

Abstract
Hemangiomas are tumors formed by hyper-proliferation of vascular endothelial cells. This is caused by elevated vascular endothelial growth factor (VEGF) signaling through VEGF receptor 2 (VEGFR2). Here we show that elevated VEGF levels produced by hemangioma endothelial cells are reduced by the mTOR inhibitor rapamycin. mTOR activates p70S6K, which controls translation of mRNA to generate proteins such as hypoxia inducible factor-1 (HIF-1). VEGF is a known HIF-1 target gene, and our data show that VEGF levels in hemangioma endothelial cells are reduced by HIF-1α siRNA. Over-expression of HIF-1α increases VEGF levels and endothelial cell proliferation. Furthermore, both rapamycin and HIF-1α siRNA reduce proliferation of hemangioma endothelial cells. These data suggest that mTOR and HIF-1 contribute to hemangioma endothelial cell proliferation by stimulating an autocrine loop of VEGF signaling. Furthermore, mTOR and HIF-1 may be therapeutic targets for the treatment of hemangiomas.
Introduction
Hemangiomas are the most common tumors found in children. These vascular anomalies arise via uncontrolled angiogenesis through clonal expansion of vascular endothelial cells [1]. Heterozygous single amino acid substitutions in VEGFR2 or TEM8 have been found in hemangioma patients and have been linked to reduced NFATc2-dependent expression of VEGFR1 [2]. As a decoy receptor that binds and sequesters VEGF and prevents it from activating VEGFR2 [3], the low level of VEGFR1 expression by hemangioma endothelial cells causes constitutive VEGF signaling through VEGFR2. This results in increased proliferation of the cells and tumor formation [2].
HIF-1 is a transcription factor that is activated during hypoxia by a mechanism that involves stabilization of HIF-1 protein levels [4]–[6]. However, various signaling pathways can also increase expression and activity of HIF-1 [7]. The kinase p70S6K controls translation of mRNA to protein for factors such as HIF-1 [8], [9]. Therefore, phosphorylation of p70S6K through a constitutive signaling pathway such as the VEGFR2 pathway in hemangioma endothelial cells might increase HIF-1 activity. p70S6K has been shown to be phosphorylated by mTOR [10], a downstream signaling molecule in the phosphoinositide-3 kinase (PI3K) pathway [11], [12]. We previously showed that PI3K is constitutively active as a result of enhanced VEGF-dependent VEGFR2 signaling in hemangioma endothelial cells [2]. Rapamycin, a known mTOR inhibitor [13], [14], can be used to determine the role of this pathway in regulating biochemical and physiological processes. Also, VEGF is a known HIF-1 target gene [15], [16].
Based on these findings, we sought to determine whether a HIF-1-dependent autocrine loop of VEGF signaling might contribute to the hyper-proliferation of hemangioma endothelial cells. We also asked whether rapamycin could inhibit HIF-1 expression and reduce VEGF signaling in these cells.
Results
To determine HIF-1α protein levels we performed immunoblotting using lysates from cultured normal human dermal microvascular endothelial cells (HDMEC) and hemangioma endothelial cells (EC2, EC17B, EC21A). We found that hemangioma endothelial cells show significantly higher expression of HIF-1α than in control cells (Figure 1A and S1). Immunocytochemistry showed constitutive nuclear localization of activated HIF-1α in hemangioma endothelial cells, but not in control endothelial cells (Figure 1B).
Figure 1. Elevated HIF-1 expression in hemangioma endothelial cells caused by VEGF/PI3K signaling.

A: Immunoblotting showing high expression of HIF-1α in hemangioma endothelial cells (EC2, EC17B, EC21A) compared to normal endothelial cells (HDMEC). B: Immunocytochemistry showing constitutive nuclear localization of HIF-1α in hemangioma endothelial cells. C and D: Luminex analyses demonstrating that elevated HIF-1α levels in hemangioma endothelial cells are reduced in the presence of VEGF-A165 neutralizing antibodies (VEGF Ab) or a chemical inhibitor of PI3K (LY294002). Data represent mean (n = 3)±SD; *P<0.01 compared to IgG or vehicle.
To determine whether the elevated levels of HIF-1α found in hemangioma endothelial cells is the result of their constitutive VEGF/VEGFR2 signaling as previously described [2], we treated cells for 6 hours with neutralizing antibodies specific for VEGF-A165. Luminex assays were performed to measure real-time quantitative protein expression levels of HIF-1α. The data showed that the VEGF antibodies significantly reduced the elevated HIF-1α levels in hemangioma endothelial cells (Figure 1C). We previously reported that hemangioma endothelial cells have constitutive PI3K/AKT signaling as a result of this VEGF activity [2]. Addition of a chemical inhibitor of PI3K (LY294002) was sufficient to reduce elevated HIF-1α levels in hemangioma endothelial cells almost to the levels of control endothelial cells (Figure 1D).
To assess the effects of HIF-1α siRNA in the primary control and hemangioma endothelial cells, we performed immunoblotting and Luminex assays using lysates from cells transfected with either HIF-1α siRNA or a negative control siRNA duplex. Our data show a substantial knockdown of HIF-1α protein expression by HIF-1α siRNA (Figure 2A and 2B). To establish whether the elevated VEGF levels observed in hemangioma endothelial cells are the result of HIF-1α activity, we performed immunoblotting and Luminex analysis using the same siRNA treated samples. HIF-1α siRNA significantly reduced the higher levels of VEGF-A165 in hemangioma endothelial cells (Figure 2A and 2C). These data were confirmed with a second HIF-1α siRNA duplex (Figure S2A and S2B).
Figure 2. Elevated VEGF levels in hemangioma endothelial cells are HIF-1-dependent.
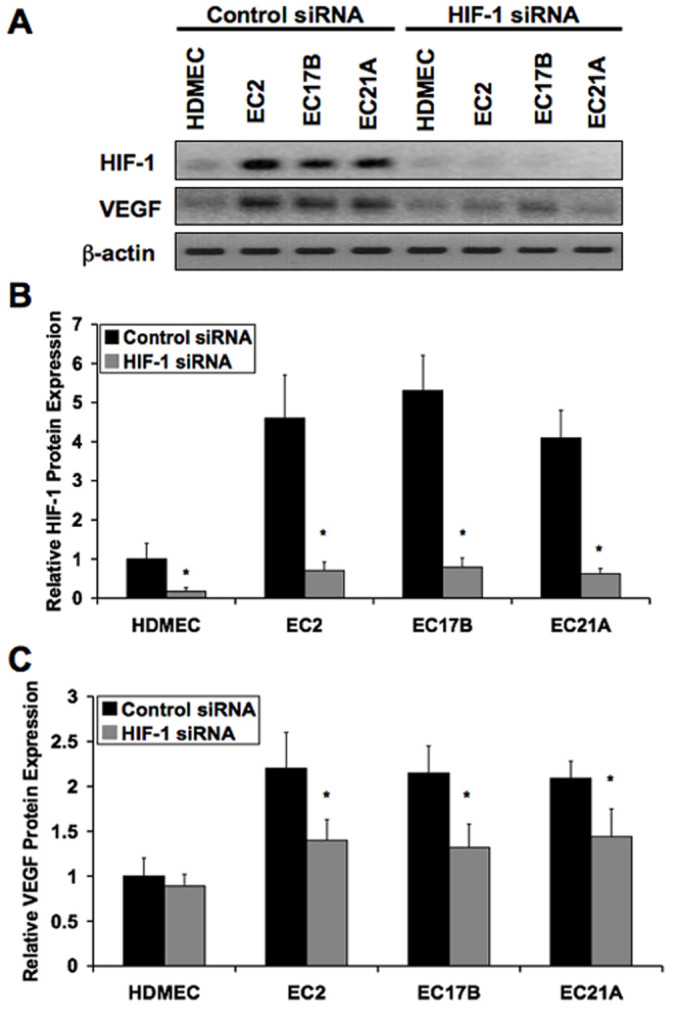
A: Immunoblotting showing the expression knockdown effects of HIF-1α siRNA on HIF-1α and VEGF-A165 in normal (HDMEC) and hemangioma (EC2, EC17B, EC21A) endothelial cells. B and C: Luminex analysis showing suppression of HIF-1α and VEGF-A165 protein levels in hemangioma endothelial cells by HIF-1α siRNA. Data represent mean (n = 3)±SD; *P<0.05 compared to control siRNA.
Next, we sought to confirm a role for HIF-1α on the proliferation rates of hemangioma endothelial cells. Cultured normal or hemangioma endothelial cells transfected with HIF-1α siRNA or negative control siRNA were stained in suspension for BrdU incorporation. Hemangioma endothelial cells showed much higher proliferation rates than control cells. HIF-1α siRNA significantly reduced the hyper-proliferation of hemangioma endothelial cells (Figure 3A and 3B). These results were confirmed by quantifying the total number of cells in culture over time (Figure S3A) and by using a second HIF-1α siRNA duplex (Figure S2C).
Figure 3. Inhibiting HIF-1 expression decreases proliferation of hemangioma endothelial cells.

A: Flow cytometry analysis of BrdU incorporation in normal (HDMEC) and hemangioma (EC2, EC17B, EC21A) endothelial cells transfected with control or HIF-1α siRNA duplexes. B: Quantification of BrdU incorporation showing decreased proliferation of hemangioma endothelial cells transfected with HIF-1α siRNA. Data represent mean (n = 3)±SD; *P<0.05 compared to control siRNA.
To determine whether HIF-1 expression increases VEGF levels we decided to over-express HIF-1α in control endothelial cells (HDMEC) using a pcDNA3-HIF-1α plasmid. Cells transfected with the HIF-1α expression plasmid showed higher levels of HIF-1α and VEGF-A165 protein by immunoblotting compared to those transfected with the pcDNA3 empty vector (Figure 4A). Flow cytometry was performed to assess cell proliferation by BrdU staining. Cells containing the HIF-1α expression plasmid showed higher BrdU incorporation compared to those with the empty vector. However, in the presence of VEGF-A165 neutralizing antibodies, the HIF-1α-dependent increase in cell proliferation was blocked (Figure 4B and 4C). These results were confirmed by quantifying the total cell numbers in culture over time (Figure S3B).
Figure 4. HIF-1 over-expression increases VEGF-dependent proliferation of endothelial cells.

A: Immunoblotting showing increased HIF-1α and VEGF-A165 levels in HDMEC containing the pcDNA3-HIF-1α expression plasmid. B: Flow cytometry analysis of BrdU incorporation showing increased proliferation of cells with the pcDNA3-HIF-1α plasmid, which is prevented by VEGF neutralizing antibodies. Non-specific IgG and pcDNA3 vector were used as negative controls. C: Quantification of flow cytometry analysis of BrdU incorporation. Data represent mean (n = 3)±SD; *P<0.01.
To confirm that VEGF signaling can induce synthesis of HIF-1α, we treated control endothelial cells with recombinant VEGF-A165. Immunoblotting confirmed increased phosphorylation of p70S6K after 15 minutes of treatment, a kinase known to promote translation of HIF-1 mRNA into protein [8], [9]. The mTOR inhibitor rapamycin successfully inhibited VEGF-dependent increases in p70S6K phosphorylation (Figure 5A). Increased HIF-1α expression was observed in cells treated with VEGF for 6 hours, and total VEGF-A165 levels themselves were also higher when stimulating cells with exogenous VEGF-A165, suggesting an autocrine loop of VEGF signaling. Exposing the cells to rapamycin inhibited VEGF-dependent expression of HIF-1α and VEGF (Figure 5B). Exposing the cells to HIF-1α siRNA inhibited VEGF-dependent increases in cell proliferation as determined by flow cytometry analysis of BrdU staining (Figure 5C and 5D) or by counting cells is culture over time (Figure S3C).
Figure 5. VEGF stimulates p70S6K phosphorylation and HIF-1 expression.

A: Immunoblotting showing that treatment of normal endothelial cells (HDMEC) with exogenous VEGF-A165 increases phosphorylation of p70S6K. B: Immunoblotting showing increased expression of HIF-1α and VEGF-A165 in HDMEC treated with recombinant VEGF-A165, suggesting an autocrine loop of signaling. Rapamycin prevents these increases. C: Flow cytometry analysis of BrdU incorporation showing that increased proliferation of HDMEC by VEGF-A165 is inhibited in the presence of HIF-1α siRNA. D: Quantification of flow cytometry analysis of BrdU incorporation. Data represent mean (n = 3)±SD; *P<0.05. The p70S6K panel in Figure 5A is excluded from this article's CC-BY license. See the accompanying retraction notice for more information.
We next attempted to assess potential therapeutic effects of rapamycin on hemangioma endothelial cells, since their elevated proliferation is caused by hyperactive VEGF signaling [2]. Immunoblotting and Luminex analysis showed that hemangioma endothelial cells have constitutively higher phosphorylation of p70S6K and higher expression of HIF-1α and VEGF-A165 compared to control cells. Treatment of these cells with rapamycin for 6 hours dramatically decreased p70S6K phosphorylation, HIF-1α expression, and VEGF-A165 expression (Figure 6A–6C).
Figure 6. Rapamycin reduces elevated VEGF and HIF-1 levels in hemangioma endothelial cells.

A: Immunoblotting showing the effects of rapamycin on p70S6K phosphorylation, HIF-1α expression and VEGF-A165 expression in normal (HDMEC) or hemangioma (EC2, EC17B, EC21A) endothelial cell lysates. B and C: Luminex analysis showing reduced expression of HIF-1α (B) and VEGF-A165 (C) in hemangioma endothelial cells treated with rapamycin. Data represent mean (n = 3)±SD; *P<0.05 compared to vehicle.
To measure proliferation rates of control and hemangioma endothelial cells, we performed BrdU staining of cells treated in culture with vehicle or rapamycin. Hemangioma endothelial cells showed a much higher rate of proliferation than normal endothelial cells. This hyper-proliferation of hemangioma endothelial cells was significantly reduced by exposure to rapamycin (Figure 7A and 7B). These data were confirmed by quantifying total cell numbers in culture over time (Figure S3D).
Figure 7. Rapamycin inhibits proliferation of hemangioma endothelial cells.

A: Flow cytometry analysis of normal (HDMEC) or hemangioma (EC2, EC17B, EC21A) endothelial cell proliferation by BrdU incorporation in the presence of rapamycin. B: Quantification of flow cytometry assessing the effects of rapamycin on BrdU incorporation. Data represent mean (n = 3)±SD; *P<0.01 compared to vehicle.
Taken together, our data suggest that constitutive VEGF-dependent mTOR signaling in hemangioma endothelial cells promotes HIF-1 activity, which increases expression of VEGF to form an autocrine loop of signaling (Figure 8). This mechanism enhances proliferation of hemangioma endothelial cells.
Figure 8. Schematic diagram of mTOR signaling in hemangioma endothelial cells.

Low levels of VEGFR1 in hemangioma endothelial cells result in constitutive VEGFR2 signaling and cell proliferation [2]. PI3K and its downstream kinase AKT are constitutively phosphorylated in hemangioma endothelial cells as previously described [2]. Phosphorylation of p70S6K, a known target of PI3K/AKT signaling, promotes translation of HIF-1 mRNA into protein, which translocates to the nucleus to regulate expression of target genes such as VEGF. This causes an autocrine loop of VEGF signaling via activation of VEGFR2. The mTOR inhibitor rapamycin prevents p70S6K phosphorylation and decreases expression of HIF-1. mTOR and HIF-1 inhibition is sufficient to reduce VEGF levels and proliferation rate of hemangioma endothelial cells.
Discussion
Our results provide novel insight into the mechanism that control hyper-proliferation of hemangioma endothelial cells. Our previous studies identified VEGF, VEGFR1, and VEGFR2 as potential targets for therapeutic treatment of hemangiomas. Treating hemangioma endothelial cells with VEGF neutralizing antibodies, recombinant soluble VEGFR1, or VEGFR2 siRNA was sufficient to reduce their proliferation [2]. Here we show that the PI3K–mTOR–p70S6K signaling pathway is constitutively active in hemangioma endothelial cells. As a result of this activity, the expression of a downstream target, HIF-1α, is up-regulated. HIF-1 has been shown to be constitutively located in the nuclei of hemangioma endothelial cells in vivo [17]. However, it has been unclear as to what potential role HIF-1 plays in promoting the hemangioma phenotype. We demonstrate that HIF-1 is a major contributor to the elevated VEGF levels produced in hemangioma endothelial cells, and that decreased expression of HIF-1 reduces proliferation of these cells.
Based on our data, it is clear that other factors besides HIF-1 are involved in hemangioma endothelial cell proliferation. VEGF signals through many pathways besides mTOR, which may play a significant role in controlling proliferation. This would explain why reduced proliferation rates after knockdown of HIF-1 with siRNA were significant, but not dramatic. Furthermore, rapamycin may have other effects on inhibiting proliferation other than regulating HIF-1-dependent VEGF expression. Rapamycin had a much greater effect on hemangioma endothelial cell proliferation than by direct knockdown of HIF-1 with siRNA. Interestingly, rapamycin also had a significant effect on the proliferation of control endothelial cells, whereas HIF-1 siRNA did not.
Rapamycin is a potent inhibitor of the mTOR pathway and treatment of hemangioma cells with rapamycin results in a significant decrease in HIF-1 and VEGF levels and reduced proliferation. Treatment with rapamycin may therefore prove effective in the clinical management of large and rapidly proliferating hemangiomas. Given the serious adverse effects of rapamycin-based drugs when administered systemically to adult and pediatric patients [18], [19], systemic treatments with rapamycin for even large hemangiomas is not advisable. However, recent efforts to develop and clinically test topical rapamycin-based therapy for cutaneous facial angiofibromas [20], the benign tumors seen in patients with tuberous sclerosis [21], may provide a basis for considering topical cutaneous rapamycin treatment also for infantile hemangiomas. The outcome of randomized double blind Phase I clinical trials for patients with tuberous sclerosis [20] will provide a useful guide for further efforts along this line.
Materials and Methods
Ethics Statement
All cells were obtained from patients with informed written consent and protocols for this study approved by the Investigational Review Board of Harvard Medical School (IRB# M10510-110). The investigation conformed to the principles outlined in the Declaration of Helsinki.
Cell Culture
Primary human dermal microvascular endothelial cells derived from foreskin (HDMEC) or facial skin (HCMEC), human umbilical vein endothelial cells (HUVEC), and primary human hemangioma endothelial cells (EC2, EC17B, EC21A) derived from three different proliferating-phase hemangioma tumors were isolated as previously described [1]. Cells were tested for purity and found to express no markers for lymphatic endothelial cells or stromal cells (pericytes, smooth muscle cells, fibroblasts, etc.) as previously described [2], [22]. Cells were grown in standard culture conditions of 5% CO2 at 37°C using EGM-2 medium (Cambrex), containing 20% FBS and 1% Penicillin/Streptomycin, followed by human endothelial serum free medium (Gibco) 24 hours prior to all experimental conditions. Rapamycin (Sigma-Aldrich) was used at a concentration of 10 nM. Recombinant VEGF-A165 (R&D Systems) was added at a concentration of 25 ng/ml. The PI3K inhibitor LY294002 (Cell Signaling Technology) was used at a concentration of 50 µM and added to the cultures 1 hour prior to experimental conditions. VEGF-A165 neutralizing antibodies (R&D Systems) were used at a concentration of 25 ng/ml. The pcDNA3-HIF-1α expression plasmid was provided by Dr. Ernestina Schipani (Indiana University School of Medicine). Cells were transfected using 1 µg of plasmid per sample with Lipofectin and PLUS reagents (Invitrogen) according to the manufacturer’s guidelines. All experiments for this study were performed at minimum in triplicate.
RNA Interference
siRNA gene expression knockdown studies were performed using the TriFECTa RNAi kit (Integrated DNA Technologies) and corresponding protocol. Each 27 mer siRNA duplex was transfected into cells using X-tremeGene siRNA transfection reagent (Roche) following the manufacturer’s guidelines. Transfections were performed in human endothelial serum free medium (Gibco) without antibiotics using 1 µg of siRNA per sample. All transfections were performed 24 hours prior to experimental conditions or analysis. siRNA was synthesized (Integrated DNA Technologies) with the following sequences: HIF-1α: 5′- ACACGCAAAUAGCUGAUGGUAAGCCUC-3′; HIF-1α (2): 5′-AUACUGUAACUGUGCUUUGAGGACUUG-3′; negative control: 5′- UCACAAGGGAGAGAAAGAGAGGAAGGA -3′.
Flow Cytometry
Cells were stained in suspension for BrdU incorporation 8 hours after all experimental conditions with the 5-Bromo-2′-deoxy-uridine Labeling and Detection Kit I (Roche). Cells were pre-labeled with BrdU for 30 minutes then fixed with ethanol. Cells were then incubated with monoclonal antibodies against BrdU mixed with nucleases, followed by fluorescein-conjugated secondary antibodies according to the manufacturer’s protocol. Flow cytometry was performed at the Harvard Medical School, Department of Pathology flow cytometry core facility using a FACSCalibur (BD Biosciences) cell sorter. Data were analyzed using WinMDI software.
Proliferation Assays
Cells were seeded in 6-well culture dishes at 2.5×104 cells using human endothelial serum-free medium (Gibco). Cell counts per cm2 were carried out 5 days after all experimental conditions.
Immunoblotting
Cell lysates were collected using RIPA buffer (Pierce) supplemented with Halt protease and phosphatase inhibitor cocktail (Pierce). Protein (20 mg) was resolved by SDS-PAGE and transferred onto immobilon-P-membranes (Millipore Corporation) then blocked with 5% dry milk in TBS-T (TBS (pH 7.6), 0.1% tween20). Primary antibodies against phospho-p70S6K, p70S6K (Cell Signaling Technology), VEGF-A165 (R&D Systems), HIF-1α (Santa Cruz Biotechnology); β-actin (Sigma-Aldrich) were used at a dilution of 1∶1000. HRP-conjugated IgG TrueBlot secondary antibodies (eBioscience) were used at a dilution of 1∶1000. Protein bands were visualized using an enhanced chemiluminescence detection system (Pierce).
Immunocytochemistry
Cells grown on glass cover slips were fixed and permeablized with cold acetone for 15 minutes then washed with PBS. Cells were blocked with 10% FBS mixed into a solution of 1% BSA for 1 hour at room temperature. HIF-1α antibodies (Santa Cruz Biotechnology) were used at a dilution of 1∶50 in a solution of 1% BSA for 2 hours at room temperature. Cells were then washed three times with PBS for 5 minutes each. AlexaFlour 488 IgG secondary antibodies (Invitrogen) were used at a dilution of 1∶200 in a solution of 1% BSA for 2 hours at room temperature. Cells were washed three times with PBS for 5 minutes each then allowed to completely dry. Vectashield (Vector Labs) fluorescent mounting medium containing DAPI was used when attaching the cover slips to glass slides. Images were acquired using a Nikon 80i fluorescent microscope.
Luminex Assays
Assays were performed using the Beadlyte Universal Cell Signaling Assay Kit and protocol (Millipore). VEGF-A165 (R&D Systems) and HIF-1α (Santa Cruz Biotechnology), and β-actin (Sigma-Aldrich) antibodies were conjugated to Bio-Plex carboxylated beads (BioRad) with unique optical codes using the Bio-Plex Amine Coupling Kit (BioRad). Cell lysates were collected using Beadlyte Cell Signaling Universal Lysis Buffer (Millipore). 25 µl of each lysate was added to wells of a 96-well filter plate, and mixed with 25 µl of each 1X bead type pre-conjugated with primary antibodies overnight shaking at 4°C. Lysates were discarded by vacuuming the buffer through the filter at the bottom of each well, while the beads remained in the wells. Beads were washed twice with Beadlyte Cell Signaling Universal Assay Buffer, followed by addition of biotinylated secondary antibodies for 1 hour at room temperature. The biotinylated reporter was then discarded followed by addition of streptavidin-PE for 15 minutes, then signal amplification buffer for 15 minutes. Amplification buffer was removed and beads were resuspended in Beadlyte Cell Signaling Universal Assay Buffer. The beads were then analyzed on a Luminex 200 multiplex testing system (Luminex). Beads were detected by two lasers; one determining the bead type (each conjugated with different antibodies) and the other determining the amount of fluorescence given off based on the amount of protein attached to each bead. VEGF and HIF-1 values were divided by the β-actin control values to provide normalized data.
Statistics
One-way analysis of variance (ANOVA) was performed and confirmed with two-tailed paired student’s t test using GraphPad Prism 4 software. P values less than 0.05 were considered significant.
Supporting Information
Elevated HIF-1α in hemangioma endothelial cells. Immunoblotting showing higher expression of HIF-1α in hemangioma endothelial cells (EC2, EC17B, EC21A) compared to control cells (HUVEC, HCMEC, HDMEC). β-actin was used as an internal control. The β-actin panel is excluded from this article's CC-BY license. See the accompanying retraction notice for more information.
(TIF)
Assessment of the effects of a second HIF-1α siRNA duplex. A and B: Luminex analyses of HIF-1α (A) and VEGF-A165 (B) protein levels showing that HIF-1α siRNA (2) reduces their expression in hemangioma endothelial cells (EC2, EC17B, EC21A). C: Proliferation assays quantifying total cell numbers in culture 5 days after treatment showing that HIF-1α siRNA (2) perturbs hyper-proliferation of hemangioma endothelial cells. Data represent mean (n = 3)±SD; *P<0.05 compared to control siRNA.
(TIF)
Proliferation assays confirming the results of BrdU incorporation experiments. A: Quantification of total cell numbers in culture after 5 days of treatment with HIF-1 siRNA showing that it inhibits proliferation of hemangioma endothelial cells. B: Total cell numbers of HDMEC 5 days after treatment with vehicle or VEGF-A165 in the presence of control siRNA or HIF-1α siRNA showing that VEGF-induced cell proliferation is partially dependent upon HIF-1 expression. C: Assessment of total cell numbers 5 days after transfection of HDMEC with pcDNA3 or pcDNA3-HIF-1α plasmids in the presence of non-specific IgG or VEGF-A165 neutralizing antibodies (VEGF Ab) showing that HIF-1α-induced proliferation is VEGF-dependent. D: Total cell numbers 5 days post treatment with vehicle or rapamycin showing that rapamycin successfully inhibits proliferation of hemangioma endothelial cells. Data represent mean (n = 3)±SD; *P<0.05.
(TIF)
Acknowledgments
We thank J. Bischoff (Children’s Hospital Boston) for providing primary cell cultures and E. Shipani (Indiana University School of Medicine) for providing the HIF-1α expression plasmid.
Funding Statement
This work was supported by a grant from the John Butler Mulliken Foundation to DM and grant P01-AR048564 from the National Institutes of Health to BRO. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Boye E, Yu Y, Paranya G, Mulliken JB, Olsen BR, et al. (2001) Clonality and altered behavior of endothelial cells from hemangiomas. J Clin Invest 107: 745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jinnin M, Medici D, Park L, Limaye N, Liu Y, et al. (2008) Suppressed NFAT-dependent VEGFR1 expression and constitutive VEGFR2 signaling in infantile hemangioma. Nat Med 14: 1236–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clauss M (1998) Functions of the VEGF receptor-1 (FLT-1) in the vasculature. Trends Cardiovasc Med 8: 241–245. [DOI] [PubMed] [Google Scholar]
- 4.Semenza GL (2000) HIF-1: mediator of physiological and pathophysiological responses to hypoxia. J Appl Physiol 88: 1474–1480. [DOI] [PubMed] [Google Scholar]
- 5.Manalo DJ, Rowan A, Lavole T, Natarajan L, Kelly BD, et al. (2005) Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 105: 659–669. [DOI] [PubMed] [Google Scholar]
- 6.Ke Q, Costa M (2006) Hypoxia-Inducible Factor-1 (HIF-1). Mol Phamacol 70: 1469–1480. [DOI] [PubMed] [Google Scholar]
- 7.Lee JW, Bae SH, Jeong JW, Kim SH, Kim KW (2004) Hypoxia-inducible factor (HIF-1)alpha: its protein stability and biological functions. Exp Mol Med 36: 1–12. [DOI] [PubMed] [Google Scholar]
- 8.Garcia-Meceira P, Mateo J (2009) Silibinin inhibits hypoxia-inducible factor-1alpha and mTOR/p70S6K/4E-BP1 signalling pathway in human cervical and hepatoma cancer cells: implications for anticancer therapy. Oncogene 28: 313–324. [DOI] [PubMed] [Google Scholar]
- 9.Bian CX, Shi Z, Meng Q, Jiang Y, Liu LZ, et al. (2010) P70S6K1 regulation of angiogenesis through VEGF and HIF-1alpha expression. Biochem Biophys Res Commun 398: 395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vignot S, Faivre S, Aguirre D, Raymond E (2005) mTOR-targeted therapy of cancer with rapamycin derivatives. Ann Oncol 16: 525–537. [DOI] [PubMed] [Google Scholar]
- 11.Cantley LC (2002) The phosphoinositide 3-kinase pathway. Science 296: 1655–1657. [DOI] [PubMed] [Google Scholar]
- 12.Wong KK, Engelman JA, Cantley LC (2010) Targeting the PI3K signaling pathway in cancer. Curr Opin Genet Dev 20: 87–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tee AR, Blenis J (2005) mTOR, translational control and human disease. Semin Cell Dev Biol 16: 29–37. [DOI] [PubMed] [Google Scholar]
- 14.Ballou LM, Lin RZ (2008) Rapamycin and mTOR kinase inhibitors. J Chem Biol 1: 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, et al. (1996) Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol 16: 4604–4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang N, Wang L, Esko J, Giordano FJ, Huang Y, et al. (2004) Loss of HIF-1alpha in endothelial cells disrupts a hypoxia-driven autocrine loop necessary for tumorigenesis. Cancer Cell 6: 485–495. [DOI] [PubMed] [Google Scholar]
- 17.Kleinmann ME, Greives MR, Churgin SS, Blechman KM, Chang EI, et al. (2007) Hypoxia-induced mediators of stem/progenitor cell trafficking are increased in childrem with hemangioma. Arterioscler Thromb Vasc Biol 27: 2664–2670. [DOI] [PubMed] [Google Scholar]
- 18.Straatman L, Coles J (2000) Pediatric utilization of rapamycin for severe cardiac allograft rejection. Transplantation 70: 541–543. [DOI] [PubMed] [Google Scholar]
- 19.Oellerich M, Armstrong VW, Streit F, Weber L, Tonshoff B (2004) Immunosuppressive drug monitoring of sirolimus and cyclosporine in pediatric patients. Clin Biochem 37: 424–428. [DOI] [PubMed] [Google Scholar]
- 20.Koenig MK, Northrup H (2009) Topical rapamycin therapy to alleviate cutaneous manifestations of tuberous sclerosis complex (TSC) and neurofibromatosis (NF1). ClinicalTrials.gov NCT01031901.
- 21.Hamilton HK, Tonkovic-Capin V (2011) Images in clinical medicine. Facial angiofibromas associated with tuberous sclerosis. 364: 1061. [DOI] [PubMed] [Google Scholar]
- 22.Medici D, Shore EM, Lounev VY, Kaplan FS, Kalluri R, et al. (2010) Conversion of vascular endothelial cells into multipotent stem-like cells. Nat Med 16: 1400–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Elevated HIF-1α in hemangioma endothelial cells. Immunoblotting showing higher expression of HIF-1α in hemangioma endothelial cells (EC2, EC17B, EC21A) compared to control cells (HUVEC, HCMEC, HDMEC). β-actin was used as an internal control. The β-actin panel is excluded from this article's CC-BY license. See the accompanying retraction notice for more information.
(TIF)
Assessment of the effects of a second HIF-1α siRNA duplex. A and B: Luminex analyses of HIF-1α (A) and VEGF-A165 (B) protein levels showing that HIF-1α siRNA (2) reduces their expression in hemangioma endothelial cells (EC2, EC17B, EC21A). C: Proliferation assays quantifying total cell numbers in culture 5 days after treatment showing that HIF-1α siRNA (2) perturbs hyper-proliferation of hemangioma endothelial cells. Data represent mean (n = 3)±SD; *P<0.05 compared to control siRNA.
(TIF)
Proliferation assays confirming the results of BrdU incorporation experiments. A: Quantification of total cell numbers in culture after 5 days of treatment with HIF-1 siRNA showing that it inhibits proliferation of hemangioma endothelial cells. B: Total cell numbers of HDMEC 5 days after treatment with vehicle or VEGF-A165 in the presence of control siRNA or HIF-1α siRNA showing that VEGF-induced cell proliferation is partially dependent upon HIF-1 expression. C: Assessment of total cell numbers 5 days after transfection of HDMEC with pcDNA3 or pcDNA3-HIF-1α plasmids in the presence of non-specific IgG or VEGF-A165 neutralizing antibodies (VEGF Ab) showing that HIF-1α-induced proliferation is VEGF-dependent. D: Total cell numbers 5 days post treatment with vehicle or rapamycin showing that rapamycin successfully inhibits proliferation of hemangioma endothelial cells. Data represent mean (n = 3)±SD; *P<0.05.
(TIF)