

Abstract
We evaluated the efficacy, pharmacokinetics, and safety of adalimumab in Japanese patients with active ankylosing spondylitis (AS) who had an inadequate response to, or who were intolerant of, treatment with ≥1 nonsteroidal anti-inflammatory drugs (NSAIDs). This phase 3, multicenter, open-label trial assessed the percentage of patients with a 20% response in the Assessment of SpondyloArthritis international society working group criteria (ASAS20) at week 12 as the primary endpoint. Secondary outcome measures included assessments of disease activity, clinical response, functionality, and spinal mobility at weeks 12 and 60. Serum trough adalimumab concentrations were summarized using descriptive statistics. The adverse event profile was summarized for patients who received at least one dose of the study drug during the assessment period. At week 12, 73.2% (30/41) achieved an ASAS20 response and nearly 40% met ASAS partial remission criteria; proportions were maintained after up to 60 weeks of therapy. Mean adalimumab concentrations reached steady-state between weeks 12 and 20. Adalimumab was generally safe and well tolerated, with approximately 90% of adverse events considered to be mild. These results support the use of adalimumab as a safe and effective therapy for Japanese patients with active AS.
Keywords: Adalimumab, Ankylosing spondylitis, ASAS, Japan, Safety
Introduction
Ankylosing spondylitis (AS) is a chronic and debilitating inflammatory disease of the axial skeleton, large peripheral joints, and entheses [1]. AS belongs to a group of diseases known as the spondyloarthritides, many of which share common features of sacroiliitis, human leukocyte antigen (HLA)-B27 positivity, extra-articular manifestations (e.g., uveitis, inflammatory bowel disease, etc.), and/or enthesitis. In Caucasians, the prevalence of AS is estimated to be as high as 0.9% [2], but the prevalence is estimated to be substantially lower in Japanese (0.0065%) [3] owing to reduced HLA-B27 positivity among Asian populations [4]. Disease onset, typically occurring by the third decade of life, is often missed in the primary care setting, as it can take several years for the chronic lower back pain to develop the hallmark of active disease, sacroiliitis [5]. In certain AS patients, the course of disease progression can lead to significant structural damage, syndesmophyte formation, functional impairment, and poor quality-of-life outcomes. As a result, individual and societal costs are high for AS [6–8].
There is a paucity of effective treatment options available to patients with AS. Traditional disease-modifying antirheumatic drugs (DMARDs) and systemic corticosteroid therapy are often ineffective in treating the signs and symptoms of AS [9]. Instead, treatment regimens often begin with physical modalities and/or nonsteroidal anti-inflammatory drugs (NSAIDs), but these may not adequately control disease symptoms in many patients and, in the case of NSAIDs, can be associated with toxicities [10]. The proinflammatory cytokine, tumor necrosis factor (TNF), has been found in increased concentrations in joints of patients with AS and has been identified in biopsies of affected sacroiliac joints [11]. In Western patients, TNF inhibition through application of biologic therapies is highly effective at improving the signs and symptoms of active AS, restoring physical function, increasing spinal mobility, and reducing the concentration of acute-phase reactants [12–19].
Adalimumab is a recombinant, fully human, anti-TNF monoclonal antibody indicated for treating active AS and has a favorable risk–benefit profile, which was established in the Adalimumab Trial Evaluating Long-Term Efficacy and Safety for Ankylosing Spondylitis (ATLAS) and M03-606 pivotal, placebo-controlled studies in Western patients [17, 18]. Because genetic, environmental, and/or cultural differences among disparate populations may result in unexpected clinical response rates, our study was designed to test adalimumab efficacy, pharmacokinetics (PK), and safety in Japanese patients with active AS who have had an inadequate response to, or intolerance of, one or more NSAIDs. The primary and secondary endpoints of this study were designed to mirror those obtained in the ATLAS and M03-606 studies [17, 18].
Methods
Patients
Patients were at least 15 years of age at the time of informed consent and met the definition of AS based on the modified New York criteria [20]. Patients must have had active disease at the time of enrollment, as defined by the fulfillment of at least two of the following: Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) score on a visual analog scale (VAS) ≥4 cm (BASDAI was measured using a VAS to allow for a more accurate determination of changes from baseline), total back pain on a VAS ≥40 mm, or duration of morning stiffness ≥1 h. Patients must have had an inadequate response to, or intolerance of, one or more NSAIDs, as defined by the investigator. Patients could have failed one or more DMARDs. Ongoing treatment with NSAIDs, methotrexate (MTX ≤8 mg/week), sulfasalazine (SSZ ≤1 g/day), or corticosteroids (prednisone, prednisone equivalents, ≤10 mg/day) was allowed during the study if the patient was on a stable dose for at least 4 weeks prior to baseline through week 16. Patients with total spinal ankylosis (bamboo spine) were also eligible for enrollment. Patients with psoriasis, uveitis, inflammatory bowel disease (e.g. ulcerative colitis, Crohn’s disease, etc.), and reactive arthritis were allowed to enroll in the study as long as these conditions were stable and well controlled as defined by the investigator’s clinical judgment, for at least 4 weeks prior to screening.
Patients with a history of cancer, lymphoma, leukemia, or lymphoproliferative disease—other than a successfully treated nonmetastatic cutaneous squamous- or basal-cell carcinoma in situ of the cervix—were excluded from enrollment. Similarly, patients were excluded if they had a history of human immunodeficiency virus (HIV) infection or other immunosuppressive disorder, or if they had experienced a recent infection that required hospitalization or treatment with anti-infectives. Patients with a poorly controlled medical condition, such as uncontrolled diabetes, congestive heart failure, or recent cerebrovascular accident, were also excluded. Patients were screened for latent tuberculosis (TB) infection using a purified protein derivative (PPD) skin test and a chest X-ray. Patients with active TB were excluded from the study. Patients identified as having latent TB (PPD ≥5 mm of induration or abnormal chest X-ray) were eligible for the study if they began prophylactic treatment (isoniazid 300 mg/day for 9 months) at least 3 weeks prior to the first administration of study drug. Patients who received anti-TNF therapy at any time prior to study entry, or any other biological or investigational biological agent in the past 6 months or five half-lives prior to baseline (whichever was longer), were excluded. Patients were also excluded if they had neurological symptoms suggestive of central nervous system demyelinating disease or if they had received DMARD therapy other than MTX or SSZ or any other immunosuppressants within 4 weeks prior to baseline. Patients were assessed for eligibility based on all inclusion and exclusion criteria during a 2-week screening period.
This study was conducted at 19 centers in Japan. The institutional review board (IRB) of each study site reviewed the ethical, scientific, and medical appropriateness of the protocol prior to study initiation. The study was conducted in compliance with the ethical principles of the year 2000 version of the Declaration of Helsinki. Patients provided written informed consent and complied with the requirements set forth in the study protocol. For patients <20 years of age, a parent or legal guardian also must have provided written informed consent.
Study design
This was a phase 3, multicenter, open-label trial of adalimumab in Japanese patients with active AS who had an inadequate response to, or who were intolerant of, treatment with one or more NSAIDs. Enrolled patients received adalimumab 40 mg every other week (eow) until the time of adalimumab approval for the AS indication in Japan. Patient data for up to 60 weeks of adalimumab therapy are presented in this analysis. Efficacy, PK, and safety were assessed at baseline, weeks 2, 4, 8, 12, 16, 20, 24, and every 12 weeks after week 24. Patients were to be injected with the study drug within a 3-day window from baseline to week 24, and within a 7-day window on or after week 26. An interval of at least 7 days was to be taken between any consecutive adalimumab injections. Patients who received proper training on self-injection techniques and agreed to record any relevant information regarding self-injection were allowed to conduct self-injection on or after week 2. Patients who completed 16 weeks of therapy but failed to achieve a 20% response in the Assessment of SpondyloArthritis international society working group criteria (ASAS20) on or after week 16 could have their dose of adalimumab increased to 80 mg eow, which would be continued throughout the study. Patients completing the study or discontinuing prematurely were to have a 4-week follow-up after the last dose of study drug and a 70-day follow-up to assess safety.
Efficacy evaluations
The primary efficacy endpoint was the percentage of ASAS20 responders at week 12. An ASAS20 response was defined as ≥20% improvement in three of the following four domains, with no deterioration (defined as a worsening of ≥20% and a net worsening of ≥10 U) in the remaining domain: patient’s global assessment of disease activity (PaGA), total back pain (on a VAS), functionality [through the Bath Ankylosing Spondylitis Functional Index (BASFI), on a VAS scale], and inflammation (mean of BASDAI questions 5 and 6) [21]. An ASAS20 response rate of at least 40% was required to satisfy study efficacy criteria. This rate was established on the basis of the lower limit of the 95% confidence interval (CI) of normal approximation following combination of efficacy results from the ATLAS and M03-606 studies (expected response = 56.5 ± 17.7%). The effect of concomitant medications (e.g., DMARDs, corticosteroids, etc.) and total spinal ankylosis on the primary efficacy endpoint was summarized. Secondary efficacy variables by visit included the following: ASAS20/50/70 responses, change in PaGA, change in total back pain, change in BASFI, change in C-reactive protein (CRP), BASDAI50 response, change in Bath Ankylosing Spondylitis Metrology Index (BASMI), change in the Maastricht Ankylosing Spondylitis Enthesitis Score (MASES), changes in physical and mental components of the SF-36, the short-form health assessment with 36 questions, and the percentages of patients achieving ASAS40 (defined as improvement of ≥40% and absolute improvement of ≥20 U from baseline in three or more of the four domains of the ASAS20, with no worsening in the potential remaining domain) and ASAS partial remission [defined as a value <20 on a 0–100 scale in each of the four ASAS domains (PaGA, pain, function, and inflammation)] responses. At weeks 12 and 60, the percentage of patients achieving an ASAS 5/6 [defined as a ≥20% improvement in five of the six domains (BASFI, total back pain, PaGA, inflammation, BASMI, and CRP)] response was summarized. All efficacy data were summarized using last observation carried forward (LOCF).
Pharmacokinetic evaluations
Blood samples to determine concentrations of adalimumab and antiadalimumab antibodies (AAA) were collected at baseline and weeks 2, 4, 8, 12, 16, 20, 24, and every 12 weeks thereafter, prior to adalimumab injection (to obtain serum trough concentrations). Concentrations of adalimumab and AAA were quantified using a validated enzyme-linked immunosorbent assay (ELISA), based on a double-antigen technique [22]. The lower limit of quantitation (LLOQ) for adalimumab was established at 3.125 ng/mL in diluted serum; concentrations less than the LLOQ were considered to be undetectable (i.e. 0). Mean adalimumab concentrations were determined separately for patients who received concomitant MTX therapy and for those who received adalimumab without concomitant MTX therapy. Individual adalimumab concentrations were plotted for patients receiving an escalated dose of adalimumab. The LLOQ for AAA was established at 1.0 ng/mL in diluted serum. Samples with serum adalimumab concentrations <2 μg/mL were analyzed for AAA and were considered positive if the measured AAA concentration was >20 ng/mL in diluted serum. Serum trough adalimumab and AAA concentrations were summarized using descriptive statistics.
Safety evaluations
The safety assessment group consisted of all patients who received at least one dose of the study drug during the assessment period. A treatment-emergent adverse event (TEAE) was defined as an AE with onset or worsening after the patient’s first injection of the study drug and up to 70 days after the patient’s last injection. The number and percentages of patients experiencing a TEAE, as well as the number of events, were tabulated using the Medical Dictionary for Drug Regulatory Affairs (MedDRA) system organ class and MedDRA preferred terms (v11.0). In addition, a summary of AEs by severity (severe, moderate, mild) and relationship to the study drug (probably related, possibly related, probably not related, not related) as assessed by the investigator was tabulated. Severe and serious AEs, including serious infections, malignancies, tuberculosis, and death, were summarized. Any TEAE leading to study discontinuation was also summarized.
Results
Patient disposition and baseline demographics and disease characteristics
Data were collected for patients enrolled at 19 centers across Japan from 17 March 2008 to 21 November 2009. A total of 41 patients entered the study, 40 of whom completed 12 weeks of open-label adalimumab treatment. Thirty-seven patients (90.2%) received open-label adalimumab treatment through week 60. Of the four discontinuations occurring prior to week 60, two occurred as a result of AEs (please see below), one was discontinued for not satisfying entry criteria (which was determined only after the first dose of the study drug), and the fourth relocated and was no longer within proximity to the investigator’s practice. In general, baseline demographics and disease characteristics of the enrolled patients were typical of the Western study populations with active AS enrolled in pivotal trials of various anti-TNF therapies [15–17, 19] (Table 1), with a mean BASDAI score of 6.2 cm and mean total back pain of 63.0 mm on VAS. However, average disease duration at baseline (4.1 years) was considerably lower than was observed in the Western anti-TNF pivotal trials [15–17]. Further, more patients received concomitant DMARDs [n = 24 (58.5%): 16 received MTX, 12 received SSZ] and corticosteroid [n = 19 (46.3%)] therapy, albeit at lower average doses (data not shown). All 41 patients (100%) received ongoing NSAID therapy at study entry. With respect to history of extra-articular manifestations typically seen with AS, three patients had been diagnosed with psoriasis, eight had a history of uveitis, and two had preexisting inflammatory bowel disease (both were ulcerative colitis). A total of seven patients (17.1%) were deemed by their rheumatologist to have total spinal ankylosis.
Table 1.
Baseline demographics and disease characteristics
| Patient characteristics | Adalimumab (N = 41) |
|---|---|
| Age (years) | 37.2 ± 12.2 |
| Male, n (%) | 32 (78) |
| HLA-B27 positive, n (%) | 20 (48.8) |
| Duration of AS (years) | 4.1 ± 6.6 |
| Duration of AS <10 years, n (%) | 35 (85.4) |
| Total spinal ankylosis, n (%) | 7 (17.1) |
| Baseline DMARD use, n (%)a | 24 (58.5) |
| Baseline NSAID use, n (%) | 41 (100.0) |
| Baseline corticosteroid use, n (%) | 19 (46.3) |
| Global assessment (0–100 mm VAS) | 64.5 ± 17.2 |
| Total back pain (0–100 mm VAS) | 63.0 ± 17.7 |
| Inflammation (0–10 cm VAS) | 6.3 ± 2.2 |
| BASFI (0–100 mm VAS) | 37.8 ± 23.2 |
| BASDAI (0–10 cm VAS) | 6.2 ± 1.5 |
| CRP, mg/dL | 1.6 ± 1.6 |
| BASMI (0–10) | 4.0 ± 2.1 |
| Chest expansion (0–10 cm) | 2.8 ± 1.7 |
| SJC (0–44 joints) | 1.7 ± 3.3 |
| TJC (0–46 joints) | 4.8 ± 8.1 |
| Physical component of the SF-36 | 33.7 ± 8.9 |
| Mental component of the SF-36 | 40.9 ± 11.5 |
All values are mean ± standard deviation, unless otherwise indicated
HLA-B27 human leukocyte antigen-B27, AS ankylosing spondylitis, DMARD (nonbiologic) disease-modifying antirheumatic drug, NSAID nonsteroidal anti-inflammatory drug, VAS visual analog scale, BASFI Bath Ankylosing Spondylitis Functional Index, BASDAI Bath Ankylosing Spondylitis Disease Activity Index, CRP C-reactive protein, BASMI Bath Ankylosing Spondylitis Metrology Index, SJC swollen joint count, TJC tender joint count, SF-36 short-form health status survey of 36 questions
aOngoing DMARD treatment was allowed to continue for patients receiving a stable dose of methotrexate and/or sulfasalazine
Efficacy
Of the 41 patients enrolled in the study, 30 (73.2%) met the primary endpoint of ASAS20 response at week 12 (Fig. 1). This percentage surpassed the prespecified efficacy criterion of 40%. Medications taken with adalimumab did not appear to alter ASAS20 response rates. Comparable ASAS20 response rates were observed for patients treated with or without concomitant DMARD at baseline [75.0% (n = 18/24) vs. 70.6% (n = 12/17), respectively] and for patients treated with or without concomitant corticosteroid therapy [68.4% (n = 13/19) vs. 77.3% (n = 17/22), respectively]. Similarly, comparable ASAS20 response rates were observed for patients with and without total spinal ankylosis [71.4% (n = 5/7) vs. 73.5% (n = 25/34), respectively]. Further, more substantial improvements in ASAS response criteria were observed within the first 12 weeks of adalimumab therapy. A total of 23 (56.1%) and 13 (31.7%) patients achieved ASAS50 and ASAS70 responses, respectively, at week 12 (Fig. 1). These clinical improvements occurred rapidly and were observed within 2 weeks of adalimumab initiation, with rates approaching the maximum observed levels within 12 weeks. Response rate magnitude was either sustained or improved at the time points evaluated over the 60-week period of adalimumab therapy.
Fig. 1.
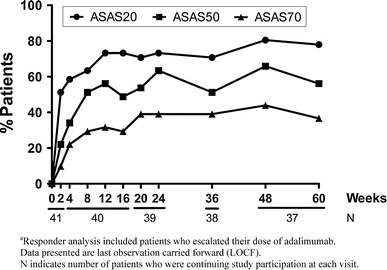
Percentages of patientsa who achieved Assessment of SpondyloArthritis international society working group criteria (ASAS)20, ASAS50, and ASAS70 responses by visit
Further, adalimumab treatment led to effective disease management, with 68.3% of patients achieving an ASAS5/6 response following 12 weeks of therapy (Fig. 2). Similarly, 63.4% of patients achieved an ASAS40 response at week 12, and nearly 40% exhibited an ASAS partial remission response. The proportions of patients achieving ASAS5/6, ASAS40, and ASAS partial remission responses were all maintained at week 60.
Fig. 2.
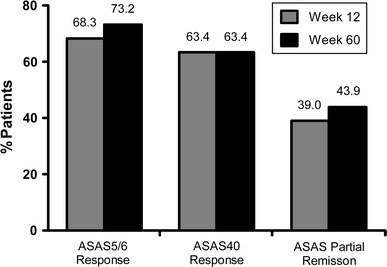
Percentages of patients who achieved Assessment of SpondyloArthritis international society working group criteria (ASAS)5/6, ASAS40, and ASAS partial remission responses following 12 and 60 weeks of adalimumab therapy
Mean improvements in the four domains of the ASAS response—PaGA, total back pain, BASFI, and inflammation—were observed within 12 weeks of adalimumab initiation (Table 2). In fact, each of these four response measures were reduced to less than half of their baseline values within 12 weeks of adalimumab treatment. These improvements were maintained at all time points evaluated throughout the 60 weeks of adalimumab therapy (Table 2, and data not shown). Similar improvements were observed across a range of AS signs and symptoms following treatment with adalimumab, including improvements in the concentration of acute-phase reactants (CRP), in spinal mobility (BASMI), and in enthesitis (MASES) scores (Table 2). In addition, adalimumab treatment led to a clinically relevant decrease (≥22.5% decrease, [23]) in the mean BASDAI score. Following 12 weeks of therapy, 27 of 41 patients (65.9%) achieved a 50% improvement in BASDAI response, and a similar proportion maintained this response at all time points evaluated through week 60. Treatment with adalimumab improved mean patient scores in both the physical and mental components of the SF-36 questionnaire in clinically meaningful increments (≥3.0 points, [24]) by week 12, and this level of response was maintained at all time points evaluated through 60 weeks.
Table 2.
Summary of mean changes in clinical signs and symptoms from baseline to week 12 and baseline to week 60
| Assessment | Baseline to week 12 | Baseline to week 60 |
|---|---|---|
| PaGA (0–100 mm VAS) | −34.6 (−42.8, −26.3) | −38.7 (−47.3, −30.2) |
| Total back pain (0–100 mm VAS) | −35.6 (−43.7, −27.6) | −37.8 (−45.9, −29.7) |
| BASFI (0–100 mm VAS) | −19.4 (−24.5, −14.3) | −21.0 (−27.1, −14.9) |
| Inflammation (mean of questions 5 and 6 on the BASDAI) (0–10 cm VAS) | −3.6 (−4.6, −2.7) | −4.0 (−4.9, −3.1) |
| BASDAI (0–10 cm VAS) | −3.4 (−4.2, −2.7) | −3.9 (−4.6, −3.2) |
| CRP, mg/dl | −1.2 (−1.6, −0.8) | −1.4 (−1.8, −1.0) |
| BASMI (range 0–10) | −0.4 (−0.8, −0.02) | −0.5 (−1.0, −0.2) |
| MASES | −1.0 (−1.7, −0.4) | −1.2 (−1.9, −0.6) |
| Physical component of the SF-36 | 9.6 (6.8, 12.4) | 11.6 (8.9, 14.4) |
| Mental component of the SF-36 | 7.0 (3.1, 10.9) | 7.3 (3.5, 11.2) |
All values are mean (95% confidence interval) change from baseline
PaGA patient’s global assessment of disease activity, VAS visual analog scale, BASFI Bath Ankylosing Spondylitis Functional Index, BASDAI Bath Ankylosing Spondylitis Disease Activity Index, CRP C-reactive protein, BASMI Bath Ankylosing Spondylitis Metrology Index, MASES Maastricht Ankylosing Spondylitis Enthesitis Score, SF-36 short form health status survey of 36 questions
A total of six patients (14.6%) failed to reach an ASAS20 response on or after week 16 and received an increased dose of adalimumab (dose escalation to 80 mg eow). Following the dose increase, two of these six patients achieved an ASAS20 response at follow-up visits and maintained this response at week 60. Although the remaining four patients either failed to achieve or maintain an ASAS20 response through week 60, each experienced improvements in at least one of the objective variables (e.g. CRP, BASMI, chest expansion, MASES).
Pharmacokinetics
In patients with RA, serum clearance of adalimumab is lower when MTX is coadministered [22, 25]. Therefore, serum trough adalimumab concentrations were evaluated separately for patients who received adalimumab with MTX (n = 15) and for those who received adalimumab without MTX (n = 23). Similarly, adalimumab concentrations were assessed separately following dose escalation. Mean (SD) concentrations for patients receiving adalimumab with and without MTX reached steady-state by week 20 at 11.44 (4.93) μg/mL and week 12 at 8.03 (4.87) μg/mL, respectively, and remained relatively constant at all time points evaluated through 60 weeks of therapy (Fig. 3a). Dose escalation was associated with a concomitant increase in serum trough adalimumab concentrations for the majority (n = 4/6, 66.7%) of patients (Fig. 3b) but had little impact on the concentration of adalimumab in the remaining two patients.
Fig. 3.
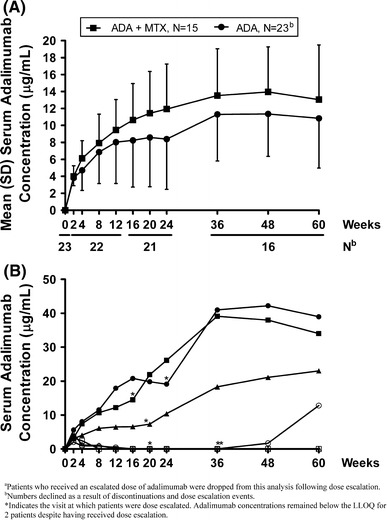
a Mean serum adalimumab (ADA) concentrations by visit in Japanese patients with ankylosing spondylitis (AS) receiving adalimumab 40 mg every other week with [+ standard deviation (SD)] and without (−SD) concomitant methotrexate (MTX) therapya; b serum adalimumab concentrations by visit in Japanese patients (n = 6) receiving adalimumab 40 mg every other week, with dose escalation to 80 mg every other week
Fewer than 10% (n = 4/41) of patients treated with adalimumab tested positive for AAA at one or more visits on or before week 24; none of the 16 patients who received concomitant MTX therapy and only one of the 12 who received concomitant SSZ therapy became AAA positive. Of the four patients who became AAA positive during the course of the study, only two remained AAA positive at week 60. Both patients had received adalimumab dose escalation, and one achieved an ASAS20 response at week 36 and maintained it at week 60. The other did not meet an ASAS20 response at any visit subsequent to the development of AAA-positive status. The remaining two patients became AAA negative prior to study end; both were ASAS20 responders at week 60.
Safety
Patients were treated with adalimumab for a mean (SD) duration of 391.6 (92.8) days. During this time and through the safety follow-up period, all 41 patients experienced at least 1 AE (Table 3), the majority of which were reported to be mild. Four patients (9.8%) experienced a total of seven serious AEs (SAEs), including one case each of thrombocytopenia, periodontitis, intervertebral discitis, osteomyelitis, pneumonia, breast cancer, and adenomyosis. The events of intervertebral discitis and osteomyelitis, the only two SAEs considered at least possibly or probably related to adalimumab, occurred in one patient, as did the events of pneumonia and breast cancer; adalimumab therapy was discontinued prior to week 60 in both of these patients as a result of these SAEs. All four of these events resolved and were not reported throughout the duration of safety follow-up. A total of 25 patients (61.0%) experienced 51 infectious AEs, with nasopharyngitis as the most frequent infectious AE to be reported. Other than the two events requiring study discontinuation (intervertebral discitis and osteomyelitis in one patient), all remaining events resolved with appropriate treatment and did not lead to early study termination. One patient developed an opportunistic infection (cytomegalovirus), which, although considered to be moderate in severity, resolved with treatment.
Table 3.
Summary of adverse events through up to 60 weeks of adalimumab therapy
| Number (%) | Events | |
|---|---|---|
| Any adverse event (AE) | 41 (100) | 250 |
| AE at least possibly drug-related | 24 (58.5) | 73 |
| Serious AE | 4 (9.8) | 7 |
| AE leading to discontinuation of study drug | 2 (4.9) | 3 |
| Infectious AE | 25 (61.0) | 51 |
| Serious infectious AE | 2 (4.9) | 3 |
| Tuberculosis (TB) | 0 (0.0) | 0 |
| Opportunistic infection (excluding TB)a | 1 (2.4) | 1 |
| All malignanciesb | 1 (2.4) | 1 |
| Injection site reaction | 9 (22.0) | 19 |
| Hepatic related AE | 13 (31.7) | 15 |
All values are the number (%) of patients. Patients and adverse events may be counted in more than one adverse event category
aCytomegalovirus infection
bBreast cancer
Injection-site reactions represented the next most common AE, with 19 events occurring in nine patients (22.0%). Of these, injection-site erythema was the most frequent (14.6% of patients). All injection-site reactions were reported to be mild, and all but one resolved without the need for treatment. A total of 15 hepatic events were reported in 13 patients (31.7%), of which abnormal liver function (n = 8, 19.5%) and hepatic steatosis (n = 3, 7.3%) had incidence rates ≥5%. In general, hepatic-related AEs were associated with alanine transaminase (ALT) or aspartate aminotransferase (AST) values that exceeded or approached 2.5 times the upper limit of normal at one or more study visits, although these abnormalities typically did not persist and were only accompanied by a concomitant increase in total bilirubin in one patient. Of the 13 patients who developed hepatic-related AEs, ten received concomitant DMARD therapy. One of the three patients who had an AE of hepatic steatosis had a preexisting medical history of the disease. However, no hepatic event was deemed to be serious by the investigators, and none led to study discontinuation.
There were no reports of tuberculosis, lymphoma, or nonmelanoma skin cancer during the safety assessment period, and no patient died during the course of the study. One malignancy (breast cancer) developed approximately 50 days after starting adalimumab treatment. This patient had surgery as treatment of the SAE and was discontinued from the study. Among the six patients who increased their adalimumab dose to 80 mg eow, there were no increased incidences of AEs, including hepatic events, nor did new events emerge as a result of dose escalation compared with the study period prior to dose escalation. Furthermore, the overall incidence of AEs among the dose-escalation group was comparable with those who did not receive an escalated dose.
Discussion
AS is a chronic and debilitating disease of the axial skeleton that can result in fusion of vertebrae and progressive functional disability in young adults. Treatment with TNF inhibitors, such as the fully human anti-TNF antibody, adalimumab, leads to marked improvement in signs and symptoms of active disease in Western patients [12–14, 17–19], but their efficacy in Japanese patients with AS has not been previously reported. The results presented in this open-label trial suggest that adalimumab is an effective and safe option for Japanese patients with AS who have had an inadequate response to, or who are intolerant of, NSAID therapy. Greater than two thirds of patients achieved the primary efficacy endpoint (ASAS20 response) within the first 12 weeks of therapy initiation, and the AE profile following up to 60 weeks of exposure was similar to the known profile of adalimumab in other inflammatory, immune-mediated diseases [26].
Open-label adalimumab treatment effectively managed the signs and symptoms of AS, with improvements occurring rapidly in all areas evaluated (e.g., physical function, back pain, inflammation, spinal mobility, enthesitis, etc.). The magnitude of responses, observed as early as the first dose (week 2), was maintained through up to 60 weeks of treatment. Further, treatment led to improvements in composite indices reflective of the full scope of disease characteristics. Specifically, ASAS40 and ASAS5/6, the latter of which includes both spinal mobility (BASMI) and acute-phase reactants (CRP), in addition to the four components of the ASAS response criteria, have been proposed to represent major clinical responses. The majority of patients experienced ASAS40 (63.4%) and ASAS5/6 (68.3%) responses within 12 weeks of adalimumab therapy initiation, and comparable proportions achieved these response rates through up to 60 weeks of therapy. Furthermore, approximately 40% of patients achieved an ASAS partial remission response at weeks 12 and 60.
A number of observations can be made about the efficacy data presented in this analysis. Use of DMARDs and/or corticosteroids with adalimumab therapy appears to have offered little added benefit to Japanese patients with AS. This is in line with the ASAS recommendations for managing AS, which indicate a lack of evidence for efficacy of these medications on axial disease [9]. Comparable rates of clinical efficacy were observed for patients treated with and without concomitant DMARD and/or corticosteroid therapy, supporting the use of adalimumab monotherapy in this population.
This study enrolled a small number of patients (n = 7) with total spinal ankylosis, a population that is often excluded from AS trials because of the limited information available regarding their response to TNF inhibition and advanced disease state. Data from this study suggest that patients with total spinal ankylosis also benefit from adalimumab therapy, a finding that is consistent with results from the ATLAS study [27]. Future studies that include more patients with total spinal ankylosis would allow further evaluation of the efficacy of adalimumab in this population.
Lastly, dose escalation of adalimumab (to 80 mg eow) may be an effective means by which to achieve the desired clinical response for adalimumab-treated patients who fail to demonstrate an adequate response within a few months of therapy initiation. Our study results suggest that dose escalation of adalimumab may be appropriate for certain AS patients who do not achieve an adequate response to the standard dose of 40 mg eow.
Serum trough adalimumab concentrations were higher in patients who received concomitant MTX therapy than for those without, as previously reported [22, 25]. However, concentrations of adalimumab were within the known therapeutic range in both groups of patients [25], and both groups demonstrated comparable rates of efficacy, as measured through ASAS20 response. Fewer than 10% of patients (n = 4) developed AAA-positive status following up to 24 weeks of treatment with adalimumab, but only two of them had detectable levels of AAA at week 60. This finding suggests that the presence of AAA is not sustained throughout long-term therapy; however, an examination into the kinetics of AAA development would require a study with larger enrollment. The percentage of AAA-positive patients in this study was lower than that reported in a 24-week study of Japanese patients with RA [28], in which approximately 40% of patients were AAA positive at some point during the study. The development of AAA-positive status during this study might be a unique feature associated with adalimumab monotherapy, as no patient receiving adalimumab with concurrent MTX became AAA positive during the course of this study. These data further emphasize the protective role that even a low dose of DMARD therapy can have on the development of antiadalimumab immunogenicity [25].
Adalimumab was shown to be generally safe and well tolerated, and the AE profile was comparable with that reported in the ATLAS and M03-606 studies of adalimumab in Western patients with AS [17, 18] and with Japanese studies of adalimumab in RA, psoriasis, and Crohn’s disease [28, 29], although the rate of infectious AEs was numerically higher in this study than was reported in the ATLAS study [17]. Only two patients discontinued due to AEs (one as a result of breast cancer and pneumonia, and one as a result of intervertebral discitis and osteomyelitis). Although abnormal liver function (19.5%) and hepatic steatosis (7.3%) had incidence rates greater than that observed in Western populations, no hepatic events were deemed to be serious by the investigators, and none led to study discontinuation. Through 60 weeks of treatment with adalimumab, there were no cases of TB, congestive heart failure, demyelinating disease, allergic reaction, lupus-like syndrome, or deaths.
The low prevalence of AS in the Japanese population precluded enrollment of a sufficiently large population necessary for a placebo-controlled trial. As a result, the strength of conclusions drawn from these data must be tempered. On the basis of comparison with the ATLAS and M03-606 studies in Western patients with AS [17, 18], it is reasonable to assume that adalimumab therapy provides similar benefits in Japanese patients with AS. In addition, the limited patient numbers available for various efficacy and safety subanalyses precluded definitive conclusions regarding concomitant DMARD therapy, dose escalation, AE rates, etc.
The results of this study support the use of adalimumab as an effective and safe therapy for treating Japanese patients with active AS. Adalimumab monotherapy and combination therapy were both effective at reducing AS signs and symptoms and improving health-related quality of life in patients aged 15 years and older. Adalimumab efficacy in this population was observed as early as after the first dose and was sustained for up to 60 weeks of therapy. The safety data presented in this study were also consistent with the known profile of adalimumab in a variety of inflammatory immune-mediated diseases [26, 30].
Acknowledgments
Abbott sponsored the study (NCT00667355). The authors acknowledge Benjamin Wolfe, PhD, of Abbott, for his assistance with the drafting and revision of this manuscript, and Jocelyn Leu, PharmD, Ph.D., and Rochelle Jurasz, BS, of Abbott, for their assistance with the pharmacokinetic data
Conflict of interest
SK has received consultant fees from Abbott Japan and Tanabe-Mitsubishi Pharmaceutical Co. MH has received research grants, consultant fees, and/or speakers’ bureau honoraria from Abbott Japan, Bristol-Myers Japan, Chugai Pharmaceutical Co. Ltd., Eisai Co. Ltd., Janssen Pharmaceutical KK, Mitsubishi Tanabe Pharma, Pfizer Japan Inc., and Takeda Pharmaceutical Co. Ltd. NMi has received research grants, consultant fees, and/or speakers’ bureau honoraria from Chugai Pharmaceutical Co., Tanabe-Mitsubishi Pharmaceutical Co., Takeda Pharmaceutical Co., Pfizer Japan, Abbott Japan, Eisai Pharmaceutical Co., Astellas Pharmaceutical Co., Bristol-Myers-Squibb, and Otsuka Pharmaceutical Co. NMo, ALP, SS, and LSB are all full-time employees of Abbott and may hold stock or stock options.
References
- 1.Rudwaleit M, Heijde D, Landewe R, Listing J, Akkoc N, Brandt J, et al. The development of Assessment of SpondyloArthritis international Society classification criteria for axial spondyloarthritis (part II): validation and final selection. Ann Rheum Dis. 2009;68:777–783. doi: 10.1136/ard.2009.108233. [DOI] [PubMed] [Google Scholar]
- 2.Braun J, Bollow M, Remlinger G, Eggens U, Rudwaleit M, Distler A, et al. Prevalence of spondylarthropathies in HLA-B27 positive and negative blood donors. Arthritis Rheum. 1998;41:58–67. doi: 10.1002/1529-0131(199801)41:1<58::AID-ART8>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 3.Hukuda S, Minami M, Saito T, Mitsui H, Matsui N, Komatsubara Y, et al. Spondyloarthropathies in Japan: nationwide questionnaire survey performed by the Japan Ankylosing Spondylitis Society. J Rheumatol. 2001;28:554–559. [PubMed] [Google Scholar]
- 4.Yamaguchi A, Tsuchiya N, Mitsui H, Shiota M, Ogawa A, Tokunaga K, et al. Association of HLA-B39 with HLA-B27-negative ankylosing spondylitis and pauciarticular juvenile rheumatoid arthritis in Japanese patients. Evidence for a role of the peptide-anchoring B pocket. Arthritis Rheum. 1995;38:1672–1677. doi: 10.1002/art.1780381120. [DOI] [PubMed] [Google Scholar]
- 5.Rudwaleit M, Khan MA, Sieper J. The challenge of diagnosis and classification in early ankylosing spondylitis: do we need new criteria? Arthritis Rheum. 2005;52:1000–1008. doi: 10.1002/art.20990. [DOI] [PubMed] [Google Scholar]
- 6.Boonen A, Heijde D, Landewe R, Spoorenberg A, Schouten H, Rutten-van Molken M, et al. Work status and productivity costs due to ankylosing spondylitis: comparison of three European countries. Ann Rheum Dis. 2002;61:429–437. doi: 10.1136/ard.61.5.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ward MM. Functional disability predicts total costs in patients with ankylosing spondylitis. Arthritis Rheum. 2002;46:223–231. doi: 10.1002/1529-0131(200201)46:1<223::AID-ART498>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 8.Boonen A, Heijde D, Landewe R, Guillemin F, Rutten-van Molken M, Dougados M, et al. Direct costs of ankylosing spondylitis and its determinants: an analysis among three European countries. Ann Rheum Dis. 2003;62:732–740. doi: 10.1136/ard.62.8.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Braun J, Berg R, Baraliakos X, Boehm H, Burgos-Vargas R, Collantes-Estevez E, et al. 2010 update of the ASAS/EULAR recommendations for the management of ankylosing spondylitis. Ann Rheum Dis. 2011;70:896–904. doi: 10.1136/ard.2011.151027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ward MM, Kuzis S. Medication toxicity among patients with ankylosing spondylitis. Arthritis Rheum. 2002;47:234–241. doi: 10.1002/art.10399. [DOI] [PubMed] [Google Scholar]
- 11.Braun J, Bollow M, Neure L, Seipelt E, Seyrekbasan F, Herbst H, et al. Use of immunohistologic and in situ hybridization techniques in the examination of sacroiliac joint biopsy specimens from patients with ankylosing spondylitis. Arthritis Rheum. 1995;38:499–505. doi: 10.1002/art.1780380407. [DOI] [PubMed] [Google Scholar]
- 12.Braun J, Brandt J, Listing J, Zink A, Alten R, Golder W, et al. Treatment of active ankylosing spondylitis with infliximab: a randomised controlled multicentre trial. Lancet. 2002;359:1187–1193. doi: 10.1016/S0140-6736(02)08215-6. [DOI] [PubMed] [Google Scholar]
- 13.Gorman JD, Sack KE, Davis JC., Jr Treatment of ankylosing spondylitis by inhibition of tumor necrosis factor alpha. N Engl J Med. 2002;346:1349–1356. doi: 10.1056/NEJMoa012664. [DOI] [PubMed] [Google Scholar]
- 14.Bosch F, Kruithof E, Baeten D, Herssens A, Keyser F, Mielants H, et al. Randomized double-blind comparison of chimeric monoclonal antibody to tumor necrosis factor alpha (infliximab) versus placebo in active spondylarthropathy. Arthritis Rheum. 2002;46:755–765. doi: 10.1002/art.511. [DOI] [PubMed] [Google Scholar]
- 15.Davis JC, Jr, Heijde D, Braun J, Dougados M, Cush J, Clegg DO, et al. Recombinant human tumor necrosis factor receptor (etanercept) for treating ankylosing spondylitis: a randomized, controlled trial. Arthritis Rheum. 2003;48:3230–3236. doi: 10.1002/art.11325. [DOI] [PubMed] [Google Scholar]
- 16.Heijde D, Dijkmans B, Geusens P, Sieper J, DeWoody K, Williamson P, et al. Efficacy and safety of infliximab in patients with ankylosing spondylitis: results of a randomized, placebo-controlled trial (ASSERT) Arthritis Rheum. 2005;52:582–591. doi: 10.1002/art.20852. [DOI] [PubMed] [Google Scholar]
- 17.Heijde D, Kivitz A, Schiff MH, Sieper J, Dijkmans BA, Braun J, et al. Efficacy and safety of adalimumab in patients with ankylosing spondylitis: results of a multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2006;54:2136–2146. doi: 10.1002/art.21913. [DOI] [PubMed] [Google Scholar]
- 18.Lambert RG, Salonen D, Rahman P, Inman RD, Wong RL, Einstein SG, et al. Adalimumab significantly reduces both spinal and sacroiliac joint inflammation in patients with ankylosing spondylitis: a multicenter, randomized, double-blind, placebo-controlled study. Arthritis Rheum. 2007;56:4005–4014. doi: 10.1002/art.23044. [DOI] [PubMed] [Google Scholar]
- 19.Inman RD, Davis JC, Jr, Heijde D, Diekman L, Sieper J, Kim SI, et al. Efficacy and safety of golimumab in patients with ankylosing spondylitis: results of a randomized, double-blind, placebo-controlled, phase III trial. Arthritis Rheum. 2008;58:3402–3412. doi: 10.1002/art.23969. [DOI] [PubMed] [Google Scholar]
- 20.van der Linden S, Valkenburg HA, Cats A. Evaluation of diagnostic criteria for ankylosing spondylitis. A proposal for modification of the New York criteria. Arthritis Rheum. 1984;27:361–8. [DOI] [PubMed]
- 21.Anderson JJ, Baron G, Heijde D, Felson DT, Dougados M. Ankylosing spondylitis assessment group preliminary definition of short-term improvement in ankylosing spondylitis. Arthritis Rheum. 2001;44:1876–1886. doi: 10.1002/1529-0131(200108)44:8<1876::AID-ART326>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 22.Weisman MH, Moreland LW, Furst DE, Weinblatt ME, Keystone EC, Paulus HE, et al. Efficacy, pharmacokinetic, and safety assessment of adalimumab, a fully human anti-tumor necrosis factor-alpha monoclonal antibody, in adults with rheumatoid arthritis receiving concomitant methotrexate: a pilot study. Clin Ther. 2003;25:1700–1721. doi: 10.1016/S0149-2918(03)80164-9. [DOI] [PubMed] [Google Scholar]
- 23.Pavy S, Brophy S, Calin A. Establishment of the minimum clinically important difference for the bath ankylosing spondylitis indices: a prospective study. J Rheumatol. 2005;32:80–85. [PubMed] [Google Scholar]
- 24.Samsa G, Edelman D, Rothman ML, Williams GR, Lipscomb J, Matchar D. Determining clinically important differences in health status measures: a general approach with illustration to the Health Utilities Index Mark II. Pharmacoeconomics. 1999;15:141–155. doi: 10.2165/00019053-199915020-00003. [DOI] [PubMed] [Google Scholar]
- 25.Humira [package insert]. In: Abbott; 2011.
- 26.Burmester GR, Mease P, Dijkmans BA, Gordon K, Lovell D, Panaccione R, et al. Adalimumab safety and mortality rates from global clinical trials of six immune-mediated inflammatory diseases. Ann Rheum Dis. 2009;68:1863–1869. doi: 10.1136/ard.2008.102103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heijde D, Pangan AL, Schiff MH, Braun J, Borofsky M, Torre J, et al. Adalimumab effectively reduces the signs and symptoms of active ankylosing spondylitis in patients with total spinal ankylosis. Ann Rheum Dis. 2008;67:1218–1221. doi: 10.1136/ard.2007.082529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miyasaka N. Clinical investigation in highly disease-affected rheumatoid arthritis patients in Japan with adalimumab applying standard and general evaluation: the CHANGE study. Mod Rheumatol. 2008;18:252–262. doi: 10.1007/s10165-008-0045-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Asahina A, Nakagawa H, Etoh T, Ohtsuki M. Adalimumab in Japanese patients with moderate to severe chronic plaque psoriasis: efficacy and safety results from a phase II/III randomized controlled study. J Dermatol. 2010;37:299–310. doi: 10.1111/j.1346-8138.2009.00748.x. [DOI] [PubMed] [Google Scholar]
- 30.Schiff MH, Burmester GR, Kent JD, Pangan AL, Kupper H, Fitzpatrick SB, et al. Safety analyses of adalimumab (HUMIRA) in global clinical trials and US postmarketing surveillance of patients with rheumatoid arthritis. Ann Rheum Dis. 2006;65:889–894. doi: 10.1136/ard.2005.043166. [DOI] [PMC free article] [PubMed] [Google Scholar]