

Abstract
In periodontitis, a common chronic inflammatory condition, gram negative-rich bacterial biofilms trigger, in susceptible individuals, perpetuating inflammation that results in extensive tissue damage of tooth supporting structures. To delineate immune cell-dependent mechanisms whereby bacterial challenge drives persistent destructive inflammation in periodontitis and other inflammatory diseases, we studied involved tissues ex vivo and investigated host cell responses to the periodontal pathogen P. gingivalis, in vitro. Diseased lesions were populated by abundant Th17 cells, linked to infection, chronic inflammation/autoimmunity and tissue pathology. In vitro, P. gingivalis, particularly the more virulent strain W83, stimulated myeloid antigen presenting cells (APC) to drive Th17 polarization. Supernatants from myeloid APC exposed to P. gingivalis were capable of enhancing Th17 but not Th1 polarization. P. gingivalis favored the generation of Th17 responses by stimulating the production of Th17 related cytokines IL-1β, IL-6 and IL-23, but not Th1 related IL-12. By inducing NFκB activation, P. gingivalis promoted IL-1β, IL-6 and IL-12p40 production, but not IRF-3 phosphorylation, connected to generation of the IL-12p35 chain, ultimately restricting formation of the intact IL-12 molecule. Promotion of Th17 lineage responses was also aided by P. gingivalis proteases, which appeared to differentially degrade pivotal cytokines. In this regard, IL-12 was largely degraded by P. gingivalis, whereas IL-1β was more resistant to proteolysis. Our data unveil multiple pathways by which P. gingivalis may orchestrate chronic inflammation, providing insights into interventional strategies.
Introduction
In the oral mucosa, immune-mediated destruction of the osseous support surrounding the dentition underlies periodontal disease, one of the most prevalent of chronic inflammatory conditions[1]. A necessary trigger for the initiation and perpetuation of the inflammatory response in this setting is the colonization of the dentition with a microbial biofilm enriched in gram-negative bacteria, belonging to the red complex of periodontal pathogens[1]. Among these, Porphyromonas gingivalis (P. gingivalis), a non-spore forming, non-motile, obligate anaerobe, has been most strongly implicated in the pathogenic sequelae of periodontal disease[2], but also in the generation of remote inflammatory responses connected to chronic disease and autoimmunity [3-5]. P. gingivalis expresses a number of virulence factors including, but not limited to its atypical LPS, its fimbriae and its cysteine proteinases (gingipains)[2], which may contribute to periodontitis pathogenesis. Considerable variation in virulence between strains of P. gingivalis has been documented based on their behavior in animal models[6], with virulent strains causing an invasive spreading type of infection and the lesser virulent strains inducing mostly a localized abscess[7, 8].
In a susceptible host, persistence of periodontal pathogens such as P. gingivalis results in aberrant and prolonged inflammation and subsequent tissue damage of tooth supporting structures. How bacteria initiate and perpetuate mechanisms of pathologic inflammation leading to tissue damage is an important research question for which periodontal disease may serve as a valuable research model.
Tissue pathology is largely the consequence of chronic release of factors by activated cells of the immune system. Among the immune cells initially responding to challenge by bacteria, including P. gingivalis, are antigen presenting cells (APC) of myeloid origin such as recruited peripheral blood monocytes and resident DCs and macrophages, poised strategically along portals of entry[9]. After recognition of pathogen associated molecular patterns (PAMPs)[10] via toll like receptors (TLR), innate immune cells initiate responses aiming to contain and/or clear the inciting agent. How distinct APC cell types may differentially initiate and coordinate responses to periodontal pathogens, such as P. gingivalis has not yet been fully elucidated. Previous work in our laboratory has shown that monocytes, macrophages and DC have both shared and distinct responses to the LPS of P. gingivalis[9]. However, these studies did not characterize responses to the intact P. gingivalis bacterium nor addressed the potential roles of the different myeloid subpopulations to support downstream development of adaptive immunity.
As coordinators, APC orchestrate the transition to the adaptive arm of the immune response, including the generation of effector T cell responses. Effector T cells were previously considered to differentiate into either a Th1 or Th2 phenotype[11], but with the recent recognition of effector T cell plasticity and the Th17 lineage[12], there has been an emphasis on characterizing the contributing cells involved in chronic mucosal lesions. IL-17 secreting Th17 cells play a protective role in antimicrobial immunity but have also been linked to inflammatory and autoimmune pathologies[13] [14]. Persistence of the Th17 population has been shown to support chronicity of inflammation and to directly mediate tissue destruction, via activation of resident matrix cells such as fibroblasts and osteoclasts[15] [16], unlike Th1/Th2 cells which can inhibit osteoclast differentiation[15][17]. Immune-mediated osseous destruction is not only implicated in periodontal disease but is the hallmark of pathology in autoimmune conditions such as rheumatoid arthritis (RA) [18] and, hence, delineating a role for Th1/Th2/Th17 lineages in chronic inflammation and tissue damage is ongoing[9], in an effort to further the understanding of disease pathologies and ultimately uncover possible targets to disrupt pathogenesis. To this end, we aimed to study the Th lineage representation within the pathologic lesions of periodontitis and to investigate possible mechanisms by which strains of the periodontal pathogen P. gingivalis which are considered more or less virulent may orchestrate T cell lineage commitment relevant to oral and remote immunopathology.
Materials and Methods
Tissue specimens
Periodontitis-involved tissue samples from patients with severe chronic periodontitis, diagnosed according to the latest American Academy of Periodontology criteria[19], were obtained during therapeutic surgery. Inclusion criteria for diseased tissues were evidence of inflammation (bleeding on probing) and advanced loss of tooth supporting structures (probing depth (PD≥6mm), clinical attachment loss (CAL>6mm)[20]. Minimally inflamed tissue samples were obtained during routine oral surgery procedures and classified as gingivitis with evidence of inflammation, and limited loss of attachment (PD/CAL≤3mm)[19]. All clinical samples were obtained from systemically healthy adult patients undergoing oral surgery procedures. Tissues were either immediately placed in 10% formalin for histology or placed in RNAlater (Ambion, Austin TX) and stored at -70°C for isolation of RNA. Research involving human subjects in this study was reviewed and approved by the Institutional Review Board of the Loma Linda University, School of Dentistry (protocol # 58034). For participation in this study all patients provided written informed consent.
Immunohistochemistry
Formalin fixed tissues were paraffin embedded and sectioned into 5μm sections, deparaffinized and rehydrated, followed by heat-induced epitope retrieval. Methanol containing 3% hydrogen peroxide was used to block the endogenous peroxidase for 15 minutes. Sections then were blocked with the corresponding pre-immune serum and incubated overnight at 4°C with primary antibody to CD3, IL-23, IL-4 (Abcam, Cambridge, MA) and IL-17, IFNγ (R&D, Minneapolis, MN) or an isotype control. After washing with phosphate buffered saline (PBS) three times, immunolabeling was detected using a biotinylated secondary antibody followed by visualization with an avidin biotin horseradish peroxidase labeling kit (Invitrogen Inc, Carlsbad, CA) and diaminobenzidine staining. Finally, the specimens were counterstained with Mayer’s hematoxylin and mounted with Permount (Fisher Scientific).
Cell preparation
Human peripheral blood mononuclear cells (PBMC) obtained from healthy volunteers at the Department of Transfusion Medicine (National Institutes of Health, Bethesda, MD) were diluted in endotoxin-free PBS without Ca2+ and Mg2+ (BioWhittaker, Walkersville, MD) and density-sedimented on lymphocyte separation medium (LSM; ICN Pharmaceuticals, Aurora, OH). Monocytes were purified from the mononuclear cell layer using centrifugal elutriation and divided into 3 groups for immediate use as monocytes or for generation of donor matched macrophages and DC, as previously described[9]. The viability of each cell type was >95%. All donor-matched cell populations were tested for their purity in these differentiated populations by flow cytometry[9] (Suppl. Figure 1).
Bacterial growth and stimulation of myeloid cells
P. gingivalis ATCC 33277 and P. gingivalis W83 were grown in brain-heart infusion medium (BHI; Becton Dickinson, Franklin Lakes, NJ) supplemented with 0.1% yeast extract, 0.5% hemin and 0.001% menadione[21]. Both strains were grown in a Bactron anaerobic (N2:CO2:H2, 90:5:5) environmental chamber (Sheldon Manufacturing Inc., Cornelius, OR) at 37°C.
Myeloid cells (at a concentration of 3×106/ml) were left untreated (medium control) or exposed to live P. gingivalis (ATCC 33277 or W83) at ratios of 1:10-1:100 (myeloid cells/P. gingivalis) in the presence or absence of gingipain specific inhibitors (KYT-1 and KYT-36, Pepnet, Louisville, KY) at a concentration of 106M each for 30min-1 hr and processed for protein or total RNA isolation. In some experiments myeloid cells were also exposed to E. coli LPS (100ng/ml (100ng/ml, Sigma, St. Louis, MO) for 30min and processed for protein isolation. P. gingivalis exposed cells were cultured further (24hr) for supernatant collection. After 24hr culture, the media were collected, spun at 12,000 × g, filtered to eliminate bacteria (0.22μm filter, Millipore, Bedford, MA), transferred to fresh tubes and stored at -70°C until further use.
PBMC exposure to myeloid cell supernatant
PBMC (at a concentration of 3×106/ml) were cultured in RPMI 1640 medium (Lonza, Walkersville, MD) supplemented with 2mM L-glutamine, 10μg/ml gentamycin and 10% FCS (Life Technologies, Gaithersburg, MD). The cells were then stimulated with anti-CD3, anti-CD28 (3μg/ml, Becton Dickson, San Jose, CA) in the presence or absence of 24hr myeloid cell supernatants at a ratio of 1:1 with media.
Flow cytometry
Myeloid cells were stained with antibodies to CD14, HLA-DR, DC-SIGN, CD68 (Becton Dickinson). For surface staining of PBMC a monoclonal antibody against CD3 was used (Becton Dickinson). For intracellular staining, a cytofix/cytoperm kit was used (Becton Dickinson) with antibodies against IL-17A and IFNγ. The antibodies were conjugated with either fluorescein isothiocyanate, phycoerythrin or allophycocyanin, PerCP-Cy5.5 or Alexa Fluor® 647. Cells were analyzed by FACScalibur system (Becton Dickinson).
RNA isolation and Real-Time PCR
Total RNA was isolated from clinical tissue samples with TRIzol Reagent (Invitrogen, Carlsbad, CA). Total RNA was isolated from myeloid cells with the RNeasy Mini Columns (Qiagen, Valencia, CA) followed by assessment of RNA integrity with the 2100 Bioanalyzer (Agilent, Foster City, CA). Total RNA (1μg) from each sample was reverse transcribed using an oligodeoxythymidylic acid primer (Invitrogen) and the resulting cDNA amplified by real-time PCR on an ABI Prism 7500 Sequence Detector (Applied Biosystems, Foster City, CA). Amplification was performed with TaqMan expression assays for HPRT (Assay ID: Hs99999909_m1), β-actin (Hs99999903_m1), GAPDH (Hs99999905_m1), IL-17 (Hs00174383_m1), IFNγ (Hs00989291_m1), IL-22 (Hs01574154_m1), IL-23p19 (Hs00372324_m1), IL-1β (Hs01555410_m1), IL-6 (Hs00985639_m1), IL-23 (Hs00372324_m1), IL-12p40 (Hs00233688_m1), IL-12p35 (Hs00168405_m1), from Applied Biosystems. Amplification parameters and conditions were set by the manufacturer. Data were analyzed using the 2-ΔCT method (2ˆ((-1)*(Mean cycle number of Target-mean cycle number of housekeeping gene)). Data between groups were compared using a two-tailed Student’s t test for correlated or non-correlated samples (http://faculty.vassar.edu/lowry/VassarStats.html). The results were considered statistically significant at p≤ 0.05.
Multiplex ELISA for detection of cytokine levels and cytokine degradation
Levels of IL-1β, IL-6, IL-12p40, IL-12p70, IL-23 were measured in cell culture supernatants, using a custom designed Panomics Procarta Cytokine Profiling Assay (Affymetrix, Fremont, CA). Levels of total TGF-β were measured using an ELISA (Promega, Madison, WI). For cytokine degradation studies, the recombinant cytokine mix used for determination of the standard curve was exposed to P. gingivalis W83 and 33277 at 105 to 107 bacteria/ml and for a time course of 10 min to 2 hr. Remaining cytokine levels were detected using the Panomics Procarta Cytokine Profiling Assay (Affymetrix). Data were expressed as mean ± SEM and compared using a two-tailed Student’s t test for correlated samples (http://faculty.vassar.edu/lowry/VassarStats.html). The results were considered statistically significant at p≤ 0.05.
Direct cytokine degradation assays
Recombinant cytokines IL-1β and IL-12 (Peptrotech, Rocky Hill, NJ) were incubated alone or with P. gingivalis (W83 and 33277 at concentrations of 105 and 107/ml) for 10-20 min. Following incubation, cytokine-bacterial mixtures were filtered (0.22μm filter, Millipore, Bedford, MA) to eliminate bacteria. Original and bacterial-exposed cytokines were subjected to Tris-glycine SDS-PAGE and blotted onto nitrocellulose membranes. Membranes were stained with Coomasie Blue (Thermo Fisher Scientific, Rockford, IL).
Western
Cell pellets were lysed with ice-cold lysis buffers (50mM Tris, pH 7.5, 1%NP-40, 0.25% sodium deoxycholate, 150mM NaCl, 1 mM EGTA, 1mM Na3VO3, 10 mM NaF, 1mM PMSF, 1mM Na4P2O7, 5μg/ml of Aprotinin, Pepstatin, Leupeptin), incubated on ice for 20 min and centrifuged at 40,000rpm for 20min at 4°C and supernatant protein was quantitated (Bio-Rad DC Protein assay). Lysates were subjected to Tris-glycine SDS-PAGE and blotted onto nitrocellulose membranes. After blocking with 5% milk in Tris-buffered saline (TBS), membranes were probed with anti phospho-IRF3, IRF3, phospho-IkB or phospho-NFκB antibody (Cell Signaling Technology, Danvers, MA) in 5% BSA in TBS with 0.05% Triton 100 (TBST) at 4°C overnight. After washing with TBST, signals were analyzed by the addition of Alexa Fluor 680 goat anti-rabbit or Alexa Fluor 750 or goat anti-mouse Abs (LI-COR) and the infrared fluorescence detected with the Odyssey infrared imaging system (LI-COR).
Results
Th17+ cells in lesions of severe periodontitis
To investigate the immune-mediated mechanisms underlying mucosal tissue and bone damage in chronic periodontal disease, we first compared the inflammatory infiltrate in tissues harvested from patients with severe periodontitis and associated tissue damage to tissues with minimal inflammation. As evident in Figure 1A, in severe periodontitis, massive numbers of inflammatory cells infiltrate the soft tissue adjacent to the tooth and bone, in contrast to evidence of fewer inflammatory cells in tissues from patients with less gingival inflammation and minimal or no periodontal destruction (Fig. 1B). The periodontal inflammatory lesions are dominated by CD3+ T cells (Fig. 1C), while APC and other immune populations are also represented in the lesion as we have shown previously[9]. Immunohistochemical analysis of the CD3 enriched T cell infiltrate revealed IFNγ staining (likely reflecting a Th1 phenotype, Fig. 1D) but minimal IL-4/Th2 staining (not shown). More prevalent were regions of IL-17 and IL-23 positive staining, suggesting heightened Th17 representation in periodontitis (Fig. 1E/F). Subsequent analysis of the transcriptional profile of severely diseased (perio) compared to minimally diseased (gingivitis) tissues was consistent with the protein evidence. mRNA expression for IL-17 and IFNγ was significantly elevated in periodontitis (Fig. 1G, p<0.05), but relative IL-17 expression was more abundant (Fig. 1G). Moreover cytokines shown to be linked to Th17 polarization, IL-22 and IL-23 were also detected at protein and mRNA levels in these diseased lesions (data not shown). Further characterization of the tissue milieu revealed evidence of additional mediators that could support Th17 lineage commitment [12]. Notably, IL-6 mRNA levels were detectable in gingivitis and periodontitis tissues, and increased IL-1β levels were evident in gingivitis and periodontitis, particularly in severe periodontitis tissues (Fig. 1H). By comparison, the Th1 driving IL-12 was minimally detectable (Fig. 1H) and IL-4 was undetected in all tissue specimens (data not shown).
Figure 1. Inflammatory infiltrate in lesions of periodontal disease.
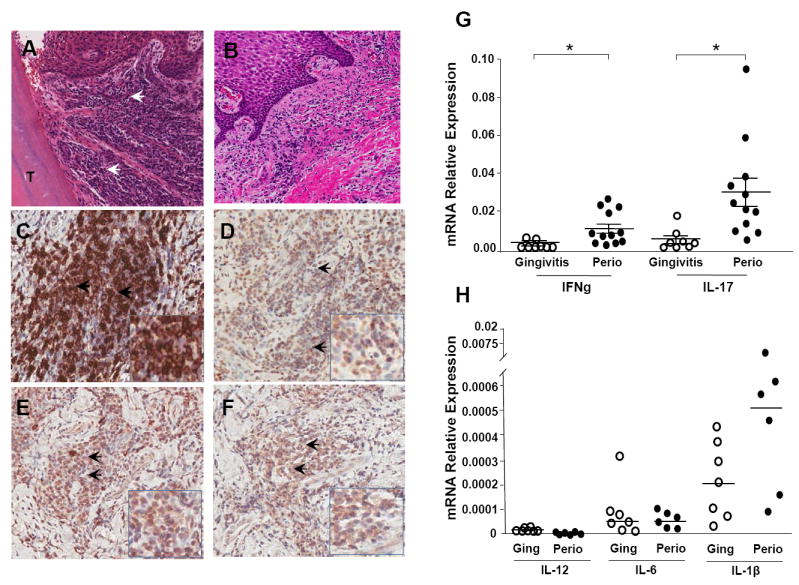
A. H&E staining reveals the dense inflammatory infiltrate (indicated by arrowheads) in areas adjacent to the tooth structure (T) in representative tissue from patients with severe periodontitis (CAL>6mm) as compared to minimally inflamed tissues from gingivitis individuals (B). C. Immunohistochemical staining using a monoclonal antibody to CD3 indicates large numbers of CD3 positive T cells (brown) in the periodontitis lesion. D-F. Immunohistochemical detection of cytokines using cytokine-specific monoclonal antibodies. Within the lesion, IFN-γ+ (D), IL-17+ (E), and IL-23+ (F) cell populations, are identified by brown staining, and indicated by arrows. All images are at original magnification 20x with inserts at magnification 40x. G. mRNA expression relative to housekeeping gene HPRT (2ˆ-ΔCT) in periodontitis (Perio, severe tissue destruction CAL>6mm, n=12) and gingivitis tissues (inflamed tissues with minimal tissue destruction, n=8) for IFN-γ and IL-17. Median ± SE indicated. *indicates significance at p<0.05, median values indicated. H. Relative mRNA expression (2ˆ-ΔCT) for IL-12, IL-6 and IL-1β in gingivitis (n=7) and periodontitis (n=6) patient tissues.
To account for the potential contribution of an increased representation of CD3 T cells in lesions of periodontitis, we have normalized our cytokine data to the expression of CD3. Our data show that periodontitis tissues have significantly higher levels of IFNγ and IL-17 than gingivitis lesions even after normalizing to CD3 expression (Fig. 2A), reflecting a bias towards the development of pro-inflammatory Th1/17 cell subsets. Interestingly, normalized levels of IL-17 expression also exceed those of IFNγ.
Figure 2. Cytokine expression in periodontitis tissues.
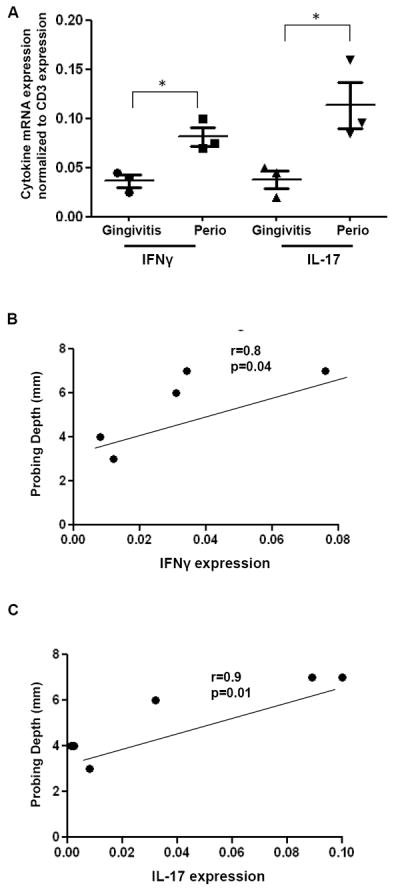
A. mRNA expression for IFN-γ and IL-17 relative to the expression of the T cell marker CD3 is shown in gingivitis tissues (inflamed tissues with minimal tissue destruction, n=3) and in periodontitis (Perio, severe tissue destruction, n=3). Median ± SE indicated. *indicates significance at p<0.05. B-C. Correlation between tissue destruction in each sample (measured in mm, Probing Depth measure) and IFN-γ and IL-17 cytokine expression. P values and Pearson r shown for each comparison.
Finally, in additional analyses, we have investigated the correlation of cytokine expression to tissue destruction/immunopathology in each patient (mm of bone loss in the specific area measured, indicated as probing depth (PD)). Our data show a significant correlation between tissue destruction and IFNγ expression (p=0.04), but an even more significant relationship to the expression of IL-17 (p=0.01) (Figure 2B, C).
In vitro differentiation of Th17 cells is driven by the periodontal pathogen P. gingivalis
To associate mechanisms driving Th lineage commitment in periodontitis to periodontal pathogens, we exposed donor-matched myeloid APCs, including monocytes (mono), macrophages (mac) and dendritic cells (DC) to two different strains of P. gingivalis, one considered more virulent (W83) and the other less virulent (ATCC 33277), based on their behavior in animal models of periodontitis [6]. We treated APC with P. gingivalis for 24h and collected their secreted products once depleted of bacteria, to test for their ability to affect T cell polarization. In all experiments APC supernatants (1:1 to media) were added to PBMC (at 3×106 cells/ml) which were concurrently stimulated by anti-CD3/CD28 and subsequently treated with PMA and ionomycin to enhance cytokine production.
As anticipated, anti-CD3 stimulation caused CD3 T cells to predominantly produce IFNγ (7-13% of PBMC) with minimal evidence of IL-17 (Fig. 3A). However, exposure to soluble factors produced by APC exposed to P. gingivalis, augmented generation of CD3+IL-17+ cells (Fig. 3A,B), without corresponding increase in IFNγ-producing cells above the level achieved by anti-CD3/CD28 stimulation (Fig. 3A, C). Supernatants from monocytes and DC exposed to P. gingivalis, particularly the W83 strain, significantly increased the percentage of IL-17+ T cells, whereas macrophage products were less effective under parallel conditions. A dual positive IL-17+/IFNγ + population was most evident following CD3+ T cell exposure to products of DC treated with the W83 strain (Fig. 3A, 0.3-2.2%).
Figure 3. Th cell lineage differentiation after exposure of PBMC to supernatants from APC treated with P. gingivalis.
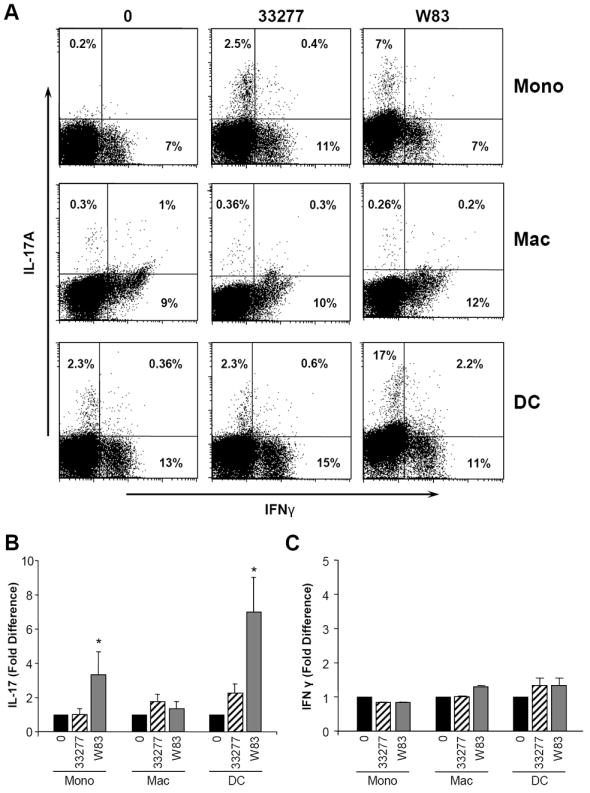
Antigen presenting cells (APC), including donor-matched monocyte, macrophage and dendritic cells were untreated or exposed to P. gingivalis strains 33277 and W83 (1:50) for 0-24h. 24h APC supernatants (media:sup ratio 1:1) were added to PBMC which were cultured for 3-5 days in the presence of anti-CD3/anti-CD28 (3μg/ml). At the end of this period, PBMC were stimulated with PMA and ionomycin for 5 hr, in the presence of Golgistop, and cells were stained for extracellular CD3 and intracellular IFNγ and IL-17 and analyzed by flow cytometry. A. Representative dot blot showing percent IFNγ+, IL-17+, IFNγ +/IL-17+ within the CD3+ population, stimulated with aCD3/CD28 in the presence of indicated APC supernatants. B. Fold difference in percent CD3+/IL-17+ in PBMC treated with supernatants from untreated APC versus P. gingivalis exposed APC (mean±SE, n=3 experiments. C. Fold difference in percent CD3+/IFN-γ+ + in PBMC treated with supernatants from untreated APC versus P. gingivalis exposed APC (mean±SE, n=3 experiments). *indicates significance at p<0.05.
Th17 supporting cytokines are generated in response to P. gingivalis
Having observed that secreted factors from APC exposed to P. gingivalis favor the generation of IL-17+ cells, we quantified levels of potential polarizing cytokines that might be contributing. In response to P. gingivalis, cytokines involved in innate immunity but also associated with promoting Th17 polarization, IL-1β and IL-6, were variably increased at the mRNA level in all cell types (Fig. 4A-B). Moreover, protein levels of IL-1β, measured by multiplex ELISA, were significantly increased in monocytes and DC (Fig. 3C), whereas in macrophages, despite increased mRNA (Fig. 4A), cytokine production was modest, likely reflecting known differences in post translational modification [22]. In comparison, IL-6 protein levels, similar to mRNA, were significantly increased in macrophages and also in DC by P. gingivalis (Fig. 4D). There was typically a higher induction of IL-1β and IL-6 in response to the W83 strain compared to the 33277 strain, consistent with the enhanced ability of W83-treated APC to support IL-17 producing cells (Fig. 3 and 4A-D).
Figure 4. Th17 polarizing cytokines produced by APC exposed to P. gingivalis.
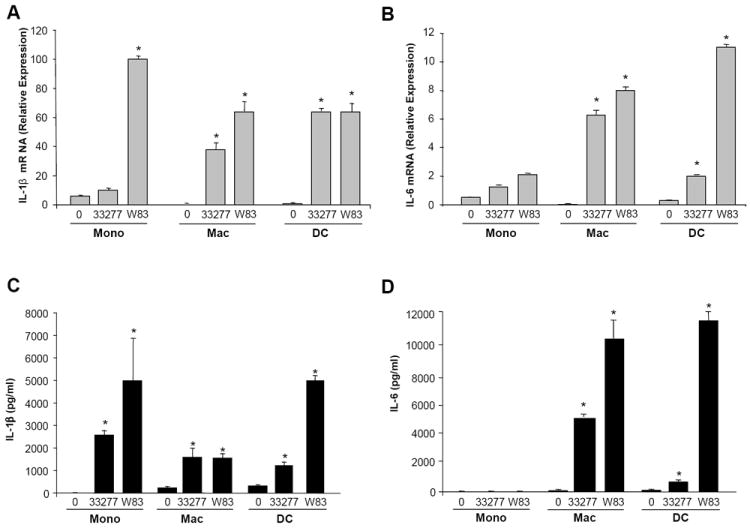
Donor matched monocyte (mono), macrophage (mac) and dendritic cells (DC) (3×106/ml) were exposed to P. gingivalis ATCC 33277 or W83 (1:50). At 1h, cells were processed for total RNA and in parallel cultures at 24h, supernatants were evaluated for cytokine levels by multiplex ELISA. A. IL-1β mRNA relative expression levels (2ˆ-ΔCT) measured by real-time PCR. B. IL-6 mRNA relative expression (2ˆ-ΔCT). C. IL-1β protein levels in 24hr culture supernatant. D. IL-6 protein levels in 24hr culture supernatant. All data representative of n=3 experiments. Significance indicated between untreated (0) and P. gingivalis exposed, * = p<0.05.
Additional cytokines linked to Th17 polarization were also evaluated. Although, TGF-β levels in the APC supernatants, were low, non-discriminating between treatment conditions and cell types, and potentially attributable to presence of serum in the culture medium (not shown), IL-23 was detectable at mRNA and protein levels. mRNA transcription of the p19 chain of IL-23 was highest in macrophages (Fig. 5A) and transcription and production of the p40 chain was evident in macrophages and DC (Fig. 5B, E), reflected in production of the IL-23 molecule (Fig. 5D).
Figure 5. IL-23 vs IL-12 production by APC exposed to P. gingivalis.
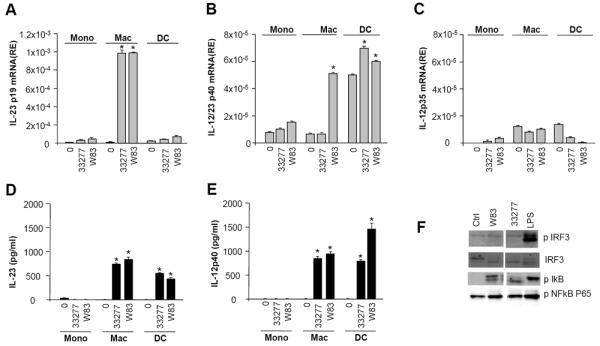
Donor matched monocyte (mono), macrophage (mac) and dendritic cells (DC) (3×106/ml) were untreated or exposed to P. gingivalis ATCC 33277 or W83 at a ratio of 1:50. At 1h cells were processed for total RNA and supernatants from 24h cultures were collected and tested for cytokine levels by multiplex ELISA. A. mRNA levels (RE= relative expression) for p19 measured by real-time PCR. B. IL-12p40 mRNA expression levels. C. IL-12p35 mRNA expression levels. D. IL-23 protein levels. E. IL-12p40 protein levels. All data representative of n=3 experiments,* p<0.05, * representing significance between untreated and P. gingivalis exposed. At 30min cells were processed for protein (signaling); F. Western blots for pIRF3, IRF, pIκB, pNFκB.
Since the p40 chain of IL-23 is shared with IL-12, we also monitored P. gingivalis-induced IL-12. In contrast to the p40 chain, transcription of p35, which is needed to form the IL-12 molecule, was minimal (Fig. 5C), indicative of differential transcriptional regulation [23] [24]. Protein production of the intact IL-12p70 heterodimer was undetectable (not shown), limiting P. gingivalis-induced Th1 polarization.
Consistent with our cytokine expression patterns, we observed abundant NFκB activation, corresponding to increased pIκB phosphorylation by P. gingivalis and E.Coli LPS (Fig. 5F, representative experiment in macrophages shown). IRF-3 phosphorylation, linked to IL-12p35 gene expression [23], was minimal in response to both strains of P. gingivalis but highly inducible by E.coli LPS (Fig. 5F), reflecting possible differential engagement of TLR pathways. Finally, cytokine levels for IFNα/β that may also facilitate IL-12p35 generation[23], were also undetectable in response to P. gingivalis (not shown).
Differential degradation of cytokine mediators by P. gingivalis
Our data show that the encounter between P. gingivalis and APC appears to preferentially trigger the release of mediators that may enrich the Th17 compartment in the inflammatory milieu. P. gingivalis also influences the immune response through its cysteine proteinases, referred to as gingipains, which degrade various cytokines[25]. To evaluate the contribution of protease degradation in the cytokine profiles elicited by P.gingivalis, we performed bacterial stimulation experiments in the presence and absence of gingipain specific inhibitors and measured levels of inducible cytokines after 24hr. Our results show that addition of gingipain inhibitors led to a modest increase in the levels of APC secreted IL-1β and a significant increase in the levels of IL-6 and IL-23. IL-12p70 was undetected even after the addition of inhibitors, suggesting an absence of cytokine production rather than increased gingipain degradation (Fig 6A, representative levels in DC shown). Similarly, addition of supernatants from APC exposed to P.gingivalis in the presence of protease inhibitors further enhanced Th17 differentiation, but did not alter Th1 responses (data not shown).
Figure 6. Cytokine degradation by P. gingivalis.

Antigen presenting cells (APC), including donor-matched monocyte, macrophage and dendritic cells were untreated or exposed to P. gingivalis strains 33277 and W83 (1:50) in the presence/absence of P. gingivalis specific protease inhibitors (gingipains KYT-1 and KYT-36, at a concentration of 106M) for 24h and supernatants were tested for concentrations of IL-1β, Il-6, IL-23 and IL-12p70.
A. Supernatant cytokine levels in 24h cultures of DC exposed to P. gingivalis (2 strains) +/- PI (protease inhibitors), representative experiment shown, *indicates significance at p<0.05. B. Recombinant cytokines (IL-1β, IL-6, IL-12p70, IL-23 at 5000pg/ml) were exposed to P. gingivalis at 105-107 bacteria/ml for 10-20min. Detected cytokine levels are shown at 20 min after P.gingivalis exposure at 105 bacteria/ml. Cytokine levels expressed as a percentage of input levels. C. Protein gel of IL-1β post P.gingivalis (2 strains) exposure at 20 min. D. Protein gel of IL-12 post P. gingivalis exposure at 20 min.
To directly assess the potential of P. gingivalis proteases to differentially target cytokine substrates, relevant recombinant cytokines at standardized concentrations were incubated with bacteria (105-107/ml) for indicated intervals. Intact and bacteria-exposed cytokine levels were then compared by multiplex ELISA (Fig. 6B). At early time points (10-20min, shown in Fig. 6B), detectable levels of IL-1β remained near input levels, although possible fragmentation/inactivation of these molecules may not always be discriminated by ELISA. By comparison, IL-6, IL-12, IL-23 and IL-22 were markedly reduced by bacterial exposure (Fig. 6B), irrespective of bacterial strain. A time course for cytokine depletion revealed that although detectable levels of IL-1β decreased over time (10min-2h, not shown), >50% of the measurable protein was retained after 2hr. In contrast IL-12 was more rapidly depleted and virtually undetectable within 10 min (only 10-20 min time points shown).
In additional experiments, gel separation and visualization of P. gingivalis-mediated IL-1β and IL-12 degradation products were performed. Consistent with the ELISA data, IL-1β appeared relatively resistant to degradation, particularly when exposed to low concentrations of bacteria (Fig. 6C). At high concentrations of bacteria IL-1β was modestly cleaved (Fig. 6C), as evident by the reduced intensity of the intact cytokine band. By comparison, IL-12 was susceptible to degradation (Fig. 6D) and at highest concentrations of bacteria IL-12 appeared completely degraded within 10-20 minutes.
Collectively, our data suggest that P. gingivalis may favor Th17 differentiation not only by triggering APC to produce Th17 supporting cytokines, but also by differentially degrading polarizing mediators.
DISCUSSION
Periodontitis, a common mucosal inflammatory condition which affects approximately 10% of the general population in its severe form with overt tissue destruction, appears to involve a Th17 polarizing milieu. These findings expand previous clinical studies demonstrating high levels of IL-17 cytokine in the sera of patients with aggressive periodontitis and elevated levels of IL-17 related cytokines in periodontitis tissues[9, 26, 27]. We show an increased expression of IL-17 in severe periodontitis as compared to other Th secreted cytokines and demonstrate that levels of IL-17 correlate with increased tissue damage in this setting. The predominance of this cytokine in severe lesions could suggest that it has a pathogenic role in driving immunopathology in this setting. Additionally, it could be speculated that shifts in microbial biofilms associated with areas of severe disease may drive the production of IL-17 and generation of pathogenic Th17 cells. How Th17 pathways become activated and dominate in this and other chronic inflammatory conditions remains a topic of increased interest[14, 28]. Our data show that the periodontal pathogen P. gingivalis may be instrumental and can support Th17 differentiation by at least two distinct mechanisms. First, exposure to P. gingivalis leads to the generation of Th17 supporting cytokines such as IL-1β, IL-6 and IL-23, but not to the induction of the Th1 related IL-12. P. gingivalis expresses a number of virulence factors, including its LPS and fimbria, which signal through TLR ligands, particularly TLR2[2], thus activating NFκB-dependent proinflammatory pathways in innate cells [9, 29, 30]. In our novel comparison of immature monocytes, differentiated macrophages and myeloid DC responses to P. gingivalis, all of which are evident in periodontal disease lesions[9], we found that these cell populations exhibited overlapping and unique responses. Monocytes and DC were most effective in driving Th17 development, relative to cultured macrophages exposed to P. gingivalis. This correlated with highest levels of IL-1β production, despite similar transcription levels, a phenomenon likely attributed to differential post-translational modifications by the inflammasome complex [22].
In our system, P. gingivalis challenge of APC did not enhance Th1 differentiation, consistent with undetectable levels of P. gingivalis inducible IL-12p70. Of particular interest was the ability of P.gingivalis to induce transcription of the p40 chain, which is shared between the IL-12 and IL-23 molecules and not the p35 chain, which is essential for the formation of functional IL-12p70. Differential regulation of transcription of the two chains of IL-12 has been previously studied in the context of microbial exposure and TLR ligation. The MyD88-dependent pathway, typically activated via TLR1,2,4,5 or 6 ligands is required for IL-12p40 gene expression, whereas TRIF-dependent signaling, typically activated by TLR4 ligands appears critical for IRF-3-dependent IL-12p35 gene expression[24] and also for the activation of an autocrine loop in which type I IFN production facilitates the generation of IL-12p35[23]. In our studies, P. gingivalis favored MyD88 pathways leading to NFκB activation over pathways necessary for IRF3 phosphorylation. Consequently, P. gingivalis did not support induction of type I IFN production, consistent with the notion that it primarily engages TLR2 related pathways [2] [9].
An additional path by which P. gingivalis may bias towards a Th17 response is through the differential proteolytic degradation of cytokine mediators. P. gingivalis possess cysteine proteinases, referred to as gingipains, which are products of three genes rgpA, rgpB, and kgp, and can cleave synthetic and natural substrates after arginine (gingipain R, RgpA or RgpB) or lysine residues (gingipain K, kgp)[25]. While the gingipains exhibit multiple activities, their ability to degrade factors of the immune system may be pivotal through inactivation or degradation of complement components, antimicrobial peptides and cytokine-chemokine mediators[25]. We demonstrate that Th1 and Th17 driving cytokines are degraded by P. gingivalis proteases to varying degrees, which may influence immunopathogenesis. Of the cytokines studied, IL-1β, capable of aiding in the differentiation of Th17 responses in humans [12], was the most resistant to P. gingivalis cleavage.
Our data also reveal differences in the ability of the two tested P. gingivalis strains to induce proinflammatory responses. A number of studies have shown considerable variation in virulence between strains of P. gingivalis in animal models, with strains W83, W50, ATCC 49417, A7A1 being classified as virulent and strains 381, 33277 and 23A4 considered less virulent, reflective, at least in part of the capsule status[6]. In this regard, W83 strain is an encapsulated strain, while 33277 does not possess a capsule[7]. Capsular types of P. gingivalis have been shown to contributed to virulence via an invasive spreading pattern of infection in experimental models, whereas the non-encapsulated strains induce mostly a localized abscess[31]. Capsular serotypes of P. gingivalis are also most efficient in inducing pro-inflammatory responses[30] and in particular W83 has most clearly associated with periodontitis status in humans[32, 33]. Accordingly, in our system W83 was most potent in inducing the transcription/production of cytokines and pathways that support downstream generation of Th17 responses.
Ultimately, the ability of P. gingivalis strains to drive inflammatory responses, particularly of a Th17 nature, may underlie tissue pathology in periodontal disease[11]. Our data show a significant correlation between IL-17 expression and bone loss in periodontitis, suggesting that the Th17 subset may drive or contribute to osseous destruction. One of the mechanisms linking Th17 with tissue destruction is the recently uncovered potential of Th17 cells to induce osteoclastogenesis and bone destruction in autoimmune arthritis[16, 18, 34]. It has been a frequent observation that in settings of autoimmunity and infection, exacerbated osteoclastic bone resorption is associated with activation of T-cell immune responses[35, 36]. Activated T cells express the osteoclast activating factor, receptor activator of nuclear factor kappa-B ligand (RANKL), that can directly act on osteoclast precursor cells to induce osteoclastogenesis[15, 18, 35]. Th17 cells are thought to exert their osteoclastic effect via IL-17 which may stimulate RANKL expression on osteoclastogenesis-supporting mesenchymal cells (fibroblasts and osteoblasts), while concurrently activating local inflammation, leading to the release of inflammatory cytokines such as IL-1β, IL-6 and TNFα, which further enhance RANKL signal transduction[15, 37]. Conversely, cytokines such as IFNγ, IL-12, IL-4 and IL-10, released by other Th cell subsets, apparently exert potent inhibitory effects on osteoclast differentiation [38][39]. The ability of P. gingivalis-exposed APC to enhance Th1 type responses, which are largely anti-osteoclastogenic, was modest and further impaired by P. gingivalis-mediated degradation. Consequently, an imbalance in these products may dictate a fate towards increased/pathologic osteoclastogenesis.
Finally, this potential of P. gingivalis to initiate inflammatory tissue-destructive chronic mechanisms may be of even greater relevance due to its ability to cross the epithelial barrier [40] and enter the systemic circulation. P. gingivalis has been detected in distant sites and has been implicated in the pathogenesis of a number of systemic inflammatory conditions. For example, titers of antibodies to P. gingivalis are high in patients with cardiovascular disease and stroke as well as rheumatoid arthritis (RA) and have been associated with disease activity measures[3] [41]. P. gingivalis DNA and antibodies have also been harbored in atheromas[3, 42, 43] and in synovial fluid of patients with RA[4, 5] where it may potentially contribute to bone resorptive activity[41, 44, 45]. Hence, uncovering mechanisms through which P. gingivalis may contribute to the generation of chronic inflammatory tissue-destructive mechanisms may be relevant not only for periodontitis, but also for a number of systemic inflammatory and autoimmune conditions.
Supplementary Material
Highlights.
Th17 cells are linked to tissue destructive inflammation in periodontitis
Th17 driving cytokines are abundant in tissues of periodontitis
The periodontal pathogen P. gingivalis may orchestrate induction of Th17 pathways
P.gingivalis drives production of Th17 not Th1 cytokines
P. gingivalis selectively degrades cytokine mediators favoring a Th17 milieu
Acknowledgments
The authors are grateful to Calley Grace for editorial assistance and Teresa Wild and Sarala Sarah for technical assistance. This research was supported in part by the Intramural Research Program of the NIH, National Institute of Dental and Craniofacial Research.
Footnotes
Disclosure
The authors have no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Darveau RP. Periodontitis: a polymicrobial disruption of host homeostasis. Nat Rev Microbiol. 8:481–90. doi: 10.1038/nrmicro2337. [DOI] [PubMed] [Google Scholar]
- 2.Hajishengallis G. Porphyromonas gingivalis-host interactions: open war or intelligent guerilla tactics? Microbes Infect. 2009;11:637–45. doi: 10.1016/j.micinf.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pussinen PJ, Alfthan G, Jousilahti P, Paju S, Tuomilehto J. Systemic exposure to Porphyromonas gingivalis predicts incident stroke. Atherosclerosis. 2007;193:222–8. doi: 10.1016/j.atherosclerosis.2006.06.027. [DOI] [PubMed] [Google Scholar]
- 4.Moen K, Brun JG, Madland TM, Tynning T, Jonsson R. Immunoglobulin G and A antibody responses to Bacteroides forsythus and Prevotella intermedia in sera and synovial fluids of arthritis patients. Clin Diagn Lab Immunol. 2003;10:1043–50. doi: 10.1128/CDLI.10.6.1043-1050.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martinez-Martinez RE, Abud-Mendoza C, Patino-Marin N, Rizo-Rodriguez JC, Little JW, Loyola-Rodriguez JP. Detection of periodontal bacterial DNA in serum and synovial fluid in refractory rheumatoid arthritis patients. J Clin Periodontol. 2009;36:1004–10. doi: 10.1111/j.1600-051X.2009.01496.x. [DOI] [PubMed] [Google Scholar]
- 6.Van Steenbergen TJ, Delemarre FG, Namavar F, De Graaff J. Differences in virulence within the species Bacteroides gingivalis. Antonie Van Leeuwenhoek. 1987;53:233–44. doi: 10.1007/BF00393930. [DOI] [PubMed] [Google Scholar]
- 7.Laine ML, Appelmelk BJ, van Winkelhoff AJ. Novel polysaccharide capsular serotypes in Porphyromonas gingivalis. J Periodontal Res. 1996;31:278–84. doi: 10.1111/j.1600-0765.1996.tb00494.x. [DOI] [PubMed] [Google Scholar]
- 8.Shelburne CE, An FY, Dholpe V, Ramamoorthy A, Lopatin DE, Lantz MS. The spectrum of antimicrobial activity of the bacteriocin subtilosin A. J Antimicrob Chemother. 2007;59:297–300. doi: 10.1093/jac/dkl495. [DOI] [PubMed] [Google Scholar]
- 9.Nares S, Moutsopoulos NM, Angelov N, Rangel ZG, Munson PJ, Sinha N, et al. Rapid myeloid cell transcriptional and proteomic responses to periodontopathogenic Porphyromonas gingivalis. Am J Pathol. 2009;174:1400–14. doi: 10.2353/ajpath.2009.080677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zeytun A, Chaudhary A, Pardington P, Cary R, Gupta G. Induction of cytokines and chemokines by Toll-like receptor signaling: strategies for control of inflammation. Crit Rev Immunol. 30:53–67. doi: 10.1615/critrevimmunol.v30.i1.40. [DOI] [PubMed] [Google Scholar]
- 11.O’Shea JJ, Paul WE. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science. 2010;327:1098–102. doi: 10.1126/science.1178334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. 2007;8:950–7. doi: 10.1038/ni1497. [DOI] [PubMed] [Google Scholar]
- 13.Miossec P, Korn T, Kuchroo VK. Interleukin-17 and type 17 helper T cells. N Engl J Med. 2009;361:888–98. doi: 10.1056/NEJMra0707449. [DOI] [PubMed] [Google Scholar]
- 14.Katsifis GE, Rekka S, Moutsopoulos NM, Pillemer S, Wahl SM. Systemic and local interleukin-17 and linked cytokines associated with Sjogren’s syndrome immunopathogenesis. Am J Pathol. 2009;175:1167–77. doi: 10.2353/ajpath.2009.090319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takayanagi H. Osteoimmunology and the effects of the immune system on bone. Nat Rev Rheumatol. 2009;5:667–76. doi: 10.1038/nrrheum.2009.217. [DOI] [PubMed] [Google Scholar]
- 16.Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med. 2006;203:2673–82. doi: 10.1084/jem.20061775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ji JD, Park-Min KH, Shen Z, Fajardo RJ, Goldring SR, McHugh KP, et al. Inhibition of RANK expression and osteoclastogenesis by TLRs and IFN-gamma in human osteoclast precursors. J Immunol. 2009;183:7223–33. doi: 10.4049/jimmunol.0900072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Okamoto K, Takayanagi H. Regulation of bone by the adaptive immune system in arthritis. Arthritis Res Ther. 13:219. doi: 10.1186/ar3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Armitage GC. Development of a classification system for periodontal diseases and conditions. Ann Periodontol. 1999;4:1–6. doi: 10.1902/annals.1999.4.1.1. [DOI] [PubMed] [Google Scholar]
- 20.Periodontology AAo. Parameter on chronic periodontitis with advanced loss of periodontal support. J Periodontal. 2000;74:856–8. doi: 10.1902/jop.2000.71.5-S.856. [DOI] [PubMed] [Google Scholar]
- 21.Periasamy S, Kolenbrander PE. Mutualistic biofilm communities develop with Porphyromonas gingivalis and initial, early, and late colonizers of enamel. J Bacteriol. 2009;191:6804–11. doi: 10.1128/JB.01006-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Franchi L, Eigenbrod T, Munoz-Planillo R, Nunez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 2009;10:241–7. doi: 10.1038/ni.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gautier G, Humbert M, Deauvieau F, Scuiller M, Hiscott J, Bates EE, et al. A type I interferon autocrine-paracrine loop is involved in Toll-like receptor-induced interleukin-12p70 secretion by dendritic cells. J Exp Med. 2005;201:1435–46. doi: 10.1084/jem.20041964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goriely S, Molle C, Nguyen M, Albarani V, Haddou NO, Lin R, et al. Interferon regulatory factor 3 is involved in Toll-like receptor 4 (TLR4)- and TLR3-induced IL-12p35 gene activation. Blood. 2006;107:1078–84. doi: 10.1182/blood-2005-06-2416. [DOI] [PubMed] [Google Scholar]
- 25.Holt SC, Ebersole JL. Porphyromonas gingivalis, Treponema denticola, and Tannerella forsythia: the “red complex”, a prototype polybacterial pathogenic consortium in periodontitis. Periodontol 2000. 2005;38:72–122. doi: 10.1111/j.1600-0757.2005.00113.x. [DOI] [PubMed] [Google Scholar]
- 26.Cardoso CR, Garlet GP, Crippa GE, Rosa AL, Junior WM, Rossi MA, et al. Evidence of the presence of T helper type 17 cells in chronic lesions of human periodontal disease. Oral Microbiol Immunol. 2009;24:1–6. doi: 10.1111/j.1399-302X.2008.00463.x. [DOI] [PubMed] [Google Scholar]
- 27.Ohyama H, Kato-Kogoe N, Kuhara A, Nishimura F, Nakasho K, Yamanegi K, et al. The involvement of IL-23 and the Th17 pathway in periodontitis. J Dent Res. 2009;88:633–8. doi: 10.1177/0022034509339889. [DOI] [PubMed] [Google Scholar]
- 28.Nell S, Suerbaum S, Josenhans C. The impact of the microbiota on the pathogenesis of IBD: lessons from mouse infection models. Nat Rev Microbiol. 8:564–77. doi: 10.1038/nrmicro2403. [DOI] [PubMed] [Google Scholar]
- 29.Stathopoulou PG, Benakanakere MR, Galicia JC, Kinane DF. Epithelial cell pro-inflammatory cytokine response differs across dental plaque bacterial species. J Clin Periodontol. 37:24–9. doi: 10.1111/j.1600-051X.2009.01505.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vernal R, Leon R, Silva A, van Winkelhoff AJ, Garcia-Sanz JA, Sanz M. Differential cytokine expression by human dendritic cells in response to different Porphyromonas gingivalis capsular serotypes. J Clin Periodontol. 2009;36:823–9. doi: 10.1111/j.1600-051X.2009.01462.x. [DOI] [PubMed] [Google Scholar]
- 31.van Steenbergen TJ, Kastelein P, Touw JJ, de Graaff J. Virulence of black-pigmented Bacteroides strains from periodontal pockets and other sites in experimentally induced skin lesions in mice. J Periodontal Res. 1982;17:41–9. doi: 10.1111/j.1600-0765.1982.tb01129.x. [DOI] [PubMed] [Google Scholar]
- 32.Griffen AL, Becker MR, Lyons SR, Moeschberger ML, Leys EJ. Prevalence of Porphyromonas gingivalis and periodontal health status. J Clin Microbiol. 1998;36:3239–42. doi: 10.1128/jcm.36.11.3239-3242.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Griffen AL, Lyons SR, Becker MR, Moeschberger ML, Leys EJ. Porphyromonas gingivalis strain variability and periodontitis. J Clin Microbiol. 1999;37:4028–33. doi: 10.1128/jcm.37.12.4028-4033.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pollinger B, Junt T, Metzler B, Walker UA, Tyndall A, Allard C, et al. Th17 cells, not IL-17+ gammadelta T cells, drive arthritic bone destruction in mice and humans. J Immunol. 186:2602–12. doi: 10.4049/jimmunol.1003370. [DOI] [PubMed] [Google Scholar]
- 35.Kong YY, Feige U, Sarosi I, Bolon B, Tafuri A, Morony S, et al. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature. 1999;402:304–9. doi: 10.1038/46303. [DOI] [PubMed] [Google Scholar]
- 36.Teng YT, Nguyen H, Gao X, Kong YY, Gorczynski RM, Singh B, et al. Functional human T-cell immunity and osteoprotegerin ligand control alveolar bone destruction in periodontal infection. J Clin Invest. 2000;106:R59–67. doi: 10.1172/jci10763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakashima T, Takayanagi H. Osteoimmunology: crosstalk between the immune and bone systems. J Clin Immunol. 2009;29:555–67. doi: 10.1007/s10875-009-9316-6. [DOI] [PubMed] [Google Scholar]
- 38.Yoshimatsu M, Kitaura H, Fujimura Y, Eguchi T, Kohara H, Morita Y, et al. IL-12 inhibits TNF-alpha induced osteoclastogenesis via a T cell-independent mechanism in vivo. Bone. 2009;45:1010–6. doi: 10.1016/j.bone.2009.07.079. [DOI] [PubMed] [Google Scholar]
- 39.Takayanagi H, Ogasawara K, Hida S, Chiba T, Murata S, Sato K, et al. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma. Nature. 2000;408:600–5. doi: 10.1038/35046102. [DOI] [PubMed] [Google Scholar]
- 40.Yilmaz O. The chronicles of Porphyromonas gingivalis: the microbium, the human oral epithelium and their interplay. Microbiology. 2008;154:2897–903. doi: 10.1099/mic.0.2008/021220-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.de Pablo P, Chapple IL, Buckley CD, Dietrich T. Periodontitis in systemic rheumatic diseases. Nat Rev Rheumatol. 2009;5:218–24. doi: 10.1038/nrrheum.2009.28. [DOI] [PubMed] [Google Scholar]
- 42.Ford PJ, Gemmell E, Timms P, Chan A, Preston FM, Seymour GJ. Anti-P. gingivalis response correlates with atherosclerosis. J Dent Res. 2007;86:35–40. doi: 10.1177/154405910708600105. [DOI] [PubMed] [Google Scholar]
- 43.Padilla C, Lobos O, Hubert E, Gonzalez C, Matus S, Pereira M, et al. Periodontal pathogens in atheromatous plaques isolated from patients with chronic periodontitis. J Periodontal Res. 2006;41:350–3. doi: 10.1111/j.1600-0765.2006.00882.x. [DOI] [PubMed] [Google Scholar]
- 44.Trombone AP, Claudino M, Colavite P, de Assis GF, Avila-Campos MJ, Silva JS, et al. Periodontitis and arthritis interaction in mice involves a shared hyper-inflammatory genotype and functional immunological interferences. Genes Immun. doi: 10.1038/gene.2010.13. [DOI] [PubMed] [Google Scholar]
- 45.Lundberg K, Wegner N, Yucel-Lindberg T, Venables PJ. Periodontitis in RA-the citrullinated enolase connection. Nat Rev Rheumatol. 6:727–30. doi: 10.1038/nrrheum.2010.139. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.