

Abstract
The bifunctional major autolysin Atl plays a key role in staphylococcal cell separation. Processing of Atl yields catalytically active amidase (AM) and glucosaminidase (GL) domains that are each fused to repeating units. The two repeats of AM (R1 and R2) target the enzyme to the septum, where it cleaves murein between dividing cells. We have determined the crystal structure of R2, which reveals that each repeat folds into two half-open β-barrel subunits. We further demonstrate that lipoteichoic acid serves as a receptor for the repeats and that this interaction depends on conserved surfaces in each subunit. Small-angle X-ray scattering of the mature amidase reveals the presence of flexible linkers separating the AM, R1, and R2 units. Different levels of flexibility for each linker provide mechanistic insights into the conformational dynamics of the full-length protein and the roles of its components in cell wall association and catalysis. Our analysis supports a model in which the repeats direct the catalytic AM domain to the septum, where it can optimally perform the final step of cell division.
INTRODUCTION
Peptidoglycan (PGN), also known as murein, is the main constituent of bacterial cell walls and must be continuously synthesized and degraded to maintain the integrity and viability of cells. The Gram-positive cell wall possesses a number of cell wall glycopolymers, among them the PGN-attached wall teichoic acid (WTA) and the membrane-bound lipoteichoic acid (LTA) (for an excellent review, see Weidenmaier and Peschel [49]). During cell growth, enzymatic cleavage of the existing PGN structure is followed by incorporation of newly synthesized PGN subunits (18). Proper maintenance of the cell wall, therefore, requires a careful balance between PGN synthesis and degradation, and bacteria employ an arsenal of enzymes that act on cell wall components. Enzymes that cleave cell walls are collectively referred to as autolysins and play central roles in cell wall turnover, cell separation, and the lysis of bacteria by β-lactam antibiotics. Autolysins are murein hydrolases and, in many cases, N-acetylmuramyl-l-alanine amidases. Some murein hydrolases are involved in cleaving PGN structures that still link daughter cells following cell division. Amidases of this type are present in Gram-negative, as well as Gram-positive, bacteria.
In Staphylococcus aureus and Staphylococcus epidermidis, the major murein hydrolase involved in cell separation is named Atl (15). atl deletion mutants form large cell clusters in which the walls of individual cells appear to be interlinked with other cells, indicating a severe defect in cell separation (6). Atl enzymes of both species have the same modular organization (signal peptide, propeptide [PP], amidase, three major repeats, and glucosaminidase). AtlA and AtlE, furthermore, share a high degree of sequence similarity and are functionally interchangeable (6). Atl is secreted in a Sec protein-dependent manner as a bifunctional precursor that is composed of two distinct murein hydrolases, an N-acetylmuramyl-l-alanine amidase (AM) (6) and a glucosaminidase (GL) (Fig. 1). The AM and GL domains are separated by three major repeats, R1 to R3. Following cleavage of the signal peptide, external Atl is proteolytically cleaved at two positions, after the PP and after repeat R2, leading to the formation of processed enzymes AM-R1-R2 and R3-GL (15). Both enzymes bind to the staphylococcal cell wall, particularly to the septum sites of dividing cells (22, 50). The repeat sequences are responsible for the targeting of the enzymes to the septum region (2, 38). A first insight into how the Atl amidase AM-R1-R2 is targeted to the septum has been described recently. It was shown that WTA, which is present in the old cell wall but not yet accessible in the septum region, prevents binding of repeats or repeats linked to the catalytic AM domain (38). This finding is in line with the results obtained with the Bacillus subtilis endopeptidase LytF, the binding of which can be prevented by the major and minor WTAs of the sidewall (51).
Fig 1.

Domain arrangement of the bifunctional Atl precursor protein. SP, signal peptide; PP, propeptide; AM/GL (cat), catalytic domains of amidase and glucosaminidase; R, repeat domains; L, linker. The arrows indicate sites of posttranslational processing. Numbers giving the positions of amino acids in pre-Atl mark the start of each domain. The starting points of R1a, R1b, R3a, R3b, and GL were deduced from sequence alignments with R2a and R2b. Structural information on the following amidase variants was obtained: R2ab (crystal structure); R1ab (model); and AM-R1ab-R2ab, AM-R1ab, and R1ab-R2ab (solution structures).
atl mutants are biofilm negative (14) on polystyrene surfaces and attenuated in virulence in a rat intravascular-catheter-associated infection model (37). Furthermore, AM-R1-R2 binds to vitronectin and fibronectin (15), and very recently, it has been shown that Atl is involved in host cell internalization by interacting with heat shock cognate protein Hsc70 receptor (16).
A crystal structure for catalytically active AM is available, and its enzymatic mechanism and substrate binding properties have been elucidated (52). AM adopts a globular fold and binds a muramyltripeptide sequence in a groove on one side of the protein. Cleavage by zinc-dependent hydrolysis occurs between the glycan and the d-alanine of the substrate (6). However, the structure-function analysis of AM did not shed light on the role and organization of the repeats R1 and R2. Regarding the surface localization of AM-R1-R2, it was only known that WTA in the old cell wall acts as a repellant for the repeats and thereby directs the amidase to the septum region (38). It was also established that AM-R1-R2 binds to PGN (6).
Nevertheless, it is highly questionable whether PGN, which is likely exposed in the septum region, can function as a receptor for the repeats. PGN is the substrate of AM-R1-R2 and would therefore permanently lose its receptor function with progressive degradation. We therefore hypothesized that R1 and R2 might interact with another cell wall structure as a receptor. In order to establish the structural organization and substrate specificity of the AM repeats, we determined the crystal structure of the repeat R2. Based on the structural analysis, we designed repeat mutants and evaluated their ability to engage staphylococcal cell wall components. Our mutagenesis data indicate that LTA interacts with the AM repeats in an early step of cell separation, thereby helping to position the amidase at future sites of cell division. Furthermore, we calculated structure-based sequence alignments that revealed the presence of linker regions between AM and R1 (L1) and between R1 and R2 (L2) (Fig. 1). Given the different functions of AM (cleavage) and R1-R2 (adherence), these linkers likely serve to facilitate movement of the individual domains with respect to each other in order to access binding partners. The structure and conformational dynamics of AM-R1-R2 in solution were further characterized by small-angle X-ray scattering (SAXS). The two linkers, L1 and L2, were indeed found to be flexible, but with different degrees of flexibility. In combination with the functional and crystallographic data, the SAXS analysis results in a model in which the individual regions of AM-R1-R2 are each able to favorably interact with LTA and PGN in the staphylococcal cell wall, thus targeting the enzyme to the proper location and maximizing cleavage efficiency.
MATERIALS AND METHODS
Bacterial strains, plasmids, media, and growth conditions.
The bacterial strains and plasmids used in this study are listed in Table S1 in the supplemental material. Unless otherwise indicated, Escherichia coli and staphylococcal strains were grown at 37°C in LB broth (1% tryptone, 0.5% yeast extract, 0.5% NaCl) and BM broth (LB broth supplemented with 0.1% K2HPO4 and 0.1% glucose), respectively. Selective media were supplemented with ampicillin (100 μg ml−1) or chloramphenicol (10 μg ml−1).
Protein expression and purification.
A DNA fragment encoding AtlE residues 516 to 845 was amplified from S. epidermidis O-47 genomic DNA using PCR and cloned into the pGEX 4T-3 expression plasmid (GE Healthcare) via the BamHI and XhoI restriction sites. These residues were predicted to encode the R1ab-R2ab segment (15). Overexpression of R1ab-R2ab fused to a thrombin-cleavable N-terminal glutathione S-transferase (GST) tag was performed in E. coli BL21(DE3). After induction of protein expression with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside), the bacteria were grown for 12 h at 22°C. Soluble fusion protein was purified by GST affinity chromatography on GSTrap FF (GE Healthcare). After cleavage of the tag with thrombin, the protein was subjected to size exclusion chromatography on Superdex 75 (GE Healthcare) to remove GST and thrombin. For crystallization, lysine residues of the purified protein were reductively methylated to improve its crystallization properties (48). For the methylation reaction, 1 ml of the protein solution (<0.5 mg ml−1) was mixed with 40 μl of 1 M formaldehyde and 20 μl of 1 M dimethylamine borane complex (DMAB) as a reducing agent. The same amounts of formaldehyde and DMAB were added twice every 2 h, while the reaction mixture was kept at 4°C. In the last step, only 10 μl of DMAB was added, and the reaction mixture was incubated for an additional 12 h. Aggregates were removed using size exclusion chromatography (Superdex 75). Successful methylation was assessed by a shift in molecular weight relative to the unmethylated protein in SDS-PAGE.
In addition to the protein used for crystallization, two longer amidase constructs were expressed and purified for SAXS measurements. DNA fragments encoding AtlE residues 309 to 681 (AM-R1ab) and 303 to 845 (AM-R1ab-R2ab) were PCR amplified from S. epidermidis O-47 genomic DNA. The AM-R1ab-encoding DNA fragment was cloned into the pET52b expression plasmid (Merck Chemicals) using SmaI and SacI restriction sites. The protein was overexpressed in E. coli BL21(DE3) fused to a human rhinovirus (HRV) 3C-cleavable Strep tag. The bacteria were grown for 3 days at 20°C in autoinduction medium (40). The soluble fusion protein was purified by Strep tag affinity chromatography on a StrepTrap HP column (GE Healthcare). In order to remove the tag, the fusion protein was applied to a GSTrap column containing immobilized PreScission Protease (GE Healthcare). Cleavage was complete after incubation for 3 h at 4°C. The final purification step was size exclusion chromatography on Superdex 75. Cloning, expression, and purification of AM-R1ab-R2ab were conducted essentially as described for R1ab-R2ab. Following affinity chromatography, the protein was further purified by two size exclusion chromatography runs on Superdex 200 (GE Healthcare).
Site-directed mutagenesis.
Point mutations were introduced into the pGEX 4T-3 expression vector encoding R1ab-R2ab, as well as into the complementation vector pRC20 (14), using the QuikChange II XL Kit (Agilent Technologies). Single mutations at corresponding residues in R1a and R2a (R1a_A578L/R3_A742L) and double mutations at corresponding locations in R1b and R2b (R1b_Y629R/R1b_S677E/R2b_H793R/R2b_S841E) were introduced. The plasmids are referred to as pGEX-mutR1aR2apGEX-mutR1bR2b, pRC20-mutR1aR2a, and pRC20-mutR1bR2b, and the mutant proteins are referred to as mutR1a/R2a and mutR1b/R2b (see Table S1 in the supplemental material). Plasmids pRC20-mutR1aR2a and pRC20-mutR1bR2b were introduced into the restriction-negative strain S. aureus RN4220 and finally into S. epidermidis mut1 (transposon insertion in atl locus [14]) by electroporation.
The purification of the overexpressed mutant proteins from E. coli was carried out similarly to that of the wild-type (wt) protein. Correct folding of wt and mutant repeat domains was confirmed by circular dichroism (CD) spectroscopy and analytical gel filtration (see Fig. S1 in the supplemental material).
Crystallization and structure determination.
Purified and methylated R1ab-R2ab was concentrated to 12 mg/ml and mixed with crystallization buffer (1.2 M NaH2PO4, 0.8 M K2HPO4, 0.2 M LiSO4, and 0.1 M CAPS [N-cyclohexyl-3-aminopropanesulfonic acid], pH 10.5) in a 1:1 ratio. Crystals grew at 20°C within 4 days using the hanging-drop method. Prior to data collection, the crystals were transferred into crystallization buffer supplemented with 20% (vol/vol) glycerol and flash cooled in liquid nitrogen. Native data collection was carried out at the European Synchrotron Radiation Facility (ESRF) beamline ID14-1 (Grenoble, France) using an ADSC Quantum Q210 detector (Area Detector Systems Corporation, San Diego, CA). To prepare two derivatives for phase determination, crystals were soaked in crystallization solution supplemented with either 10 mM HgCl2 or 5 mM K2PtCl4 prior to freezing. Derivative data sets were collected on a Rigaku Micromax 007 HF rotating-anode X-ray generator equipped with a Mar345 detector. X-ray data were indexed, integrated, and scaled using the XDS package (19). Experimental phases were determined by multiple isomorphous replacement. The programs HYSS, SOLVE, and RESOLVE implemented in the AutoSol routine of the PHENIX software suite (1) were used for heavy-atom site identification, initial phase calculation, and density modification. A preliminary model was then manually built into the experimental electron density map using Coot (11). Crystallographic refinement at 2.9-Å resolution was carried out using PHENIX (simulated annealing and rigid-body refinement) and REFMAC5/BUSTER (individual coordinate and B-factor refinement) (7, 8).
SAXS data collection and processing.
SAXS data were collected at the EMBL X33 beamline of the storage ring DORIS III (DESY, Hamburg, Germany) (33) using a robotic sample changer (34). Different amidase variants were measured at 10°C at concentrations ranging from 1 to 14 mg/ml. Data were recorded using a Pilatus 1 M pixel detector (DECTRIS) at a sample-detector distance of 2.7 m and a wavelength of 1.5 Å, covering the range of momentum transfer 0.012 < s < 0.6 Å−1 (here, s = 4π sinθ/λ, where 2θ is the scattering angle). The data were processed with the ATSAS package (23, 29) using standard procedures, corrected for buffer contribution, and extrapolated to infinite dilution using the program PRIMUS (24). The radius of gyration, Rg, and forward scattering, I(0); the maximum particle dimension, Dmax; and the distance distribution function, p(r), were evaluated using the program GNOM (42). The excluded volume of the hydrated particle (the Porod volume, Vp) was computed using the Porod invariant (32).
Scattering data analysis.
Particle shapes at low resolution were reconstructed ab initio by the bead-modeling programs DAMMIN (43) and GASBOR (45). The scattering amplitudes of the individual subunits were calculated from their atomic coordinates using the program CRYSOL (44). Rigid-body modeling was performed by the simulated annealing program BUNCH (30) to identify optimal spatial configurations of the domains/subunits fitting the scattering data from the complex. In BUNCH, flexible interdomain linkers were modeled as chains of dummy residues. Flexibility was quantitatively assessed using the Ensemble Optimization Method (EOM) package (5).
We performed an ab initio analysis of the overall shapes of the proteins, as well as combined ab initio/rigid-body modeling using the available crystal structures of AM and R2ab and a model of R1ab. Ten independently generated ab initio reconstructions converged to similar shapes, as judged by low normalized spatial discrepancy (NSD) values (see the supplemental material). Assuming that isolated R1ab-R2ab would closely resemble its conformation in AM-R1ab-R2ab, two scattering curves were fitted simultaneously, utilizing the available high-resolution structures, by the rigid-body modeling program BUNCH (see the supplemental material). The resulting models displayed misfits in the low- and high-angle regions. The fit to the R1ab-R2ab data was especially poor, suggesting that the conformation of isolated R1ab-R2ab is different from its conformation in AM-R1ab-R2ab. We then generated 10 BUNCH models for each protein by fitting the single scattering curve. In order to assess whether the isolated repeats R1ab-R2ab are similar in overall conformation to AM-R1ab-R2ab, 10 BUNCH models were edited to contain only R1ab-R2ab. The scattering from these portions was computed using CRYSOL (44) and compared to the experimental data for R1ab-R2ab. These models were used as a single body in the modeling of AM-R1ab-R2ab with BUNCH. The latter program was run as described above, with the exception that only two rigid bodies (AM and R1ab-R2ab) were used. The data could not be fitted well with either CRYSOL in the case of R1ab-R2ab or BUNCH in the case of AM-R1ab-R2ab. This indicates that the conformation of isolated R1ab-R2ab is indeed different from its conformation in the AM-R1ab-R2ab protein. (See the supplemental material for details about SAXS-based modeling.)
Dynamic light scattering.
Dynamic light scattering (Malvern Instruments; Zetasizer Nano-ZS) was performed to determine the monodispersity and hydrodynamic radii (Rh) of amidase variants AM-R1ab, R1ab-R2ab, and AM-R1ab-R2ab. All samples were measured in buffer containing 50 mM Tris-HCl, 150 mM NaCl, pH 8.0, at concentrations of 0.5, 1, and 2 mg/ml.
PGN-LTA competition assays.
To investigate the influence of LTA on the ability of the Atl amidase to lyse PGN, 5 μg purified amidase with (AM-R1ab-R2ab) and without (AM) repeats was incubated with increasing concentrations of LTA (Sigma-Aldrich; 0.01 to 5 μg/ml plus PBS control) prior to addition of PGN (optical density at 578 nm [OD578] = 0.3). PGN lysis was assayed by a decrease of turbidity. The effect of increasing LTA concentrations on the PGN binding capacity of the repeats was also investigated using fluorescence-labeled R1ab-R2ab. LTA in different concentrations (0.05 to 1 μg/ml plus PBS control) was preincubated with purified Cy5-R1ab-R2ab prior to addition of PGN. After incubation, the PGN was centrifuged, and fluorescence (630/670 nm) was determined in the pellet and in the supernatant.
PGN binding assay.
PGN was purified according to protocols described previously (38). To obtain PGN with decreased cross-linking, penicillin G (PCG) was added to growing cells at an OD578 of 0.2 in sub-MIC (0.02-μg/ml) dilutions. PGN binding was determined by pulldown assays. Wt repeats (R1ab-R2ab) and mutants (mutR1a/R2a and mutR1b/R2b) were each incubated with wt PGN, PGN lacking O acetylation, and PGN with decreased cross-linking induced by PCG. Insoluble PGN was washed twice with PBS and sedimented by centrifugation at 8,000 × g. Bound proteins were pulled down, together with insoluble PGN, and separated by SDS-PAGE. In order to rule out precipitation of misfolded mutant proteins, the same experiment was performed without PGN. Band intensities in stained SDS-PAGE were determined with Gelscan ProC software.
Western blotting.
To prove the presence of the amidase in the S. epidermids atlE mutant complemented with pRC20-mutR1a/R2a and pRC20-mutR1b/R2b, Western blots were performed, and 1 ml of cells was harvested at an OD578 of 1. The cell pellets were treated with 6% SDS to release cell wall-bound proteins. Amidase was detected with a mouse-derived antibody directed against the repeat domains visualized by goat anti-mouse IgG-biotin conjugate (Sigma-Aldrich), followed by treatment with streptavidin-biotinylated horseradish peroxidase conjugate RPN 1051 (Amersham Life Science). The Western blots were developed in a Bio-Rad ChemiDoc XRS ECL device.
LTA-ELISA.
Binding of LTA to R1ab-R2ab, mutR1a/R2a, and mutR1b/R2b was measured by enzyme-linked immunosorbent assay (ELISA). Absorbing 96-well microtiter plates (Nunc PolySorp; Thermo Fisher Scientific) were coated with 5 μg of protein overnight at 4°C. An LTA-ELISA was carried out according to a previously described protocol (47). Briefly, the plate was blocked for 1 h with phosphate-buffered saline containing 0.05% Tween 20 and 0.5% bovine serum albumin. Then, every sample was incubated for 10 min with 25 ng LTA (Sigma-Aldrich). After washing, the wells were incubated for 1 h with monoclonal anti-LTA antibody, clone 55 (mouse IgG3; Dunn Labortechnik GmbH, Asbach, Germany) at a final concentration of 400 ng/ml, followed by incubation with goat anti-mouse IgG peroxidase conjugate (Calbiochem) at a final concentration of 500 ng/ml for 1 h. Subsequently, 100 μl of substrate solution (substrate reagent pack; R&D Systems) was added, and the reaction was stopped by adding 50 μl 1 M H2SO4. Absorption was determined at 450 nm. Apart from the coating of the plates, all steps were carried out at room temperature.
Microscopic studies.
For localization studies, purified R1ab-R2ab was labeled with the fluorescent dye Cy5, as described previously (38). For preparation of cells, BM broth was inoculated with an overnight culture of SA113 to an OD578 of 0.1, and the cells were grown to an OD578 of 0.6. Next, 500 μl of the cell suspension was incubated with 4′,6-diamidino-2-phenylindole (DAPI) and DNase I (Sigma-Aldrich) for 10 min prior to addition of 4 μg of Cy5-labeled repeats. Unbound substances were removed by centrifugation and washing with PBS. The cells were fixed on agarose-coated slides and applied to fluorescence microscopy or light microscopy, respectively.
Protein structure accession number.
Coordinates and structure factors have been deposited with the Protein Data Bank under accession number 4EPC.
RESULTS
Overall structure of the Atl amidase repeat R2ab.
The Atl amidase of S. epidermidis is composed of the catalytic AM domain and repeats R1 and R2. These three modules are separated by two linkers, L1 and L2 (Fig. 1). Inspection of sequence alignments confirmed earlier predictions that repeats R1 and R2 can each be further divided into an a-type and a b-type subunit (25), giving rise to the R1ab and R2ab nomenclature used here. Both subunit types carry a highly conserved glycine-tryptophan (GW) motif. In order to understand their structural and functional properties, we expressed, purified, and crystallized repeats R1ab and R2ab of Atl (residues 516 to 845; see Materials and Methods). The purified protein also included the L1 linker at the N terminus of R1ab.
Although the crystallized protein contained repeats R1ab and R2ab, only a model for R2ab could be built into the electron density map, which is sufficient to explain the crystal lattice (see Fig. S2 in the supplemental material). R1ab projects into a large solvent channel via the L2 linker and is not visible. Using multiple isomorphous replacement, we performed a structural analysis to a resolution of 2.9 Å (see Table S2 in the supplemental material). The final electron density for the R2ab tandem repeat has excellent quality and confirms the unambiguous placement of all main chain and most side chain atoms, including many methylated lysine residues (see Fig. S3 in the supplemental material).
The R2ab repeat folds into two small, similar structures that correspond to the a-type and b-type subunits (Fig. 2). Each subunit resembles a half-open β-barrel formed by a semicircular, four-stranded β-sheet (strands β1 to β4 in R2a and strands β6 and β9 to β11 in R2b). This half-barrel is capped in each case with an elaborate loop that bends inward and packs against the concave face of the β-barrel. In the case of R2b, this loop contains an additional β-hairpin structure formed by strands β7 and β8. The two subunits are arranged in similar orientations and feature a large interface in which one side of R2a packs tightly against the β-hairpin and loop of R2b. Thus, the structure indicates that R2ab forms a rigid entity. Comparison of R2a and R2b shows that the two subunits have similar overall structures that can be superimposed with a root mean square deviation (RMSD) of 2.3 Å for all Cα atoms (Fig. 2C). However, significant structural differences are present in the loop regions.
Fig 2.

Crystal structure of the Atl amidase repeat R2ab. (A) Cartoon representation of the crystal structure of R2ab in two orientations. For clarity, loop regions have been smoothed. (B) B-factor distribution in the R2ab structure. B factors decrease from red to blue. (C) Structural comparison of repeats R2a and R2b. The superimposed structures have an RMSD of 2.3 Å (all Cα atoms).
A search for homologs using the DALI server (17) confirmed that R2a and R2b belong to the SH3b family (for bacterial SH3) domains (25). With a Z score of 10.3, the two-domain targeting sequence of Listeria monocytogenes InlB (Protein Data Bank [PDB], 1m9s) (25) was identified as the closest homolog of R2ab. SH3b domains in turn possess structural homology to the well-studied eukaryotic SH3 (Src homology 3) domains. SH3 domains contain between 50 and 80 amino acids and recognize proline-rich consensus sequences in signal transduction pathways (21). The SH3 fold has also been implicated in binding to DNA and RNA (13, 21, 27). Although SH3b domains are larger than their eukaryotic counterparts, they share common structural features, such as an elaborate RT loop, the distal loop, and the n-src loop. The loops of R2a and R2b were named after their corresponding regions in eukaryotic SH3 domains. In addition, the loop region that connects the C terminus to the RT loop and is not present in SH3 domains has been named the SU loop (Fig. 2C).
Loop structure and putative binding regions of R2ab.
The two subunits of R2ab each contain several loops that likely contribute to ligand interaction and specificity. The RT loops are especially prominent, establishing surface properties, as well as contacts between the two domains. The distal and SU loops are located on the other sides of the subunit, distant from the RT loop (Fig. 2). The RT loop of R2a is a highly dynamic structural element. With the exception of the very N terminus of R2ab, the loop exhibits the highest thermal mobility in the entire structure (Fig. 2B). The almost parallel orientation of this loop relative to the central sheet creates a shallow hydrophobic groove, the bottom of which is formed by the side chains of A742 in β2 and V721 in the RT loop. Residues in strands β1 and β3 and the tip of the covering RT loop form the walls of this groove. The RT loop of R2b is wedged between the two half-open β-barrels and, in contrast to the R2a RT loop, is stabilized by an extensive hydrogen-bonding network with residues of the neighboring SU loop/strand β2 and the R2a distal loop, which is significantly longer than in R2b. This results in the R2b RT loop having the lowest B factors in the entire crystal structure (Fig. 2B). The tip of the R2b RT loop is bent 90° downwards and packs against the outside the barrel-like β-sheet formed by strands β9 to β11. The twisted β-sheet in R2b creates a deep cavity on the opposite side of the protein. The bottom of the cavity is formed by W839 and is walled by the tip of the RT loop, strands β10 and β11, and the short 310 helix. Unlike all other loop regions of R2a and R2b, the n-src loops are rather short in both subunits and are not engaged in any contacts.
Implications for the structure of R1ab-R2ab.
Sequence comparisons show that all four subunits of the Atl amidase repeats possess common features, such as the GW motifs, which can also be found in several other staphylococcal amidases (Fig. 3). The sequence identities between R1a and R2a and between R1b and R2b are especially high (68.8% and 83.6%, respectively), while R1a and R1b (or R2a and R2b) are clearly less similar to each other (sequence identity, 21.0% and 22.9%, respectively). This indicates that each tandem repeat (R1ab or R2ab) can be regarded as a structural unit, an arrangement that has also been reported for the SH3b domains of InlB, a surface-localized protein of L. monocytogenes (25). Since R1ab is not visible in the electron density, the software MODELLER 9.10 (12) was used to calculate a model based on the high sequence identity (76.2%) to R2ab. The evaluation routine implemented in MODELLER reports a zDOPE (discrete optimized protein energy) score of −1.3. zDOPE scores give a normalized atomic-distance-dependent potential relative to a known structure. Generally, a score of less than −1 indicates a reliable model (39). The accuracy of the calculated model is further supported by a low RMSD (1.2 Å) and a native overlap of 0.95 (the fraction of Cα atoms within 3.5 Å of the native structure). The model features GW motifs at the centers of the central β-barrels, a tight interface between the two subunits, and elaborate RT loops in the same orientation.
Fig 3.

Sequence alignments of R1a/R2a and R2a/R2b with repeat sequences of different staphylococcal amidases. The alignments were calculated with the programs ClustalW2 (46), MUSCLE (10), and MAFFT (20) and combined into a single output using COMBINE (31). Conserved amino acids are color coded according to their degree of conservation as follows: gray, <80%; pink, 80 to 90%; red, 100%. Linker regions at the N termini of repeats R1a and R2a are omitted in the alignment and shown separately in Fig. 7D.
Analysis of conservation and design of mutants.
We hypothesized that R1ab-R2ab residues, involved in binding to cell wall components, should be largely conserved in repeat sequences of other staphylococci. We therefore aligned the sequence of the Atl amidase repeats with repeats from seven other representative staphylococcal species (Fig. 3) and mapped conserved residues onto the surface of R2ab and the model of R1ab (Fig. 4A and C). In each subunit, most of the strictly conserved residues cluster in a recessed area around the GW motif. Interestingly, these areas are located on opposite sides and show significantly different topologies in the a-type and b-type subunits. While the conserved cluster in the R1a/R2a groove is wide and shallow, the conserved region in R1b/R2b forms a deep cavity. Both conserved regions lie at the contact points between the tip of the RT loop and the conserved tryptophan in the GW motif (R1a, amino acids [aa] 598 to 599; R1b, aa 674 to 675; R2a, aa 762 to 763; R2b, aa 838 to 839). The tryptophan residues are likely to serve as integral parts of the putative binding areas. The GW dipeptide represents the consensus motif of all staphylococcal autolysin repeats in the alignment and is also present in the InlB targeting domain of L. monocytogenes (25), the closest structural homolog of R2ab. In addition to the putative binding grooves, conserved residues are also present at the interface between the two subunits in each repeat, where they participate in a hydrogen-bonding network that tightly links the two subunits.
Fig 4.

Conserved residues cluster in two distinct regions on opposite sides of R2ab. (A) Conservation pattern on the surface of R2ab, shown in two different views. Amino acids are colored according to their degree of conservation using the color scheme of Fig. 3. A distinct patch of conserved residues is present on the surface of each subunit in the tandem. Amino acids labeled in green were mutated. R2ab has the same orientation as in Fig. 2. (B) Close-up views of the putative substrate binding sites, with mutated amino acids colored green. (C) Conservation pattern on the surface of R1ab. The R1ab model shown here was generated using MODELLER (12).
To study the interaction properties of R1ab-R2ab with putative ligands, we designed binding-impaired mutants based on the R2ab crystal structure. Due to the different topologies and positions of the putative binding grooves in R1a/R2a and R1b/R2b, two different mutants with inaccessible grooves were generated. They carry the same mutations in each of the sequence and structurally related subunits R1a/R2a and R1b/R2b, respectively. In the first mutant, two conserved alanine residues that form the bottom of the putative binding groove in R1a (A578) and R2a (A742) were mutated to leucines (mutR1a/R2a). These mutations aim to create a shallower groove with a conformation that would be unfavorable for the accommodation of a substrate. The second mutant carries four point mutations, two in R1b and two in R2b (mutR1b/R2b). Each of the mutated residue pairs, Y629R/S677E and H793R/S841E, is predicted to form a salt bridge that closes the binding pocket in the respective subunit and thereby constricts its accessibility.
Repeats R1ab-R2ab interact with LTA.
Previously, we demonstrated that repeats R1ab-R2ab-R3ab exhibit a higher affinity for PGN than the AM domain (6). We also found that WTA, which protrudes from the thick murein layer in staphylococci, prevents binding of the repeats, thus allowing rebinding of AM-R1ab-R2ab or R3ab-GL only in the septum region, where WTA is apparently not yet fully matured (38). However, it was unclear whether the repeats use a receptor other than PGN at the septum. Since it has been shown that addition of LTA prevents penicillin-induced autolysis in S. aureus (41), we hypothesized that LTA could display such a receptor and therefore investigated the interaction of R1ab-R2ab with LTA in competitive assays. We analyzed the influence of LTA on the PGN binding capacity of the repeats by monitoring the catalytic activity of the AM domain. Purified amidase with and without repeats was therefore incubated with decreasing concentrations of LTA (5 to 0.01 μg/ml). PGN was added, and the amidase activity was determined using the decrease in turbidity of polymeric PGN as an assay. The activity of the amidase alone (AM), lacking the repeats, was not affected by any LTA concentration tested (Fig. 5A). However, the activity of the amidase with repeats (AM-R1ab-R2ab) was gradually decreased with increasing LTA content (>0.1 μg/ml) (Fig. 5B). In a similar assay, LTA in different concentrations was preincubated with purified, Cy5-labeled R1ab-R2ab prior to addition of PGN. After incubation, the PGN was spun down, and fluorescence was determined in the insoluble PGN fraction and the supernatant. Increasing concentrations of LTA inhibited PGN binding completely, and fluorescence was therefore measured mostly in the supernatant (Fig. 5C). At lower LTA concentrations, fluorescent protein was found attached to the PGN pellet. These results imply that the repeats (R1ab-R2ab) use LTA as a binding partner. An interaction with LTA might contribute to fastening AM-R1ab-R2ab at the septum, particularly during the process of murein hydrolysis.
Fig 5.

Interaction of the Atl amidase with PGN and LTA. (A and B) Five micrograms of AM (A) or AM-R1ab-R2ab (B) was incubated with increasing concentrations of LTA (5 to 0.01 μg/ml) prior to addition of PGN. PGN lysis was measured as the decrease of turbidity. AM was not affected by any concentration of LTA tested, while the activity of AM-R1ab-R2ab was decreased at concentrations above 0.1 μg/ml. ×, positive control (PBS); ■, negative control; ●, 1 μg/ml; ♦, 0.5 μg/ml; ○, 0.1 μg/ml; □, 0.05 μg/ml; ♢, 0.01 μg/ml. (C) LTA in different concentrations (0.05 to 1 μg/ml) was preincubated with purified Cy5-R1ab-R2ab prior to addition of PGN. After incubation, PGN was centrifuged, and fluorescence was determined in the pellet and in the supernatant. Increasing concentrations of LTA lead to a decrease in binding of Cy5-R1ab-R2ab to purified PGN. (D) R1ab-R2ab, mutR1a/R2a, and mutR1b/R2b were immobilized on 96-well plates and incubated with purified LTA. Bound LTA was detected by anti-LTA-IgG. Wt repeats are able to bind LTA in high concentrations. Mutation in R1a/R2a leads to reduced binding of LTA, while a mutation in R1b/R2b almost completely prevents binding of LTA. The number of asterisks indicates the degree of significance. (E) Amidase-specific Western blot and light microscopic pictures of S. epidermidis O-47, S. epidermidis mut1, S. epidermidis mut1(pRC20), S. epidermidis mut1(pRCmutR1a/R2a), and S. epidermidis mut1(pRCmutR1b/R2b). Amidase was released from the cell wall by 6% sodium dodecyl sulfate (SDS) and visualized by amidase-specific antibody. The molecular masses correspond to amidase with (87 kDa) and without (62 kDa) propeptide. The error bars indicate standard deviations.
Mutations in R1ab-R2ab affect LTA but not PGN binding.
As the regions that mediate targeting were unknown, we hypothesized that the conserved grooves in R2ab might be involved in the process. Due to the high sequence identity between R1ab and R2ab (Fig. 3), we predicted the presence of similar conserved patches in R1ab (Fig. 4C). In order to associate the conserved areas with either PGN or LTA binding, binding assays with repeat mutants mutR1a/R2a and mutR1b/R2b were performed.
PGN pulldown assays did not display a significant difference in binding affinity between wt and mutant repeats, indicating that the mutations had no effect on PGN-binding and therefore did not result in larger structural changes of the protein (see Fig. S4 in the supplemental material, upper left panel). Similar experiments were carried out using PGN with decreased cross-linking (penicillin G treatment) or lack of O-acetylation (3); these structural PGN alterations also did not affect repeat binding (see Fig. S4 in the supplemental material, upper right and lower left panels).
The interaction of mutant repeats with LTA was analyzed in an ELISA using αLTA-IgG (Fig. 5D). Wt and mutant repeats were immobilized on 96-well plates and incubated with purified LTA; bound LTA was detected by αLTA-IgG (mouse) and goat anti-mouse IgG–peroxide conjugate.
The mutations in both types of subunits resulted in a significantly reduced affinity for LTA. Interestingly, repeats with mutations in the a subunits showed binding behavior different from that of repeats with mutations in the b subunits. While mutR1a/R2a was already significantly affected in LTA binding, binding of mutR1b/R2b to LTA was almost abolished. These results suggest that the interaction of the repeats with LTA is more specific than their interaction with PGN.
Mutations in R1ab-R2ab prevent cell wall binding in vivo.
In earlier studies, it could be shown that the amidase portion of atlE, carried on the plasmid pRC20, is sufficient to rescue the cell aggregation phenotype in transposon insertion and deletion mutants of atl (14). We introduced the mutations described above into the complementation vector pRC20 in order to examine the cell division phenotype of a lipoteichoic acid binding-deficient but peptidoglycan binding-proficient mutant. In both mutated complementation strains, only small amounts of autolysin could be detected in cell wall fractions with an amidase-specific Western blot, while in the wt and the complemented mutant, bands for amidase with (87 kDa) and without (60 kDa) propeptide were visible (2, 14, 38). While complementation by pRC20 led to proper cell division, as described previously (14), mutation in both the a-type and the b-type subunits of the repeats resulted in macroscopic aggregates resembling the atl deletion phenotype, as shown by light microscopy (Fig. 5E).
Absence of LTA in S. aureus RN4220 (4S5) prevents surface localization of R1ab-R2ab.
A recently described LTA-deficient S. aureus suppressor strain, RN4220 (4S5), is capable of growing almost normally in the absence of LTA (9). However, misplacement of division septa still occurs in some cells. This strain is more susceptible to autolysis and has fewer autolysins bound to the cell wall but more secreted autolysins. If LTA serves as an anchor for the Atl repeats, one should not expect binding of the repeats to the septum region of the suppressor strain. In order to verify this assumption, we investigated the ability of RN4220 (4S5) to bind externally applied Cy5-labeled R1ab-R2ab repeats. While in the wt strain, RN4220, fluorescent repeats accumulated in the septum region and putative new sites of cell division, as described previously (50), no binding could be observed in the LTA-deficient suppressor mutant (Fig. 6). This result suggests that LTA might indeed serve as an important receptor for Atl repeats.
Fig 6.

Distribution of fluorescence-labeled repeats on the surfaces of S. aureus cells. Cells of S. aureus wt and the LTA-deficient strain RN4220 (4S5) were incubated with Cy5-labeled repeats, and the surface distribution was examined by fluorescence microscopy. Chromosomal DNA was visualized by DAPI staining. R1ab-R2ab binding could be observed at the interface between separating cells (top). RN4220 (4S5) did not show fluorescent repeats on the surface (bottom).
Solution structure of AM-R1ab-R2ab.
Although we could not resolve the R1ab unit in our crystals, the position of R2ab in the crystal lattice (see Fig. S2 in the supplemental material) indicates that a linker (L2) exists between R1b and R2a, consistent with sequence alignments. As repeat R1ab is highly homologous to R2ab and therefore predicted to fold into a similar structure, the L2 linker likely allows considerable movement between the two repeats. An additional linker, L1, is present between the catalytic domain AM (52) and R1a (Fig. 1). These linkers likely create mobility between the catalytic AM domain and the cell wall anchoring units R1ab-R2ab. In order to gain insight into the overall conformation of AM-R1ab-R2ab and to characterize the mobility of its substructures, we performed SAXS experiments in solution on AM-R1ab-R2ab, AM-R1ab, and R1ab-R2ab. The overall parameters and the SAXS modeling results are summarized in Table 1. The models agree well with the scattering data (s range, 0.020 to 5 nm−1) (Table 1 and Fig. 7A). A comparison of the models obtained is presented in Fig. S5 in the supplemental material. The Kratky plots [I(s) · s2 versus s], elevated values of the radius of gyration (Rg), and skewed shapes of p(r) indicated that the proteins have elongated structures and exhibit some flexibility (Fig. 7B and C; see Fig. S6 in the supplemental material).
Table 1.
Overall parameters and modeling summary for SAXS data
| Sample | Rg (nm)a | RgGNOM (nm)a | Rh (nm)b | Rg/Rhc | Dmax (nm)d | MWmone | MWexcf | χsg | χgh | χbi | χb_mcj | χfk |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AM-R1ab | 3.8 ± 0.1 | 4.2 ± 0.2 | 3.6 ± 0.5 | 1.2 | 15.5 ± 0.5 | 44,000 | 50,000 ± 5,000 | 1.0 | 1.0 | 1.2 | 1.1 | |
| R1ab-R2ab | 3.9 ± 0.1 | 4.2 ± 0.2 | 3.7 ± 0.5 | 1.1 | 14.5 ± 0.5 | 35,000 | 35,000 ± 5,000 | 1.0 | 1.0 | 1.1 | 0.9 | |
| AM-R1ab-R2ab | 4.2 ± 0.1 | 4.6 ± 0.2 | 4.3 ± 0.5 | 1.1 | 16.0 ± 0.5 | 61,000 | 59,000 ± 5,000 | 1.4 | 1.1 | 1.3 | 1.4 | 1.2 |
Rg, radius of gyration calculated using either Guinier approximation or the indirect Fourier transform package GNOM.
Rh, hydrodynamic radius measured by dynamic light scattering.
An Rg/R[infi]h ratio higher than 1 indicates flexibility in the system (35).
Dmax, maximum size, estimated using GNOM.
MWmon, expected molecular weight of monomeric proteins.
MWexc, concentration-independent estimation of MW from DAMMIN volume.
χs, discrepancy from average refined ab initio model generated by DAMMIN.
χg, discrepancy from reference GASBOR ab initio model.
χb, discrepancy from best BUNCH model.
χb_mc, discrepancy from multicurve BUNCH model.
χf, discrepancy from EOM ensemble model.
Fig 7.

Summary of SAXS modeling. (A) Scattering profiles of analyzed amidase variants. Experimental data fitted to ab initio, rigid-body, and EOM models are shown as open circles, yellow dotted line, and blue and red dashed lines, respectively. Experimental SAXS profiles were appropriately displaced along the logarithmic axis for better visualization and overlaid with corresponding fits. The inset shows linear Guinier plots confirming monodispersity of the samples. (B) Distance distribution function estimated for the studied amidase variants using GNOM. The p(r) of R1ab-R2ab (green line) displays rather pronounced shoulders, typical of rigid structures (4) at greater distances corresponding to the interdomain correlation peaks. p(r) functions of AM-R1ab (red line) and AM-R1ab-R2ab (blue line) contain fewer features, suggesting that the variants including the catalytic domain AmiE are more flexible. (C) Radius of gyration distribution for the EOM models of AM-R1ab (top), R1ab-R2ab(middle), and AM-R1ab-R2ab (bottom). The distribution for the pools of models is shown by the area filled with gray; dashed lines of different colors correspond to selected ensembles. The representative conformations, as well as discrepancies, are shown near the distributions: compact conformation (χc) on the left and extended conformation (χe) on the right; discrepancy from an ensemble model is indicated by χeom. Folded domains of AM-R1ab-R2ab are depicted as cartoons (AM in red and R1ab and R2ab in blue); linker L1 (between AM and R1ab) is represented by gray spheres, while linker L2 (between R1ab and R2ab) is represented by green spheres. (D) Sequence alignment of the two linkers. Glycine residues are highlighted in yellow and prolines in gray.
As the proteins are expected to possess flexibility in solution due to the presence of linker regions L1 and L2, the data were further analyzed, accounting for the possible coexistence of different conformers using EOM (see Materials and Methods). The use of EOM allowed us to neatly fit the data for all amidase variants (Fig. 7C and Table 1). The Rg distribution of the selected ensemble for AM-R1ab shows two distinct peaks, corresponding to extended and compact subpopulations (Fig. 7C, top). In the case of the R1ab-R2ab protein, the selected ensemble displays a relatively narrow distribution of extended conformations, with the major peak for the Rg values between 3.5 and 4.5 nm (Fig. 7C, middle). AM-R1ab-R2ab has broad Rg distributions for both the pool and the selected ensemble (Fig. 7C, bottom).
Comparison of the Rg distributions indicates that L1 can sample a broad range of extended and compact conformations, revealing two peaks in Fig. 7C (top). Inclusion of L2 shifts this equilibrium to more extended subpopulations (Fig. 7C, middle). The flexibility of R1ab-R2ab is largely defined by L2, which separates R1ab and R2ab. The presence of two linkers in AM-R1ab-R2ab allows sampling of a broader range of conformations, similar to those of the random pool (Fig. 7C, middle).
The EOM results show that L1 is more flexible than L2. This finding can be corroborated by analysis of the primary sequences. The two linkers (L1, 19 residues, and L2, 17 residues) have 63.2% and 78.9% sequence identity and similarity, respectively. However, L1 contains two extra consecutive glycine residues at the C terminus, and furthermore, it has a glycine at its N terminus, while L2 has a leucine at this position (Fig. 7D). The three glycines likely increase the flexibility of L1. Our results suggest different roles of the two linkers. We therefore propose a tentative model of AM-R1ab-R2ab (Fig. 8) with different conformational states. In this model, L1 is able to sample a broad range of conformations, while the less flexible linker L2 primarily serves to separate R2a from R1b.
Fig 8.
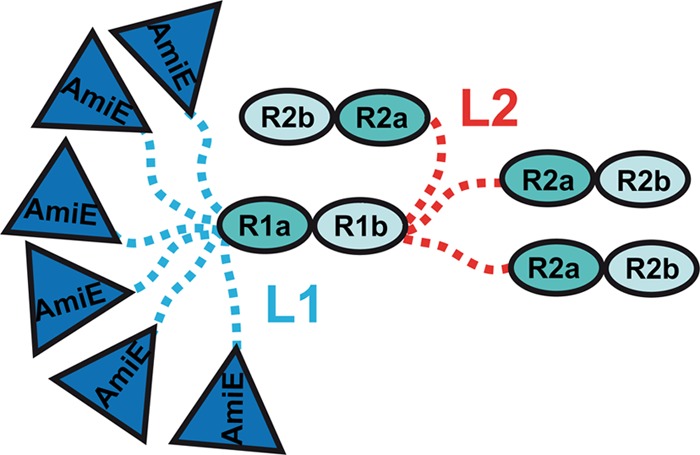
Conformational-state model of AM-R1ab-R2ab. Different geometrical shapes filled with colors represent domains with an available high-resolution structure or a predicted homology model (R1ab). The dashed lines, blue and red, represent flexible linkers. The linker between AM and R1ab (L1) is highly flexible and adopts a broad range of compact as well as extended conformations. The linker between R1ab and R2ab (L2) is mostly extended but may also sample a few compact conformations.
DISCUSSION
Structural information about full-length autolysins is currently limited to LytC, a pneumococcal lysozyme (28). The catalytic domain and the repeats of LytC are arranged in an unusual hook-shaped structure that allows the enzyme to hydrolyze only specific PGN segments. LytC consists of an N-terminal choline-binding module possessing 11 homologous repeats that specifically recognize the choline moieties in pneumococcal WTA and LTA and a C-terminal catalytic module that belongs to the GH25 family of glycosyl hydrolases (26). The mode of LytC targeting in pneumococci is likely unique, as cholinated teichoic acids do not occur in most other Gram-positive bacteria. Moreover, pneumococcal and S. aureus LTAs have completely different chemical structures. Nevertheless, the available structural and functional data on LytC render it likely that staphylococcal Atl proteins have also evolved a mechanism to target glycerolphosphate-containing LTA via their repeats. Based on electron microscopic examination of ultrathin sections of protoplasts, Yamada et al. proposed in 1996 that the 62-kDa amidase (AM-R1ab-R2ab) associates with cellular components protruding from the membranes of S. aureus protoplasts, such as LTA (50).
In order to provide a structural basis for the interaction of Atl with LTA, we solved the crystal structure of the R2ab repeat. Consistent with earlier predictions (25), the Atl repeats fold into two tightly packed SH3b domains that each feature a small hydrophobic core centered around a conserved GW motif. We also provide several lines of evidence that staphylococcal LTA interacts with the Atl repeats and likely serves as a receptor for Atl in the septum (cross-wall) region (i) Pulldown assays with purified LTA from S. aureus and fluorescence-labeled Atl repeats (R1ab-R2ab) demonstrated that the repeats bind to LTA (Fig. 5C). (ii) Moreover, the amidase activity of AM-R1ab-R2ab gradually decreases in the presence of increasing concentrations of LTA, in contrast to the amidase activity of the AM domain alone. The most likely interpretation of this finding is that the repeats preferentially bind LTA. Such an interaction would restrict the mobility of AM in the full-length protein, limiting its ability to engage PGN and thus leading to a decrease in PGN hydrolysis. On the other hand, AM lacking the repeats would not be hindered in its movement and could access and hydrolyze PGN in solution (Fig. 5A and B). (iii) Finally, analysis of S. aureus suppressor mutants that lack LTA (9, 27) showed that Cy5-labeled R1ab-R2ab repeats do not bind to the septum, in contrast to the wt strain, RN4220 (Fig. 6). Taken together, these results strongly suggest that LTA is the receptor for rebinding of Atl repeats in the septum region. This is further corroborated by the finding that the conserved grooves in a- and b-type subunits exhibit a positive electrostatic potential that would complement the negative charge of the LTA phosphate backbone (see Fig. S7 in the supplemental material). Currently, it is unclear whether LTA at the septum is mature and therefore long enough to be accessible to the repeats. However, even if LTA is hidden in the murein layer, the repeats are able to bind to PGN (6), and in that case, the AM domain would likely be able to cleave enough of the murein structure to render LTA accessible. Additional support for the hypothesis that LTA serves as a receptor for the Atl repeats is provided by the observation that, in the ΔltaS (4S5) mutant, the majority of the autolytic enzymes were not attached to the cell wall but remained in the supernatant (9).
To address the question of whether the observed interaction between repeats and LTA is specific, structure-guided binding studies with repeats carrying defined mutations in conserved regions were performed. Mutations were inserted in either the two a-type or the two b-type subunits of the repeats. While the a-type mutants were already significantly impaired in LTA binding, mutations in the b type led to negligible LTA binding (Fig. 5D). Furthermore, mutations in both the a-type and the b-type subunits also led to a loss of function in vivo, as complementation plasmids encoding amidases with mutant repeats did not result in reestablishment of proper cell division in S. epidermidis mut1. In contrast, binding to PGN was not affected in any of the mutants (see Fig. S4 in the supplemental material). These results suggest that the binding of the repeats to LTA is specific and that the mutations also did not introduce larger structural changes in the protein.
The Atl repeats most likely interact with the glycerolphosphate backbone of LTA, as its lipid anchor is buried in the cytoplasmic membrane and would therefore not be accessible. As the putative LTA binding grooves in the a-type and b-type subunits exhibit rather different shapes and the respective mutants show different affinities, it is possible that the grooves are designed to also recognize different motifs on the glycerol phosphate backbone. The glycerol phosphate repeating unit of S. aureus LTA contains 15% 2-hydroxyl groups, 15% N-acetylglucosamine glycosylation, and 70% d-alanyl esterification (36); sugar compositions may vary among strains. It would certainly be important to carry out binding studies with substituted glycerol phosphate repeating units. However, no such structures are currently available.
Our data show that the Atl repeats bind to PGN and probably more specifically to LTA. They do not bind to WTA, which is structurally related to LTA. WTA also has d-alanyl esterifications, but in contrast to LTA, it is formed by a ribitolphosphate backbone that is supplemented with different sugar substituents. Indeed, WTA would probably act as a repellent for Atl. The presence of WTA at the septum prevents uncontrolled autolysin-mediated cell lysis, as demonstrated recently by thorough investigation of a WTA mutant (38). On the other hand, the absence of WTA at the cross-wall would allow the Atl repeats to engage its receptor LTA.
In addition to their interaction with cell wall components, Atl repeats have been reported to interact with proteins of the extracellular matrix (15), as well as Hsc70 (16). However, only one patch of conserved residues could be detected on each repeat subunit. It is therefore conceivable that the repeats, which adopt an SH3b fold, have a substrate promiscuity that resembles that of the structurally related eukaryotic SH3 domains (13, 21, 27). Multifunctionality in ligand binding would perhaps be best associated with the a-type subunits. The a-type subunits have highly mobile RT loops that could modulate substrate specificity through movement relative to the central β-sheet. This is consistent with the elevated temperature factors of the R2a RT loop in the crystal structure. The presence of linker regions L1 and L2, which precede R1a and R2a, allows conformational changes to occur in the RT loops of a-type subunits. Particularly in the case of L2, which is significantly less flexible than L1, such a spacer role could be a central part of the function.
In order to advance our understanding of the conformational dynamics of AM-R1ab-R2ab in solution, SAXS data were collected. Analyses of these data show that AM, R1ab, and R2ab form separate, stable entities that are flexibly linked to each other. The levels of flexibility clearly vary between the two linker regions. Combining the insights from SAXS analyses with our crystallographic and functional data enabled us to postulate a tentative but plausible model describing the likely functions of amidase domains and linker regions in cell division (Fig. 9). In this model, the Atl amidase is anchored to LTA in the septum via the repeats, where it would be able to assume several preferred conformational states that would facilitate cell wall cleavage. The highly flexible linker L1 would allow the catalytic domain to access a large number of cleavage sites, thereby increasing the enzymatic efficiency significantly. The second, less flexible linker L2, on the other hand, would hold R1ab-R2ab in a nearly extended conformation while anchored to LTA in the cell wall. Small changes in localization could be achieved by switching between nearby LTA molecules through independent movements of R1ab and R2ab. However, SAXS data indicate that a switch to a compact conformation is also possible. Such a switch would position the catalytic domain in an inverted orientation in the PGN layer and thereby further increase the radius of action of AM. In this sense, the amidase could be guided through the PGN layer in a coordinated manner.
Fig 9.

Hypothetical model showing the orientation of the Atl amidase in the septum.
In conclusion, the structural analyses that we present here provide new insights into the interplay between the different domains of the staphylococcal major autolysin and the cell wall. Our results also advance an understanding of the targeting of staphylococcal major autolysins to the cross-wall, where they can trigger cell separation using LTA as the most likely candidate.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge access to EMBL beamline X33 at the DORIS storage ring, DESY, Hamburg, Germany, and beamline ID14-1 at ESRF, Grenoble, France. We thank the beamline personnel at ESRF for their support. We thank Angelika Gründling (Imperial College London, Section of Microbiology) for providing the LTA-deficient S. aureus strain RN4220 (4S5).
This project was funded by the German Research Foundation (SFB-TRR34 to S.Z., T.S., and F.G.). A.V.S. and D.I.S. further acknowledge Bundesministerium für Bildung und Forschung (grant SYNC-LIFE, contract number 05K10YEA) and European Union FP7 e-Infrastructure (grant WeNMR, contract number 261572). The research leading to these results received funding from the European Community's Seventh Framework Programme (FP7/2007-2013) under grant agreement 226716.
S.Z., M.S., and A.V.S. conceived, designed, and performed the experiments. M.R. conducted the LTA-ELISA. S.Z., M.S., A.V.S., T.S., F.G., and D.I.S. analyzed the data. S.Z., T.S., and F.G. wrote the manuscript. M.S. and A.V.S. also wrote part of the manuscript.
Footnotes
Published ahead of print 18 May 2012
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1. Adams PD, et al. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66:213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baba T, Schneewind O. 1998. Targeting of muralytic enzymes to the cell division site of Gram-positive bacteria: repeat domains direct autolysin to the equatorial surface ring of Staphylococcus aureus. EMBO J. 17:4639–4646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bera A, Biswas R, Herbert S, Götz F. 2006. The presence of peptidoglycan O-acetyltransferase in various staphylococcal species correlates with lysozyme resistance and pathogenicity. Infect. Immun. 74:4598–4604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bernado P. 2010. Effect of interdomain dynamics on the structure determination of modular proteins by small-angle scattering. Eur. Biophys. J. 39:769–780 [DOI] [PubMed] [Google Scholar]
- 5. Bernado P, Mylonas E, Petoukhov MV, Blackledge M, Svergun DI. 2007. Structural characterization of flexible proteins using small-angle X-ray scattering. J. Am. Chem. Soc. 129:5656–5664 [DOI] [PubMed] [Google Scholar]
- 6. Biswas R, et al. 2006. Activity of the major staphylococcal autolysin Atl. FEMS Microbiol. Lett. 259:260–268 [DOI] [PubMed] [Google Scholar]
- 7. Bricogne G, et al. 2011. BUSTER, 2.10.0 ed Global Phasing Ltd., Cambridge, United Kingdom [Google Scholar]
- 8. Collaborative Computational Project Number 4 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50:760–763 [DOI] [PubMed] [Google Scholar]
- 9. Corrigan RM, Abbott JC, Burhenne H, Kaever V, Gründling A. 2011. c-di-AMP is a new second messenger in Staphylococcus aureus with a role in controlling cell size and envelope stress. PLoS Pathog. 7:e1002217 doi:10.1371/journal.ppat.1002217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66:486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Eswar N, et al. 2007. Comparative protein structure modeling using MODELLER. Curr. Protoc. Protein Sci. Chapter 2:Unit 2.9 [DOI] [PubMed] [Google Scholar]
- 13. Gao YG, et al. 1998. The crystal structure of the hyperthermophile chromosomal protein Sso7d bound to DNA. Nat. Struct. Biol. 5:782–786 [DOI] [PubMed] [Google Scholar]
- 14. Heilmann C, Gerke C, Perdreau-Remington F, Götz F. 1996. Characterization of Tn917 insertion mutants of Staphylococcus epidermidis affected in biofilm formation. Infect. Immun. 64:277–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Heilmann C, Hussain M, Peters G, Götz F. 1997. Evidence for autolysin-mediated primary attachment of Staphylococcus epidermidis to a polystyrene surface. Mol. Microbiol. 24:1013–1024 [DOI] [PubMed] [Google Scholar]
- 16. Hirschhausen N, et al. 2010. A novel staphylococcal internalization mechanism involves the major autolysin Atl and heat shock cognate protein Hsc70 as host cell receptor. Cell Microbiol. 12:1746–1764 [DOI] [PubMed] [Google Scholar]
- 17. Holm L, Rosenström KSP, Schenkel A. 2008. Searching protein structure databases with DaliLite v. 3. Bioinformatics 24:2780–2781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Höltje JV. 1995. From growth to autolysis: the murein hydrolases in Escherichia coli. Arch. Microbiol. 164:243–254 [DOI] [PubMed] [Google Scholar]
- 19. Kabsch W. 2010. XDS. Acta Crystallogr. D Biol. Crystallogr. 66:125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Katoh K, Misawa K, Kuma K, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30:3059–3066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kishan KVR, Agrawal V. 2005. SH3-like fold proteins are structurally conserved and functionally divergent. Curr. Protein Pept. Sci. 6:143–150 [DOI] [PubMed] [Google Scholar]
- 22. Komatsuzawa H, et al. 1997. Subcellular localization of the major autolysin, ATL and its processed proteins in Staphylococcus aureus. Microbiol. Immunol. 41:469–479 [DOI] [PubMed] [Google Scholar]
- 23. Konarev PV, Petoukhov MV, Volkov VV, Svergun DI. 2006. ATSAS 2.1, a program package for small-angle scattering data analysis. J. Appl. Crystallogr. 39:277–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. 2003. PRIMUS—a Windows-PC based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36:1277–1282 [Google Scholar]
- 25. Marino M, Banerjee M, Jonquières R, Cossart P, Ghosh P. 2002. GW domains of the Listeria monocytogenes invasion protein InlB are SH3-like and mediate binding to host ligands. EMBO J. 21:5623–5634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Monterroso B, et al. 2005. Unravelling the structure of the pneumococcal autolytic lysozyme. Biochem. J. 391:41–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nakagawa A, et al. 1999. The three-dimensional structure of the RNA-binding domain of ribosomal protein L2; a protein at the peptidyl transferase center of the ribosome. EMBO J. 18:1459–1467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Perez-Dorado I, et al. 2010. Insights into pneumococcal fratricide from the crystal structures of the modular killing factor LytC. Nat. Struct. Mol. Biol. 17:576–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Petoukhov MV, et al. 2012. New developments in the ATSAS program package for small-angle scattering data analysis. Appl. Crystallogr. 45:342–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Petoukhov MV, Svergun DI. 2005. Global rigid body modelling of macromolecular complexes against small-angle scattering data. Biophys. J. 89:1237–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Poirot O, O'Toole E, Notredame C. 2003. Tcoffee@igs: a web server for computing, evaluating and combining multiple sequence alignments. Nucleic Acids Res. 31:3503–3506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Porod G. 1982. General theory, p 17–51 In Glatter O, Kratky O. (ed), Small-angle X-ray scattering. Academic Press, London, United Kingdom [Google Scholar]
- 33. Roessle MW, et al. 2007. Upgrade of the small-angle X-ray scattering beamline X33 at the European Molecular Biology Laboratory, Hamburg. J. Appl. Crystallogr. 40:s190–s194 [Google Scholar]
- 34. Round AR, et al. 2008. Automated sample-changing robot for solution scattering experiments at the EMBL Hamburg SAXS station X33. J. Appl. Crystallogr. 41:913–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rubinstein M, Colby RH. 2003. Polymer physics, 1st ed Oxford University Press, Oxford, United Kingdom [Google Scholar]
- 36. Ruhland GJ, Fiedler F. 1990. Occurrence and structure of lipoteichoic acids in the genus Staphylococcus. Arch. Microbiol. 154:375–379 [DOI] [PubMed] [Google Scholar]
- 37. Rupp ME, Fey PD, Heilmann C, Götz F. 2001. Characterization of the importance of Staphylococcus epidermidis autolysin and polysaccharide intercellular adhesin in the pathogenesis of intravascular catheter-associated infection in a rat model. J. Infect. Dis. 183:1038–1042 [DOI] [PubMed] [Google Scholar]
- 38. Schlag M, et al. 2010. Role of staphylococcal wall teichoic acid in targeting the major autolysin Atl. Mol. Microbiol. 75:864–873 [DOI] [PubMed] [Google Scholar]
- 39. Shen MY, Sali A. 2006. Statistical potential for assessment and prediction of protein structures. Protein Sci. 15:2507–2524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Studier FW. 2005. Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41:207–234 [DOI] [PubMed] [Google Scholar]
- 41. Suginaka H, Shimatani M, Ogawa M, Kotani S. 1979. Prevention of penicillin-induced lysis of Staphylococcus aureus by cellular lipoteichoic acid. J. Antibiot. (Tokyo) 32:73–77 [DOI] [PubMed] [Google Scholar]
- 42. Svergun DI. 1992. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25:495–503 [Google Scholar]
- 43. Svergun DI. 1999. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 76:2879–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Svergun DI, Barberato C, Koch MHJ. 1995. CRYSOL: a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28:768–773 [Google Scholar]
- 45. Svergun DI, Petoukhov MV, Koch MHJ. 2001. Determination of domain structure of proteins from X-ray solution scattering. Biophys. J. 80:2946–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. van Langevelde P, et al. 1998. Antibiotic-induced release of lipoteichoic acid and peptidoglycan from Staphylococcus aureus: quantitative measurements and biological reactivities. Antimicrob. Agents Chemother. 42:3073–3078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Walter TS, et al. 2006. Lysine methylation as a routine rescue strategy for protein crystallization. Structure 14:1617–1622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Weidenmaier C, Peschel A. 2008. Teichoic acids and related cell-wall glycopolymers in Gram-positive physiology and host interactions. Nat. Rev. Microbiol. 6:276–287 [DOI] [PubMed] [Google Scholar]
- 50. Yamada S, et al. 1996. An autolysin ring associated with cell separation of Staphylococcus aureus. J. Bacteriol. 178:1565–1571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yamamoto H, Miyake Y, Hisaoka M, Kurosawa S, Sekiguchi J. 2008. The major and minor wall teichoic acids prevent the sidewall localization of vegetative DL-endopeptidase LytF in Bacillus subtilis. Mol. Microbiol. 70:297–310 [DOI] [PubMed] [Google Scholar]
- 52. Zoll S, et al. 2010. Structural basis of cell wall cleavage by a staphylococcal autolysin. PLoS Pathog. 6:e1000807 doi:10.1371/journal.ppat.1000807 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.