

Abstract
Caffeic acid is a biologically active molecule that has various beneficial properties, including antioxidant, anticancer, and anti-inflammatory activities. In this study, we explored the catalytic potential of a bacterial cytochrome P450, CYP199A2, for the biotechnological production of caffeic acid. When the CYP199A2 enzyme was reacted with p-coumaric acid, it stoichiometrically produced caffeic acid. The crystal structure of CYP199A2 shows that Phe at position 185 is situated directly above, and only 6.35 Å from, the heme iron. This F185 residue was replaced with hydrophobic or hydroxylated amino acids using site-directed mutagenesis to create mutants with novel and improved catalytic properties. In whole-cell assays with the known substrate of CYP199A2, 2-naphthoic acid, only the wild-type enzyme hydroxylated 2-naphthoic acid at the C-7 and C-8 positions, whereas all of the active F185 mutants exhibited a preference for C-5 hydroxylation. Interestingly, several F185 mutants (F185V, F185L, F185I, F185G, and F185A mutants) also acquired the ability to hydroxylate cinnamic acid, which was not hydroxylated by the wild-type enzyme. These results demonstrate that F185 is an important residue that controls the regioselectivity and the substrate specificity of CYP199A2. Furthermore, Escherichia coli cells expressing the F185L mutant exhibited 5.5 times higher hydroxylation activity for p-coumaric acid than those expressing the wild-type enzyme. By using the F185L whole-cell catalyst, the production of caffeic acid reached 15 mM (2.8 g/liter), which is the highest level so far attained in biotechnological production of this compound.
INTRODUCTION
Phenolics are the most widespread dietary antioxidants. The evidence for a role for dietary intake of these compounds in the maintenance of health and protection from disease is accumulating (18, 25). Not only flavonoids but also simple phenolic acids have attracted much attention. Notably, caffeic acid is a powerful antioxidant. Caffeic acid exhibits higher antioxidant activity for human low-density lipoprotein oxidation than p-coumaric acid and ferulic acid and has the same level of antioxidant activity as chlorogenic acid (caffeoyl quinic acid), which is a well-known antioxidant present in plants (6, 22, 31). A recent report suggested that caffeic acid produced from chlorogenic acid in the intestine might play a major role in the antioxidative effect of chlorogenic acid in vivo (33). In addition to having the scavenging activity of a reactive oxygen species, caffeic acid has been reported to have various beneficial properties, including anticancer, anti-inflammatory, and antiviral activities (9, 14, 39). The antioxidant activity of this compound has also been shown to protect skin cells when exposed to UV radiation (35). These properties encourage the application of caffeic acid in health or functional foods and pharmaceuticals (1, 18, 23). Furthermore, caffeic acid is an important scaffold for the synthesis of a variety of biologically active compounds (36).
Cytochrome P450 monooxygenases (P450s) are a superfamily of heme-containing proteins that introduce one oxygen atom derived from molecular oxygen into an organic molecule. P450s catalyze the direct oxidation of a variety of compounds in a regio- and stereoselective manner under ambient conditions. Thus, P450s are promising catalysts for use in the oxyfunctionalization of chemicals (4, 15, 27, 28, 37). p-Coumaric acid is a relatively inexpensive chemical that is commercially available. Furthermore, this compound is widely distributed in plant tissues as a free form or as conjugated forms (10). For example, it was estimated that olive oil residues contain 200 mg/kg p-coumaric acid (19). Wheat straw after mild acid and alkaline peroxide treatment contains 4 g/kg p-coumaric acid (29). Also, p-coumaric acid was produced from glucose using metabolically engineered microorganisms (24, 38). P450s that efficiently catalyze the hydroxylation of p-coumaric acid might provide an easy and environmentally friendly synthetic approach to production of caffeic acid. In particular, biotechnological processes that use natural p-coumaric acid as a starting material could provide high-added-value natural caffeic acid. So far, however, there has been no report concerning bacterial P450s that exhibit such an activity. It was suggested that p-coumaric acid mainly undergoes hydroxylation after esterification in plants. In other words, although CYP98 in the model plant Arabidopsis efficiently hydroxylated p-coumaroyl esters of shikimic acid and quinic acid to the corresponding caffeoyl esters, this enzyme exhibited no or very low hydroxylation activity for free p-coumaric acid (21, 34).
In this study, we found that a bacterial P450 CYP199A2 had hydroxylation activity for p-coumaric acid. CYP199A2 was recently discovered in Rhodopseudomonas palustris and has been shown to exhibit oxidation activity for aromatic carboxylic acids, including 2-naphthoic acid, 4-ethylbenzoic acid, and indole- and quinolinecarboxylic acids (3, 11, 12, 13). We also subjected CYP199A2 to site-directed mutagenesis based on its crystal structure to create mutants with novel and improved catalytic properties. Herein, we examined the oxidation activities of the wild-type CYP199A2 and its mutants for 2-naphthoic acid, coumaric acids, and cinnamic acid and investigated the application of these enzymes to the production of caffeic acid.
MATERIALS AND METHODS
Chemicals.
Cinnamic acid, m-coumaric acid, p-coumaric acid, and caffeic acid were purchased from Wako Pure Chemicals (Osaka, Japan). All other chemicals were of analytical grade.
Site-directed mutagenesis.
The CYP199A2 gene was amplified by PCR using the oligonucleotide primers TTC CAT ATG CCG GTT ACC ACG CCG TCC CAA (the NdeI restriction site is underlined, and the initiation codon is in bold) and CAC AAG CTT AGC AGG CGT CAG TTG GAT CGG (the HindIII restriction site is underlined) and was cloned into the vector pET21a (Novagen, San Diego, CA), as previously reported (11). The F185 residue of CYP199A2 was replaced with Tyr (F185Y), Trp (F185W), Val (F185V), Leu (F185L), Ile (F185I), Gly (F185G), Ala (F185A), Ser (F185S), or Thr (F185T) using a QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) according to the instruction manual. Mutations were introduced into the CYP199A2 gene in pET21a using the oligonucleotide primers listed in Table 1. The correct generation of the desired mutations was confirmed by DNA sequencing.
Table 1.
Oligonucleotides used to generate CYP199A2 mutants
| Mutation | Oligonucleotide (5′ to 3′)a |
|---|---|
| F185Y | GCGGGTCTCGTGTATAACGCGTTTGGGCCGCCGAAC |
| CCCAAACGCGTTATACACGAGACCCGCATAGGGCAG | |
| F185W | GCGGGTCTCGTGTGGAACGCGTTTGGGCCGCCGAAC |
| CCCAAACGCGTTCCACACGAGACCCGCATAGGGCAG | |
| F185V | GCGGGTCTCGTGGTTAACGCGTTTGGGCCGCCGAAC |
| CCCAAACGCGTTAACCACGAGACCCGCATAGGGCAG | |
| F185L | GCGGGTCTCGTGTTAAACGCGTTTGGGCCGCCGAAC |
| CCCAAACGCGTTTAACACGAGACCCGCATAGGGCAG | |
| F185I | GCGGGTCTCGTGATTAACGCGTTTGGGCCGCCGAAC |
| CCCAAACGCGTTAATCACGAGACCCGCATAGGGCAG | |
| F185G | GCGGGTCTCGTGGGGAACGCGTTTGGGCCGCCGAAC |
| CCCAAACGCGTTCCCCACGAGACCCGCATAGGGCAG | |
| F185A | GCGGGTCTCGTGGCGAACGCGTTTGGGCCGCCGAAC |
| CCCAAACGCGTTCGCCACGAGACCCGCATAGGGCAG | |
| F185S | GCGGGTCTCGTGAGCAACGCGTTTGGGCCGCCGAAC |
| CCCAAACGCGTTGCTCACGAGACCCGCATAGGGCAG | |
| F185T | GCGGGTCTCGTGACCAACGCGTTTGGGCCGCCGAAC |
| CCCAAACGCGTTGGTCACGAGACCCGCATAGGGCAG |
Mutated nucleotides are underlined.
Preparation of whole cells and cell extracts.
The wild-type or mutant CYP199A2 gene was coexpressed with the putidaredoxin reductase gene (pdR) from Pseudomonas putida (30) and the palustrisredoxin gene (pux) from R. palustris (2) to provide the redox partners of CYP199A2 in Escherichia coli. pET21a, carrying the wild-type or mutant CYP199A2 gene, and pMW218, carrying the pdR and pux genes, which was previously constructed (12), were simultaneously introduced into E. coli BL21(DE3) cells (Novagen). These recombinant E. coli cells were cultivated in Luria-Bertani medium (1% Bacto tryptone, 0.5% Bacto yeast extract, 1% NaCl [pH 7.0]) supplemented with ampicillin (100 μg/ml) and kanamycin (100 μg/ml) at 25°C. After cultivation for 12 h, isopropyl-β-d-thiogalactopyranoside (1 mM), 5-aminolevulinic acid (0.5 mM), and FeSO4 (0.5 mM) were added to the medium, and cultivation was continued for an additional 12 h. Cells were harvested by centrifugation, washed with potassium phosphate buffer (50 mM, pH 7.5) containing glycerol (10%, vol/vol), and stored at −80°C until use. When cell extracts were prepared, frozen cells were suspended in the buffer and were disrupted using an ultraoscillator. After centrifugation at 15,000 × g for 30 min at 4°C and filtration through a 0.20-μm-pore-size cellulose acetate membrane (Advantec, Tokyo, Japan), the resulting supernatant was used as the cell extract.
Protein and P450 analyses.
The cell extracts were used to analyze protein levels and P450 expression. Protein concentration was measured using a Coomassie protein assay kit (Pierce, Rockford, IL) with a bovine serum albumin standard (5). The expression levels of P450s were examined by sodium dodecyl sulfate-polyacrylamide gel electrophoretic (SDS-PAGE) analysis. P450s were also analyzed based on their CO-reduced difference spectra (26). Before analysis of these spectra, the protein concentration of the cell extracts was adjusted to 2 g/liter.
Reactions using whole cells and cell extracts.
The reaction mixture (250 μl) contained cells of the recombinant E. coli strain (9.0 g [dry cell weight {DCW}] per liter), a substrate compound (1 mM), dimethyl sulfoxide (1%, vol/vol), and potassium phosphate buffer (50 mM, pH 7.5) containing glycerol (10%, vol/vol). Nine grams of DCW corresponded to 50 g of wet weight. When cell extracts were used, the reaction mixture contained cell extracts (2 g of protein per liter), 2-naphthoic acid (1 mM), 5 mM NADH, dimethyl sulfoxide (1%, vol/vol), and potassium phosphate buffer (50 mM, pH 7.5) containing glycerol (10%, vol/vol). The reactions were performed at 30°C with shaking.
Production of caffeic acid on a large scale.
The reaction was performed in a 500-ml flask that contained cells of the recombinant E. coli strain (9.0 g DCW of per liter), caffeic acid (20 mM), and potassium phosphate buffer (50 mM, pH 7.5) in a volume of 50 ml. Glucose (40 mM) or glycerol (10%, vol/vol) was added to the reaction mixture as an energy source to regenerate NADH from NAD+ in E. coli. The reactions were carried out at 30°C with rotary shaking at 180 rpm.
Product analysis.
High-performance liquid chromatography (HPLC) analysis was performed using an HPLC system (1100 series; Agilent, Palo Alto, CA) with an XTerra MS C18 IS column (4.6 mm by 20 mm; particle size, 3.5 μm; Waters, Milford, MA), as described previously (11). The reaction mixture was acidified by the addition of HCl (pH 2 to 3) and was extracted with ethyl acetate (1 ml). The extract (800 μl) was evaporated, and the resulting residue was dissolved in a water-methanol mixture at a ratio of 50:50 (400 μl). The sample (10 μl) was injected into the HPLC system. Mobile phases A and B were composed of a mixture of acetonitrile-methanol-potassium phosphate buffer (10 mM, pH 2.7) at a ratio of 2.5:2.5:95 and of acetonitrile, respectively. The samples were eluted with 0% B for 3 min, followed by a linear gradient of 0% to 70% B for 9 min at a flow rate of 1 ml/min. Compounds were detected spectrophotometrically at a wavelength of 220 nm. Mass analysis was performed using a Thermo Finnigan LCQ (Waltham, MA) with an electrospray ionization (ESI). Before nuclear magnetic resonance (NMR) analysis, the reaction product was isolated using the HPLC system with a fraction collector (1200 series, Agilent) and an XTerra MS C18 column (4.6 mm by 250 mm; particle size, 3.5 μm; Waters), as described previously (11). NMR analysis was performed using a Bruker ADVANCE600 (Billerica, MA).
5-Hydroxy-2-naphthoic acid: 1H NMR (600 MHz, [D6]DMSO-d6): δ = 7.07 (d, J = 7.5 Hz, 1H; H-6), 7.41 (dd, J = 8.2, 7.5 Hz, 1H; H-7), 7.55 (d, J = 8.2 Hz, 1H; H-8), 8.01 (dd, J = 8.6, 1.3 Hz, 1H; H-3), 8.31 (d, J = 8.6 Hz, 1H; H-4), 8.57 (d, J = 1.3 Hz, 1H; H-1); MS (ESI) (m/z): calculated for C11H7O3 [M − H]−, 187.0; found, 187.0.
RESULTS
Oxidation activity of wild-type CYP199A2 for coumaric and cinnamic acids.
We explored the catalytic potential of CYP199A2 for the oxidation of coumaric and cinnamic acids. Whole-cell assays were performed using E. coli cells that coexpressed CYP199A2 with putidaredoxin reductase from P. putida and palustrisredoxin from R. palustris (12). HPLC analysis of the reaction of CYP199A2 with p-coumaric acid showed a major peak (retention time, 4.2 min) in addition to the substrate peak (5.7 min) (see Fig. S1 in the supplemental material). This new peak was not detected following reaction with E. coli cells carrying the empty vector without the CYP199A2 gene (data note shown). The compound corresponding to this peak was confirmed to be a monooxygenation product of p-coumaric acid based on the determination of its mass value. Furthermore, the retention time of this product coincided with that of an authentic sample of caffeic acid. Based on these observations, this product was identified as caffeic acid. We examined the time course of p-coumaric acid hydroxylation by E. coli cells expressing CYP199A2. As shown in Fig. 1, the whole cells (9.0 g [DCW] per liter) stoichiometrically converted 1 mM p-coumaric acid to caffeic acid in 240 min. The initial rate of p-coumaric acid hydroxylation by the whole cells was estimated as 4.9 mol (mol P450)−1 min−1 (615 nmol g [DCW]−1 min−1). This rate was approximately three times lower than that of 2-naphthoic acid hydroxylation by the whole cells, which was 15 mol (mol P450)−1 min−1 (1,840 nmol g [DCW]−1 min−1) (12). In contrast, as shown in later experiments, CYP199A2 exhibited very low or no oxidation activity for m-coumaric acid or cinnamic acid (see Fig. 4B and C, respectively).
Fig 1.
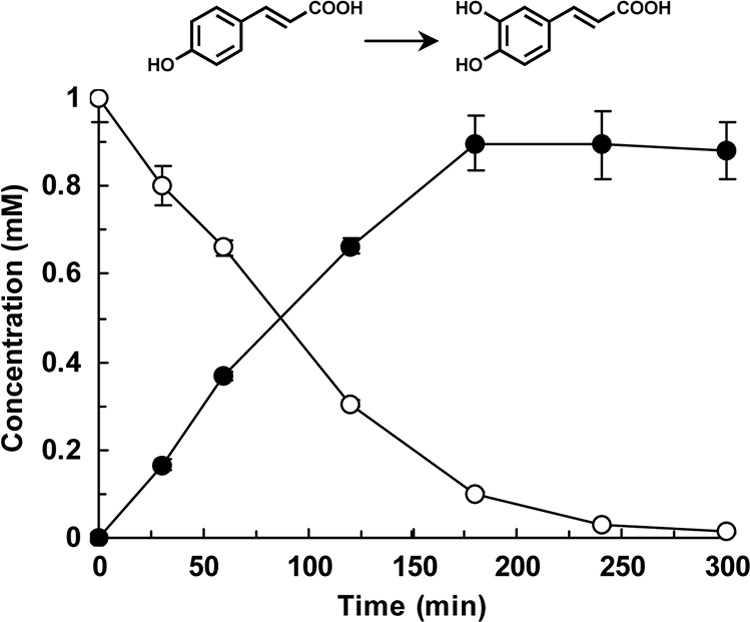
p-Coumaric acid hydroxylation by E. coli cells expressing wild-type CYP199A2. The time courses of p-coumaric acid consumption (white circles) and of caffeic acid production (black circles) are shown. Data are averages from three independent experiments, and error bars indicate standard deviations from the means. The molecular structures are shown above the graph.
Fig 4.

Oxidation activities of E. coli cells expressing the wild-type CYP199A2 or its F185 mutants for coumaric and cinnamic acids. Whole cells were reacted with p-coumaric acid (A), m-coumaric acid (B), or cinnamic acid (C). (A and B) Initial rate of caffeic acid production, which was estimated for the first 30 min of the reaction; (C) initial rates of m-coumaric, p-coumaric, and caffeic acid production (white, black, and gray bars, respectively). Data are averages from three independent experiments, and error bars indicate standard deviations from the means. The molecular structures are shown as insets.
Construction of CYP199A2 mutants.
The crystal structure of CYP199A2 determined by Bell and coworkers (3) revealed that the substrate binding pocket is composed of hydrophobic amino acids except for Ser at the 97 position (S97) and Ser at the 247 position (S247) (Fig. 2). They also performed computer docking of 4-ethylbenzoic acid as a substrate into the active site (3). The docking model suggested that the substrate is fixed in the active site through interaction between the substrate carboxyl group and the S97 and S247 residues, which leads to situation of the substrate 4-ethyl group over the heme iron. Furthermore, Phe at the 185 position (F185) is situated directly above, and only 6.35 Å from, the heme iron and is likely to play a key role in enzyme-substrate interactions. We focused our study on this F185 residue, and investigated its role in the activity and the selectivity of CYP199A2 using site-directed mutagenesis.
Fig 2.
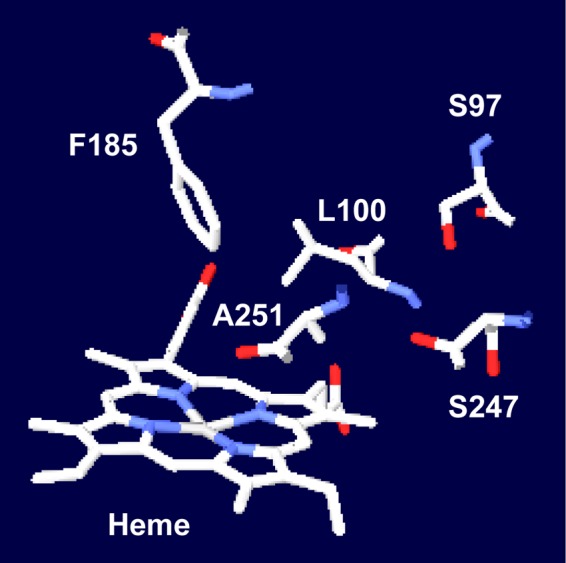
Model of the active site of CYP199A2. The structure (Protein Data Bank code 2FR7) was visualized using the Swiss-PdbViewer program.
The F185 residue of CYP199A2 was substituted with Tyr (F185Y), Trp (F185W), Val (F185V), Leu (F185L), Ile (F185I), Gly (F185G), Ala (F185A), Ser (F185S), or Thr (F185T), which have hydrophobic or hydroxylated side chains of differing sizes. Using SDS-PAGE analysis, we confirmed that recombinant wild-type CYP199A2 and all of its recombinant mutants were expressed in soluble fractions of E. coli cells (see Fig. S2 in the supplemental material). The expression levels of the F185V, F185L, and F185I mutants were relatively low, compared with those of the wild type and the other F185 mutants. We also determined the CO-reduced difference spectra of the wild type and its F185 mutants, which is a measure of the formation of an adduct of the reduced heme in the enzyme with CO (see Fig. S3). The wild type and the F185G, F185A, F185S, and F185T mutants showed only Soret absorption peaks at around 450 nm, whereas mutants with aromatic amino acid substitutions (F185Y and F185W) or with branched-chain amino acid substitutions (F185V, F185L, and F185I) showed large peaks at around 420 nm. Unlike the other F185 mutants, the F185V and F185I mutants showed no detectable Soret absorption peaks.
Oxidation activities of CYP199A2 mutants for 2-naphthoic acid.
We first examined the oxidation activities of the CYP199A2 F185 mutants for 2-naphthoic acid, which is a known substrate of CYP199A2. Whole-cell assays were performed using E. coli cells that coexpressed the wild-type or mutant CYP199A2 with the redox partners. E. coli cells carrying the empty vector without the CYP199A2 gene exhibited no oxidation activity for 2-naphthoic acid, whereas E. coli cells expressing the wild-type enzyme oxidized 2-naphthoic acid to 7-hydroxy-2-naphthoic acid and 8-hydroxy-2-naphthoic acid, as previously reported (Fig. 3A) (11). When the F185 residue of CYP199A2 was substituted with other aromatic amino acids, these mutants (F185Y and F185W) exhibited no oxidation activity. Interestingly, when the other F185 mutants were reacted with 2-naphthoic acid, all of these mutants generated a new peak (retention time 6.9 min) in HPLC analysis (see Fig. S4). The compound corresponding to this new peak was confirmed to be a monooxygenation product of 2-naphthoic acid based on the determination of its mass value. Furthermore, the 1H NMR spectrum of this product coincided with that of 5-hydroxy-2-naphthoic acid that had been previously determined (7). Based on these observations, this new product was identified as 5-hydroxy-2-naphthoic acid.
Fig 3.

Oxidation activities of E. coli cells expressing the wild-type CYP199A2 or its F185 mutants for 2-naphthoic acid. Whole cells (A) and cell extracts (B) were reacted with 2-naphthoic acid. The initial rates of 7-, 8-, and 5-hydroxy-2-naphthoic acid production (white, black, and gray bars, respectively), which were estimated for the first 10 min of the reaction, are shown. Data are averages from three independent experiments, and error bars indicate standard deviations from the means.
The wild-type CYP199A2 produced 7-hydroxy-2-naphthoic acid and 8-hydroxy-2-naphthoic acid, but not 5-hydroxy-2-naphthoic acid, from 2-naphthoic acid, whereas its F185 mutants predominantly produced 5-hydroxy-2-naphthoic acid (Fig. 3A). The F185L mutant exhibited the highest activity, followed by F185G, F185V, and F185A. Intriguingly, the rate of 2-naphthoic acid hydroxylation by E. coli cells expressing the F185L mutant was 4.3 times higher than that of its hydroxylation by E. coli cells expressing the wild-type enzyme. The whole cells expressing the F185L mutant converted 1 mM 2-naphthoic acid to 0.72 mM 5-hydroxy-2-naphthoic acid in only 20 min (see Fig. S5).
Intact cells that expressed CYP199A2 mutants with branched-chain amino acid substitutions (F185V, F185L, and F185I) exhibited high oxidation activity for 2-naphthoic acid, even though these mutants showed small or no Soret absorption peaks (see Fig. S3 in the supplemental material). These results suggested that these mutants might be unstable in the cell extracts that were used to determine the CO-reduced difference spectra. We therefore examined the oxidation activities of these mutants using the same cell extracts that were used for spectral analysis. As shown in Fig. 3B, the F185V, F185L, and F185I mutants in the cell extracts also exhibited high oxidation activity for 2-naphthoic acid. The profiles of the activities of the wild type and its F185 mutants were similar when whole cells and cell extracts were used (Fig. 3). These results indicated that these mutants were also catalytically active in the cell extracts. We also confirmed that the insoluble fractions of E. coli cells expressing these mutants exhibited no oxidation activity for 2-naphthoic acid (data not shown).
Oxidation activities of CYP199A2 mutants for coumaric and cinnamic acids.
We then examined the oxidation activities of the CYP199A2 F185 mutants for p-coumaric acid. Interestingly, besides the wild-type CYP199A2, only the F185L mutant hydroxylated p-coumaric acid to caffeic acid (Fig. 4A). The initial rate of p-coumaric acid hydroxylation by E. coli cells expressing the F185L mutant was estimated as 3,380 nmol g (DCW)−1 min−1. This rate was 5.5 times higher than that of p-coumaric acid hydroxylation by E. coli cells expressing the wild-type enzyme, which was 615 nmol g (DCW)−1 min−1. The whole cells expressing the F185L mutant stoichiometrically converted 1 mM p-coumaric acid to caffeic acid in only 40 min (see Fig. S6 in the supplemental material).
We also evaluated the oxidation activities of the CYP199A2 F185 mutants for m-coumaric acid. As shown in Fig. 4B, the wild-type enzyme exhibited very low hydroxylation activity, whereas the F185L, F185G, and F185A mutants efficiently hydroxylated m-coumaric acid to caffeic acid. The F185L mutant exhibited the highest activity, followed by F185G, F185A, and F185V. The whole cells expressing the F185L mutant converted 1 mM m-coumaric acid to 0.76 mM caffeic acid in 40 min (see Fig. S7 in the supplemental material). The conversion yield was relatively low. We detected an additional minor peak (retention time, 3.0 min) following reaction of the F185L mutant with m-coumaric acid in HPLC analysis (see Fig. S8 in the supplemental material). The compound corresponding to this peak was confirmed to be a monooxygenation product of m-coumaric acid based on the determination of its mass value, suggesting that this minor product might be 3,5-dihydroxycinnamic acid.
Intriguingly, several F185 mutants also acquired the ability to hydroxylate cinnamic acid, which was not hydroxylated by the wild-type enzyme (Fig. 4C). The F185V, F185L, F185I, F185G, and F185A mutants predominantly produced m-coumaric acid from cinnamic acid. Low levels of p-coumaric acid were also detected following reaction of these mutants with cinnamic acid. Furthermore, the F185L, F185G, and F185A mutants produced caffeic acid from cinnamic acid via coumaric acids. We examined the time course of cinnamic acid oxidation by E. coli cells expressing the F185L mutant. As shown in Fig. 5, the whole cells expressing the F185L mutant converted 1 mM cinnamic acid to 0.72 mM caffeic acid in 90 min, mainly via m-coumaric acid.
Fig 5.
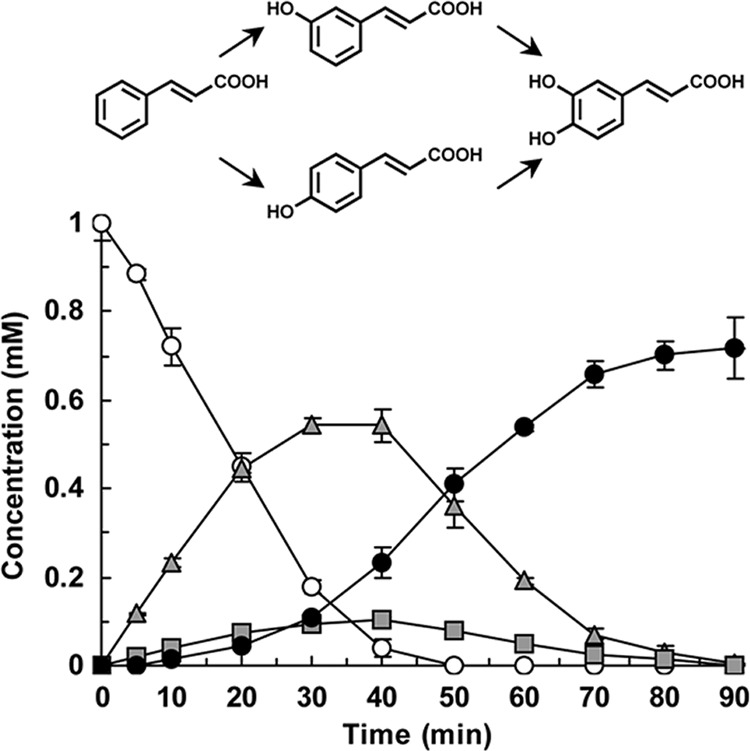
Cinnamic acid hydroxylation by E. coli cells expressing the CYP199A2 F185L mutant. The time courses of cinnamic acid consumption (white circles) and of m-coumaric, p-coumaric, and caffeic acid production (gray triangles, gray squares, and black circles, respectively) are shown. Data are averages from three independent experiments, and error bars indicate standard deviations from the means. The molecular structures are shown above the graph.
Production of caffeic acid.
We attempted to produce caffeic acid from 20 mM p-coumaric acid using E. coli cells expressing the wild-type CYP199A2 or the F185L mutant as biocatalysts in a 500-ml flask that contained 50 ml of the reaction mixture. The reaction mixture was supplemented with glucose or glycerol as an energy source to regenerate NADH from NAD+ in E. coli. E. coli cells expressing the wild-type enzyme produced 2.4 mM caffeic acid in 24 h in the absence of the energy sources, whereas the amount of this product increased to 4.3 mM and 3.4 mM in the presence of glucose and glycerol, respectively (Fig. 6). Productivity was strongly enhanced by using the F185L mutant instead of the wild-type enzyme. E. coli cells expressing the F185 mutant produced 5.0 mM caffeic acid in 15 h in the absence of the energy sources, following which there was no further increase in this product. In contrast, in the presence of glucose and glycerol, the production of caffeic acid continued to increase after 15 h and reached 9.1 mM (1.6 g/liter) and 15 mM (2.8 g/liter), respectively, in 24 h (Fig. 6).
Fig 6.
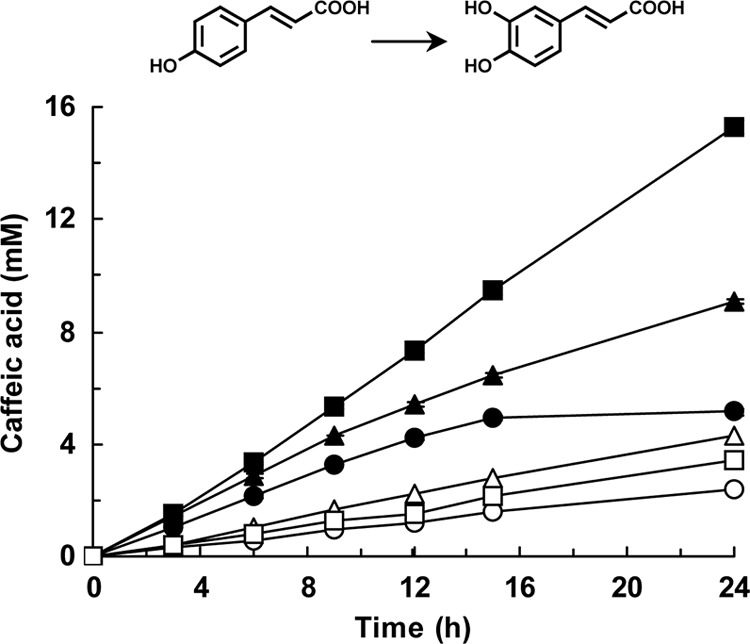
Biotechnological production of caffeic acid by E. coli cells expressing the wild-type CYP199A2 or its F185L mutant. The wild-type CYP199A2 (white symbols) and its F185L mutant (black symbols) were reacted with p-coumaric acid in the absence (circles) or in the presence of glucose (triangles) or glycerol (squares). Data are averages from three independent experiments, and error bars indicate standard deviations from the means. The molecular structures are shown above the graph.
DISCUSSION
In this study, we found that the bacterial P450 CYP199A2 has hydroxylation activity for p-coumaric acid. To our knowledge, CYP199A2 is the first bacterial P450 that has been shown to catalyze the hydroxylation of p-coumaric acid to caffeic acid. CYP199A2 was previously reported to exhibit oxidation activity for aromatic carboxylic acids, including 2-naphthoic acid, 4-ethylbenzoic acid, and indole- and quinolinecarboxylic acids (13). The similarity of their molecular sizes and the presence of the carboxyl group seem to enable these compounds to fit the active site. These results indicate that CYP199A2 is a unique enzyme that specializes in the oxidation of aromatic carboxylic acids.
We experimentally demonstrated that the F185 residue in the active site of CYP199A2 plays important roles in enzyme-substrate interactions and controls the hydroxylation regioselectivity of this enzyme for 2-naphthoic acid. As shown in Fig. 7A, only the wild-type enzyme hydroxylated 2-naphthoic acid at the C-7 and C-8 positions, whereas all of the active F185 mutants exhibited a preference for C-5 hydroxylation. The aromatic ring of the F185 residue might generate specific interactions such as π-π stacking with the 2-naphthoic acid molecule, which could result in an orientation of this substrate that facilitates C-7 and C-8 hydroxylation (Fig. 2). We reasoned that substitution of the F185 residue with aliphatic amino acids would allow 2-naphthoic acid to rotate, with the substrate carboxylate oxygen atoms being anchored by the S97 and S247 side chains. Cell extracts as well as intact cells of the E. coli strains that expressed CYP199A2 mutants with branched-chain amino acid substitutions (F185V, F185L, and F185I) exhibited high oxidation activity for 2-naphthoic acid (Fig. 3), even though these mutants showed small or no Soret absorption peaks. One possible explanation for these data is that these mutants might exhibit extremely high activity, even though their expression levels are very low. A second possibility is that it may be difficult for CO to coordinate with the heme iron center at the active site of these mutants. A phenomenon similar to that proposed in this hypothesis was observed during the analysis of the spectrum of the catalytically active human CYP19 (8, 16, 20). The introduced branched-chain amino acids might hamper the formation of a CO-enzyme complex. Elucidation of the relationship between the spectra and the activities awaits detailed characterization.
Fig 7.
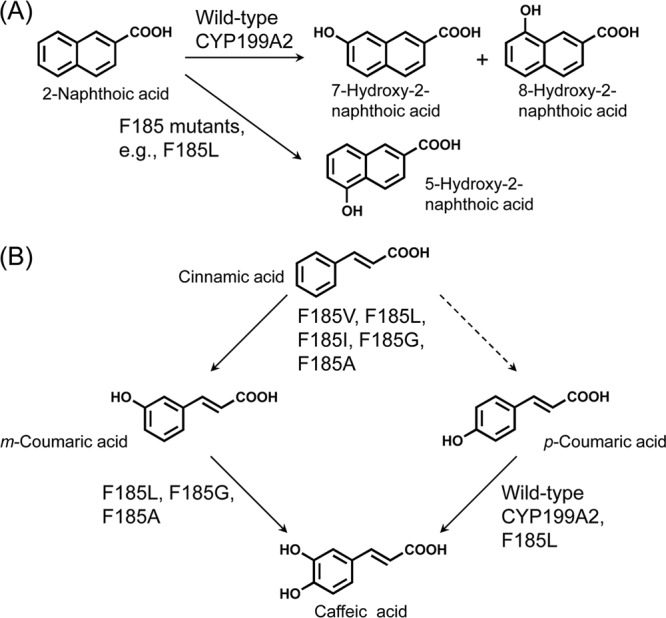
Reactions catalyzed by the wild-type CYP199A2 and its F185 mutants. (A) 2-Naphthoic acid conversion; (B) cinnamic and coumaric acid conversion.
The F185 residue of CYP199A2 also controls the substrate specificity of this enzyme. Several F185 mutants acquired the ability to hydroxylate cinnamic acid (Fig. 4C). In plants, cinnamic acid is hydroxylated to p-coumaric acid by CYP73, which is the second enzyme and first P450 in the phenylpropanoid pathway (40). The resulting p-coumaric acid is further modified to generate structurally diverse phenolics. In contrast, the F185 mutants catalyzed two-step hydroxylation of cinnamic acid to produce caffeic acid via coumaric acids. We summarize the abilities of the wild-type CYP199A2 and its F185 mutants to hydroxylate cinnamic and coumaric acids in Fig. 7B. The F185V, F185L, F185I, F185G, and F185A mutants first hydroxylated cinnamic acid predominantly at the meta position. Of these mutants, the F185V and F185I mutants accumulated m-coumaric acid, whereas the F185L, F185G, and F185A mutants, which were confirmed to exhibit hydroxylation activity for m-coumaric acid (Fig. 4B), was further able to convert m-coumaric acid to caffeic acid.
Finally, we achieved gram-per-liter-scale production of caffeic acid. Caffeic acid has great potential for pharmaceutical applications, as described above. Furthermore, caffeic acid has recently attracted attention as a “phytomonomer,” a polymerizable plant-derived chemical, for the preparation of environmentally degradable, liquid-crystalline, wholly aromatic polymers, although the amount of caffeic acid produced by plants is still limited (17). To date, several attempts have been made to biotechnologically produce caffeic acid from p-coumaric acid using microorganisms. For example, it was reported that Pycnoporus cinnabarinus and Streptomyces caeruleus catalyzed the hydroxylation of p-coumaric acid to produce 257 mg/liter and 150 mg/liter caffeic acid, respectively (1, 32). In the present study, we succeeded in creating a CYP199A2 F185L mutant that exhibited 5.5 times higher hydroxylation activity for p-coumaric acid than the wild-type enzyme (Fig. 4A). Furthermore, we found that glycerol was more effective than glucose in supporting the hydroxylation carried out by E. coli cells expressing the F185L mutant (Fig. 6). By using the F185L whole-cell catalyst in the presence of glycerol, the production of caffeic acid from p-coumaric acid reached 15 mM (2.8 g/liter), which is the highest level so far attained in biotechnological production of this compound. The F185 mutant genes might also be helpful in constructing metabolically engineered microorganisms that accumulate caffeic acid from renewable carbon sources such as glucose, and transgenic plants with increased levels of caffeic acid and its derivatives.
Supplementary Material
ACKNOWLEDGMENT
This research work was supported in part by a Waseda University Grant for Special Research Projects (project number 2011A-038).
Footnotes
Published ahead of print 22 June 2012
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1. Alvarado IE, Navarro D, Rocord E, Asther M, Asther M. 2003. Fungal biotransformation of p-coumaric acid into caffeic acid by Pycnoporus cinnabarinus: an alternative for producing a strong natural oxidant. World J. Microbiol. Biotechnol. 19: 157–160 [Google Scholar]
- 2. Bell SG, et al. 2006. Cytochrome P450 enzymes from the metabolically diverse bacterium Rhodopseudomonas palustris. Biochem. Biophys. Res. Commun. 342: 191–196 [DOI] [PubMed] [Google Scholar]
- 3. Bell SG, et al. 2008. Crystal structure of CYP199A2, a para-substituted benzoic acid oxidizing cytochrome P450 from Rhodopseudomonas palustris. J. Mol. Biol. 383: 561–574 [DOI] [PubMed] [Google Scholar]
- 4. Bernhardt R. 2006. Cytochromes P450 as versatile biocatalysts. J. Biotechnol. 124: 128–145 [DOI] [PubMed] [Google Scholar]
- 5. Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72: 248–254 [DOI] [PubMed] [Google Scholar]
- 6. Castelluccio C, et al. 1995. Antioxidant potential of intermediates in phenylpropanoid metabolism in higher plants. FEBS Lett. 368: 188–192 [DOI] [PubMed] [Google Scholar]
- 7. Cerniglia CE, Lambert KJ, Miller DW, Freeman JP. 1984. Transformation of 1- and 2-methylnaphthalene by Cunninghamella elegans. Appl. Environ. Microbiol. 47: 111–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chakraborty J, Hopkins R, Parke DV. 1972. Inhibition studies on the aromatization of androst-4-ene-3,17-dione by human placental microsomal preparations. Biochem. J. 130: 19P–20P [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chung TW, et al. 2004. Novel and therapeutic effect of caffeic acid and caffeic acid phenyl ester on hepatocarcinoma cells: complete regression of hepatoma growth and metastasis by dual mechanism. FASEB J. 18: 1670–1681 [DOI] [PubMed] [Google Scholar]
- 10. Clifford MN. 1999. Chlorogenic acids and other cinnamates—nature, occurrence and dietary burden. J. Sci. Food Agric. 79: 362–372 [Google Scholar]
- 11. Furuya T, Kino K. 2009. Discovery of 2-naphthoic acid monooxygenases by genome mining and their use as biocatalysts. ChemSusChem 2: 645–649 [DOI] [PubMed] [Google Scholar]
- 12. Furuya T, Kino K. 2010. Regioselective oxidation of indole- and quinolinecarboxylic acids by cytochrome P450 CYP199A2. Appl. Microbiol. Biotechnol. 85: 1861–1868 [DOI] [PubMed] [Google Scholar]
- 13. Furuya T, Kino K. 2010. Genome mining approach for the discovery of novel cytochrome P450 biocatalysts. Appl. Microbiol. Biotechnol. 86: 991–1002 [DOI] [PubMed] [Google Scholar]
- 14. Gomes CA, et al. 2003. Anticancer activity of phenolic acids of natural or synthetic origin: a structure-activity study. J. Med. Chem. 46: 5395–5401 [DOI] [PubMed] [Google Scholar]
- 15. Grogan G. 2011. Cytochromes P450: exploiting diversity and enabling application as biocatalysts. Curr. Opin. Chem. Biol. 15: 241–248 [DOI] [PubMed] [Google Scholar]
- 16. Harada N. 1988. Novel properties of human placental aromatase as cytochrome P-450: purification and characterization of a unique form of aromatase. J. Biochem. 103: 106–113 [DOI] [PubMed] [Google Scholar]
- 17. Kaneko T, Thi TH, Shi DJ, Akashi M. 2006. Environmentally degradable, high-performance thermoplastics from phenolic phytomonomers. Nat. Mater. 5: 966–970 [DOI] [PubMed] [Google Scholar]
- 18. Kroon PA, Williamson G. 1999. Hydroxycinnamates in plants and food: current and future perspectives. J. Sci. Food Agric. 79: 355–361 [Google Scholar]
- 19. Lesage-Meessen L, et al. 2001. Simple phenolic content in olive oil residues as a function of extraction systems. Food Chem. 75: 501–507 [Google Scholar]
- 20. Meigs RA, Ryan KJ. 1971. Enzymatic aromatization of steroids. I. Effects of oxygen and carbon monoxide on the intermediate steps of estrogen biosynthesis. J. Biol. Chem. 246: 83–87 [PubMed] [Google Scholar]
- 21. Nair RB, et al. 2002. Arabidopsis CYP98A3 mediating aromatic 3-hydroxylation. Developmental regulation of the gene, and expression in yeast. Plant Physiol. 130: 210–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nardini M, et al. 1995. Inhibition of human low-density lipoprotein oxidation by caffeic acid and other hydroxycinnamic acid derivatives. Free Radic. Biol. Med. 19: 541–552 [DOI] [PubMed] [Google Scholar]
- 23. Niggeweg R, Michael AJ, Martin C. 2004. Engineering plants with increased levels of the antioxidant chlorogenic acid. Nat. Biotechnol. 22: 746–754 [DOI] [PubMed] [Google Scholar]
- 24. Nijkamp K, Westerhof RG, Ballerstedt H, de Bont JA, Wery J. 2007. Optimization of the solvent-tolerant Pseudomonas putida S12 as host for the production of p-coumarate from glucose. Appl. Microbiol. Biotechnol. 74: 617–624 [DOI] [PubMed] [Google Scholar]
- 25. Olthof MR, Hollman PC, Katan MB. 2001. Chlorogenic acid and caffeic acid are absorbed in humans. J. Nutr. 131: 66–71 [DOI] [PubMed] [Google Scholar]
- 26. Omura T, Sato R. 1964. The carbon monoxide-binding pigment of liver microsomes. I. Evidence for its hemoprotein nature. J. Biol. Chem. 239: 2370–2378 [PubMed] [Google Scholar]
- 27. O'Reilly E, Köhler V, Flitsch SL, Turner NJ. 2011. Cytochromes P450 as useful biocatalysts: addressing the limitations. Chem. Commun. (Camb.) 47: 2490–2501 [DOI] [PubMed] [Google Scholar]
- 28. Ortiz de Montellano PR. 2010. Hydrocarbon hydroxylation by cytochrome P450 enzymes. Chem. Rev. 110: 932–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pan GX, Bolton JL, Leary GJ. 1998. Determination of ferulic and p-coumaric acids in wheat straw and the amounts released by mild acid and alkaline peroxide treatment. J. Agric. Food Chem. 46: 5283–5288 [Google Scholar]
- 30. Peterson JA, Lorence MC, Amarneh B. 1990. Putidaredoxin reductase and putidaredoxin. Cloning, sequence determination, and heterologous expression of the proteins. J. Biol. Chem. 265: 6066–6073 [PubMed] [Google Scholar]
- 31. Rice-Evans CA, Miller NJ, Paganga G. 1997. Antioxidant properties of phenolic compounds. Trends Plant Sci. 2: 152–159 [Google Scholar]
- 32. Sachan A, Ghosh S, Sen SK, Mitra A. 2006. Co-production of caffeic acid and p-hydroxybenzoic acid from p-coumaric acid by Streptomyces caeruleus MTCC 6638. Appl. Microbiol. Biotechnol. 71: 720–727 [DOI] [PubMed] [Google Scholar]
- 33. Sato Y, et al. 2011. In vitro and in vivo antioxidant properties of chlorogenic acid and caffeic acid. Int. J. Pharm. 403: 136–138 [DOI] [PubMed] [Google Scholar]
- 34. Schoch G, et al. 2001. CYP98A3 from Arabidopsis thaliana is a 3′-hydroxylase of phenolic esters, a missing link in the phenylpropanoid pathway. J. Biol. Chem. 276: 36566–36574 [DOI] [PubMed] [Google Scholar]
- 35. Staniforth V, Chiu LT, Yang NS. 2006. Caffeic acid suppresses UVB radiation-induced expression of interleukin-10 and activation of mitogen-activated protein kinases in mouse. Carcinogenesis 27: 1803–1811 [DOI] [PubMed] [Google Scholar]
- 36. Touaibia M, Jean-François J, Doiron J. 2011. Caffeic acid, a versatile pharmacophore: an overview. Mini Rev. Med. Chem. 11: 695–713 [DOI] [PubMed] [Google Scholar]
- 37. Urlacher VB, Girhard M. 2012. Cytochrome P450 monooxygenases: an update on perspectives for synthetic application. Trends Biotechnol. 30: 26–36 [DOI] [PubMed] [Google Scholar]
- 38. Vannellia T, Qib WW, Sweigardc J, Gatenbyd AA, Sariaslanid FS. 2007. Production of p-hydroxycinnamic acid from glucose in Saccharomyces cerevisiae and Escherichia coli by expression of heterologous genes from plants and fungi. Metab. Eng. 9: 142–151 [DOI] [PubMed] [Google Scholar]
- 39. Wang GF, et al. 2009. Anti-hepatitis B virus activity of chlorogenic acid, quinic acid and caffeic acid in vivo and in vitro. Antiviral Res. 83: 186–190 [DOI] [PubMed] [Google Scholar]
- 40. Werck-Reichhart D. 1995. Cytochromes P450 in phenylpropanoid metabolism. Drug Metab. Drug Interact. 12: 221–243 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.