

Abstract
In the noninfectious soil saprophyte Mycobacterium smegmatis, intracellular levels of the stress alarmones guanosine tetraphosphate and guanosine pentaphosphate, together termed (p)ppGpp, are regulated by the enzyme RelMsm. This enzyme consists of a single, bifunctional polypeptide chain that is capable of both synthesizing and hydrolyzing (p)ppGpp. The relMsm knockout strain of M. smegmatis (ΔrelMsm) is expected to show a (p)ppGpp null [(p)ppGpp0] phenotype. Contrary to this expectation, the strain is capable of synthesizing (p)ppGpp in vivo. In this study, we identify and functionally characterize the open reading frame (ORF), MSMEG_5849, that encodes a second functional (p)ppGpp synthetase in M. smegmatis. In addition to (p)ppGpp synthesis, the 567-amino-acid-long protein encoded by this gene is capable of hydrolyzing RNA·DNA hybrids and bears similarity to the conventional RNase HII enzymes. We have classified this protein as actRelMsm in accordance with the recent nomenclature proposed and have named it MS_RHII-RSD, indicating the two enzymatic activities present [RHII, RNase HII domain, originally identified as domain of unknown function 429 (DUF429), and RSD, RelA_SpoT nucleotidyl transferase domain, the SYNTH domain responsible for (p)ppGpp synthesis activity]. MS_RHII-RSD is expressed and is constitutively active in vivo and behaves like a monofunctional (p)ppGpp synthetase in vitro. The occurrence of the RNase HII and (p)ppGpp synthetase domains together on the same polypeptide chain is suggestive of an in vivo role for this novel protein as a link connecting the essential life processes of DNA replication, repair, and transcription to the highly conserved stress survival pathway, the stringent response.
INTRODUCTION
In their natural habitat, bacteria face a variety of nutritional and environmental stresses, like the depletion of nutrients and oxygen, excess salinity, extremes of temperature and pH, and other such conditions nonconducive to growth. In order to survive this adversity, most bacteria activate a highly conserved physiological response, termed the stringent response, that consists of global gene regulatory changes that aid bacterial survival under stress (9, 28, 40, 48, 58). The key players in this pathway are the RelA-SpoT homolog (RSH) proteins, the second messenger (p)ppGpp, and the RNA polymerase enzyme complex. The hyperphosphorylated guanosine derivatives, guanosine tetraphosphate (ppGpp) and guanosine pentaphosphate (pppGpp), together termed (p)ppGpp or magic spots (7), are synthesized by proteins belonging to the RSH family (4, 31). In the case of Gram-negative bacteria, the long RSH proteins (4) are comprised of monofunctional RelA, which synthesizes (p)ppGpp, and bifunctional SpoT, which both synthesizes and degrades (p)ppGpp (28, 48). On the other hand, in Gram-positive bacteria, the synthesis {SYNTH or RSD [RelA_SpoT nucleotidyl transferase domain, the SYNTH domain responsible for (p)ppGpp synthesis activity]} and degradation (HD) activities reside on a single polypeptide chain (4). A reciprocal regulation exists between these opposing activities such that futile cycles of (p)ppGpp synthesis and hydrolysis are prevented (17, 19, 20). Increased intracellular levels of (p)ppGpp facilitate the entry of bacterial cells into the growth-inhibitory dormant state, characterized by an immediate downshift in stable RNA synthesis, DNA replication, and protein synthesis and an upshift in macromolecular degradation and amino acid biosynthesis (9, 28, 40, 48, 58). Upon restoration of conditions favorable for growth, (p)ppGpp levels are brought down and the cells resume normal growth. The downstream effects of (p)ppGpp signaling are chiefly mediated by the RNA polymerase enzyme complex (9, 28, 40, 48, 58), although recent studies have shown (p)ppGpp to affect the synthesis, stability, and activity of various other proteins and regulatory RNA molecules (reference 12 and references therein). Upon binding to RNA polymerase (2, 8, 41, 55), (p)ppGpp shifts the transcriptional balance from one favoring growth to one favoring survival under stress. Given its central role in the stringent response, it is not surprising that (p)ppGpp affects all three essential processes of life, i.e., DNA replication, transcription, and translation (reference 48 and references therein).
The stringent response in the genus Mycobacterium was first reported by Ojha et al., in a carbon-starved culture of the soil saprophyte Mycobacterium smegmatis (39). In this bacterium, as in other Gram-positive bacteria (17), turnover of (p)ppGpp is maintained by a single bifunctional protein, RelMsm, which both synthesizes and hydrolyzes (p)ppGpp (19, 20). In addition to the negative coordination of the synthetic and hydrolytic abilities of this enzyme, a second layer of control is exerted by the carboxy-terminal domain of RelMsm on the amino-terminal catalytic activities (20). This double-layered control mechanism efficiently regulates intracellular levels of (p)ppGpp in M. smegmatis under various conditions of growth. Consequently, a rel-deficient strain of M. smegmatis (ΔrelMsm) (11, 30) was expected to show a (p)ppGpp null [(p)ppGpp0] phenotype and was found to be compromised in its long-term survival (30). The serendipitous observation that the ΔrelMsm strain is capable of synthesizing (p)ppGpp in vivo led to the discovery of a second, monofunctional (p)ppGpp synthetase in this bacterium, encoded by the MSMEG_5849 gene. Orthologs of this protein could be identified in other mycobacterial species with completely sequenced genomes (see Table S1 in the supplemental material) (4). In addition to conventional Rel proteins, the existence of short alarmone synthetases (SAS) in Bacillus subtilis (33, 34), Streptococcus mutans (23, 26), and Vibrio cholerae (13) has been reported previously. In this study, we report the biochemical and functional characterization of the MSMEG_5849 gene product, the novel (p)ppGpp synthetase from M. smegmatis, henceforth termed MS_RHII-RSD. In addition to (p)ppGpp synthesis activity, MS_RHII-RSD possesses a functional RNase HII activity (RHII, RNase HII domain, originally identified as domain of unknown function 429 [DUF429]). The occurrence of these two activities on the same polypeptide chain and the importance of (p)ppGpp in bacterial survival under stress lead us to propose an in vivo role for MS_RHII-RSD as a link connecting the stringent response to the essential life processes of DNA replication, repair, and transcription.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
A list of the bacterial strains and plasmids used in this study is given in Table 1. Escherichia coli strains DH5α and BL21(DE3) were grown in Luria-Bertani (LB) broth at 37°C with agitation. The temperature-sensitive E. coli strain GJ12500 (a gift from J. Gowrishankar, CDFD, India) was grown at 30°C with agitation in LB broth containing kanamycin (30 μg/ml). E. coli GJ12055 complemented with plasmids was grown at 42°C for temperature selection. All LB plate cultures contained 1.5% (wt/vol) agar. Ampicillin (100 μg/ml) was added to the growth medium for E. coli cultures harboring pET21b derivatives and pSK760 plasmids. M. smegmatis mc2155 wild type (WT) (47) and ΔrelMsm strains (relMsm::hyg) (30) were grown in Middlebrook 7H9 broth (MB7H9) (Difco) supplemented with glucose to a final concentration of either 0.02% (wt/vol) for glucose-starved or 2% (wt/vol) for glucose-enriched medium. Tween 80 to a final concentration of 0.05% (vol/vol) was added to all mycobacterial broth cultures. MB7H9 plate cultures contained 1.5% (wt/vol) agar and lacked Tween 80. Hygromycin (100 μg/ml) was added to the growth medium for the ΔrelMsm strain. To detect in vivo levels of (p)ppGpp, M. smegmatis cultures were grown in 1× MOPS (morpholinepropanesulfonic acid) medium supplemented with 80 μg/ml Casamino acids, 0.05% (vol/vol) Tween 80, and either 0.02% (wt/vol) or 2% (wt/vol) glucose. The composition of 10× MOPS medium was 400 mM MOPS (Sigma), 40 mM Tricine (Sigma), 500 mM KCl, 100 mM NH4Cl, 2 mM KH2PO4, 5 mM MgSO4·7H2O, 0.1 mM FeCl3·6 H2O. The pH of the medium was adjusted to 7.4 with Tris-Cl. The solution was made in autoclaved deionized water, filter sterilized, and stored at 4°C in a light-tight container. The 1-mg/ml stock solution of Casamino acids (Difco) adjusted to pH 7.4 with Tris-Cl was filter sterilized and stored at 4°C.
Table 1.
List of strains and plasmids used in this study
| Strain | Description | Source and/or reference |
|---|---|---|
| E. coli DH5α | E. coli host strain for DNA cloning | Laboratory stock |
| E. coli BL21(DE3) | E. coli host strain for protein expression | Laboratory stock |
| E. coli GJ12055 | E. coli K-12 temperature-sensitive strain, chromosomal knockout for RNase HI and RNase HII genes, rnhA::kan rnhB::cm(Ts) | J. Gowrishankar, CDFD, India |
| M. smegmatis mc2155 | Wild-type strain | Laboratory stock |
| M. smegmatis ΔrelMsm | M. smegmatis chromosomal knockout for relMsm gene, relMsm::hyg | Laboratory stock; 30 |
| Plasmid | ||
| pET21b | E. coli expression vector encoding carboxy-terminal hexahistidine tag, Ampr | Novagen |
| pET-MS_RHII-RSD | Full-length MS_RHII-RSD gene (nucleotides 1 to 1704) cloned into pET21b, Ampr | This study |
| pET-RHII | Nucleotides 1 to 840 of MS_RHII-RSD gene corresponding to RHII domain (amino acids 1 to 280) cloned into pET21b, Ampr | This study |
| pET-RSD | Nucleotides 841 to 1704 of MS_RHII-RSD gene corresponding to RSD domain (amino acids 281 to 567) cloned into pET21b, Ampr | This study |
| pSK760 | pBR322-derived plasmid containing full-length E. coli rnhA gene, Ampr | J. Gowrishankar, CDFD, India; 21 |
Bioinformatics analyses.
Protein amino acid sequences and gene nucleotide sequences were obtained from NCBI (http://www.ncbi.nlm.nih.gov), CMR (http://cmr.jcvi.org), and MycoDB (http://www.xbase.ac.uk/mycodb/) online databases. The protein BLAST searches (1) available on these websites were used to identify MS_RHII-RSD orthologs in other mycobacterial species. Conserved domains were identified using the NCBI Conserved Domain Database search (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) (29). ClustalW (http://www.ebi.ac.uk/clustalw) (53) was used to generate multiple sequence alignments and corresponding phylograms. Protein fold recognition was carried out using the Phyre2 server (22) (http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index). Default parameters, as suggested by the respective programs, were used in all analyses. Theoretical values of protein parameters were obtained using the ProtParam tool at the ExPASy website (http://web.expasy.org/protparam/).
Cloning of full-length MS_RHII-RSD gene and its domain variants.
The full-length MS_RHII-RSD gene (1,704 nucleotides), corresponding to the open reading frame (ORF) MSMEG_5849, was PCR amplified from M. smegmatis mc2155 wild-type genomic DNA using primers MssF (CAGGCTGGACATATGCACCACCCCCCGTAC) and MssR (GACCGACGCAAGCTTGGCCTGCAGCTTCTC). DNA sequences corresponding to the RNase HII domain (nucleotides 1 to 840) and the (p)ppGpp synthetase domain (rsd, nucleotides 841 to 1704) were PCR amplified in a similar manner from genomic DNA using primers MssF and RnhR (GTCGTCGAGCAGAAGCTTCACGAGGTCGTG) and RsdF (GCGCGCTATCATATGCTCGTGACGGGCCTG) and MssR, respectively. The PCR amplicons obtained were cloned into plasmid pET21b (Novagen) between the NdeI and HindIII sites to generate plasmids pET-RHII-RSD (7 kb), pET-RHII (6.2 kb), and pET-RSD (6.2 kb). Positive clones were confirmed by DNA sequencing (Eurofins MWG). All PCRs were carried out using Dynazyme EXT polymerase (Finnzyme) according to the manufacturer's instructions. The enzymes used for cloning were procured from New England BioLabs. Plasmid pET-RelWT (7.8 kb) (19) was used as the source of the full-length RelMsm protein.
Protein expression and purification.
Full-length MS_RHII-RSD, its RNase HII domain variant (RHII), and full-length RelMsm, all with carboxy-terminal hexahistidine tags, were overexpressed and purified as described previously (19). Briefly, E. coli BL21(DE3) cells transformed with the respective plasmids were grown in LB broth at 37°C with agitation, induced with 1 mM isopropyl-β-thio-galactopyranoside (IPTG) at an optical density at 600 nm (OD600) of 0.6, and grown for a further 3 h. Cells were harvested and lysed by sonication on ice in lysis buffer (50 mM Tris-Cl, pH 7.9 at 4°C, 500 mM NaCl, 1 mM phenylmethylsulfonyl fluoride [PMSF]), and the cleared lysate was loaded onto a nickel-nitrilotriacetic acid (Ni-NTA) column preequilibrated with equilibration buffer (50 mM Tris-Cl, pH 7.9 at 4°C, 500 mM NaCl, 10 mM imidazole). The column was washed with 100 column volumes of equilibration buffer, and the protein was eluted in elution buffer (50 mM Tris-Cl, pH 7.9 at 4°C, 500 mM NaCl, 500 mM imidazole). The eluted proteins were dialyzed against dialysis buffer (50 mM Tris-Cl, pH 7.9 at 4°C, 150 mM NaCl) and stored at 4°C. Dialysis buffer for MS_RHII-RSD and its domain variants additionally contained 100 mM imidazole, as the complete removal of imidazole resulted in protein aggregation and precipitation. The RSD domain variant of MS_RHII-RSD was purified by denaturing Ni-NTA purification, as it was seen to enrich into inclusion bodies when overexpressed. E. coli BL21(DE3) cells harboring pET-RSD plasmid were grown in LB broth at 37°C with agitation to an OD600 of 0.6, cooled to 17°C, induced with IPTG to a final concentration of 0.5 mM, and grown for a further 12 h at 17°C with agitation. Harvested cells were lysed in lysis buffer (50 mM Tris-Cl, pH 7.9 at 4°C, 500 mM NaCl, 1 mM PMSF) by sonication on ice. The clarified supernatant was discarded, and the pellet was solubilized overnight in extraction buffer (50 mM Tris-Cl, pH 7.9 at room temperature [RT], 1 mM EDTA, 10 mM MgCl2, 10 mM dithiothreitol [DTT], 0.2 M KCl, 20% glycerol, 6 M urea). After centrifugation, the clarified supernatant was applied to a Ni-NTA column preequilibrated with equilibration buffer (50 mM Tris-Cl, pH 7.9 at RT, 0.1 mM EDTA, 5% glycerol, 10 mM imidazole, 6 M urea) and washed with a further 50 volumes of equilibration buffer. The protein was eluted in elution buffer (50 mM Tris-Cl, pH 7.9 at RT, 0.1 mM EDTA, 5% glycerol, 200 mM imidazole, 6 M urea). The purified, denatured protein was renatured by stepwise dialysis in buffers containing sequentially decreasing concentrations of urea (50 mM Tris-Cl, pH 7.9 at 4°C, 150 mM NaCl, 100 mM imidazole, and 4 M, 2 M, and 0 M urea, in that order). The circular dichroism (CD) spectrum of the protein was measured after each dialysis step in order to determine the extent of refolding. Dialysis was stopped when no further change in the CD spectrum was observed. The mass of all purified proteins was determined by matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) and electrospray ionization (ESI) mass spectrometry. Protein identity was further confirmed by MALDI-TOF mass spectrometric analysis of trypsin digests, followed by a Mascot database search (Matrix Science Ltd.).
In vivo (p)ppGpp synthesis assay.
M. smegmatis mc2155 and ΔrelMsm strains were grown to an OD600 of 0.2 in 5 ml 1× MOPS medium supplemented with 80 μg/ml Casamino acids, 0.05% (vol/vol) Tween 80, and either 0.02% (wt/vol) or 2% (wt/vol) glucose at 37°C with agitation. The growth medium for the ΔrelMsm strain additionally contained 100 μg/ml hygromycin. Cultures were radiolabeled by adding [o-32P]phosphoric acid (specific activity, >3,000 mCi/mmol; BRIT) directly to the growth medium to a final concentration of 100 μCi/ml. Cells were harvested at 12, 18, 24, 36, 48, 72, 96, and 120 h of growth, washed, resuspended in 1× MOPS, lysed by adding an equal volume of 12 N formic acid, and stored on ice for 20 min. After centrifugation at 13,000 rpm, 4°C, for 10 min, 5 μl of supernatant, normalized to an OD260 of 2.0, was spotted onto a polyethylenimine (PEI)-cellulose thin-layer chromatography (TLC) sheet (Merck) and developed in one dimension in 1.5 M KH2PO4 (pH 3.4). The TLC sheets were then air dried and phosphor imaged (Fujifilm FLA5000), and spots corresponding to (p)ppGpp were analyzed by densitometry using MultiGauge software, version 2.3, provided by Fuji Film. The identity of the spots was further confirmed by MALDI-TOF mass spectrometry.
Mass spectrometric identification of (p)ppGpp.
Spots corresponding to ppGpp and pppGpp were cored out from the TLC sheets and extracted on ice in 50 mM Tris-Cl, pH 7.9 at RT, 1 mM MgCl2. One microliter of the extracted compound was mixed with 1 μl of the saturated matrix solution and spotted onto the mass spectrometric target plate. Mass spectra were acquired on a Bruker Daltonics Ultraflex TOF-TOF instrument equipped with a nitrogen laser in the positive ion, reflectron mode. The matrix used was dihydrobenzoic acid (DHB) in 1:1 acetonitrile-water and 0.1% trifluoroacetic acid (TFA). Standard calibrants provided by the instrument manufacturers were used for external calibration of the mass spectrometer.
In vitro (p)ppGpp synthesis assay using purified proteins.
To determine the substrate specificity of the (p)ppGpp synthetases under study, 10-μl reaction mixtures were set up containing 50 mM HEPES, pH 7.5, 1 mM DTT, 1 mM ATP, 1 μCi/μmol [γ-32P]ATP (specific activity, >3,000 mCi/mmol; Perkin Elmer), 1 μM desired protein, and either 1 mM GDP or 1 mM GTP, with either the absence or presence of 1 mM MgCl2. A reaction mixture lacking any protein was used as the negative control. Reactions were allowed to proceed for 30 min at 37°C, after which they were stopped by adding 1 μl of 12 N formic acid. After centrifugation, 5 μl of the supernatant was spotted onto a PEI-coated TLC sheet, developed, scanned, and analyzed by densitometry as described above. In a separate experiment, to assess the effect of Mg(II) ions on (p)ppGpp synthesis, the MgCl2 concentration was varied from 0 to 25 mM, keeping all other parameters constant. The kinetic constants for the enzymes under study were determined by keeping the protein concentration constant at 1 μM and the ATP concentration saturating at 20 mM. The concentrations of GDP and GTP were varied between 10 μM and 5 mM. In all cases mentioned, purified protein concentrations were measured spectrophotometrically (OD280) by using the extinction coefficient at 280 nm obtained from the ProtParam tool on the ExPASy website. All assays were performed at least thrice, and the average values obtained were used for further analyses.
In vitro (p)ppGpp synthesis assay using crude cell lysates.
To determine the (p)ppGpp synthetic activity of the RSD domain variant of MS_RHII-RSD, crude cell lysates were added directly to the reaction mixture instead of the purified proteins. E. coli BL21(DE3) cultures transformed with pET21b empty vector (negative control), pET-MS_RHII-RSD (positive control), pET-RHII, and pET-RSD were grown, induced, harvested, and lysed as mentioned above. The total protein concentration in the clarified lysates was determined, and 200 μg/ml of each lysate was added to the reaction mixture without further purification. To test for trans complementation and interdomain interactions, the RSD and RHII lysates were added into the same reaction mixture in a total protein ratio of 5:1, taking into consideration that only 20% of RSD was obtained in the soluble fraction, while 80% was present in inclusion bodies. Trans complementation was also tested using 1 μM (each) purified RHII and purified and renatured RSD. In this case, a 1:1 molar ratio of the purified proteins was used, based on the fact that the two domains occurred together, in a single copy, on the full-length MS_RHII-RSD polypeptide chain.
In vitro ppGpp hydrolysis assay.
A 100-μl reaction mixture for ppGpp synthesis was set up using purified MS_RHII-RSD as described earlier. The reaction was stopped after 1 h at 37°C with 10 μl of 12 N formic acid. After centrifugation, the supernatant containing radiolabeled ppGpp was spotted onto PEI-cellulose TLC sheets and developed in one dimension in 1.5 M KH2PO4 (pH 3.4). Air-dried TLC sheets were then phosphor imaged (Fujifilm FLA5000), and spots corresponding to ppGpp were cored out and extracted on ice in 50 mM Tris-Cl, pH 7.9 at RT, and 1 mM MgCl2. Fifty-microliter hydrolysis reaction mixtures were set up containing 50 mM Tris-Cl, pH 7.9 at RT, 10 mM MnCl2, 1 mM DTT, 1 mM purified ppGpp, and 1 μM either RelMsm or MS_RHII-RSD. Reaction mixtures were incubated at 37°C, and 10-μl aliquots were collected at 10, 20, 30, and 60 min after the start of the reaction. Reactions were stopped at the said time points by adding 1 μl of 12 N formic acid to precipitate the proteins. Pure pyrophosphate (PPi) and ppGpp, cored out and extracted in a similar manner from the TLC sheets, were used as controls. After centrifugation, 5-μl amounts of the supernatants were spotted onto a PEI-cellulose TLC sheet and analyzed as described above.
In vivo complementation assay for RNase HII activity.
RNase HII activity was tested in vivo in E. coli ΔrnhA ΔrnhB(Ts) strain GJ12055, which lacks the genes encoding the RNase HI and RNase HII enzymes (18). This strain can survive at the permissive temperature of 30°C but not at the restrictive temperature of 42°C. When complemented with a plasmid coding for functional RNase HI or RNase HII enzymatic activity, it is able to overcome the growth defect and survive at 42°C. E. coli GJ12055 competent cells were electrotransformed with plasmids pSK760 (positive control harboring full-length rnhA gene from E. coli) (21), pET21b (negative control, empty vector with no insert), pET-MS_RHII-RSD, and pET-RHII. Plate titrations were performed in duplicates on LB agar plates containing 100 μg/ml ampicillin, with one set incubated overnight at 30°C and the duplicate set at 42°C.
In vitro fluorescence assay for RNase HII activity.
RNase HII activity was monitored in vitro by measuring the increase in fluorescence resulting from the degradation of an artificially synthesized RNA·DNA hybrid as described earlier (57). Concentrations of 100 μM (each) custom-synthesized 12-mers, comprising 5′-fluorescein-poly(A) ribooligonucleotide and 3′-dabsyl-poly(dT) deoxyribooligonucleotide (Bioneer, South Korea), were hybridized in annealing buffer (50 mM Tris-Cl, pH 7.5 at RT, 60 mM KCl) by heating the mixture to 95°C for 5 min, followed by slow cooling to room temperature. Aliquots of the RNA·DNA annealed hybrid (AH) thus formed were stored at −70°C till further use. RNase HII activity was monitored by measuring the increase in fluorescence after 30 min at 37°C in a reaction mixture containing 50 mM Tris-Cl, pH 7.5 at RT, 60 mM KCl, 5 mM MnCl2, 500 nM AH, and 100 nM respective enzyme (full-length MS_RHII-RSD, domain variant RHII, and E. coli RNase HII [NEB]). All reagents, apparatus, and containers were treated with diethylpyrocarbonate (DEPC) to remove external RNase contamination.
Western blot analysis for in vivo protein expression.
Polyserum against MS_RHII-RSD was used to detect the presence of this protein in cell lysates from M. smegmatis mc2155 and ΔrelMsm strains grown to 12, 24, 48, 72, 96, and 120 h in both 0.02% glucose-starved and 2% glucose-enriched medium. The expression of this protein under a variety of stress conditions, including DNA damage (20 min UV exposure), oxidative stress (36 mM H2O2), acidic stress (pH 5 with HCl), alkaline stress (pH 10 with NaCl), and osmotic stress (4 M NaCl), was tested as previously described (46). An amount of 100 μg of total protein was loaded in each case. M. smegmatis RNA polymerase α-subunit was used as the internal loading control (15). Antibodies against RNA polymerase α-subunit (15), RelMsm (19), and MS_RHII-RSD were raised in New Zealand White rabbits at the Indian Institute of Science (IISc) in-house Central Animal Facility.
RESULTS
M. smegmatis ΔrelMsm synthesizes (p)ppGpp in vivo.
Until recently, the long RSH protein, RelMsm, was the only known (p)ppGpp synthetase in M. smegmatis (11, 19, 30). This bifunctional protein is expressed under stress and in the stationary phase of growth. It regulates the intracellular levels of (p)ppGpp in M. smegmatis by negatively coordinating (p)ppGpp synthesis and hydrolysis. Consequently, a relMsm knockout strain (ΔrelMsm) was expected to be deficient in (p)ppGpp formation and exhibit a (p)ppGpp0 phenotype (11, 19, 30). The converse observation that M. smegmatis ΔrelMsm is capable of synthesizing (p)ppGpp in vivo (Fig. 1a and b) led us to propose the existence of a second (p)ppGpp synthetase in this bacterium. This proposition is not entirely out of place, as the occurrence of short alarmone synthetases (SAS), in addition to the conventional Rel protein, has been reported previously (13, 23, 26, 33, 34). The SAS proteins are known to fine-tune the stringent response to a variety of environmental stresses in Streptococcus mutans (23, 26), Bacillus subtilis (33, 34), and Vibrio cholerae (13). In a recent bioinformatics study (4), several heretofore-unknown SAS and short alarmone hydrolases (SAH) have been identified across the bacterial kingdom. To test for the presence of a novel (p)ppGpp synthetase in M. smegmatis, we performed a systematic study in which M. smegmatis WT and ΔrelMsm strains were grown in glucose-starved (0.02% glucose) and glucose-enriched (2% glucose) media supplemented with radiolabeled [o-32P]phosphoric acid. Total nucleotide extracts were prepared from these cultures and subjected to one-dimensional thin-layer chromatography (TLC). The results clearly indicate the presence of basal levels of ppGpp from early stages of growth to the late stationary phase in both strains (Fig. 1a, b, c, and d, spots marked by an asterisk). The M. smegmatis ΔrelMsm strain constitutively produced ppGpp alone (Fig. 1a and b, spots marked by an asterisk), while the WT strain produced pppGpp (Fig. 1c and d, spots marked by a double asterisk) in addition to ppGpp (Fig. 1c and d, spots marked by an asterisk) between 72 and 120 h of growth. The identity of these spots was confirmed by MALDI-TOF mass spectrometry as described in Materials and Methods (Fig. 2a and b; also see Fig. S1a to d in the supplemental material). The formation of pppGpp by the WT strain at the later time points can be attributed to the expression of conventional RelMsm in the stationary phase of growth. The densitometric analysis performed showed that the levels of ppGpp remained uniform in the ΔrelMsm strain throughout various growth stages, irrespective of whether the culture was grown in glucose-starved or glucose-enriched medium (Fig. 2c). In the WT strain, ppGpp levels were enhanced in the stationary and late stationary phases (72 to 120 h) due to the added synthesis by RelMsm. However, a drop in ppGpp levels was observed for M. smegmatis cultures grown to the late stationary phase (72 to 120 h) in 0.02% glucose-starved medium (Fig. 1a and c). This is due to the increased cell death and reduction in cell number caused by the rapid depletion of the already scarce carbon source at these late time points. On the other hand, the amount of glucose available at the same time point is much higher in the 2% glucose-enriched medium, and hence, a fall in ppGpp levels is not observed in this case. Furthermore, ppGpp was not detected visibly at the 12-h time point. Decreased cell numbers at this early stage of growth may be the reason for the nondetection of ppGpp at this point. This observation is consistent with previous studies on the M. smegmatis ΔrelMsm strain (19, 30, 39), wherein cells were harvested 12 h after the addition of [o-32P]phosphoric acid (OD600 of ∼0.8) and processed for chromatographic purposes. These observations, taken together, clearly indicated the presence of a second (p)ppGpp synthetase in M. smegmatis, distinct from the conventional RelMsm.
Fig 1.

In vivo (p)ppGpp synthesis assay. Thin-layer chromatograms showing total nucleotide extracts from M. smegmatis cultures harvested at different stages of growth. (a) ΔrelMsm, 0.02% glucose; (b) ΔrelMsm, 2% glucose; (c) WT, 0.02% glucose; and (d) WT, 2% glucose. A basal level of ppGpp synthesis is seen from 12 up to 120 h of growth. * and ** indicate the spots corresponding to ppGpp and pppGpp, respectively, which were cored out, extracted from the TLC sheets, and used for mass spectrometric identification of (p)ppGpp.
Fig 2.

M. smegmatis ΔrelMsm strain produces ppGpp in vivo. (a and b) Mass spectrometric identification of ppGpp synthesized by M. smegmatis ΔrelMsm in 0.02% glucose-starved medium (a) and 2% glucose-enriched medium (b). Peaks with m/z values of 603, 626, and 642 correspond to the monoisotopic, sodiated, and potassiated mass of ppGpp. (c) Graphical representation of the relative amounts of ppGpp synthesized in vivo in M. smegmatis WT and ΔrelMsm strains under 0.02% and 2% glucose (Glc) conditions at various time points. The amount of ppGpp synthesized by the M. smegmatis WT strain grown to 120 h in 2% glucose is taken as 100%, and ppGpp synthesized at all other time points has been normalized to this value. a.u., arbitrary units. Error bars show standard deviations.
In silico analyses identify MSMEG_5849 as the second (p)ppGpp synthetase in M. smegmatis.
A bioinformatics approach was used to identify the ORF that encoded the novel (p)ppGpp synthetase in M. smegmatis. A protein BLAST search (1) was performed against the M. smegmatis mc2155 genome available at the CMR website, using the B. subtilis (33, 34) and S. mutans SAS (26) protein sequences as the query. The BLAST search results identified MSMEG_5849 as the putative ORF, showing significant amino acid sequence similarity to the SAS query sequences. This ORF codes for a 567-amino-acid-long conserved hypothetical protein in M. smegmatis. In accordance with the nomenclature for RSH proteins proposed recently by Atkinson et al. (4), we have categorized MSMEG_5849 as actRelMsm, the novel SAS protein from M. smegmatis.
A Conserved Domain Database (CDD) search (29) at the NCBI website predicted the presence of two domains in MSMEG_5849 (Fig. 3a). The amino-terminal domain was initially identified as belonging to Clusters of Orthologous Groups COG4328, now identified as domain of unknown function DUF429. COG4328 comprises proteins belonging to the RNase H superfamily. Multiple sequence alignments using EBI ClustalW (53) suggested that the amino-terminal domain of MSMEG_5849 was closely related to the RNase HII enzymes, with the key Asp (D) and Glu (E) residues being conserved (see Fig. S2a in the supplemental material). On the other hand, the carboxy-terminal domain showed distinct similarity to the RelA_SpoT nucleotidyl transferase (NT) superfamily (Fig. 3a). Proteins belonging to this superfamily possess the RSD or SYNTH domain (4) that is responsible for (p)ppGpp synthetic ability. The RSD domain of MSMEG_5849 aligns with the RSD domain of RelMsm and those of the SAS proteins from other bacterial species (see Fig. S2b in the supplemental material). It possesses an acidic conserved domain motif, DDRD. Protein fold recognition using the Phyre2 server (22) yielded similar results, with the amino-terminal domain being modeled after the RNase H-like motif and the carboxy-terminal domain after the (p)ppGpp synthetase (RSD) domain of GTP pyrophosphokinase SMU1046c, a SAS protein from S. mutans. The full-length MSMEG_5849 protein was also modeled after SMU1046c. Taking into consideration these structural similarities, MSMEG_5849 has been renamed MS_RHII-RSD, an abbreviation for Mycobacterium smegmatis RNase HII-RSD domain protein. Subsequently, the amino-terminal domain (amino acids 1 to 280) of MS_RHII-RSD has been termed RHII and the carboxy-terminal domain (amino acids 281 to 567) RSD (Fig. 3a). This nomenclature has been used throughout this study.
Fig 3.
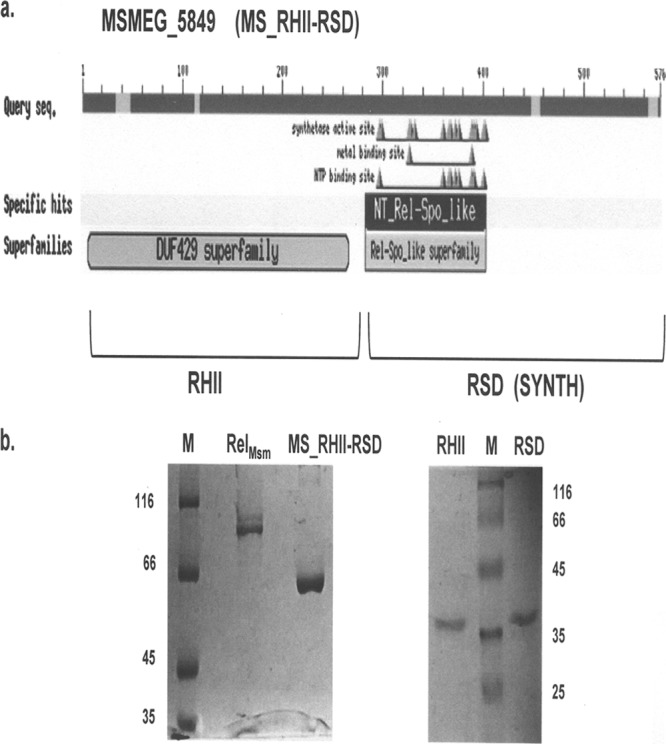
(a) Conserved domain architecture of MS_RHII-RSD obtained from NCBI CDD search identifies the presence of a domain of unknown function, DUF429, in the amino-terminal sequence and a RelA_SpoT nucleotidyl transferase (NT) domain responsible for (p)ppGpp synthesis in the carboxy-terminal sequence of the protein. Based on the experimentally proved activities of the two domains, they have been renamed RHII for RNase HII activity and RSD for the (p)ppGpp synthesis activity. (b) SDS-polyacrylamide gels showing purified His-tagged proteins used in this study. M, protein molecular size marker. Numbers indicate molecular mass in kDa.
Another notable feature of MS_RHII-RSD was revealed by the NCBI CDD search: it lacks the (p)ppGpp hydrolysis (HD) domain and the regulatory ACT and TGS domains. This is unlike RelMsm, which possesses all four domains. In this respect, MS_RHII-RSD is similar to the actinobacterial monofunctional SAS proteins that possess the (p)ppGpp synthetase domain (RSD or SYNTH) alone and have been classified as actRel in a recent study (4). In silico analyses thus identify MS_RHII-RSD as a novel dual-function RNase HII-(p)ppGpp synthetase in M. smegmatis. In the following sections, we provide experimental evidence for the same.
(p)ppGpp synthesis by MS_RHII-RSD preferentially utilizes GDP as the substrate and is unaffected by Mg(II) ion concentration.
It is known that Gram-positive bacteria, including mycobacteria, predominantly use pppGpp as the stringent factor, while Gram-negative bacteria use ppGpp (40, 58). This is attributed to the fact that bifunctional (p)ppGpp synthetases prefer GTP as a substrate over GDP, synthesizing pppGpp as the major product, unlike the monofunctional synthetases that prefer GDP as their substrate and, consequently, produce more ppGpp (43, 44). The presence of the enzyme pppGpp 5′-γ-phosphohydrolase in Gram-negative bacteria (16) is another factor that contributes to the differential utilization of (p)ppGpp observed in the two classes of bacteria. This enzyme efficiently converts pppGpp to ppGpp (16), making ppGpp the major effector alarmone in Gram-negative bacteria.
In order to study the differential substrate utilization of MS_RHII-RSD vis-à-vis that of RelMsm in vitro, recombinant forms of these two proteins were purified from E. coli BL21(DE3) cultures using nickel-histidine affinity chromatography (Fig. 3b). Purified full-length MS_RHII-RSD has a molecular mass of 64.5 kDa, possesses a predominantly alpha-helical structure, and exists as a hexamer in solution (data not shown). Purified RelMsm has a molecular mass of 89 kDa, is also predominantly alpha-helical, and elutes out as a trimer in size exclusion chromatography (data not shown). To determine the substrate specificities of the two proteins, in vitro (p)ppGpp synthesis reactions were set up using either GDP or GTP as the substrate, in the absence or presence of Mg(II) ions. It is evident from the result (Fig. 4a) that RelMsm efficiently utilizes GTP both in the absence and presence of Mg(II) ions (Fig. 4a, RelMsm, lanes 5 and 6) and forms pppGpp as the major product. It utilizes GDP less efficiently than GTP (Fig. 4a, RelMsm, lanes 2 and 3). In contrast, MS_RHII-RSD specifically utilizes GDP as a substrate, leading to the formation of ppGpp as the major product (Fig. 4a, MS_RHII-RSD, lanes 2 and 3). It utilizes GTP comparatively less efficiently than GDP (Fig. 4a, MS_RHII-RSD, lanes 5 and 6). (p)ppGpp synthesis by MS_RHII-RSD is free from the Mg(II) ion requirement, with the same amount of (p)ppGpp being formed in both the absence and presence of Mg(II) ions (Fig. 4a, MS_RHII-RSD, compare lane 2 with 3 and lane 5 with 6). The kinetic constants for (p)ppGpp synthesis reaction, mentioned in Table 2, further explain the differential substrate utilization by RelMsm and MS_RHII-RSD. The Km values obtained for both GDP and GTP are comparable to those reported for E. coli and Mycobacterium tuberculosis Rel in the absence of any cofactors (5, 6, 43, 44). It is clear from these data that MS_RHII-RSD utilizes GDP 5-fold better than GTP. Full-length RelMsm uses GTP only approximately twice as well as GDP.
Fig 4.
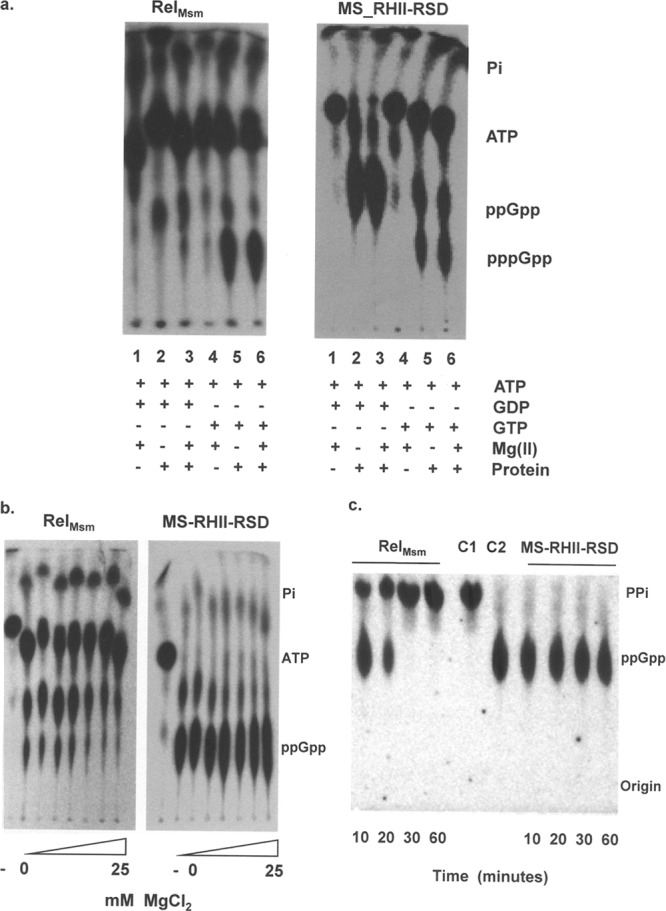
In vitro (p)ppGpp synthesis and hydrolysis assays using full-length MS_RHII-RSD and RelMsm. (a) Thin-layer chromatograms showing in vitro (p)ppGpp synthesis, performed to demonstrate the substrate specificity toward GDP and GTP in the absence and presence of Mg(II) ions. Lanes 1 and 4, negative control, no protein added to reaction mixture; lane 2, 1 mM GDP, no Mg(II); lane 3, 1 mM GDP, 1 mM Mg(II); lane 5, 1 mM GTP, no Mg(II); lane 6, 1 mM GTP, 1 mM Mg(II); +, presence of reaction component; −, absence of reaction component. (b) Thin-layer chromatogram showing effect of increasing Mg(II) ion concentration on ppGpp synthesis activity. Mg(II) ion concentration was varied between 0 and 25 mM. −, negative control wherein no protein was added to the reaction mixture. (c) Thin-layer chromatogram showing in vitro Mn(II)-dependent ppGpp hydrolysis activity. A concentration of 1 mM ppGpp was used as the substrate. Numbers indicate the time in minutes. C1 and C2 indicate control reaction mixtures containing purified radioactive PPi and ppGpp, respectively, and lacking any protein.
Table 2.
Kinetic constants for in vitro (p)ppGpp synthesis by RelMsm and MS_RHII-RSD
| Protein | Km (GDP) μM | Km (GTP) μM | Vmax (GDP) μM ppGpp formed/min | Vmax (GTP) μM pppGpp formed/min | kcat (GDP) min−1 | kcat (GTP) min−1 |
|---|---|---|---|---|---|---|
| RelMsma | 1,532.2 ± 52 | 1,162.8 ± 241 | 112.7 ± 12 | 147.2 ± 7.5 | 73 | 127 |
| MS_RHII-RSD | 983.7 ± 34 | 968.52 ± 64 | 105.6 ± 10 | 20.2 ± 9.7 | 107 | 20 |
RelMsm shows allosteric behavior both for GDP and GTP, and hence, Km values for this protein may be read as Km(apparent).
The (p)ppGpp synthetase domain (RSD or SYNTH) of bifunctional Rel proteins shares structural similarities with the synthesis domain of DNA polymerase β (17). Akin to the polymerase, bifunctional Rel proteins synthesize (p)ppGpp by exploiting a single Mg(II) ion (17). An excess of Mg(II) ions inhibits the synthetic activity by interacting with the basic RXKD motif present in the RSD domain and causing a loop-to-helix structural transition that closes the substrate binding channel (43, 44). Such an inhibition is not observed in the case of monofunctional Rel proteins that possess the acidic DXED motif, with the additional Mg(II) ions stabilizing substrate nucleotide binding. The effect of increasing Mg(II) ion concentrations on the ppGpp synthetic ability of RelMsm and MS_RHII-RSD was studied. As expected, RelMsm showed a marked decrease in ppGpp synthesis activity with increasing Mg(II) ion concentration, while MS_RHII-RSD displayed no such inhibition (Fig. 4b). The change in the CD profiles of the two proteins in the presence of increasing concentrations of Mg(II) ions confirms the loop-to-helix transition in RelMsm, while the reverse trend is observed for MS_RHII-RSD (data not shown). From these results, it may be concluded that RelMsm exploits a single Mg(II) ion to catalyze (p)ppGpp synthesis, while MS_RHII-RSD does not. The mechanistic details of (p)ppGpp production by MS_RHII-RSD need further investigation.
MS_RHII-RSD behaves like a monofunctional (p)ppGpp synthetase that can synthesize (p)ppGpp but not hydrolyze it.
In silico protein fold recognition programs and conserved domain searches had predicted the absence of the (p)ppGpp hydrolysis domain (HD) from MS_RHII-RSD (Fig. 3a). This prediction was tested experimentally by performing an in vitro ppGpp hydrolysis assay. Purified ppGpp was used as a substrate, and the in vitro enzymatic hydrolysis to GDP and PPi in the presence of Mn(II) ions was monitored as a function of time. As expected, RelMsm was seen to possess a potent Mn(II)-dependent ppGpp hydrolysis activity, with complete hydrolysis to GDP and PPi taking place within 30 min from the start of the reaction (Fig. 4c, RelMsm). MS_RHII-RSD, on the other hand, did not hydrolyze ppGpp even 1 h after the start of the reaction (Fig. 4c, MS_RHII-RSD). It is clear from the results that MS_RHII-RSD lacks the Mn(II)-dependent hydrolysis activity present in conventional Rel proteins. The GDP substrate specificity, the presence of a Mg(II)-independent (p)ppGpp synthetic activity, and the absence of a (p)ppGpp hydrolytic activity, taken together, strongly suggest that MS_RHII-RSD behaves like a monofunctional (p)ppGpp synthetase in vitro.
DUF429 in full-length MS_RHII-RSD is a functional RNase HII.
Enzymes belonging to the RNase H family degrade the RNA moiety of an RNA·DNA heteroduplex (10, 37, 51). Protein sequence comparisons showed that the DUF429 domain present at the amino terminus of MS_RHII-RSD bore similarities to RNase HII proteins (see Fig. S2a in the supplemental material). To test whether this domain was functionally active, an in vivo complementation assay was performed in the ΔrnhA ΔrnhB(Ts) E. coli strain GJ12055 that had both the RNase H genes deleted (18). This strain, lacking the RNase HI and RNase HII enzymes, grows well at the permissive temperature of 30°C but not at the restrictive temperature of 42°C unless complemented with a functional RNase H activity. It was observed that E. coli GJ12055 cultures electrotransformed with pET-MS_RHII-RSD overcame the growth defect and survived at 42°C, although not as robustly as the cultures harboring the plasmid pSK760 (21) that encoded the full-length E. coli rnhA gene (Fig. 5a). Empty pET21b vector, lacking an RNase HII insert, was unable to effect this rescue and acted as a negative control.
Fig 5.
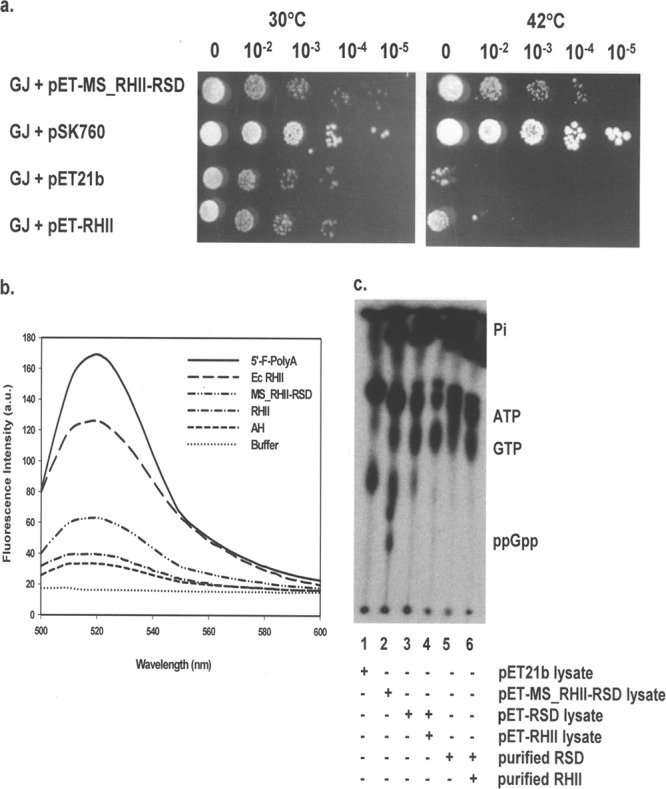
RNase HII activity assays and ppGpp synthesis assay using crude cell lysates. (a) In vivo complementation assay for RNase HII activity using E. coli ΔrnhA ΔrnhB(Ts) strain GJ12055 (GJ) electrotransformed with plasmids pSK760 (plasmid containing full-length E. coli rnhA gene, positive control), pET21b (empty vector, negative control), pET-MS_RHII-RSD, and pET-RHII. Plate titrations were performed in duplicates, and plates were incubated overnight at 30°C and 42°C as indicated. Rescue of growth defect at 42°C indicates the presence of a functional RNase H activity. Exponents above the image refer to the dilutions that were spotted. (b) In vitro fluorescence resonance energy transfer (FRET)-based assay for RNase HII activity. Increase in fluorescence caused by the hydrolysis of a 12-mer RNA·DNA hybrid [5′-fluorescein-poly(A)·3′-dabsyl-poly(dT)] was monitored at the end of 30 min of incubation at 37°C [λ(excitation) = 490 nm, λmax(emission) = 520 nm]. Commercially procured E. coli RNase HII (Ec RHII) served as the positive control. The dashed line labeled AH shows the basal fluorescence spectrum of the annealed RNA·DNA hybrid, and the solid line labeled 5′-F-polyA indicates the fluorescence of free, unhybridized 12-mer 5′-fluorescein poly(A) RNA in the absence of the dabsyl quencher DNA. a.u., arbitrary units. (c) In vitro ppGpp synthesis assay performed using crude cell lysates and purified proteins. Lane 1, pET21b lysate, negative control; lane 2, MS_RHII-RSD lysate, positive control; lane 3, RSD lysate; lane 4, RSD/RHII lysates at a total protein ratio of 5:1; lane 5, purified, renatured RSD; lane 6, purified, renatured RSD and purified RHII. Cell lysates were added at a total protein concentration of 200 μg/ml. For the reaction mixtures with purified proteins, 1 μM each protein was used. +, presence of reaction component; −, absence of reaction component.
The RNase HII activity was tested for in vitro using a sensitive fluorescence assay (57). 5′-Fluorescein-labeled-poly(A) ribooligonucleotide was hybridized to 3′-dabsyl-labeled-poly(dT) deoxyribooligonucleotide, resulting in the quenching of fluorescence by the dabsyl moiety. Hydrolysis of this RNA·DNA hybrid by an RNase H enzyme would lead to the release of free fluorescein with a concomitant increase in fluorescence that can be measured. The results of this in vitro assay showed that MS_RHII-RSD possessed a weak Mn(II)-dependent RNase HII activity (Fig. 5b). This activity was only about 33% of that shown by the commercially procured E. coli RNase HII enzyme (NEB) (Fig. 5b). It is clear from the results of the above-described assays that the DUF429 domain of full-length MS_RHII-RSD possesses a functional RNase HII activity, both in vivo and in vitro. Based on these experimental data and the predictions from in silico analyses, we have assigned domain of unknown function 429 (DUF429) as a functional RNase HII enzyme.
Domain variants of MS_RHII-RSD are functionally inactive.
Studies with full-length MS_RHII-RSD indicated that the protein was a dual-function RNase HII-(p)ppGpp synthetase. To test whether interdomain interactions were necessary for protein activity, the RNase HII (RHII) and (p)ppGpp synthetase (RSD) domains of MS_RHII-RSD were cloned, expressed, and purified separately with a carboxy-terminal hexahistidine tag (Fig. 3b). Data relating to the purified proteins used in this study have been summarized in Table 3. RHII was obtained in the soluble fraction, had a molecular mass of 32 kDa, and was present as a monomer in solution (data not shown). The in vivo complementation assay using the temperature-sensitive E. coli GJ12055 strain electrotransformed with pET-RHII indicated that the RHII domain in isolation could not rescue the growth defect of the strain at 42°C (Fig. 5a). Similar results were obtained in the in vitro fluorescence assay. Purified RHII did not show any significant level of RNA·DNA heteroduplex hydrolysis activity over that of the basal fluorescence shown by the annealed hybrid (AH) (Fig. 5b). These results indicate that the RHII domain alone, in isolation, was inactive.
Table 3.
Summary of protein parameters and activity of proteins studied
| Protein or domain | Length (aa)a | Mass (kDa) | Oligomeric status | Theoretical pI | Activityb |
||
|---|---|---|---|---|---|---|---|
| (p)ppGpp synthesis | (p)ppGpp hydrolysis | RNase HII | |||||
| RelMsm | 797 | 89 | 3 | 8.09 | Y | Y | N |
| MS_RHII-RSD | 567 | 64.5 | 6 | 5.35 | Y | N | Y |
| RHII | 280 | 32 | 1 | 6.15 | N | N | N |
| RSD | 288 | 34 | 4 | 5.11 | N | N | N |
aa, amino acids.
Y, yes, activity present; N, no, activity absent.
When E. coli BL21(DE3) cultures transformed with the plasmid pET-RSD were induced with IPTG for protein production, approximately 80% of RSD was seen to enrich into inclusion bodies. RSD was purified from the inclusion bodies by denaturing nickel-histidine affinity chromatography in the presence of 6 M urea as described in Materials and Methods. Purified, denatured RSD was renatured by stepwise dialysis in buffers containing decreasing concentrations of urea. The dialysis was continued in a buffer lacking urea until the CD spectrum of RSD stabilized and showed no further change. The secondary structure of RSD underwent a change from a distinct random coil structure in the presence of urea to a helical structure in its absence (data not shown). The refolded RSD protein was then used in ppGpp synthesis assays. This protein had a molecular mass of 35 kDa, possessed an alpha-helical structure when renatured, and existed as a tetramer in solution (data not shown). Purified, renatured RSD in isolation could not synthesize ppGpp in vitro (Fig. 5c, lane 5). It lacked the synthetic ability even when supplied with the purified RHII protein in trans (Fig. 5c, lane 6). To ensure that RSD activity was not lost during the purification and refolding processes, the (p)ppGpp synthesis assays were performed using E. coli culture lysates expressing the respective proteins. E. coli BL21(DE3) cultures transformed with pET-MS_RHII-RSD, pET-RSD, pET-RHII, and pET21b empty vector were grown as described above, induced with IPTG, and lysed, and the clarified supernatant containing 200 μg/ml of total protein was added directly to the (p)ppGpp synthesis reaction mixture. The MS_RHII-RSD and pET21b cell lysates served as the positive and negative controls, respectively. The RSD cell lysate contained approximately 20% soluble RSD protein. However, this did not show any (p)ppGpp synthesis (Fig. 5c, lane 3). No synthetic activity was observed even on complementation with the RHII cell lysate in trans (Fig. 5c, lane 4). The complementation was carried out at a total protein ratio of 5:1 RSD/RHII lysate, taking into consideration that only 20% of RSD was available in the soluble form in the lysate, compared to 100% of RHII. ppGpp formation was not detected in the reaction mixture containing pET21b lysate, probably because of very small amounts of ppGpp synthesis in this strain under the given assay conditions (Fig. 5c, lane 1). The reaction mixture containing the same concentration of MS_RHII-RSD cell lysate, however, was able to synthesize ppGpp efficiently (Fig. 5c, lane 2). These results confirm the inability of the RSD domain protein to synthesize ppGpp, whether in isolation or when complemented in trans with RHII.
Taken together, these data indicate interdependence between the RHII and RSD domains of MS_RHII-RSD, such that neither domain, in isolation or on complementation in trans, can function independently. Their presence in cis on full-length MS_RHII-RSD renders this protein functionally active. The extent of interdomain interactions, whether structural, functional, or both, is the subject of on-going investigations.
MS_RHII-RSD is expressed constitutively in vivo.
The conventional Rel proteins are known to express under conditions of nutrient or environmental stress and in the stationary phase of growth (9, 28, 40, 48, 58). The B. subtilis YwaC (33) and V. cholerae RelV (13) SAS proteins also are overexpressed under alkaline and osmotic stress, respectively. Microarray data available at the TBDB website (http://www.tbdb.org/) showed that Rv1366, the M. tuberculosis ortholog of MS_RHII-RSD, is overexpressed under conditions of oxidative stress and during DNA damage by UV irradiation. We therefore investigated the expression of MS_RHII-RSD by M. smegmatis WT and ΔrelMsm cultures at different stages of growth and under a variety of stress conditions. Contrary to the above-described observations, Western blot analyses using anti-MS_RHII-RSD polysera showed that MS_RHII-RSD was expressed constitutively in vivo, under both normal growth conditions and a variety of stress conditions (Fig. 6). No change in expression levels was observed between the WT and ΔrelMsm strains, irrespective of whether the strains were grown in 0.02% glucose-starved or 2% glucose-enriched medium. MS_RHII-RSD was expressed at the same level from 12 to 120 h of growth. The constitutive protein expression corroborates the constitutive ppGpp synthetic activity of MS_RHII-RSD (Fig. 1a to d). The slight discrepancy between observed protein expression at the 12-h time point and the nondetectability of (p)ppGpp at this stage is explained above. The small numbers of cells present at this early stage of growth synthesize extremely low levels of ppGpp that cannot be visibly detected at this stage, although a significant level of protein expression can be detected in vivo. The expression of MS_RHII-RSD under a variety of stress conditions remained the same. A basal level of protein expression was observed under conditions of DNA damage (20 min of UV exposure), oxidative stress (36 mM H2O2), acidic stress (pH 5 with HCl), alkaline stress (pH 10 with NaCl), and osmotic stress (4 M NaCl). M. smegmatis RNA polymerase α-subunit was used as the internal loading control (15) for the above analyses, and the expression levels of this protein were seen to remain unchanged under all the conditions tested.
Fig 6.
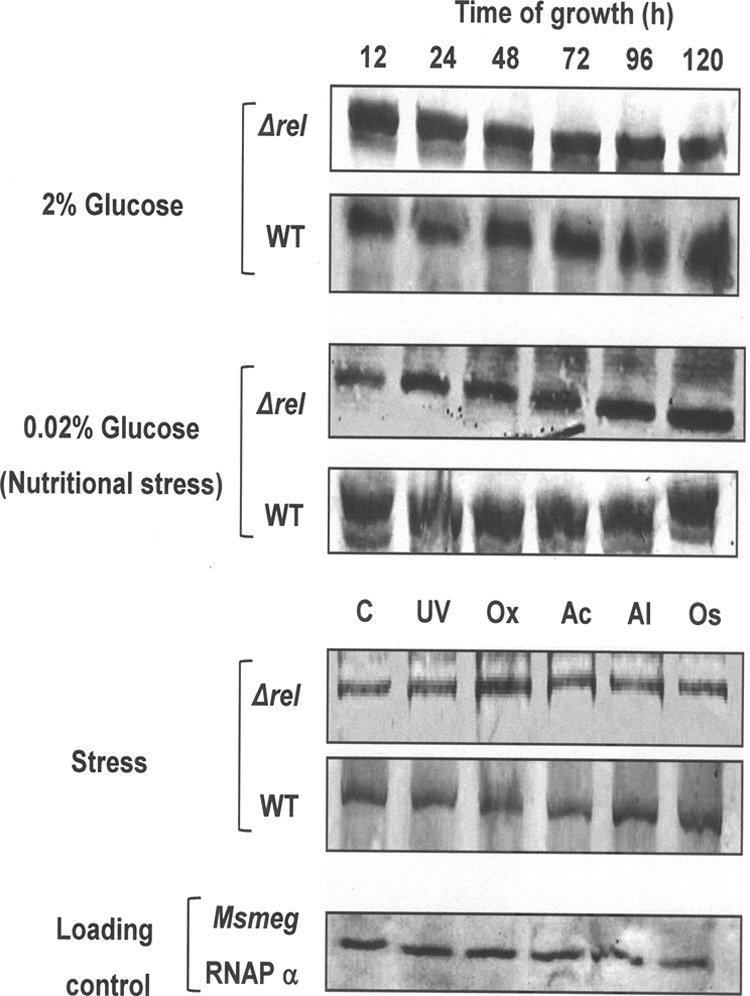
In vivo expression profile of MS_RHII-RSD. Western blots showing the in vivo expression of MS_RHII-RSD at different growth stages and under various conditions of stress, detected using anti-MS_RHII-RSD polysera. Analyses were performed on 100 μg of total protein from M. smegmatis WT and ΔrelMsm culture lysates. M. smegmatis RNA polymerase α-subunit (Msmeg RNAP α) was used as the internal loading control. Stress conditions tested for were 0.02% glucose (Nutritional stress), UV exposure (UV; 20 min), oxidative stress (Ox; 36 mM H2O2), acid stress (Ac; pH 5 with HCl), alkali stress (Al; pH 10 with NaOH), and osmolar stress (Os; 4 M NaCl).
The constitutive expression of MS_RHII-RSD in vivo, unlike the expression of Rel and SAS proteins under general or specific stress conditions, points toward a more basic function for this protein within the cell. We propose a role for MS_RHII-RSD that is secondary to that of conventional RelMsm, the protein chiefly responsible for mounting the stringent response in M. smegmatis. The importance of domain coevolution (24), with the RNase HII (RHII) and (p)ppGpp synthetase (RSD) domains occurring on the same polypeptide chain, opens up interesting possibilities of heretofore-unknown functions carried out by this dual-function enzyme in vivo.
DISCUSSION
The stringent response is a highly conserved physiological response mounted by bacteria under stress (9, 28, 40, 48, 58). Until recently, the only known players in this pathway were the (p)ppGpp-synthesizing and -hydrolyzing long RSH enzymes (4) RelA and SpoT in Gram-negative bacteria and the bifunctional Rel in Gram-positive bacteria. The existence of short alarmone synthetases (SAS) (4, 13, 23, 26, 33, 34) and short alarmone hydrolases (SAH) (4, 49), small proteins possessing a single functional (p)ppGpp synthetase or hydrolase domain, respectively, is a recent discovery that has modified this paradigm. Around the same time that the presence of the SAS proteins was reported, we chanced upon such small (p)ppGpp synthetases in the genus Mycobacterium. In this study, we report the cloning, purification, and functional characterization of a second (p)ppGpp synthetase from Mycobacterium smegmatis, distinct from RelMsm, which we term MS_RHII-RSD, indicating the RNase HII and (p)ppGpp synthesis activities displayed by it. Our present work shows that MS_RHII-RSD behaves like a monofunctional (p)ppGpp synthetase with regard to its GDP substrate preference, Mg(II) ion-independent (p)ppGpp synthesis, and lack of (p)ppGpp hydrolysis activity. Interestingly, unlike the Rel and SAS proteins, MS_RHII-RSD possesses the ability to hydrolyze the RNA moiety of RNA·DNA heteroduplexes in the presence of Mn(II) ions, and its amino-terminal domain bears structural similarity to those of the bacterial RNase HII proteins. We have experimentally proved and ascribed the function of RNase HII to the amino-terminal domain of MS_RHII-RSD, initially identified as domain of unknown function DUF429. We have characterized in detail the (p)ppGpp synthetase activity present in the carboxy-terminal domain of this protein. Domain interdependence for functional activity has also been demonstrated. In conclusion, we undisputedly identify MS_RHII-RSD as the second monofunctional (p)ppGpp synthetase in M. smegmatis, differing from bifunctional RelMsm in possessing an RNase HII activity and lacking the (p)ppGpp hydrolytic capability.
MS_RHII-RSD orthologs can be identified in silico in other mycobacterial species with completely sequenced genomes. They are found in close proximity to the pur genes that are involved in purine biosynthesis. These proteins occur either as RHII (DUF429)-RSD fusion proteins (collectively termed MS_RHII-RSD) or, alternatively, as proteins containing the RHII (DUF429) domain alone (collectively termed RHII) or the RSD domain alone (collectively termed SAS) (see Fig. S3a and b in the supplemental material). These are structurally distinct from the Rel proteins of mycobacteria, which are multidomain proteins possessing the (p)ppGpp hydrolysis (HD), (p)ppGpp synthesis (RSD), and TGS and ACT regulatory domains (see Fig. S3a). Phylogenetic analysis of mycobacterial MS_RHII-RSD, RHII, SAS (RSD), and Rel proteins shows that they form evolutionarily distinct groups (see Fig. S3b). Furthermore, the MS_RHII-RSD orthologs from nonpathogenic mycobacterial strains (see Fig. S3b, highlighted box) cluster differently from the pathogenic ones. It is interesting to note that SAH were identified in BLAST searches against mycobacterial genomes (see Table S1 in the supplemental material) (4). There are several of this wide variety of small stringent response proteins present within the genome of a single mycobacterium, indicating a certain degree of redundancy in protein function. It implies that these proteins moonlight in other pathways and have more than one function. The recent discovery of a functional SpoT homolog in the eukaryote Drosophila melanogaster (49) lends credence to this fact.
There are instances in the literature of an RNase H domain fused to a functionally distinct non-RNase H domain, like the RNase HI-CobC acid phosphatase fusion proteins (38, 57). Protein BLAST search shows that the M. smegmatis genome encodes an RNase HI-CobC fusion protein (MSMEG_4305). However, an RNase HII-(p)ppGpp synthetase fusion protein has not yet been reported. Given the central importance of (p)ppGpp in bacterial survival under stress, it seems inevitable that the stringent response should, in some way, be able to regulate the three processes essential to life, i.e., DNA replication, transcription, and translation, and this is found to be true (48). Previous studies (27, 56) have shown that (p)ppGpp directly affects the activity of DnaG, the DNA primase involved in DNA replication. It is tempting to speculate that MS_RHII-RSD, with its RNase HII and (p)ppGpp synthesis activities, as well as its constitutive expression, acts at the level of DNA replication and transcription, further coupling these two processes to the stringent response.
Enzymes belonging to the RNase H family are known to degrade the RNA moiety of an RNA·DNA heteroduplex, with profound implications in DNA replication, repair, and transcription (10, 18, 50, 51). The possibility of the involvement of the RHII domain of MS_RHII-RSD in the process of DNA replication is strengthened by arguments that RNase HII enzymes play an important role in maintaining genome integrity (35, 42, 45) by the removal of ribonucleotides mistakenly incorporated into DNA. This happens in conjugation with DNA mismatch repair (45) and junction RNase H enzymes like yeast FenI (42). DNA-damaging agents are known to have a profound effect on RNase HII activity (3, 36). Although RNase H genes have evolved to avoid functional redundancy (25), it has been observed that the location and/or the activity of RNase H proteins may be influenced by other domains present on the same protein chain (10), indicating a possible interrelation between the RHII and RSD activities of MS_RHII-RSD. A basal level of (p)ppGpp is essential to destabilize RNA polymerases and maintain genomic integrity during DNA replication (54). This is especially the case when RNA polymerases stall at DNA lesions under conditions of DNA damage (54). A direct link between in vivo (p)ppGpp levels and survival under UV stress has been reported previously (52, 54). Furthermore, the levels of RNase H are known to go down under these conditions to facilitate DNA replication from cryptic origins of replication (54). Since the expression and activity of MS_RHII-RSD remain unaffected under UV stress, we propose that this protein takes over the functions of the conventional RNase HI (MSMEG_5562) (14, 32) and RNase HII (MSMEG_2442) of M. smegmatis under UV damage conditions. The RHII domain of MS_RHII-RSD would then function in DNA replication from cryptic origins, while the RSD domain would destabilize RNA polymerases stalled at DNA lesions by constitutively synthesizing (p)ppGpp. On the other hand, during normal growth, the RHII domain might facilitate the removal of R-loops formed during high transcriptional activity that may otherwise lead to mutagenesis (48). The possibility of RHII domain involvement in the copy number control of mycobacterial plasmids also poses an interesting line of investigation. Insights into the in vivo role, mechanism of action, and regulation of MS_RHII-RSD activity will be revealed by the detailed genetic, biochemical, and crystallographic analyses of this novel enzyme.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to J. Gowrishankar, CDFD, India, for the kind gift of the temperature-sensitive E. coli strain GJ12055 and plasmid pSK760 and to J. Krishna Leela for helpful discussions. M.S.M. thanks T. Dhanaraman for help with protein purifications, as well as Sunita Prakash of the proteomics facility and P. Sreedhar and M. Sandeep of the phosphor-imaging facility and the staff of the IISc Central Animal Facility for antibody generation.
This work was supported by a grant from the Department of Biotechnology, Government of India, to D.C., and a Senior Research fellowship awarded to M.S.M. by the Council of Scientific and Industrial Research, India.
Footnotes
Published ahead of print 25 May 2012
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403–410 [DOI] [PubMed] [Google Scholar]
- 2. Artsimovitch I, et al. 2004. Structural basis for transcription regulation by alarmone ppGpp. Cell 117:299–310 [DOI] [PubMed] [Google Scholar]
- 3. Arudchandran A, et al. 2000. The absence of ribonuclease H1 or H2 alters the sensitivity of Saccharomyces cerevisiae to hydroxyurea, caffeine and ethyl methanesulphonate: implications for roles of RNases H in DNA replication and repair. Genes Cells 5:789–802 [DOI] [PubMed] [Google Scholar]
- 4. Atkinson GC, Tenson T, Hauryliuk V. 2011. The RelA/SpoT homolog (RSH) superfamily: distribution and functional evolution of ppGpp synthetases and hydrolases across the tree of life. PLoS One 6:e23479 doi:10.1371/journal.pone.0023479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Avarbock D, Avarbock A, Rubin H. 2000. Differential regulation of opposing RelMtb activities by the aminoacylation state of a tRNA·ribosome·mRNA·RelMtb complex. Biochemistry 39:11640–11648 [DOI] [PubMed] [Google Scholar]
- 6. Avarbock A, et al. 2005. Functional regulation of the opposing (p)ppGpp synthetase/hydrolase activities of RelMtb from Mycobacterium tuberculosis. Biochemistry 44:9913–9923 [DOI] [PubMed] [Google Scholar]
- 7. Cashel M. 1969. The control of ribonucleic acid synthesis in Escherichia coli. IV. Relevance of unusual phosphorylated compounds from amino acid-starved stringent strains. J. Biol. Chem. 244:3133–3141 [PubMed] [Google Scholar]
- 8. Chatterji D, Fujita N, Ishihama A. 1998. The mediator for stringent control, ppGpp, binds to the beta-subunit of Escherichia coli RNA polymerase. Genes Cells 3:279–287 [DOI] [PubMed] [Google Scholar]
- 9. Chatterji D, Ojha AK. 2001. Revisiting the stringent response, ppGpp and starvation signaling. Curr. Opin. Microbiol. 4:160–165 [DOI] [PubMed] [Google Scholar]
- 10. Crouch RJ. 1990. Ribonuclease H: from discovery to 3D structure. New Biol. 9:771–777 [PubMed] [Google Scholar]
- 11. Dahl JH, et al. 2005. The relA homolog of Mycobacterium smegmatis affects cell appearance, viability, and gene expression. J. Bacteriol. 187:2439–2447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dalebroux ZD, Swanson MS. 2012. ppGpp: magic beyond RNA polymerase. Nat. Rev. Microbiol. 10:203–212 [DOI] [PubMed] [Google Scholar]
- 13. Das B, Pal RR, Bag S, Bhadra RK. 2009. Stringent response in Vibrio cholerae: genetic analysis of spoT gene function and identification of a novel (p)ppGpp synthetase gene. Mol. Microbiol. 72:380–398 [DOI] [PubMed] [Google Scholar]
- 14. Dawes SS, Crouch RJ, Morris SL, Mizrahi V. 1995. Cloning, sequence analysis, overproduction in Escherichia coli and enzymatic characterization of the RNase HI from Mycobacterium smegmatis. Gene 165:71–75 [DOI] [PubMed] [Google Scholar]
- 15. Dey A, Verma AK, Chatterji D. 2010. Role of an RNA polymerase interacting protein, MsRbpA, from Mycobacterium smegmatis in phenotypic tolerance to rifampicin. Microbiology 156:873–883 [DOI] [PubMed] [Google Scholar]
- 16. Hara A, Sy J. 1983. Guanosine 5′-triphosphate, 3′-diphosphate 5′-phosphohydrolase. Purification and substrate specificity. J. Biol. Chem. 258:1678–1683 [PubMed] [Google Scholar]
- 17. Hogg T, Mechold U, Malke H, Cashel M, Hilgenfeld R. 2004. Conformational antagonism between opposing active sites in a bifunctional RelA/SpoT homolog modulates (p)ppGpp metabolism during the stringent response. Cell 117:57–68 [DOI] [PubMed] [Google Scholar]
- 18. Itaya M, et al. 1999. Isolation of RNase H genes that are essential for growth of Bacillus subtilis 168. J. Bacteriol. 181:2118–2123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jain V, Saleem-Batcha R, China A, Chatterji D. 2006. Molecular dissection of the mycobacterial stringent response protein Rel. Prot. Sci. 15:1449–1464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jain V, Saleem-Batcha R, Chatterji D. 2007. Synthesis and hydrolysis of pppGpp in mycobacteria: a ligand mediated conformational switch in Rel. Biophys. Chem. 127:41–50 [DOI] [PubMed] [Google Scholar]
- 21. Kanaya S, Crouch RJ. 1983. DNA sequence of the gene coding for Escherichia coli ribonuclease H. J. Biol. Chem. 258:1276–1281 [PubMed] [Google Scholar]
- 22. Kelley LA, Sternberg MJE. 2009. Protein structure prediction on the web: a case study using the Phyre server. Nat. Protoc. 4:363–371 [DOI] [PubMed] [Google Scholar]
- 23. Kim JN, Ahn SJ, Seaton K, Garrett S, Burne RA. 2012. Transcriptional organization and physiological contributions of the relQ operon of Streptococcus mutans. J. Bacteriol. 194:1968–1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim Y, Koyutürk M, Topkara U, Grama A, Subramaniam S. 2006. Inferring functional information from domain co-evolution. Bioinformatics 22:40–49 [DOI] [PubMed] [Google Scholar]
- 25. Kochiwa H, Tomita H, Kanai A. 2007. Evolution of ribonuclease H genes in prokaryotes to avoid inheritance of redundant genes. BMC Evol. Biol. 7:128–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lemos JA, Lin VK, Nascimento MM, Abranches J, Burne RA. 2007. Three gene products govern (p)ppGpp production by Streptococcus mutans. Mol. Microbiol. 65:1568–1581 [DOI] [PubMed] [Google Scholar]
- 27. Maciag M, Kochanowska M, Lyzén R, Wegrzyn G, Szalewska-Pałasz A. 2010. ppGpp inhibits the activity of Escherichia coli DnaG primase. Plasmid 63:61–67 [DOI] [PubMed] [Google Scholar]
- 28. Magnusson LU, Farewell A, Nyström T. 2005. ppGpp: a global regulator in Escherichia coli. Trends Microbiol. 13:236–242 [DOI] [PubMed] [Google Scholar]
- 29. Marchler-Bauer A, et al. 2011. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 39(D):D225–D229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mathew R, Ojha AK, Karande A, Chatterji D. 2004. Deletion of the rel gene in Mycobacterium smegmatis reduces its stationary phase survival without altering the cell-surface associated properties. Curr. Sci. 86:149–153 [Google Scholar]
- 31. Mittenhuber GJ. 2001. Comparative genomics and evolution of genes encoding bacterial (p)ppGpp synthetases/hydrolases (the Rel, RelA and SpoT Proteins). Mol. Microbiol. Biotechnol. 3:585–600 [PubMed] [Google Scholar]
- 32. Mizrahi V, Huberts P, Dawes SS, Dudding LR. 1993. A PCR method for the sequence analysis of the gyrA, polA and rnhA gene segments from mycobacteria. Gene 136:287–290 [DOI] [PubMed] [Google Scholar]
- 33. Nanamiya H, et al. 2008. Identification and functional analysis of novel (p)ppGpp synthetase genes in Bacillus subtilis. Mol. Microbiol. 67:291–304 [DOI] [PubMed] [Google Scholar]
- 34. Natori Y, et al. 2009. Transcription activity of individual rrn operons in Bacillus subtilis mutants deficient of (p)ppGpp synthetase genes relA, yjbM, and ywaC. J. Bacteriol. 191:4555–4561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nick McElhinny SA, et al. 2010. Genome instability due to ribonucleotide incorporation into DNA. Nat. Chem. Biol. 6:774–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ohtani N, et al. 1999. Identification of the genes encoding Mn2+-dependent RNase HII and Mg2+-dependent RNase HIII from Bacillus subtilis: classification of RNases H into three families. Biochemistry 38:605–618 [DOI] [PubMed] [Google Scholar]
- 37. Ohtani N, Haruki M, Morikawa M, Kanaya S. 1999. Molecular diversities of RNases H. J. Biosci. Bioeng. 88:12–19 [DOI] [PubMed] [Google Scholar]
- 38. Ohtani N, Saito N, Tomita M, Itaya Itoh A. 2005. The SCO2299 gene from Streptomyces coelicolor A3(2) encodes a bifunctional enzyme consisting of an RNase H domain and an acid phosphatase domain. FEBS J. 272:2828–2837 [DOI] [PubMed] [Google Scholar]
- 39. Ojha AK, Mukherjee TK, Chatterji D. 2000. High intracellular level of guanosine tetraphosphate in Mycobacterium smegmatis changes the morphology of the bacterium. Infect. Immun. 68:4084–4091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Potrykus K, Cashel M. 2008. (p)ppGpp: still magical? Annu. Rev. Microbiol. 62:35–51 [DOI] [PubMed] [Google Scholar]
- 41. Reddy PS, Raghavan A, Chatterji D. 1995. Evidence for a ppGpp-binding site on Escherichia coli RNA polymerase: proximity relationship with the rifampicin-binding domain. Mol. Microbiol. 15:255–265 [DOI] [PubMed] [Google Scholar]
- 42. Rydberg B, Game J. 2002. Excision of misincorporated ribonucleotides in DNA by RNase H (type 2) and FEN-1 in cell-free extracts. Proc. Natl. Acad. Sci. U. S. A. 99:16654–16659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sajish M, Tiwari D, Rananaware D, Nandicoori VK, Prakash B. 2007. A charge reversal differentiates (p)ppGpp synthesis by monofunctional and bifunctional Rel proteins. J. Biol. Chem. 282:34977–34983 [DOI] [PubMed] [Google Scholar]
- 44. Sajish M, Kalayil S, Verma SK, Nandicoori VK, Prakash B. 2009. The significance of ExDD and RxKD motif conservation in Rel proteins. J. Biol. Chem. 284:9115–9123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shen Y, Koh KD, Weiss B, Storici F. 2012. Mispaired rNMPs in DNA are mutagenic and are targets of mismatch repair and RNases H. Nat. Struct. Mol. Biol. 19:98–104 [DOI] [PubMed] [Google Scholar]
- 46. Smeulders MJ, Richard JK, Speight A, Williams HD. 1999. Adaptation of Mycobacterium smegmatis to stationary phase. J. Bacteriol. 181:270–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Snapper SB, Melton RE, Mustafa S, Kieser T, Jacobs WR., Jr 1990. Isolation and characterization of efficient plasmid transformation mutants of Mycobacterium smegmatis. Mol. Microbiol. 4:1911–1919 [DOI] [PubMed] [Google Scholar]
- 48. Srivatsan A, Wang JD. 2008. Control of bacterial transcription, translation and replication by (p)ppGpp. Curr. Opin. Microbiol. 11:100–105 [DOI] [PubMed] [Google Scholar]
- 49. Sun D, et al. 2010. A metazoan ortholog of SpoT hydrolyses ppGpp and functions in starvation responses. Nat. Struct. Mol. Biol. 17:1188–1194 [DOI] [PubMed] [Google Scholar]
- 50. Svejstrup JQ. 2010. The interface between transcription and mechanisms maintaining genome integrity. Trends Biochem. Sci. 35:333–338 [DOI] [PubMed] [Google Scholar]
- 51. Tadokoro T, Kanaya S. 2009. Ribonuclease H: molecular diversities, substrate binding domains, and catalytic mechanism of the prokaryotic enzymes. FEBS J. 276:1482–1493 [DOI] [PubMed] [Google Scholar]
- 52. Thiam K, Favre A. 1984. Role of the stringent response in the expression and mechanism of near-ultraviolet induced growth delay. Eur. J. Biochem. 145:137–142 [DOI] [PubMed] [Google Scholar]
- 53. Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Trautinger BW, Jaktaji RP, Rusakova E, Llyod RG. 2005. RNA polymerase modulators and DNA repair activities resolve conflicts between DNA replication and transcription. Mol. Cell 19:247–258 [DOI] [PubMed] [Google Scholar]
- 55. Vrentas CE, et al. 2008. Still looking for the magic spot: the crystallographically defined binding site for ppGpp on RNA polymerase is unlikely to be responsible for rRNA transcription regulation. J. Mol. Biol. 377:551–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang JD, Sanders GM, Grossman AD. 2007. Nutritional control of elongation of DNA replication by (p)ppGpp. Cell 128:865–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Watkins HA, Baker EN. 2010. Structural and functional characterization of an RNase HI domain from the bifunctional protein Rv2228c from Mycobacterium tuberculosis. J. Bacteriol. 192:2878–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wu J, Xie J. 2009. Magic spot: (p)ppGpp. J. Cell. Physiol. 220:297–302 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.