

Abstract
Earlier, we reported on the design of sulfated benzofuran dimers (SBDs) as allosteric inhibitors of thrombin (Sidhu et al. (2011) J Med Chem 54: 5522-5531). To identify the site of binding of SBDs, we studied thrombin inhibition in the presence of exosite 1 and 2 ligands. Whereas hirudin peptide and heparin octasaccharide did not affect the IC50 of thrombin inhibition by a high affinity SBD, the presence of full-length heparin reduced inhibition potency by 4-fold. The presence of γ’ fibrinogen peptide, which recognizes Arg93, Arg97, Arg173, Arg175 and other residues, resulted in a loss of affinity that correlated with the ideal Dixon-Webb competitive profile. Replacement of several arginines and lysines of exosite 2 with alanine did not affect thrombin inhibition potency, except for Arg173, which displayed a 22-fold reduction in IC50. Docking studies suggested a hydrophobic patch around Arg173 as a plausible site of SBD binding to thrombin. Absence of Arg173-like residue in factor Xa supported the observed selectivity of inhibition by SBDs. Cellular toxicity studies indicated that SBDs are essentially non-toxic to cells at concentrations as high as 250 mg/kg. Overall, the work presents the localization of the SBD binding site, which could lead to allosteric modulators of thrombin that are completely different from all clinically used anticoagulants.
INTRODUCTION
The coagulation cascade is a rich assembly of homologous serine proteases. Each enzymatic factor of the cascade recognizes a P-1 arginine residue in its target, which sets up possible cross-over reactivity with enzymes of other systems too. Nature avoids these cross-over reactivities, especially of macromolecular substrates, through clever engineering of the environment around the enzyme active sites. For example, thrombin contains the 60-insertion loop, the 149-insertion loop and the bulky Trp215 residue to restrict access to its active site.1-3 Such stringent steric and/or electronic natural ‘gating’ assists in the design of small molecule, active site inhibitors. In fact, a number of scaffolds that selectively inhibit thrombin have been designed, such the pyrazole, napthylamidine, or benzimidazole scaffolds.4-7 Yet, the process remains challenging and is threatened by cross-reactivity with closely related enzymes, e.g., factor Xa, or with enzymes that exhibit too broad substrate specificity, e.g., trypsin. A significant advance in the design of thrombin inhibitors was the use of hydrophobic P-1 substituents, e.g., a halophenyl group, that interact with the Tyr228 in the S-1 pocket.8-10 This strategy has also been exploited in the design of rivaroxaban, a non-amidine or guanidine based factor Xa inhibitor, approved for clinical use in the EU.11
In addition to steric or electronic ‘gating’, an alternative strategy that nature exploits for engineering high selectivity is the use of exosites. A classic example of this phenomenon is thrombin cleavage of fibrinogen. In this process, the substrate binds to exosite 1 that enables its efficient cleavage.1-3,12 Likewise, binding of full-length heparin in exosite 2 enables a much faster inhibition of thrombin by antithrombin – heparin complex.13,14 In addition to this exquisite dual recognition, exosites afford a fine opportunity of allosteric modulation of catalytic machinery. Both exosites 1 and 2 of thrombin as well as exosites of many other coagulation enzymes are coupled with the active site. Although the intricate mechanism of this coupling is not fully understood, it is known that it may involve alteration of structure of catalytic triad and/or of neighboring residues. For example, sodium binding is known to allosterically alter the conformation of the catalytic triad.15 Likewise, heparin binding in exosite 2 is known to change the electrostatics around the active site with minimal change in catalytic activity.16-18
Although allosteric modulation of thrombin’s catalytic function by macromolecules is well established, its exploitation in the design of drug-like molecules is still in its infancy. Allosteric regulation is likely to offer a delicate control over thrombin’s procoagulant activity, which is difficult to achieve with competitive, active site inhibitors because of the drive to achieve very high potency. At a fundamental level, small molecules may be designed so that the allosteric conformational change can exhibit tailored balance between pro- and anti- coagulant activities. Another advantage of allosteric regulation is the possibility of greater specificity of recognition arising from greater differences in exosite geometries as compared to active sites. In effect, allosteric regulation promises to afford exquisite control over both specificity of recognition and efficacy of inhibition.
To develop such regulators of thrombin, we started with the design of sulfated low molecular weight lignins (LMWLs), which were found to potently inhibit thrombin, factor Xa and plasmin.19-22 The oligomeric molecules targeted exosite 2 of thrombin exclusively and were the first molecules in the class of exclusive exosite 2-based allosteric modulators of thrombin.20 Sulfated LMWLs prevented human blood from clotting in ex vivo assays with potency comparable to low molecular weight heparins (LMWHs) and thus represented molecules of considerable interest.21 Yet, the oligomeric nature of these molecules did not bode well for clinically potential. To address this issue, we designed sulfated benzofuran monomers from sulfated LMWLs, which were found to allosterically inhibit thrombin and factor Xa, albeit with poor potency (IC50 in mM range).23 Subsequent advance in the design led to sulfated benzofuran dimers (SBDs) that exhibited a wide range of inhibition potencies (μM to mM) and efficacies (20 – 60%).24 Michaelis-Menten kinetic studies revealed that the SBDs (Figure 1), reduced the maximal velocity of substrate hydrolysis without affecting the substrate’s Michaelis constant (KM), a classic case of non-competitive allosteric modulation of thrombin activity.24
Figure 1.
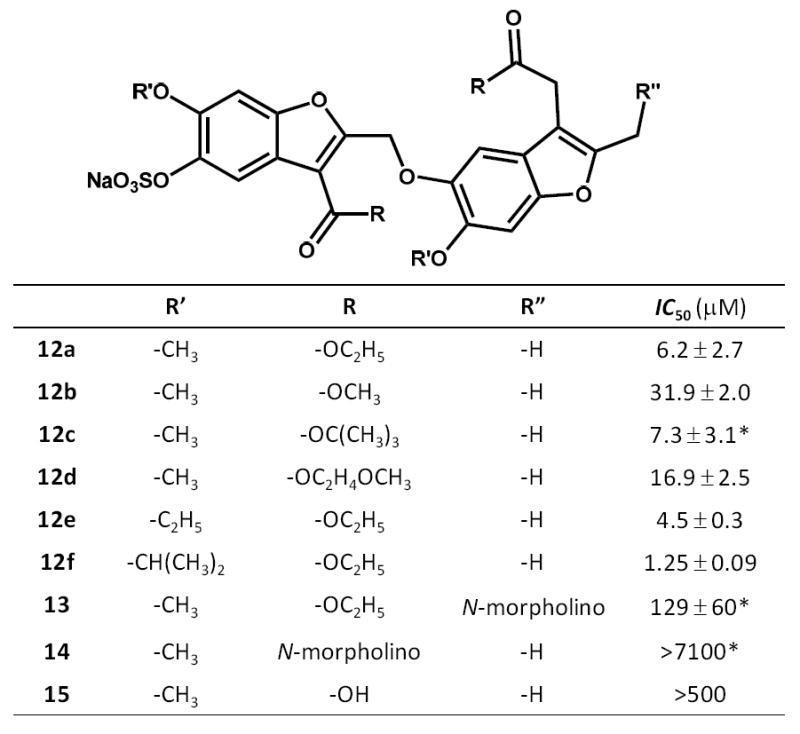
Structure of sulfated benzofuran dimers 12a-f and 13-15 found to potently inhibit human thrombin. The selected dimers correspond to those identified in our previous work24 and their labels (12a-12f and 13-15) have been retained here to maintain consistency and ease of comparison. Most IC50 values being reported here are lower than those reported earlier24 and possibly arise from differences in glycosylation pattern of human α-thrombin. Asterisk corresponds to IC50 values taken from the earlier report.24
In this work, we present detailed biochemical studies on the interaction of one of the potent SBDs with thrombin with a goal of identifying the site of binding for further rational structure-based drug design. Our work reveals that the SBD interacts with exosite 2 of thrombin but in a manner dramatically different from all known exosite 2 ligands including full-length heparin, heparin octasaccharide, sucrose octasulfate, γ’-fibrinogen peptide, and sulfated LMWLs. The localization of the binding region on thrombin, the allosteric nature of inhibition and the cellular toxicity profile suggests that clinically relevant, allosteric modulation of thrombin based on the SBD scaffold may be achievable.
Experimental Procedures
SBD 12a, Thrombin, Fibrinogen Peptide, Chemicals and Reagents
SBD 12a was synthesized as described in our earlier report24 and stored at -80 °C in solid form until use. Human α-thrombin and factor Xa were from Haematologic Technologies (Essex Junction, VT). Stock solutions of the enzymes were prepared in 20 mM sodium phosphate buffer, pH 7.4, containing 100 mM NaCl. Chromogenic substrate Spectrozyme TH (H-d-hexahydrotyrosol-Ala-Arg-p-nitroanilide) was purchased from American Diagnostica (Greenwich, CT). A peptide corresponding to the 20 amino acids at the carboxyl terminal of γ’-fibrinogen chain (VRPEHPAETE-Y(PO3)-DSL-Y(PO3)-PEDDL) was obtained from Dr. David Farrell.25 All other chemicals were analytical reagent grade from either Sigma Chemicals (St. Louis, MO) or Fisher (Pittsburgh, PA) and used as such. Tyr63-sulfated hirudin-(54-65), labeled as HirP in this work and labeled as 5-(carboxy)fluorescein ([5F]-Hir[54-65](SO3-)) elsewhere, was a gift from Dr. Paul Bock.26
Recombinant Thrombin Mutants
Recombinant wild-type and mutant thrombins were prepared in the Rezaie laboratory, as described earlier.27,28 Briefly, Lys169Ala, Arg173Ala, Arg175Ala, Lys235Ala, or Arg93,97,101Ala thrombin was prepared in prothrombin-1 form by PCR mutagenesis and expression in baby hamster kidney cells (BHK) using the pNUT-PL2 expression/purification vector system. The mutants were purified to homogeneity by immunoaffinity chromatography using the Ca2+-dependent monoclonal antibody, HPC4, and activated to thrombin. The active-site concentrations of thrombin mutants were determined by an amidolytic acivity assay and stoichiometric titrations with antithrombin.27,28 These concentrations were within 90-100% of those expected on the basis of their absorbance at 280 nm.
Competitive Studies with Exosite 1 and Exosite 2 ligands
The inhibition effect of sulfated benzofuran diethyl ester dimer (12a) on wild-type thrombin was studied in the presence of exosite 1 ligand, a twelve residue hirudin peptide containing fluorescein at its N-terminus, i.e., [5F]-Hir[54-65](SO3-)20,26 and abbreviated in this paper as HirP. Inhibition experiments were also performed in the presence of exosite 2 ligands, heparin octasaccharide H8, bovine heparin (UFH) and γ’-fibrinogen peptide (FibP) in a manner similar to that described above for direct thrombin inhibition. Briefly, a solution of 10 μL of the dimer (0–1000 μM) and 10 μL thrombin (4 – 10 nM) was mixed at 25 °C with 10 μL of the competitor at appropriate stock concentration in either 20 mM Tris-HCl buffer, pH 7.4, containing 100 mM NaCl, 2.5 mM CaCl2 and 0.1 % PEG 8000 or 50 mM Tris-HCl buffer, pH 7.4 containing 150 mM NaCl, 0.1 % PEG 8000 and 0.02% Tween 80. The solution was incubated for 10 min followed by addition of Spectrozyme TH and monitoring the increase in absorbance at 405 nm. The dose-dependence of the fractional residual thrombin activity at each concentration of the competitor was fitted using equation 1 to obtain the apparent concentration of the dimer required to inhibit thrombin activity by 50% (IC50,app) and the efficacy of inhibition (ΔY = YM − Y0).
| Eq. 1 |
In this equation, Y is the ratio of residual thrombin activity in the presence of inhibitor to that in its absence (fractional residual activity), YM and YO are the maximum and minimum possible values of the fractional residual proteinase activity, IC50 is the concentration of the inhibitor that results in 50% inhibition of enzyme activity, and HS is the Hill Slope. A current version of SigmaPlot (SPSS, Inc. Chicago, IL) was used to perform non-linear curve fitting in which IC50, YM, YO and HS were allowed to float.
Inhibition of Recombinant Thrombin Mutants and Factor Xa
Direct inhibition of thrombin mutants and factor Xa by sulfated benzofuran derivatives was measured through a chromogenic substrate hydrolysis assay, as reported earlier.19,20 The buffer used in these experiments was 20 mM Tris-HCl buffer, pH 7.4, containing 100 mM NaCl, 2.5 mM CaCl2, and 0.1% polyethylene glycol (PEG) 8000. Sulfated benzofuran derivatives (2 to 30 μL) at concentrations ranging from 0.25 to 12.5 mM were diluted with appropriate volume of assay buffer in PEG 20,000-coated acrylic cuvettes at 25 °C. To this solution, 5 μL of thrombin solution was added to give approximately 5 nM thrombin or 4 nM factor Xa final concentration. Following addition of thrombin, 20 μL (thrombin) or 100 μL (factor Xa) of 1 mM Spectrozyme TH was added after 10 min incubation. The residual thrombin activity was then measured from the initial rate of increase in absorbance at 405 nm. Relative residual thrombin activity at each concentration of the inhibitor was calculated from the ratio of thrombin activity in the presence and absence of inhibitor. Logistic equation 1 was used to fit the dose-dependence of residual proteinase activity to obtain the apparent IC50, HS and the efficacy ΔY (= YM − Y0) of inhibition.
Molecular Modeling Studies
The crystal structure of thrombin bound to its allosteric exosite 1 ligand hirugen at 1.53 Å resolution was obtained from the Protein Data Bank (PDB ID = 3EQ0), as well as two additional thrombin structures (PDB IDs = 1H8D and 2UUF).29,30 All the residues in region of Arg173 were manually inspected to ensure that the side chains were completely resolved. Each of the acidic and basic residues was modeled in its most predominant ionization state at physiological pH (i.e. anionic Asp and Glu; cationic Arg and Lys). The inorganic salts and water molecules were deleted from the crystal structure, and hydrogen atoms were added using SYBYL 8.1 (Tripos International, St. Louis, MO). To prepare the structure for docking and scoring protocol, the newly-added hydrogen atoms were energy-minimized using the Tripos Force Field (Gasteiger–Hückel charges; termination criterion of 0.05 kcal/(mol×Å) or 1 × 105 iterations; distance-dependent dielectric constant ε = 4.0).
Substituted SBDs 12a–f and 13–15, each synthesized and studied previously,24 were prepared in SYBYL for virtual library screening. Using in-house SYBYL Programming Language (SPL) scripts, compounds contained in the resulting virtual library were post-processed to (a) assign appropriate atom types to the sulfate groups (sulfur atom = S.o2; terminal oxygen atoms = O.co2), (b) assign an initial set of 3D coordinates using the CONCORD module within SYBYL, (c) add missing hydrogen atoms and (d) energy-minimize the resulting structures using the Tripos Force Field as described for the protein (vide supra).
Docking of the synthesized inhibitors onto the defined binding site of the thrombin was performed using GOLD 4.1 (Cambridge Crystallographic Data Centre, Cambridge, UK). The binding site was defined to include all atoms within 22 Å around the Arg173 Cα atom. Default parameters were employed during the GOLD docking runs with the following exceptions: A protein hydrogen bonding constraint was added such that the score was reduced by 10.0 GoldScore units if the ligand did not form an H-bond with the guanidinium group of Arg173. Amide bonds were allowed to flip. The number of GA runs was increased to 30 to more accurately screen all possible binding geometries, and early termination was disabled. The docking was driven by the GoldScore fitness function and the docked solutions were ranked based on the unmodified GoldScore obtained. To assess the reproducibility of the docked poses, the docking runs were performed in triplicate.
Cellular Toxicity Studies
Human A549 cells were obtained from American Type Culture Collection (ATCC) and maintained in growth medium at 37 °C in 5% CO2 atmosphere. The growth medium was Dulbecco’s Modified Eagle Medium (Invitrogen, CA) supplemented with 10% heat-inactivated fetal bovine serum enriched with 2 mM L-glutamine, 0.1 mM nonessential amino acids, 25 mM HEPES buffer, 50 U/mL penicillin, and 50 μg/mL streptomycin. Stock solutions of 12a were prepared in deionized water and stored at -20 °C. The MTT solution was prepared at 5 mg/mL in 50 mM phosphate buffered saline, pH 7.4, filtered, and stored at 4 °C. The MTT lysis buffer was prepared by dissolving 25 g of SDS in 100 mL of 50% DMF in water, and the pH was adjusted to 4.7 with a solution of 2.5% HCl in 80% acetic acid. The MTT cell viability assay was carried out as described earlier.31,32 Briefly, A549 cells were seeded at 30,000 cells/well on a 96-well plate for 4 h before the treatment with either medium or SBDs. After incubation for 72 h, the MTT solution (10 μL) was added to each well followed by incubation for 10 h. The medium was then replaced with the lysis buffer (100 μL). After 10 h, the A570 was read with a 318C-plate reader (Shanghai Sanke Instrumental Corp. Ltd., China). Background correction was performed using A690 values. Statistical analyses (oneway ANOVA with Dunnett’s test) were performed by GraphPad Prism (version 4.00, GraphPad Software, San Diego, CA). Each SBD concentration was tested at least three times.
RESULTS
SBDs Do Not Bind in the Anion-Binding Exosite 1 of Thrombin
To test whether SBDs bind in exosite 1 of thrombin, we selected 12a (Figure 1), a potent SBD designed in our earlier work.24 Structurally, 12a is a monosulfated benzofuran dimer containing two ethyl ester groups and is a prototypical SBD. This scaffold is dramatically different from the thousands of thrombin inhibitors being studied for improved pharmacological and safety profile.4-11 It is the only small molecule scaffold that has been found to allosterically inhibit thrombin.24 Interestingly, the unsulfated precursor of 12a was completely inactive suggesting that both the anionic group and the hydrophobic backbone are important for thrombin inhibition.24 Considering that anion-binding exosites 1 and 2 of thrombin recognize charged ligands, SBDs were hypothesized to induce inhibition through one of these sites. In fact, sulfated tyrosine containing hirudin, a 65 amino acid long polypeptide that binds in exosite 1 as well as active site, is known to be the most potent thrombin inhibitor.33 On the other hand, exosite 2 of thrombin prefers highly sulfated oligosaccharides.1,34 The anionic density of these highly sulfated oligosaccharides is far greater than that of molecules recognizing exosite 1. A priori, this indicated that SBDs may prefer to interact with exosite 1 of thrombin.
To test with 12a binds in exosite 1, its inhibition potency was measured in the presence of a hirudin-based peptide [5F]-Hir[54-65](SO3-), abbreviated as HirP, a molecule known to engage exosite 1 of thrombin. HirP binds with a dissociation constant of 28 nM under similar conditions26,35 and its presence was expected to significantly reduce the potency of 12a, if the two molecules competed for the same binding site. Our earlier work on the effect of HirP on thrombin’s hydrolysis of Spectrozyme TH had shown that the peptide did not affect the Michaelis constant of the substrate, while increasing the catalytic efficiency of the enzyme nearly 50%.20 This implied that the two molecules, HirP and 12a, were expected to induce opposing catalytic influence (VMAX effect), which afforded a convenient setup to study exosite 1 competition.
The IC50 was measured by quantifying thrombin’s hydrolysis of Spectrozyme TH in the presence of fixed concentration of HirP at pH 7.4 and 25 °C, as described earlier.20 A standard sigmoidal dose-response profile was observed for 12a inhibition of thrombin at all concentrations of HirP, which could be fitted by the logistic equation 1 to obtain the potency (IC50) and efficacy (ΔY = YM − Y0) of inhibition (Figure 2). In the absence of HirP, 12a inhibited thrombin with an IC50 of 6.2 ± 2.7 μM. In our earlier work, the IC50 was found to be 61 μM.24 The primary difference between our two studies was a new batch of human plasma α-thrombin. The change also resulted in a slightly lower efficacy of inhibition (from 70% to 50%) and an increased Hill slope (from ~1.2 to ~2.5, see Table S1 in Supplementary Material). We also tested whether the ability of other SBDs to inhibit thrombin is similarly affected and found a significant decrease in the IC50 of majority of inhibitors (Figure 1 and Supplementary Material Figure S1). It is possible that commercial human plasma α-thrombin contains minor proportions of forms other than just α-thrombin, e.g., differentially glycosylated or partially lysed, resulting in altered inhibition potential of 12a and other SBDs. The difference in Hill slope could also arise from changes in glycosylation pattern of the protein. However, the substantial Hill slope of 2.5 raised a possibility that SBDs were aggregating under the assay conditions. Yet, the UV-Vis absorption profile of 12a showed a red shift of 40–50 nm only at a concentration 100-fold the IC50 value (not shown) suggesting that aggregation is not likely to be significant under the assay conditions. Thus, for the purpose of identifying changes in the inhibition potency as a function of the competitor concentration, the IC50 in the absence of 12a served as a reasonable reference point.
Figure 2.
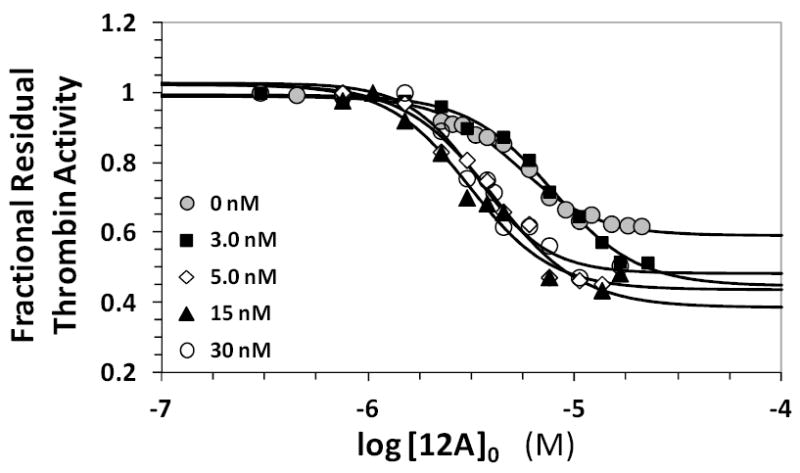
Competitive effect of the hirudin peptide HirP on the inhibition of human plasma thrombin by 12a. Residual thrombin activity was measured through Spectrozyme TH hydrolysis in 20 mM Tris-HCl buffer, pH 7.4, containing 100 mM NaCl, 2.5 mM CaCl2 and 0.1 % polyethylene glycol (PEG) 8000 at 25 °C in the presence of 0 to 103 nM of HirP. Solid lines represent fits using the logistic equation 1 to obtain the apparent IC50, as described in ‘Experimental Methods’.
Varying the concentration of HirP from 0 to 30 nM resulted in the no apparent change in IC50 (Figure 2 & Table S1). The efficacy of inhibition increased slightly with increasing concentrations of HirP (5–10%), which may imply a coupling between exosite 1 and the SBD binding site. Together the two results suggest that HirP and 12a do not directly compete with each other, but the presence of HirP makes 12a a slightly better inhibitor of thrombin. Thus, 12a does not appear to bind in anion-binding exosite 1 of thrombin.
SBD 12a Does Not Compete with Heparin Octasaccharide For Binding in Exosite 2
Highly sulfated polysaccharide chains including heparin octasaccharide H8, low-molecular weight heparin, full-length heparin, and chondroitin sulfate utilize anion-binding exosite 2 to bind to thrombin.1-3 The major difference in how these polysaccharide ligands recognize thrombin is their span of interaction domain. Whereas H8 recognizes primarily Arg233, Lys235, Lys236 and Lys240, the full-length polymer recognizes in addition Arg93, Arg101, and Arg165 residues. None of these ligands affect the proteolytic activity of thrombin (not shown), which implies that inhibition assays could be used for exosite 2 competitive studies. Figure 3A shows the dose-response curves of 12a inhibiting thrombin in the presence of H8 at pH 7.4 and 25 °C. As the concentration of H8 was increased from 0 to 10 μM, the IC50 of thrombin inhibition increased from 2.7 to 3.7 μM (Table S1). This inconsequential change in potency was accompanied by no noticeable change in the efficacy of inhibition in the presence of H8. Thus, H8 does not compete with 12a for binding to thrombin suggesting that the sulfated benzofuran does not recognize Arg233, Lys235, Lys236, or Lys240. This is not too unexpected considering the widely different structural features of the two molecules. H8 is a highly anionic polysaccharide, while 12a is a fairly hydrophobic small molecule with one negative charge. But interestingly, the results suggest that while exosite 1 and 12a binding site appear to be slightly coupled (described above), similar increase in efficacy was not detected in H8 and 12a competition experiments.
Figure 3.
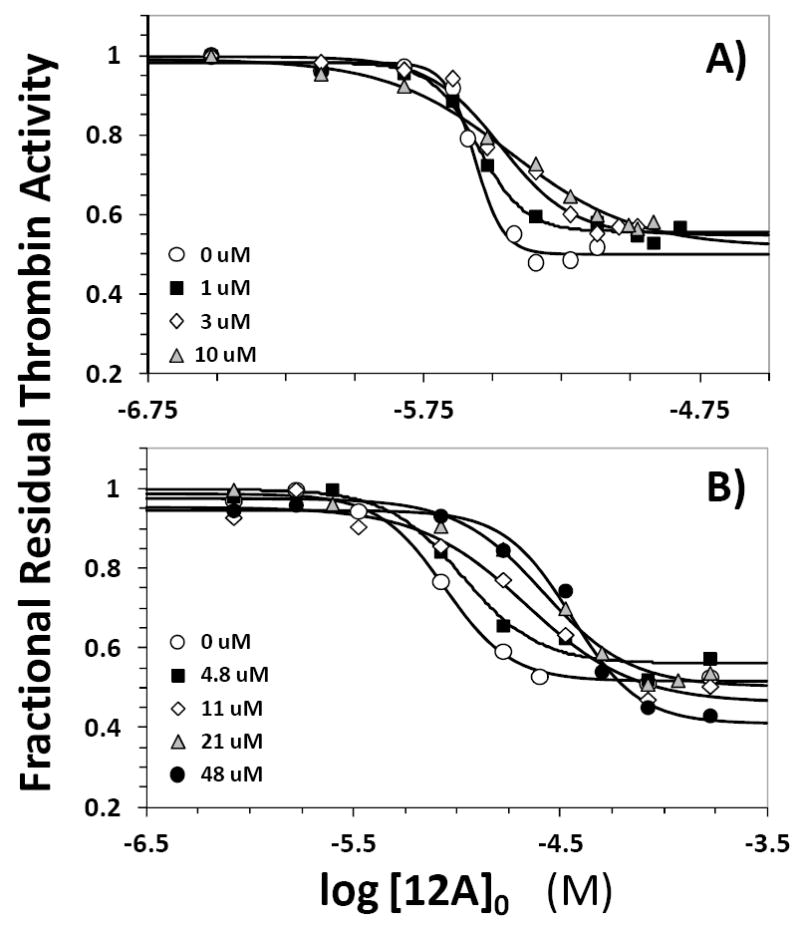
Competitive effect of heparin octasaccharide H8 (A) and unfractionated heparin UFH (B) on the inhibition of human plasma thrombin by 12a in 20 mM Tris-HCl buffer, pH 7.4, containing 100 mM NaCl, 2.5 mM CaCl2 and 0.1 % polyethylene glycol (PEG) 8000 at 25 °C in the presence of 0 to 23 μM H8 and 0 to 48 μM UFH. Solid lines represent fits using the logistic equation 1 to obtain the apparent IC50, as described in ‘Experimental Methods’.
SDB 12a Competes Partially with Full-length Heparin
To assess whether 12a interacts with other exosite 2 residues, e.g., Arg93, Arg101, and Arg165, we studied competition with the full-length heparin. Figure 3B shows the inhibition profile in the presence of 0 to 48 μM UFH. In contrast to H8, the sigmoidal dose-response profiles displayed a slight shift to the right with increasing concentration of UFH suggesting a noticeable competitive phenomenon. The IC50 of 12a inhibition of thrombin at pH 7.4 and 25 °C was found to increase from 8.8 μM in the absence of UFH to 36.1 μM in the presence of 48 μM UFH (Table S1). This represents a maximal decrease in potency of 4.1-fold at a UFH concentration ~5-times higher than the KD of thrombin–UFH complex.34 This suggests that although UFH competes with SBD, the competition is inefficient or partial.
SBD 12a Competes with γ’-Fibrinogen Peptide for Binding to Thrombin
The absence of competition for binding to thrombin with H8 eliminated Lys236 and Lys240 as site of interaction for 12a. Yet, partial competition observed with UFH suggested recognition of one or more electropositive residues of exosite 2, which is a fairly extensive domain with a large number of basic residues. To further narrow the domain of recognition, competition with γ’-fibrinogen peptide (FibP) was studied. FibP is known to bind in exosite 2 by interacting with residues that bind to H8 (Arg126, Lys235, Lys236 and Lys240),36 while also interacting with Arg93, Arg97, Arg173 and Arg175 as demonstrated by NMR studies.37 This makes HirP a useful probe for studying interaction with this group of arginines that lie beyond the H8 binding site. Figure 4A shows the dose-response profiles of 12a inhibition of thrombin in the presence of 0 to 6.5 μM FibP. A distinct shift in the inhibition profiles was observed as the concentration of FibP increased suggesting strong competition between the two ligands for thrombin binding. The apparent IC50 increased from 7.9 μM in the absence to 118 μM in the presence of 6.5 μM FibP (Table S1).
Figure 4.
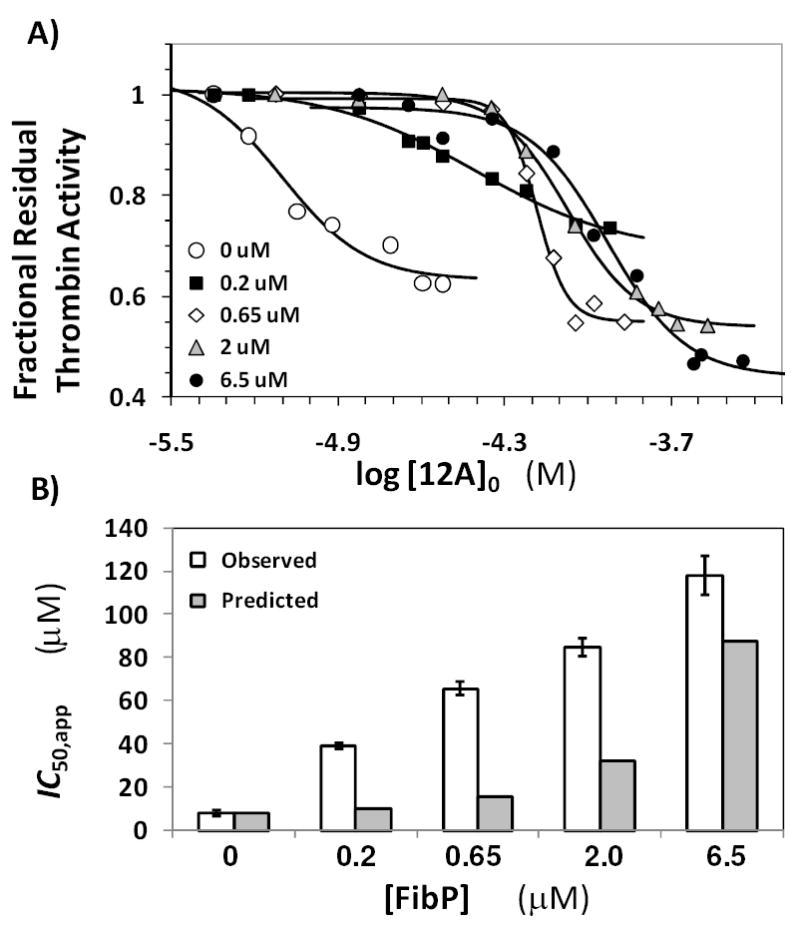
(A) Competitive effect of the γ’-fibrinogen peptide on the inhibition of human plasma thrombin by 12a in 20 mM Tris-HCl buffer, pH 7.4, containing 100 mM NaCl, 2.5 mM CaCl2 and 0.1 % polyethylene glycol (PEG) 8000 at 25 °C in the presence of 0 to 6.5 μM of the peptide. Solid lines represent fits using the logistic equation 1 to obtain the apparent IC50, as described in ‘Experimental Procedures’. (B) Comparison of the predicted (shaded bars) and observed (unshaded bars) IC50,app. Error bars represent ±2 S.E.
A more quantitative test of competitive binding is the Dixon-Webb relationship (Eq. II), which predicts the effect of competition on a measured equilibrium parameter (KD or IC50). In this equation, KFibP is the dissociation constant of thrombin–FibP complex, which was earlier measured to be 0.63 μM under similar conditions.37
| Eq. II |
The IC50,app predicted using equation II for 12a inhibition of thrombin in the presence of varying concentrations of FibP are listed in Table S2 and Figure 4B shows a comparison of the observed and predicted IC50,app. The measured IC50,app is consistently higher than those predicted for FibP competition suggesting a strong competitive effect. The reason for the more-than-predicted competitive effect of FibP is not clear, however it is possible that a small change in the affinity of FibP for thrombin under the current experimental conditions is the cause. Yet overall, competition with FibP indicates that 12a binds in exosite 2 in a region away from the H8 binding residues and most probably with one or more of Arg93, Arg97, Arg173 and Arg175.
Sulfated Benzofuran 12a Interacts with a Single Arginine, Arg173, of Exosite 2
To identify the basic residues that might play an important role in the dimer recognition, we studied the inhibition of five thrombin mutants containing single and triple replacement of Arg and/or Lys to Ala. The preparation of these recombinant thrombins has been described earlier27,28 and each mutant was screened for direct inhibition by 12a in a manner similar to that used for the wild-type form. The dose – response profiles of three of the four single point mutants studied in this work were essentially identical to the recombinant wild type enzyme (Figure 5). The IC50 measured for Arg175Ala, Lys169Ala, and Lys235Ala thrombins ranged from 4.9 μM to 9.0 μM, while that measured for the recombinant wild-type enzyme was 5.5 μM (Table S2). The triple point mutant Arg93,97,101Ala had a comparable IC50 of 5.0 μM suggesting none of the three amino acids contribute to 12a binding. This was quite different from our results with macromolecular sulfated LMWLs, which primarily relied on Arg93 and Arg175 for mediating their inhibitory role.38 Although the above mentioned basic residues do not contribute to 12a recognition, the site-directed thrombin mutants were inhibited more than the wild-type enzyme. For example, the efficacy of 12a inhibition (i.e., ΔY) increased to ~70% for the triple mutant in comparison to ~30% for wild-type recombinant enzyme (see Table 2).
Figure 5.
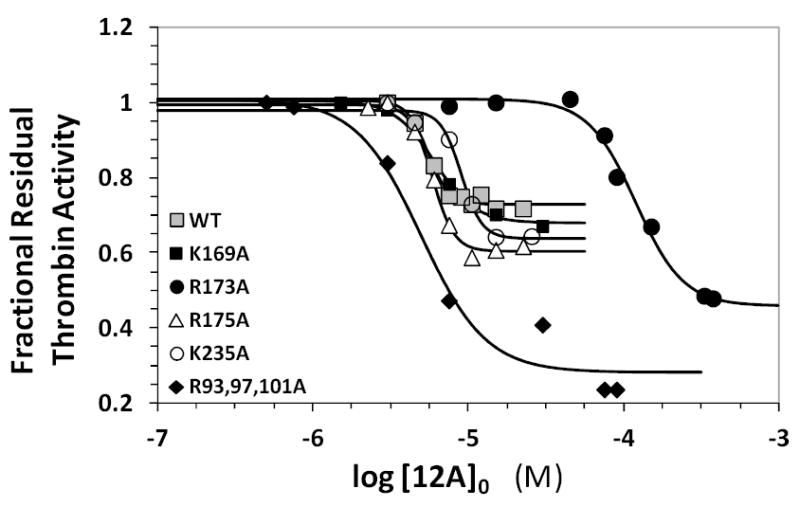
Dose-response profiles for 12a inhibition of recombinant wild type and mutant thrombins in 20 mM Tris-HCl buffer, pH 7.4, containing 100 mM NaCl, 2.5 mM CaCl2 and 0.1 % polyethylene glycol (PEG) 8000 at 25 °C. Solid lines represent sigmoidal dose-response fits of equation 1.
One particular thrombin mutant, Arg173Ala, was dramatically different. It displayed a 22-fold increase in IC50 compared to the wild-type enzyme (Figure 5, Table S2). This suggested that Arg173 was essential for thrombin binding and inhibition by 12a.24 It is striking that a single residue, of the 12 electropositive residues present in exosite 2 of thrombin, was found to be critical for 12a interaction. This explains the critical role of the 5-sulfate, observed earlier,24 in mediating the inhibitory effect of all SBDs. Further, 12a inhibition of human factor Xa was found to be impaired by at least an order of magnitude (IC50,fXa >300 μM, data not shown). This result is readily explainable considering that residue 173 in factor Xa is a serine, instead of an arginine. Likewise, factor VIIa contains a serine at position 173 and was also not targeted by 12a (data not shown). In combination, the results indicate a high level of specificity that supports a strong possibility of developing allosteric modulators.
Molecular Docking Identifies a Single Plausible Binding Geometry for 12a on Thrombin
To identify a plausible binding geometry of sulfated benzofuran dimers on thrombin, we employed a molecular docking and scoring approach. We have previously utilized a genetic algorithm (GA)-based strategy relying on the automated docking routine GOLD to understand how highly sulfated oligosaccharides recognize proteins.39,40 This approach was also exploited to study the recognition of antithrombin by sulfated LMWLs,41 which are the parent molecules for the SBDs being studied here. Hence, we chose to employ GOLD-based identification of plausible binding site and geometry of 12a on human plasma thrombin.
The GOLD-based docking and scoring approach utilizes a GA to iteratively derive the best binding geometry for each ligand in a pre-defined binding site. Our recent work on the recognition of sulfated LMWLs binding to thrombin mutants revealed that hydrophobic patches in the vicinity of Arg93 may be involved.38 Considering this, we hypothesized that hydrophobic region(s) near Arg173, which has been implicated by the Arg173Ala thrombin mutant studies presented here, would be important for recognition of 12a and related SBDs. Several GA docking runs were thus performed to assess how well 12a could be recognized by the binding site regions surrounding Arg173. Each GA docking run was performed using a constraint that encouraged the guanidine group of Arg173 to hydrogen bond with the sulfate group of SBD (see Experimental Methods). GOLD-based docking suggested that the region around Arg173 of thrombin was strikingly complementary to 12a. In this site, 12a bound in an extended, flat conformation. Supporting this interaction is the hydrophobic area of thrombin located between Arg173 and its active site (Figure 6), which recognizes 12a’s benzofuran rings and their substituents (see Supplemental Figures S2 and S3). To cross-check whether the localization remains independent of thrombin’s crystal structure, we performed GOLD-based docking studies on 3EQ0, 1H8D and 2UUF29,30 structures that had been acquired with high resolution (1.5–2.5 Å) and displayed well-defined side-chains around Arg173. In each of these docking runs, 12a bound in an essentially identical orientation.
Figure 6.
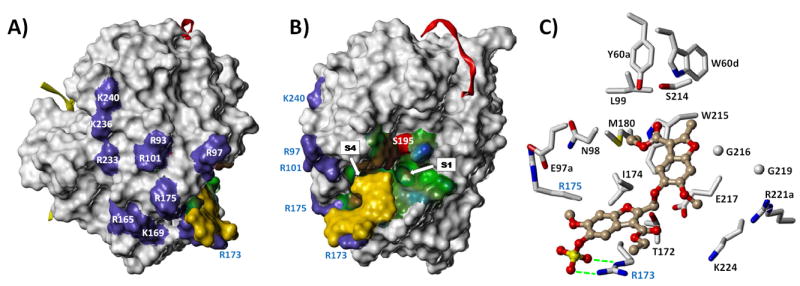
A) Connolly surface of thrombin (PDB ID = 3EQ0), showing the location of several electropositive basic residues, including those contributing to exosite-II and the γ‘-fibrinogen peptide binding site (purple patches). The putative binding site and mode of the lead sulfated benzofuran dimer 12a is rendered as a yellow surface and shows the high degree of shape complementarity with its putative binding site. The thrombin light chain (yellow ribbon) and hirudin peptide denoting the location of exosite-1 (red ribbon) are also shown. B) The same 3D image rotated by 90°, further highlighting the high degree of shape complementarity between the ligand dimer and its proposed binding site. The active site is displayed with a hydropathic potential map (blue = hydrophilic, green = amphipathic, brown = hydrophobic) with the nucleophilic serine S195 shown as a red patch. The S1 and S4 substrate specificity pockets are indicated; the hydrophobic S4 pocket is occluded because it is occupied by the ethyl group of the 3’-ethyl ester of 12a. C) Expanded view of the interaction of 12a with it putative binding site as shown in Fig. 6B. The ligand is rendered as a ball-and-stick figure and binding site residues rendered as capped sticks. All binding site side chains within 5 Å of 12a are displayed. Hydrogen bonds between Arg173 and the ligand sulfate group are shown as green dotted lines.
Sulfated Benzofuran 12a Does Not Inhibit Factor Xa
To further support the site of SBD binding on thrombin, we compared the crystal structures of thrombin and factor Xa, especially exosite II, to identify similarities and differences. Factor Xa does not contain an Arg 173-like residue in this region suggesting that 12a can be expected to not inhibit this highly homologous serine protease. Direct inhibition of human factor Xa was performed in a manner similar to thrombin. The dose–response profile showed essentially no decrease in factor Xa activity (Figure 7). These results strongly support the allosteric, hydrophobic binding site capped with Arg173 as a site for the small SBD binding to thrombin.
Figure 7.
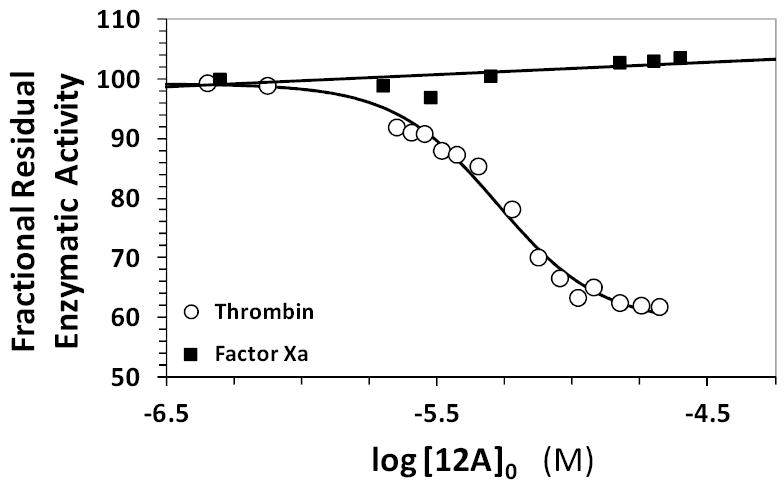
Dose-response profiles for 12a inhibition of human plasma alpha-thrombin and human plasma factor Xa in 20 mM Tris-HCl buffer, pH 7.4, containing 100 mM NaCl, 2.5 mM CaCl2 and 0.1 % polyethylene glycol (PEG) 8000 at 25 °C. Solid lines show sigmoidal dose-response fit of equation 1 to thrombin data or linear trendline fit to factor Xa data. See text for details.
SBDs are Essentially Non-Toxic to Cells
The above work highlights that SBDs are pharmacologically novel entities, the fundamental basis for which is their unique structure. This implied that for further pharmaceutical development a critical question to address was whether the SBD scaffold is tolerated well by cells. To address this, the effect of 12a on human lung A549 cell line was studied. Human A549 cells were cultured in a 96-well microplate in the presence of varying concentrations of 12a. Following exposure of cells to 12a for 120 h, MTT assay for cell viability was performed according to literature reports.31,32 The proportion of viable cells remaining at the end of this exposure was obtained spectrophotometrically (A570) using a reference of viable cells in the wells containing medium alone. The viabilities of human A549 cells in the presence of 12a were not significantly different by one way ANOVA statistical analysis with Dunnet’s test (P>0.05) (Figure 8). These results indicate that 12a, and most probably other SBDs, do not induce any significant toxicity up to ~420 μM, a concentration more than 80-fold higher than the IC50 of thrombin inhibition. This concentration roughly corresponds to a dose of 250 mg/kg, a fairly high dosage level for a pharmaceutical agent.
Figure 8.
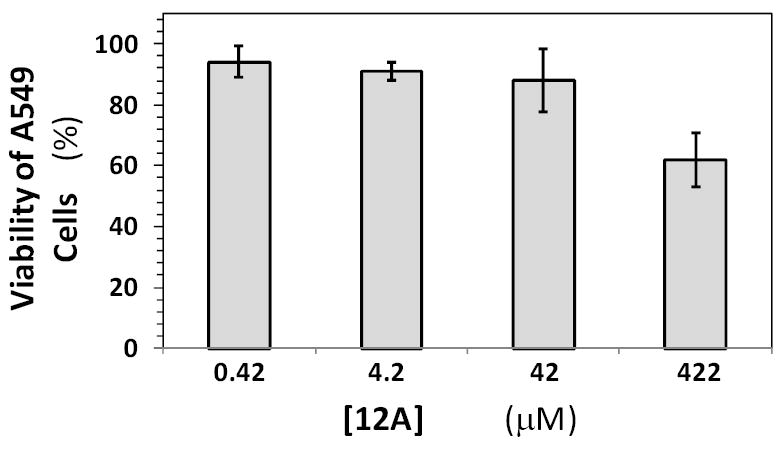
Cell toxicity studies with 12a. The viability of human A549 after 120 h of incubation at 37 °C with varying concentrations of 12a was studied using MTT cell viability assay. Cell viability was measured spectrophotometrically using absorbance at 570 nm, as described in Experimental Procedures. Error bars represent standard deviation measured in three experiments at each concentration studied.
DISCUSSION
No small, drug-like molecules have been designed as yet that function as allosteric modulators of thrombin. Our SBDs, which were designed as mimetics of heparin, are the first small hydrophobic molecules that allosterically reduce thrombin’s catalytic activity.23,24 Discovering allosteric modulators is challenging because identifying exosites that are energetically coupled to the active site in an agonistic or antagonistic manner is not routinely possible. More importantly, even if such an exosite was known, designing molecules that fit the site ‘snugly’ is challenging because the classical, allosteric induced-fit mechanism would require good recognition of both the native and conformationally altered states.
The advantage with thrombin is that it is a highly pliable enzyme. The presence of multiple exosites in thrombin enhances the likelihood of discovering small molecule allosteric regulators. However, such probabilistic advantage does not imply automatic translation into drug-like molecule design and thrombin is no exception. The scenario with other coagulation enzymes, such as factors Xa, IXa and XIa, is also similar. Yet, allosteric regulation of these enzymes offers phenomenal opportunity to induce tailored conformational change so as to maintain a delicate balance between bleeding and clotting tendencies, especially in the disordered state. With this goal in mind, we designed sulfated LMWLs from which were designed SBDs.19-24
This work suggests that SBDs appear to utilize a hydrophobic region between around Arg173 and thrombin’s active site (Figure 7). Although designed as mimetics of heparin, SBDs do not engage the traditional exosite 2 ligand residues including Arg101, Arg126, Lys235, Lys236 and Lys240. SBDs recognize Arg173, a heparin-binding residue. Yet, the predicted binding mode and orientation of 12a is likely to be dramatically different from that of polymeric heparin with its chain oriented along the highly electropositive surface.34 In fact, 12a binds in a region different from that predicted for sulfated LMWLs, which appear to bind in a linear hydrophobic segment present in exosite 2 on either sides of residues Arg93 and Arg101.38
SBD 12a does not compete with the chromogenic substrate and yet reduces the rate of substrate hydrolysis24 suggesting that the new binding site is energetically coupled to active site. The affinity of 12a for thrombin at pH 7.4 and 25 °C is approximately 6 μM, which implies a reasonably high affinity interaction. In fact, other than the antithrombin-binding heparin pentasaccharide, which binds with an affinity of ~50 nM,14,42 no small, sulfated molecule is known to bind with such high affinity.43,44 Recent study of a group of synthetic heparin octasaccharides that target herpes virus glycoproteins with ~20 μM affinity further support this observation.45 The reason for the very high affinity of antithrombin-binding heparin pentasaccharide is marvelous engineering by nature over millions of years. Thus, the ~6 μM affinity of 12a, which is ~1000-fold higher than that of its monomer precursor,23 is a good lead. In fact, the putative binding site presents excellent opportunities advancing the design. For example, Lys224 and Arg221A are within 8 Å of the carboxylic acid ester (Figure 7). Likewise, several hydrophobic residues are also available for targeting.
Molecule 12a reduces thrombin’s catalytic activity by only about 50% in comparison to other active site and exosite 1 inhibitors that display efficacy of nearly 100%.5-7 This implies that 12a does not completely inhibit the procoagulant signal. Whereas this could be traditionally considered as a defect of design, considering the problems of over-anticoagulation induced by thrombin inhibitors, the less than perfect reduction in catalytic activity of thrombin may serve to create the fine balance between procoagulant and anticoagulant signals. More importantly, detailed inhibition profiles reveal that some SBDs exhibit efficacies of greater than 50%, while for others it is between 10 and 20%.24 The variability in the efficacy of thrombin inhibition suggests that the equilibrium between the native and conformationally altered state is ‘tunable’ and optimally designed molecule(s) may be effective in treating varying levels of pro-coagulant tendencies.
A feature of critical interest for developing effective anticoagulants is the extent of their adverse effects, e.g., organ toxicity, in addition to bleeding complications.46,47 A major advantage of SBDs appears to be the low in vitro cellular toxicity, which may translate into high efficacy to toxicity ratio. The reason for the low cellular toxicity noted with SBD is not clear at present, but it is likely to be the considerable water solubility of the monosulfated molecule. We predict that an advanced derivative of 12a that is designed to interact with Lys224, Arg221A and other polar amino acids will possess even better safety profile.
The discovery of the new allosteric binding pocket and the benzofuran scaffold that is able to bind tightly and regulate thrombin activity raises many questions before a clinically viable candidate can be realized. For example, it is well established that presence of FibP does not affect thrombin cleavage of fibrinogen,48 but reduces the cleavage of other thrombin substrates such as factor V, factor VIII, platelet glycoprotein 1 and PAR1. Our earlier work has shown that SBDs inhibit clotting of human plasma, which implies that presence of this molecule reduces the rate of fibrinogen cleavage by thrombin.24 But if these molecules also reduce platelet glycoprotein 1 and PAR1 cleavage, SBDs may function as dual anti-platelet and anti-coagulant agents. This does not necessarily imply a distinct advantage because of the possibility of introducing bleeding risk. Future studies will clarify these involved aspects.
Overall, this work presents a new specific, hydrophobic binding pocket near Arg173 that is energetically coupled to the active site and a group of small, synthetic, hydrophobic molecules that recognize this site with reasonably high affinity. An appropriately designed allosteric SBD inhibitor could prevent the hemorrhagic complications and avoid narrow dosing regimens that are characteristic of thrombin active site inhibitors.49
Supplementary Material
Acknowledgments
This work was supported by grants HL090586, HL099420, and HL107152 from the National Institutes of Health. We thank Drs. Paul Bock of Vanderbilt University for providing HirP and H. Tonie Wright of Virginia Commonwealth University for helpful discussions. We thank Dr. Alireza Rezaie of St. Louis School of Medicine for supplying recombinant thrombin mutants.
Abbreviations
- LMWL
low molecular weight lignin
- LMWH
low molecular weight heparin
- SBD
sulfated benzofuran dimer
- PEG
polyethylene glycol
Footnotes
Supporting Information Available: This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Huntington JA. Molecular recognition mechanisms of thrombin. J Thromb Haemost. 2005;3:1861–1872. doi: 10.1111/j.1538-7836.2005.01363.x. [DOI] [PubMed] [Google Scholar]
- 2.Bode W. The structure of thrombin: A Janus-headed proteinase. Semin Thromb Hemost. 2006;32:16–31. doi: 10.1055/s-2006-939551. [DOI] [PubMed] [Google Scholar]
- 3.Di Cera E. Thrombin. Mol Aspects Med. 2008;29:203–254. doi: 10.1016/j.mam.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Steinmetzer T, Sturzebecher J. Progress in the development of synthetic thrombin inhibitors as new orally active anticoagulants. Curr Med Chem. 2004;11:2297–2321. doi: 10.2174/0929867043364540. [DOI] [PubMed] [Google Scholar]
- 5.Saiah E, Soares C. Small molecule coagulation cascade inhibitors in the clinic. Curr Top Med Chem. 2005;5:1677–1695. doi: 10.2174/156802605775009702. [DOI] [PubMed] [Google Scholar]
- 6.Smallheer JM, Quan ML. Recent advances in coagulation serine protease inhibitors. Ann Rep Med Chem. 2009;44:189–208. [Google Scholar]
- 7.Straub A, Roehrig S, Hillisch A. Oral, direct thrombin and factor Xa inhibitors: The replacement of warfarin, leeches and pig intestines? Angew Chem Int Ed. 2011;50:4574–4590. doi: 10.1002/anie.201004575. [DOI] [PubMed] [Google Scholar]
- 8.Tucker TJ, Brady SF, Lumma WC, Lewis SD, Gardell SJ, Naylor-Olsen AM, Yan YW, Sisko JT, Stauffer KJ, Lucas BJ, Lynch JJ, Cook JJ, Stranieri MT, Holahan MA, Lyle EA, Baskin EP, Chen IW, Dancheck KB, Krueger JA, Cooper CM, Vacca JP. Design and synthesis of a series of potent and orally bioavailable noncovalent thrombin inhibitors that utilize nonbasic groups in the P1 position. J Med Chem. 1998;41:3210–3219. doi: 10.1021/jm9801713. [DOI] [PubMed] [Google Scholar]
- 9.Burgey CS, Robinson KA, Lyle TA, Nantermet PG, Selnick HG, Isaacs RCA, Lewis SD, Lucas BJ, Krueger JA, Singh R, Miller-Stein C, White RB, Wong B, Lyle EA, Stranieri MT, Cook JJ, McMasters DR, Pellicore JM, Pal S, Wallace AA, Clayton FC, Bohn D, Welsh DC, Lynch JJ, Yan YW, Chen ZG, Kuo L, Gardell SJ, Shafer JA, Vacca JP. Pharmacokinetic optimization of 3-amino-6-chloropyrazinone acetamide thrombin inhibitors. Implementation of P3 pyridine N-oxides to deliver an orally bioavailable series containing P1N-benzylamides. Bioorg Med Chem Lett. 2003;13:1353–1357. doi: 10.1016/s0960-894x(03)00099-4. [DOI] [PubMed] [Google Scholar]
- 10.Isaacs RCA, Newton CL, Cutrona KJ, Mercer SP, Dorsey BD, McDonough CM, Cook JJ, Krueger JA, Lewis SD, Lucas BJ, Lyle EA, Lynch JJ, Miller-Stein C, Michener MT, Wallace AA, White RB, Wong BK. P3 optimization of functional potency, in vivo efficacy and oral bioavailability in 3-aminopyrazinone thrombin inhibitors bearing non-charged groups at the P1 position. Bioorg Med Chem Lett. 2011;21:1532–1535. doi: 10.1016/j.bmcl.2010.12.108. [DOI] [PubMed] [Google Scholar]
- 11.Straub A, Roehrig S, Hillisch A. Entering the era of non-basic P1 site groups: Discovery of Xarelto (TM) (Rivaroxaban) Curr Top Med Chem. 2010;10:257–269. doi: 10.2174/156802610790725506. [DOI] [PubMed] [Google Scholar]
- 12.Bock PE, Panizzi P, Verhamme IMA. Exosites in the substrate specificity of blood coagulation reactions. J Thromb Haemost. 2007;5(Suppl. 1):81–94. doi: 10.1111/j.1538-7836.2007.02496.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olson ST, Swanson R, Raub-Segall E, Bedsted T, Sadri M, Petitou M, Hérault JP, Herbert JM, Björk I. Accelerating ability of synthetic oligosaccharides on antithrombin inhibition of proteinases of the clotting and fibrinolytic systems. Comparison with heparin and low-molecular-weight heparin. Thromb Haemost. 2004;92:929–939. doi: 10.1160/TH04-06-0384. [DOI] [PubMed] [Google Scholar]
- 14.Desai UR. New antithrombin–based anticoagulants. Med Res Rev. 2004;24:151–181. doi: 10.1002/med.10058. [DOI] [PubMed] [Google Scholar]
- 15.Di Cera E. Thrombin: A paradigm for enzymes allosterically activated by monovalent cations. C R Biol. 2004;327:1065–1076. doi: 10.1016/j.crvi.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 16.Evans SA, Olson ST, Shore JD. p-Aminobenzamidine as a fluorescent probe for the active site of serine proteases. J Biol Chem. 1982;257:3014–3017. [PubMed] [Google Scholar]
- 17.Ng NM, Quinsey NS, Matthews AY, Kaiserman D, Wijeyewickrema LC, Bird PI, Thompson PE, Pike RN. The effects of exosite occupancy on the substrate specificity of thrombin. Arch Biochem Biophys. 2009;489:48–54. doi: 10.1016/j.abb.2009.07.012. [DOI] [PubMed] [Google Scholar]
- 18.Desai BJ, Boothello RS, Mehta AY, Scarsdale JN, Wright HT, Desai UR. Interaction of thrombin with sucrose octasulfate. Biochemistry. 2011;50:6973–6982. doi: 10.1021/bi2004526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Monien BH, Henry BL, Raghuraman A, Hindle M, Desai UR. Novel chemo-enzymatic oligomers of cinnamic acids as direct and indirect inhibitors of coagulation proteinases. Bioorg Med Chem. 2006;14:7988–7998. doi: 10.1016/j.bmc.2006.07.066. [DOI] [PubMed] [Google Scholar]
- 20.Henry BL, Monien BH, Bock PE, Desai UR. A novel allosteric pathway of thrombin inhibition. Exosite II mediated potent inhibition of thrombin by chemo-enzymatic, sulfated dehydropolymers of 4-hydroxycinnamic acids. J Biol Chem. 2007;282:31891–31899. doi: 10.1074/jbc.M704257200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Henry BL, Thakkar JN, Martin EJ, Brophy DF, Desai UR. Characterization of the plasma and blood anticoagulant potential of structurally and mechanistically novel oligomers of 4-hydroxycinnamic acids. Blood Coag Fibrinol. 2009;20:27–34. doi: 10.1097/MBC.0b013e328304e077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Henry BL, Abdel Aziz M, Zhou Q, Desai UR. Sulfated, low molecular weight lignins are potent inhibitors of plasmin, in addition to thrombin and factor Xa : Novel opportunity for controlling complex pathologies. Thromb Haemost. 2010;103:507–515. doi: 10.1160/TH09-07-0454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Verghese J, Liang A, Sidhu PS, Hindle M, Zhou Q, Desai UR. First steps in the direction of synthetic, allosteric, direct inhibitors of thrombin and factor Xa. Bioorg Med Chem Lett. 2009;19:4126–4129. doi: 10.1016/j.bmcl.2009.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sidhu PS, Liang A, Mehta AY, Abdel Aziz MH, Zhou Q, Desai UR. Rational design of potent, small, synthetic allosteric inhibitors of thrombin. J Med Chem. 2011;54:5522–5531. doi: 10.1021/jm2005767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lovely RS, Moaddel M, Farrell DH. Fibrinogen gamma’ chain binds thrombin exosite II. J Thromb Haemost. 2003;1:124–131. doi: 10.1046/j.1538-7836.2003.00027.x. [DOI] [PubMed] [Google Scholar]
- 26.Bock PE, Olson ST, Bjork I. Inactivation of thrombin by antithrombin is accompanied by inactivation of regulatory exosite I. J Biol Chem. 1997;272:19837–19845. doi: 10.1074/jbc.272.32.19837. [DOI] [PubMed] [Google Scholar]
- 27.He X, Ye J, Esmon CT, Rezaie AR. Influence of arginines 93, 97, and 101 of thrombin to its functional specificity. Biochemistry. 1997;36:8969–8976. doi: 10.1021/bi9704717. [DOI] [PubMed] [Google Scholar]
- 28.Yang L, Rezaie AR. Calcium-binding sites of the thrombin-thrombomodulin-protein C complex: Possible implications for the effect of platelet factor 4 on the activation of vitamin K-dependent coagulation factors. Thromb Haemost. 2007;97:899–906. doi: 10.1160/th06-12-0697. [DOI] [PubMed] [Google Scholar]
- 29.Skordalakes E, Dodson GG, St Clair-Green D, Goodwin CA, Scully MF, Husdson HR, Kakkar VV, Deadman JJ. Inhibition of human α-thrombin by a phosphonate tripeptide proceeds via a metastable pentacoordinated phosphorus intermediate. J Mol Biol. 2001;311:549–555. doi: 10.1006/jmbi.2001.4872. [DOI] [PubMed] [Google Scholar]
- 30.Ahmed HU, Blakeley MP, Cianci M, Cruickshank DWJ, Hubbard JA, Helliwell JR. The determination of protonation states in proteins. Acta Crystallogr Sect D. 2007;63:906–922. doi: 10.1107/S0907444907029976. [DOI] [PubMed] [Google Scholar]
- 31.Zhou Q, Zhang L, Zuniga MA, Tombes RM, Stewart JK. Mixed inhibition of P450 3A4 as a chemoprotective mechanism against aflatoxin B1-induced cytotoxicity with cis-terpenones. Chem Res Toxicol. 2008;21:732–738. doi: 10.1021/tx700363s. [DOI] [PubMed] [Google Scholar]
- 32.Zhou Q, Xie H, Zhang L, Stewart JK, Gu XX, Ryan JJ. cis-Terpenones as an effective chemopreventive agent against aflatoxin B1-induced cytotoxicity and TCDD-induced P450 1A/B activity in HepG2 cells. Chem Res Toxicol. 2006;19:1415–1419. doi: 10.1021/tx0601307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu CC, Brustad E, Liu W, Schultz PG. Crystal structure of a biosynthetic sulfo-hirudin complexed to thrombin. J Am Chem Soc. 2007;129:10648–10649. doi: 10.1021/ja0735002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carter WJ, Cama E, Huntington JA. Crystal structure of thrombin bound to heparin. J Biol Chem. 2005;280:2745–2749. doi: 10.1074/jbc.M411606200. [DOI] [PubMed] [Google Scholar]
- 35.Verhamme IM, Olson ST, Tollefsen DM, Bock PE. Binding of exosite ligands to human thrombin. Re-evaluation of allosteric linkage between thrombin exosites I and II. J Biol Chem. 2002;277:6788–6798. doi: 10.1074/jbc.M110257200. [DOI] [PubMed] [Google Scholar]
- 36.Pineda AO, Chen ZW, Marino F, Mathews FS, Mosesson MW, Di Cera E. Crystal structure of thrombin in complex with fibrinogen gamma’ peptide. Biophys Chem. 2007;125:556–559. doi: 10.1016/j.bpc.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 37.Sabo TM, Farrell DH, Maurer MC. Conformational analysis of gamma’ peptide (410-427) interactions with thrombin anion binding exosite II. Biochemistry. 2006;45:7434–7445. doi: 10.1021/bi060360k. [DOI] [PubMed] [Google Scholar]
- 38.Abdel Aziz MH, Mosier PD, Desai UR. Identification of the site of binding of sulfated, low molecular weight lignins on thrombin. Biochem Biophys Res Commun. 2011;413:348–352. doi: 10.1016/j.bbrc.2011.08.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Raghuraman A, Mosier PD, Desai UR. Understanding dermatan sulfate-heparin cofactor II interaction through virtual library screening. ACS Med Chem Lett. 2010;1:281–285. doi: 10.1021/ml100048y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Raghuraman A, Mosier PD, Desai UR. Finding a needle in a haystack: Development of a combinatorial virtual screening approach for identifying high specificity heparin/heparan sulfate sequence(s) J Med Chem. 2006;49:3553–3562. doi: 10.1021/jm060092o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Henry BL, Connell J, Liang A, Krishnasamy C, Desai UR. Interaction of antithrombin with sulfated, low molecular weight lignins: Opportunities for potent, selective modulation of antithrombin function. J Biol Chem. 2009;284:20897–20908. doi: 10.1074/jbc.M109.013359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Desai UR, Petitou M, Björk I, Olson ST. Mechanism of heparin activation of antithrombin. Role of individual residues of the pentasaccharide activating sequence in the recognition of native and activated states of antithrombin. J Biol Chem. 1998;273:7478–7487. doi: 10.1074/jbc.273.13.7478. [DOI] [PubMed] [Google Scholar]
- 43.Capila I, Linhardt RJ. Heparin-protein interactions. Angew Chem Int Ed Engl. 2002;41:391–412. doi: 10.1002/1521-3773(20020201)41:3<390::aid-anie390>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 44.Gandhi NS, Mancera RL. The structure of glycosaminoglycans and their interactions with proteins. Chem Biol Drug Des. 2008;72:455–482. doi: 10.1111/j.1747-0285.2008.00741.x. [DOI] [PubMed] [Google Scholar]
- 45.Hu YP, Lin SY, Huang CY, Zulueta MM, Liu JY, Chang W, Hung SC. Synthesis of 3-O-sulfonated heparan sulfate octasaccharides that inhibit the herpes simplex virus type 1 host-cell interaction. Nat Chem. 2011;3:557–563. doi: 10.1038/nchem.1073. [DOI] [PubMed] [Google Scholar]
- 46.Keisu M, Andersson TB. Drug-induced liver injury in humans: the case of ximelagatran. Handb Exp Pharmacol. 2010:407–418. doi: 10.1007/978-3-642-00663-0_13. [DOI] [PubMed] [Google Scholar]
- 47.Stewart RA. Clinical trials of direct thrombin and factor Xa inhibitors in atrial fibrillation. Curr Opin Cardiol. 2011;26:294–299. doi: 10.1097/HCO.0b013e3283477dbc. [DOI] [PubMed] [Google Scholar]
- 48.Lovely RS, Rein CM, White TC, Jouihan SA, Boshkov LK, Bakke AC, McCarty OJ, Farrell DH. GammaA/gamma’ fibrinogen inhibits thrombin-induced platelet aggregation. Thromb Haemost. 2008;100:837–846. [PubMed] [Google Scholar]
- 49.Adams TE, Everse SJ, Mann KG. Predicting the pharmacology of thrombin inhibitors. J Thromb Haemost. 2003;1:1024–1027. doi: 10.1046/j.1538-7836.2003.00127.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.