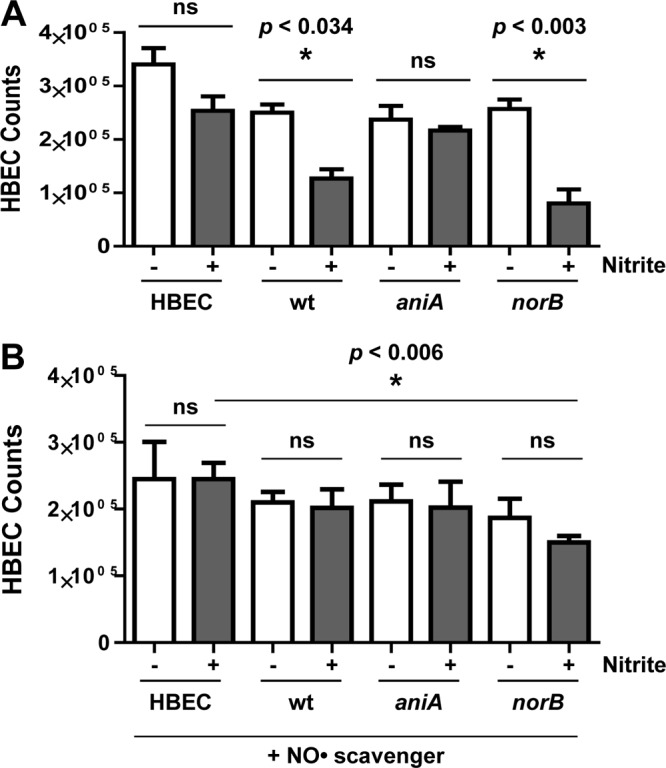
M. catarrhalis AniA-generated NO· reduces host cell viability. Viable HBE cell counts were determined in HBE cell culture (HBEC) or in cocultures with the wild-type M. catarrhalis O35E (wt) or O35E aniA mutant or norB mutant for 24 h in the following media: MEM* with (+) or without (−) 5 mM nitrite (A) or MEM* containing human hemoglobin (1 mg/ml) with (+) or without (−) 5 mM nitrite (B). ns, not significant.
To confirm that NO· is responsible for the AniA-mediated nitrite-dependent cytotoxicity, we added human hemoglobin as a major NO· scavenger (at a final concentration of 1 mg/ml) into the growth medium. As shown (Fig. 2B), supplementing with human hemoglobin was able to reverse most of the nitrite-dependent cytotoxicity to HBE cells (Fig. 2B), except that the HBE cell viability was still significantly lower (P < 0.006) when cocultured with O35E norB mutant cells (Fig. 2B, norB bar with nitrite) compared with HBE cells only in MEM* containing 5 mM nitrite. This study confirms that bacterium-generated NO· is responsible for observed nitrite-dependent pathological effects.
Furthermore, by utilizing cell culture inserts (Millipore) to physically separate M. catarrhalis cells from HBE cells but allowing NO· to diffuse into HBE cell culture, we observed reduced nitrite-dependent disruption of the host cell monolayer (data not shown), strongly suggesting that the colonization of M. catarrhalis on host cells is important for NO·-mediated pathological effects.
Bacterium-generated NO· stalls host cell division and induces the host cell to undergo programmed death.
One interesting observation that was overlooked initially may be important for understanding why NO·-mediated pathogenesis causes more severe damage on fast growing cells such as those in a newly developed monolayer. A well-developed HBE cell monolayer seemed to be more resistant to the disruption by bacterium-generated NO· (Fig. 3A, indicated by a solid black arrow), whereas the less-confluent and newly developed HBE cell monolayer (Fig. 3A, indicated by an open arrow) was severely damaged by bacterium-generated NO·.
Fig 3.

Dividing cells are more susceptible to the cytotoxicity of bacterium-generated NO·. (A) A photo of HBE cells cocultured with M. catarrhalis O35E cells in a T25 tissue culture flask and microscopic (20× objective) images of Giemsa-stained HBE cells in MEM* containing 1 mM nitrite. A fully confluent HBE cell monolayer (indicated by a solid black arrow) was not disrupted, whereas the newly developed HBE cell monolayer (indicated by an open arrow) was severely damaged by M. catarrhalis O35E. (B) Confocal microscopy live-cell images showed a commensal-like relationship of M. catarrhalis and HBE cells cocultured in MEM*. The infection of M. catarrhalis did not have apparent effects on HBE cell division. (C) Confocal microscopy live-cell images show M. catarrhalis exerting pathogenic effects on HBE cells in MEM* containing 1 mM nitrite. Dividing HBE cells failed to divide into two cells and became apoptotic (pseudocolored red) at 3 to 5 h postinfection (panel 1). HBE cells underwent secondary necrosis and released cellular contents, including DNA (YOYO-1 stained DNA green in panels 2 and 3). NO·-induced HBE cell death was associated with apoptotic-cell-like morphological changes (panels 2 and 3). The images in panels B and C were representative images from two replicate experiments. The confocal microscopic images in panels B and C were captured using a 63× objective at different time points after infection. Bar, 20 μm.
To explore whether dividing host cells are more susceptible to NO· and to investigate host cell death mechanisms, we performed a confocal microscopy live-cell image study to monitor the effects of bacterium-generated NO· on host cell morphological changes using immunofluorescent dyes to distinguish apoptotic cells from necrotic cells. The live-cell image study during infection is important, because it is known that apoptotic cells can eventually undergo secondary necrosis in tissue cultures due to the absence of a mechanism to remove apoptotic cells (10).
There were several interesting observations from the confocal microscopy live-cell image studies. When cocultured with wild-type M. catarrhalis O35E cells in MEM*, host cell division was apparently not affected, and dividing HBE cells were able to form two daughter cells (Fig. 3B). Interestingly, in medium containing 1 mM nitrite, dividing host cells moving around could not form daughter cells and became apoptotic (annexin V stains apoptotic cells red) at approximately 3 to 5 h postinfection (Fig. 3C, panel 1). At ∼8 h postinfection, the apoptotic host cells underwent secondary necrosis and released cellular contents, including nuclear DNA (YOYO-1 iodide stains nuclear DNA green [Fig. 3C, panels 2 and 3]). The released cellular contents were observed to be internalized by adjacent host cells and seemed to trigger death of adjacent cells (Fig. 3C, panels 2 and 3). During the different stages of infection, the confocal microscopy live-cell images captured the progression of host cell anoikis, a term for adherent cell apoptosis (27). Dying host cells underwent sequential morphological changes of apoptosis, such as detaching from adjacent cells and rounding, membrane blebbing, losing membrane asymmetry, and eventually losing membrane integrity (Fig. 3B, panels 2 and 3).
Bacterium-generated NO· induces host cell secretion of proinflammatory cytokines TNF-α and IL-1α.
To elucidate the possible molecular basis of observed pathogenic roles of bacterium-generated NO·, we investigated whether bacterium-generated NO· could affect host gene expression. We first utilized a Proteome Profiler human cytokine antibody array (R&D Systems) to examine the secretion of inflammatory cytokines by host HBE cells in MEM* with or without 1 mM nitrite.
In MEM* containing nitrite, the secretion of granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-8 was significantly elevated by host cells that were cocultured with wild-type M. catarrhalis O35E or aniA mutant cells at 24 h postinfection (Fig. 4A, black and gray GM-CSF and IL-8 bars [P values of 0.0011 and 0.0001]) compared with HBE cell culture alone (Fig. 4A, white GM-CSF and IL-8 bars). The secretion of GM-CSF and IL-8 was also increased at least 2-fold in MEM* without the supplementation of nitrite (data not shown), confirming that the secretion of GM-CSF and IL-8 by HBE cells can be stimulated by M. catarrhalis infection independent of bacterium-generated NO· (as reported previously [55]).
Fig 4.

Bacterium-generated NO· induces host cell secretion of proinflammatory cytokines TNF-α and IL-1α. (A) Media of 24-h cultures of HBE cells alone (HBEC) (white bars) or of HBE cells cocultured with wild-type M. catarrhalis O35E (black bars) or aniA mutant cells (gray bars) were used for detecting the levels of secreted cytokines by host cells using a human cytokine array in MEM* containing 1 mM nitrite. Values are means plus standard deviations (error bars) from two repeated experiments (each in duplicate). (B) Detection of the secreted IL-1α by host cells using LEGEND MAX human IL-1α ELISA kit in MEM* with (+) or without (−) 1 mM nitrite. (C) Detection of the secreted TNF-α by host cells using LEGEND MAX human TNF-α ELISA kit in MEM* with (+) or without (−) 1 mM nitrite. Values in panels B and C are means plus standard deviations from two repeated experiments (each in triplicate).
More importantly, our study revealed that the secretion of TNF-α and IL-1α was significantly elevated only in host cells cocultured with M. catarrhalis O35E in response to bacterium-generated NO· (Fig. 4A, black IL-1α and TNF-α bars, [P values of 0.0002 and 0.0008]). The infection with M. catarrhalis aniA mutant cells did not increase host cell secretion of IL-1α and TNF-α under the same coculturing condition (Fig. 4A, gray IL-1α and TNF-α bars).
To confirm that the bacterium-generated NO· stimulates the secretion of TNF-α and IL-1α, we further measured the levels of TNF-α and IL-1α using the LEGEND MAX Human TNF-α and IL-1α ELISA kits (BioLegend, Inc.). Again, the levels of secreted TNF-α and IL-1α were shown to be significantly elevated only under coculturing conditions when bacterial cells produced NO· from nitrite (Fig. 4B and C, black bars with nitrite). The secretion of IL-1α and TNF-α by host cells was not affected by M. catarrhalis infection in MEM* without the supplementation of nitrite (Fig. 4B and C, gray and black bars without nitrite). It should be noted that both IL-1α and TNF-α are the most potent NF-κB activators and major endogenous pyrogens (21). This study suggests that bacterium-generated NO· may be involved in increasing inflammation and producing fever by stimulating the production of proinflammatory cytokines IL-1α and TNF-α.
M. catarrhalis AniA-generated NO· affects the levels of host proteins that play key roles in programmed death.
To investigate M. catarrhalis O35E-induced nitrite-dependent host cell death mechanisms, we utilized a Proteome Profiler human apoptosis array kit (R&D Systems) to detect the levels of apoptotic proteins in HBE cells cocultured with wild-type O35E (Fig. 5, black bars) or O35E aniA mutant cells (Fig. 5, gray bars) in MEM* containing 1 mM nitrite. In HBE cells cocultured with wild-type O35E, the balance of proapoptotic and anti-apoptotic proteins in the extrinsic death pathway was shifted in favor of apoptosis. The relative level of activated caspase-3, one of the critical executioners of apoptosis (39, 41), was significantly increased (Fig. 5, black Casp-3* bar) (P < 0.0041), while the levels of anti-apoptotic proteins (reviewed in reference 2) cIAP1/2, XIAP, and survivin were significantly reduced (Fig. 5, black cIAP1, cIAP2, XIAP, and survivin bars [P values of 0.0001, 0.0002, 0.0009, and 0.0007]). The inactivated caspase-3 precursor was also increased significantly (Fig. 5, black pCasp3 bar) (P < 0.0064) in HBE cells cocultured with wild-type O35E. It should be noted that survivin is an essential regulator of cell division (reviewed in reference 2) and that cIAPs and XIAP can inhibit either apoptosis or necroptosis (25, 34).
Fig 5.

M. catarrhalis AniA-generated NO· affects the levels of host proteins that play key roles in programmed death. HBE cells were cocultured with wild-type M. catarrhalis O35E (black bars) or aniA mutant cells (gray bars) in MEM* containing 1 mM nitrite for 24 h. Cell lysates were used to determine the relative levels of apoptotic proteins using a human apoptosis protein array kit. Values are means plus standard deviations from at least two repeated experiments (each in duplicate). pCasp3, precursor of caspase-3; PC, positive control.
On the other hand, the expression of a cytoprotector heme oxygenase-1 (HO-1) (reviewed in references 22 and 28) was significantly increased (Fig. 5, black HO-1 bar) (P < 0.0074), and the level of Bad (Bcl-associated death promoter) was significantly reduced (Fig. 5, black Bad bar) (P < 0.0001) by M. catarrhalis AniA-generated NO·. These data demonstrate that NO· is able to mediate controversial functions simultaneously, promoting cell survival and inducing cell death.
The cytotoxicity of bacterium-generated NO· can be partially inhibited by a pancaspase inhibitor.
We further evaluated the effects of caspase inhibitors on NO·-mediated cytotoxicity. The addition of caspase-3 fmk inhibitor Z-DEVD-fmk at a final concentration of 100 μM provided little or no protection against bacterium-generated NO· cytotoxicity compared with the caspase fmk inhibitor control Z-FA-fmk (Fig. 6A, Z-FA and Z-DEVD bars). This result suggested that caspase-3 may not be the only apoptosis executioner in this study. The levels of other apoptosis executioners such as caspase-6 (16, 23) and caspase-7 (41, 62) were not examined in this study. Importantly, the addition of pancaspase fmk inhibitor Z-VAD-fmk at a final concentration of 100 μM resulted in a modest but statistically significant (P < 0.0128) increase in host cell viability (Fig. 6A, Z-VAD bar). This study confirms that bacterium-generated NO· reduces host cell viability by stalling cell division and inducing (at least a small population of) host cells to undergo caspase-dependent apoptosis.
Fig 6.

The cytotoxicity of bacterium-generated NO· can be partially inhibited by a pancaspase inhibitor. (A) HBE cells were cocultured with wild-type O35E in MEM* containing 1 mM nitrite and 100 μM caspase inhibitor control Z-FA-fmk, caspase-3 inhibitor Z-DEVD-fmk, or pancaspase inhibitor Z-VAD-fmk. The results shown are representative of data from three repeated experiments (each in triplicate). (B) A current working model of NO·-mediated bacterial pathogenesis. Bacterium-generated NO· may induce caspase-dependent and caspase-independent host cell death and modulate host cell gene expression through TNF-α signaling. The confocal microscopy images were captured using a 100× objective.
DISCUSSION
Nitric oxide (NO·) is a highly reactive and diffusible radical that modulates various physiological and pathological processes in mammalian cells. In order to mediate normal physiological processes, the endogenous level of NO· must be precisely maintained through the production and scavenging of NO·. Recently, bacterial denitrification has been investigated for biofilm dispersal (5) and possible involvement in virulence (reviewed in reference 50). However, the possible pathogenic involvement of bacterium-generated NO· remained to be elucidated.
Our study suggests that bacterium-generated NO· can change the bacterium-host cell interaction from commensal-like to pathogenic, mostly exacerbated by the presence of pathophysiological levels of nitrite. Our study also suggested that M. catarrhalis colonization is important for NO·-mediated pathogenesis. A current working model for M. catarrhalis AniA-mediated nitrite-dependent pathogenesis is depicted in Fig. 6B. In the absence of pathogenic levels of nitrite, bacterial cells (Fig. 6B, M. catarrhalis cells were stained green) colonize on the surfaces of host HBE cells (Fig. 6B, HBE cells were stained red) in biofilms without apparent disease symptoms (Fig. 6B, HBEC & wt panel). It is possible that when bacterial cells form biofilms directly on the surfaces of human airway epithelial cells, bacterium-generated NO· could reach a deleterious level in localized microenvironments. Previous studies showed that the higher levels of NO· generated by chemicals or by iNOS can induce apoptosis or necrosis (reviewed in references 6 and 36). Higher levels of NO· result in XIAP S-nitrosylation which consequently abolishes XIAP's anticaspase-3 activity (58).
Our data show that bacterium-generated NO· stimulates host cell secretion of TNF-α (Fig. 4A and C). TNF-α is one of the most potent activators of NF-κB and endogenous pyrogens (21). The increased levels of TNF-α or reduced IAPs can induce either apoptosis or programmed necrosis, which is newly characterized as a caspase-independent necroptotic death (selected publications and reviews in references 13, 25, 34, 51, and 60). Our studies showed that the addition of pancaspase inhibitor Z-VAD-fmk had only a modest inhibitory effect on NO·-induced host cell death, even though the confocal microscopy live-cell image study seemed to indicate that the levels of apoptotic death might be higher. It is possible that NO· also induces caspase-independent apoptosis (Fig. 6B) by damaging host DNA (7). Importantly, it should be noted that, while bacterium-generated NO· continually stimulates the secretion of elevated levels of TNF-α, the inhibition of caspase-dependent apoptosis with Z-VAD-fmk may induce a switch toward caspase-independent necroptosis (60).
Importantly, our data show that bacterium-generated NO· reduces the level of survivin (Fig. 5). It was also shown that chemical-generated NO· was able to reduce the level of survivin in lung cancer cells and induce apoptosis in vitro (13). Survivin has been shown to be a physiological regulator of cell division, and an increased level of this protein at mitosis is essential for cell division (reviewed in reference 2).
Clinical studies showed that higher levels of NO· were observed in the intestines of patients with infectious gastroenteritis (30) and in premature infants with necrotizing enterocolitis (NEC) (26). While infectious gastroenteritis is mostly acute and self-eliminating in adult patients, NEC is responsible for 1.8 million deaths in young children each year (reviewed in reference 24). NEC is a leading fatal intestinal disease in premature newborns, and NO· was suspected to play a key role in disruption of the gut epithelial barrier in NEC (14, 59). It is tempting to compare a fast developing monolayer of HBE cells with a fast developing epithelial barrier (for example, the length of the Eustachian tube doubles during the first few years of infancy) in young children and to assume that dividing epithelial cells in these populations are more vulnerable to NO·-mediated cytotoxicity. It might be understandable that higher levels of NO· (either iNOS or generated by bacteria) can exert more-noxious damage in infants and young children. In addition, this study may provide a novel insight into the molecular basis of NO·-releasing chemicals (to a certain degree) being able to differentially target the fast growing tumor cells from normal human cells in vitro (44).
Clinical studies reported that the M. catarrhalis infection in COPD patients is associated with increased levels of IL-8 and TNF-α (49, 54). The knowledge about how M. catarrhalis affects host secretion of TNF-α was previously absent. Studies showed that the secretion of IL-8 and GM-CSF by human bronchial epithelial cells can be stimulated by either wild-type M. catarrhalis O35E cells or by the addition of both purified TNF-α and IL-1β (55). The M. catarrhalis lipooligosaccharides (LOS) (68) and the outer membrane protein UspA1 might be involved in stimulating the secretion of IL-8 in vitro (57), and MID/Hag might be involved in B-cell activation (47, 67). Importantly, serum samples from COPD patients who had recently cleared M. catarrhalis infection are reactive to M. catarrhalis LOS (53) and MID/Hag (40), indicating that M. catarrhalis extracellular components may contribute to the production of inflammatory disease. To the best of our knowledge, this is the first study to reveal that the M. catarrhalis AniA-generated NO· stimulates host cell secretion of TNF-α and IL-1α. Both TNF-α and IL-1α are potent NF-κB activators and major endogenous pyrogens (19). It has been shown that subnanomolar concentrations of IL-1 could produce fever in animals and humans (20). Our study suggests that M. catarrhalis AniA-generated NO· may be involved in increasing inflammation and producing fever in M. catarrhalis diseases.
Collectively, our data indicate that, in addition to stalling host cell division, bacterium-generated NO· induces host cell programmed death through TNF-α signaling with increased levels of proapoptotic proteins (including TNF-α and activated caspase-3) as well as decreased levels of antiapoptotic proteins (including XIAP, Survivin, and cIAP1/2). This study suggests that, in the presence of pathophysiological levels of nitrite, M. catarrhalis may employ a simple yet effective NO· production mechanism to fulfill contact-efficient virulence factor delivery and interkingdom signaling.
Our study also revealed an opposite function of NO·, promoting host cell survival by inducing the expression of the cytoprotector HO-1 and reducing the expression of Bad (Fig. 2B). Studies showed that chemical-generated NO· stimulates HO-1 expression in smooth muscle cells to promote cell survival (42), and HO-1 protects endothelial cells from TNF-α-induced apoptosis (11). It is important to further explore potential beneficial effects of bacterium-generated NO· on bacterium-host cell interaction. We are actively investigating how M. catarrhalis denitrification gene expression is regulated and whether the M. catarrhalis AniA protein is a potential vaccine candidate.
ACKNOWLEDGMENTS
This study was supported by FDA/CBER operation fund to W. Wang.
We thank Eric J. Hansen for providing plasmid pWW161 and monoclonal antibody 24B5 and supporting the early stage of M. catarrhalis denitrification study. We thank Ferric C. Fang for contributions to the concept of NO· scavenging and studying the possible host cell death mechanisms as well as critical discussions during the manuscript preparation. We thank Dieter C. Gruenert for providing HBE cells and John Nelson, Anthony Campagnari, and Steven Berk for providing clinical isolates of M. catarrhalis. We thank Kazuyo Takeda at the FDA/CBER confocal microscope facility for capturing confocal microscopic images and Sandra Small and Flora Lichaa for technical support.
Footnotes
Published ahead of print 25 May 2012
REFERENCES
- 1. Albina JE, Reichner JS. 1998. Role of nitric oxide in mediation of macrophage cytotoxicity and apoptosis. Cancer Metastasis Rev. 17:39–53 [DOI] [PubMed] [Google Scholar]
- 2. Altieri DC. 2010. Survivin and IAP proteins in cell-death mechanisms. Biochem. J. 430:199–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Anjum MF, Stevanin TM, Read RC, Moir JW. 2002. Nitric oxide metabolism in Neisseria meningitidis. J. Bacteriol. 184:2987–2993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bakaletz LO, Murwin DM, Billy JM. 1995. Adenovirus serotype 1 does not act synergistically with Moraxella (Branhamella) catarrhalis to induce otitis media in the chinchilla. Infect. Immun. 63:4188–4190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Barraud N, et al. 2006. Involvement of nitric oxide in biofilm dispersal of Pseudomonas aeruginosa. J. Bacteriol. 188:7344–7353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Benhar M, Stamler JS. 2005. A central role for S-nitrosylation in apoptosis. Nat. Cell Biol. 7:645–646 [DOI] [PubMed] [Google Scholar]
- 7. Bentz BG, Hammer ND, Radosevich JA, Haines GK. 2004. Nitrosative stress induces DNA strand breaks but not caspase mediated apoptosis in a lung cancer cell line. J. Carcinog. 3:16 doi:10.1186/1477-3163-3-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Blaise GA, Gauvin D, Gangal M, Authier S. 2005. Nitric oxide, cell signaling and cell death. Toxicology 208:177–192 [DOI] [PubMed] [Google Scholar]
- 9. Blantz RC, Munger K. 2002. Role of nitric oxide in inflammatory conditions. Nephron 90:373–378 [DOI] [PubMed] [Google Scholar]
- 10. Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA. 1995. Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-d-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc. Natl. Acad. Sci. U. S. A. 92:7162–7166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brouard S, et al. 2002. Heme oxygenase-1-derived carbon monoxide requires the activation of transcription factor NF-kappa B to protect endothelial cells from tumor necrosis factor-alpha-mediated apoptosis. J. Biol. Chem. 277:17950–17961 [DOI] [PubMed] [Google Scholar]
- 12. Brown GC. 2010. Nitric oxide and neuronal death. Nitric Oxide 23:153–165 [DOI] [PubMed] [Google Scholar]
- 13. Chao JI, Kuo PC, Hsu TS. 2004. Down-regulation of survivin in nitric oxide-induced cell growth inhibition and apoptosis of the human lung carcinoma cells. J. Biol. Chem. 279:20267–20276 [DOI] [PubMed] [Google Scholar]
- 14. Chokshi NK, et al. 2008. The role of nitric oxide in intestinal epithelial injury and restitution in neonatal necrotizing enterocolitis. Semin. Perinatol. 32:92–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cope LD, et al. 1999. Characterization of the Moraxella catarrhalis uspA1 and uspA2 genes and their encoded products. J. Bacteriol. 181:4026–4034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cowling V, Downward J. 2002. Caspase-6 is the direct activator of caspase-8 in the cytochrome c-induced apoptosis pathway: absolute requirement for removal of caspase-6 prodomain. Cell Death Differ. 9:1046–1056 [DOI] [PubMed] [Google Scholar]
- 17. Cozens AL, et al. 1994. CFTR expression and chloride secretion in polarized immortal human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 10:38–47 [DOI] [PubMed] [Google Scholar]
- 18. Cronauer MV, et al. 2007. Nitric oxide-mediated inhibition of androgen receptor activity: possible implications for prostate cancer progression. Oncogene 26:1875–1884 [DOI] [PubMed] [Google Scholar]
- 19. Dinarello CA. 1999. Cytokines as endogenous pyrogens. J. Infect. Dis. 179(Suppl 2):S294–S304 [DOI] [PubMed] [Google Scholar]
- 20. Dinarello CA. 2004. Infection, fever, and exogenous and endogenous pyrogens: some concepts have changed. J. Endotoxin Res. 10:201–222 [DOI] [PubMed] [Google Scholar]
- 21. Dinarello CA, et al. 1986. Tumor necrosis factor (cachectin) is an endogenous pyrogen and induces production of interleukin 1. J. Exp. Med. 163:1433–1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Durante W. 2011. Protective role of heme oxygenase-1 against inflammation in atherosclerosis. Front. Biosci. 17:2372–2388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Eguchi R, et al. 2009. Possible involvement of caspase-6 and -7 but not caspase-3 in the regulation of hypoxia-induced apoptosis in tube-forming endothelial cells. Exp. Cell Res. 315:327–335 [DOI] [PubMed] [Google Scholar]
- 24. Elliott EJ. 2007. Acute gastroenteritis in children. BMJ 334:35–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Feoktistova M, et al. 2011. cIAPs block ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol. Cell 43:449–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ford H, Watkins S, Reblock K, Rowe M. 1997. The role of inflammatory cytokines and nitric oxide in the pathogenesis of necrotizing enterocolitis. J. Pediatr. Surg. 32:275–282 [DOI] [PubMed] [Google Scholar]
- 27. Frisch SM, Francis H. 1994. Disruption of epithelial cell-matrix interactions induces apoptosis. J. Cell Biol. 124:619–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gozzelino R, Jeney V, Soares MP. 2010. Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 50:323–354 [DOI] [PubMed] [Google Scholar]
- 29. Hall-Stoodley L, et al. 2006. Direct detection of bacterial biofilms on the middle-ear mucosa of children with chronic otitis media. JAMA 296:202–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hausladen A, Gow AJ, Stamler JS. 1998. Nitrosative stress: metabolic pathway involving the flavohemoglobin. Proc. Natl. Acad. Sci. U. S. A. 95:14100–14105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Helminen ME, et al. 1993. A major outer membrane protein of Moraxella catarrhalis is a target for antibodies that enhance pulmonary clearance of the pathogen in an animal model. Infect. Immun. 61:2003–2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hoopman TC, et al. 2012. Use of the chinchilla model for nasopharyngeal colonization to study gene expression by Moraxella catarrhalis. Infect. Immun. 80:982–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hussain SP, et al. 2008. Nitric oxide is a key component in inflammation-accelerated tumorigenesis. Cancer Res. 68:7130–7136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Imre G, Larisch S, Rajalingam K. 2011. Ripoptosome: a novel IAP-regulated cell death-signalling platform. J. Mol. Cell Biol. 3:324–326 [DOI] [PubMed] [Google Scholar]
- 35. John EO, Russell PT, Nam BH, Jinn TH, Jung TT. 2001. Concentration of nitric oxide metabolites in middle ear effusion. Int. J. Pediatr. Otorhinolaryngol. 60:55–58 [DOI] [PubMed] [Google Scholar]
- 36. Kim YM, Bombeck CA, Billiar TR. 1999. Nitric oxide as a bifunctional regulator of apoptosis. Circ. Res. 84:253–256 [DOI] [PubMed] [Google Scholar]
- 37. Kiziltepe T, et al. 2007. JS-K, a GST-activated nitric oxide generator, induces DNA double-strand breaks, activates DNA damage response pathways, and induces apoptosis in vitro and in vivo in human multiple myeloma cells. Blood 110:709–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Koopman G, et al. 1994. Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood 84:1415–1420 [PubMed] [Google Scholar]
- 39. Kothakota S, et al. 1997. Caspase-3-generated fragment of gelsolin: effector of morphological change in apoptosis. Science 278:294–298 [DOI] [PubMed] [Google Scholar]
- 40. LaFontaine ER, et al. 2009. Identification of domains of the Hag/MID surface protein recognized by systemic and mucosal antibodies in adults with chronic obstructive pulmonary disease following clearance of Moraxella catarrhalis. Clin. Vaccine Immunol. 16:653–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lakhani SA, et al. 2006. Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science 311:847–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu XM, et al. 2007. Nitric oxide stimulates heme oxygenase-1 gene transcription via the Nrf2/ARE complex to promote vascular smooth muscle cell survival. Cardiovasc. Res. 75:381–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Luke NR, Jurcisek JA, Bakaletz LO, Campagnari AA. 2007. Contribution of Moraxella catarrhalis type IV pili to nasopharyngeal colonization and biofilm formation. Infect. Immun. 75:5559–5564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McMurtry V, et al. 2011. JS-K, a nitric oxide-releasing prodrug, induces breast cancer cell death while sparing normal mammary epithelial cells. Int. J. Oncol. 38:963–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Murphy TF. 2004. Moraxella (Branhamella) catarrhalis and other gram-negative cocci, p 2529 In Mandell GL, Bennett JE, Dolin R. (ed), Mandell, Douglas, Bennett's principles and practice of infectious diseases, 6th ed Elsevier Inc, Philadelphia, PA [Google Scholar]
- 46. Murphy TF, Brauer AL, Grant BJ, Sethi S. 2005. Moraxella catarrhalis in chronic obstructive pulmonary disease: burden of disease and immune response. Am. J. Respir. Crit. Care Med. 172:195–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nordstrom T, Jendholm J, Samuelsson M, Forsgren A, Riesbeck K. 2006. The IgD-binding domain of the Moraxella IgD-binding protein MID (MID962-1200) activates human B cells in the presence of T cell cytokines. J. Leukoc. Biol. 79:319–329 [DOI] [PubMed] [Google Scholar]
- 48. Overton TW, et al. 2006. Coordinated regulation of the Neisseria gonorrhoeae-truncated denitrification pathway by the nitric oxide-sensitive repressor, NsrR, and nitrite-insensitive NarQ-NarP. J. Biol. Chem. 281:33115–33126 [DOI] [PubMed] [Google Scholar]
- 49. Parameswaran GI, Wrona CT, Murphy TF, Sethi S. 2009. Moraxella catarrhalis acquisition, airway inflammation and protease-antiprotease balance in chronic obstructive pulmonary disease. BMC Infect. Dis. 9:178 doi:10.1186/1471-2334-9-178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Philippot L. 2005. Denitrification in pathogenic bacteria: for better or worst? Trends Microbiol. 13:191–192 [DOI] [PubMed] [Google Scholar]
- 51. Rath PC, Aggarwal BB. 1999. TNF-induced signaling in apoptosis. J. Clin. Immunol. 19:350–364 [DOI] [PubMed] [Google Scholar]
- 52. Ruckdeschel EA, Kirkham C, Lesse AJ, Hu Z, Murphy TF. 2008. Mining the Moraxella catarrhalis genome: identification of potential vaccine antigens expressed during human infection. Infect. Immun. 76:1599–1607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schwingel JM, et al. 2009. The use of Moraxella catarrhalis lipooligosaccharide mutants to identify specific oligosaccharide epitopes recognized by human serum antibodies. Infect. Immun. 77:4548–4558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sethi S, et al. 2000. Airway inflammation and etiology of acute exacerbations of chronic bronchitis. Chest 118:1557–1565 [DOI] [PubMed] [Google Scholar]
- 55. Slevogt H, et al. 2006. Moraxella catarrhalis induces inflammatory response of bronchial epithelial cells via MAPK and NF-kappaB activation and histone deacetylase activity reduction. Am. J. Physiol. Lung Cell. Mol. Physiol. 290:L818–L826 [DOI] [PubMed] [Google Scholar]
- 56. Sohaskey CD, Wayne LG. 2003. Role of narK2X and narGHJI in hypoxic upregulation of nitrate reduction by Mycobacterium tuberculosis. J. Bacteriol. 185:7247–7256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Spaniol V, Troller R, Aebi C. 2009. Physiologic cold shock increases adherence of Moraxella catarrhalis to and secretion of interleukin 8 in human upper respiratory tract epithelial cells. J. Infect. Dis. 200:1593–1601 [DOI] [PubMed] [Google Scholar]
- 58. Tsang AH, et al. 2009. S-nitrosylation of XIAP compromises neuronal survival in Parkinson's disease. Proc. Natl. Acad. Sci. U. S. A. 106:4900–4905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Upperman JS, et al. 2005. Mechanisms of nitric oxide-mediated intestinal barrier failure in necrotizing enterocolitis. Semin. Pediatr. Surg. 14:159–166 [DOI] [PubMed] [Google Scholar]
- 60. Vanlangenakker N, Bertrand MJ, Bogaert P, Vandenabeele P, Berghe TV. 2011. TNF-induced necroptosis in L929 cells is tightly regulated by multiple TNFR1 complex I and II members. Cell Death Dis. 2:e230 doi:10.1038/cddis.2011.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. 1995. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J. Immunol. Methods 184:39–51 [DOI] [PubMed] [Google Scholar]
- 62. Walsh JG, et al. 2008. Executioner caspase-3 and caspase-7 are functionally distinct proteases. Proc. Natl. Acad. Sci. U. S. A. 105:12815–12819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wang W, Hansen EJ. 2006. Plasmid pWW115, a cloning vector for use with Moraxella catarrhalis. Plasmid 56:133–137 [DOI] [PubMed] [Google Scholar]
- 64. Wang W, et al. 2011. The Moraxella catarrhalis nitric oxide reductase is essential for nitric oxide detoxification. J. Bacteriol. 193:2804–2813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wang W, et al. 2007. Metabolic analysis of Moraxella catarrhalis and the effect of selected in vitro growth conditions on global gene expression. Infect. Immun. 75:4959–4971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wang W, et al. 2008. Identification of a repressor of a truncated denitrification pathway in Moraxella catarrhalis. J. Bacteriol. 190:7762–7772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wingren AG, Hadzic R, Forsgren A, Riesbeck K. 2002. The novel IgD binding protein from Moraxella catarrhalis induces human B lymphocyte activation and Ig secretion in the presence of Th2 cytokines. J. Immunol. 168:5582–5588 [DOI] [PubMed] [Google Scholar]
- 68. Xie H, Gu XX. 2008. Moraxella catarrhalis lipooligosaccharide selectively upregulates ICAM-1 expression on human monocytes and stimulates adjacent naive monocytes to produce TNF-alpha through cellular cross-talk. Cell. Microbiol. 10:1453–1467 [DOI] [PubMed] [Google Scholar]
- 69. Zumft WG. 1997. Cell biology and molecular basis of denitrification. Microbiol. Mol. Biol. Rev. 61:533–616 [DOI] [PMC free article] [PubMed] [Google Scholar]