

Abstract
Chronic drug users account for a third of all cases of AIDS in the United States and the progression to AIDS dementia is accelerated in opiate drug abusers. Clinically, microglial activation better correlates with HIV-associated neurocognitive disorders (HAND) than productive HIV-1 infection in the CNS. Moreover, pneumococcal pneumonia is the most common opportunistic infection in individuals with HAND. We show that coinfection with Streptococcus pneumoniae may be a contributing factor in the increased prevalence of HAND in the opioid-dependent population. To date, there have been no studies published implicating the Toll-like receptors (TLR) in the neurocognitive disorders associated with NeuroAIDS in the context of opportunistic infection. Our studies show for the first time, in a morphine-dependent model, synergistic increase and activation of TLR expression in the presence of HIV-1 protein TAT and S. pneumoniae with a significant increase in proinflammatory cytokines (IL-6, TNF-α) levels. Furthermore, concurrent increases in reactive oxygen species and nitric oxide production leading to increased caspase 3 activation are also observed in both murine and human microglial cells. These effects are recapitulated with TLR 2, 4, and 9 cognate ligands (Pam3CSK4, LPS, and CpG) and significantly attenuated in TLR 2 and 4 knock-out mice and TLR2/4 double knock-out mice. Therefor, our findings clearly suggest for the first time that activation of TLRs on microglia cells by morphine and TAT in the context of S. pneumoniae infection may be a potential mechanism for the increased prevalence of HAND in HIV-infected opioid-dependent patients.
Introduction
HIV-1 can induce severe and debilitating neurological problems, including behavioral abnormalities, motor dysfunction, and dementia (Mattson et al., 2005; Kaul and Lipton, 2006). Although effective highly active antiretroviral therapy significantly reduces systemic viral load, there is no effective therapy for HIV-1-associated neurocognitive disorder (HAND) and, more significantly, the lifetime prevalence of HAND seems to be on the rise (Kopnisky et al., 2007). Chronic drug users account for approximately a third of all cases of AIDS in the United States and recent data indicate that progression to AIDS-associated dementia is markedly accelerated in this population (Bell et al., 1998; Ayuso-Mateos et al., 2000; Shor-Posner, 2000; Nath, 2002; Royal et al., 2003; Hauser et al., 2006). A frequent occurrence in the HIV-positive intravenous-drug-abusing population is the incidence of infection with secondary opportunistic pathogens and concurrent bacterial meningitis. Interestingly, Streptococcus pneumoniae is the most common community-acquired pneumonia in these patients (Gordon et al., 2000; Nuorti et al., 2000; Payeras et al., 2002; Gebo et al., 2005; Shen et al., 2005; Wang et al., 2005; Le Moing et al., 2006; Caro-Murillo et al., 2007; Clatts et al., 2007; Klugman et al., 2007). In chronic neurodegenerative diseases such as Alzheimer's, frequent episodes or persistent systemic infection lead to progressive decline in cognitive function and accelerate the process of neurodegeneration through activation of proinflammatory cytokines (Kreutzberg, 1996; Cunningham et al., 2005). Furthermore, in HIV patients, clinical studies reveal that elevated proinflammatory cytokines and activated microglia consistently correlated with HAND, more so than viral load or viral protein (Rock and Peterson, 2006).
In recent years, important progress has been made in understanding how specific receptors of the immune system recognize pathogen-associated molecular patterns to induce immune response (Randhawa and Hawn, 2008). A highly relevant class of pattern recognition receptors is the family of Toll-like receptors (TLRs). Ligation of distinct TLRs by different pathogen-associated molecules has the capacity to engage specific downstream intracellular signaling cascades, thus tailoring the innate response to the activation stimulus (Randhawa and Hawn, 2008). Recent studies demonstrate the expression of TLRs in the CNS (McKimmie and Fazakerley, 2005; Bailey et al., 2006). Although there is some evidence that S. pneumoniae activates Toll-like receptors 2, 4, and 9 in peripheral immune cells (Wang et al., 2011), there are no studies clearly delineating the role of TLRs in S. pneumoniae-induced glial activation and its contribution to HIV neuropathogenesis. In this report, we demonstrate for the first time in a morphine-dependent model a significant increase in the susceptibility to S. pneumoniae infection with increased dissemination of bacteria into the CNS in the presence of HIV-1 TAT. In addition, we observed a synergistic increase in proinflammatory cytokine response with a concurrent increase in neuronal apoptosis. We hypothesize that although opioid drug abuse and HIV proteins can either independently or synergistically modulate neuropathogenesis, coinfection with S. pneumoniae reduces the threshold for proinflammatory cytokines synthesis and significantly accelerates the neuropathogenic process. We further hypothesize that activation of Toll-like receptors may be a potential mechanism for the synergistic increase in neuropathogenesis in opioid drug abusers that are coinfected with HIV and S. pneumoniae. In this study, we tested the hypothesis that coinfection with S. pneumoniae leading to activation of Toll-like receptors is a major contributing factor in the increased prevalence of HAND observed in the chronic opioid-abusing HIV-infected population.
Materials and Methods
Animals.
Male mice 8- to 10-weeks-old were used. Wild-type B6CBAF1 (WT), Toll-like receptor 2-deficient (TLR2KO), and Toll-like receptor 4-defective lipopolysaccharide response (TLR4KO) were from Jackson Laboratories; TLR2/4 double knock-out mice were generated by crossing TLR2KO with TLR4KO; mu-opioid receptor knock-out (MORKO) was produced as described previously (Wang et al., 2011); and TAT transgenic (TATtg) were from Dr. Om Prakash (LSU Health Sciences Center, New Orleans, LA). Animals were housed four animals per cage under controlled conditions of temperature and lighting (12 h light/dark) and given ad libitum access to standard food and tap water. All animals were allowed to acclimate to their environment for at least 7 d before any experimental manipulations. Discomfort, distress, and injury to the animals were minimized. The Institutional Animal Care and Use Committee at the University of Minnesota have approved all protocols in use, and all procedures are in agreement with the guidelines set forth by the National Institute of Health Guide for the Care and Use of Laboratory Animals.
Treatment with morphine, S. pneumoniae, and HIV-1 TAT protein.
Mice were lightly anesthetized with isoflurane (Halocarbon Products) and were implanted with either a morphine pellet (75 or 25 mg) or a placebo pellet (controls) to achieve a well characterized opioid-dependent model. All animals, except the controls, received an intravenous dose of 10 μg/kg recombinant full-length HIV-1 TAT (82 aa; Immuno Diagnostics). Five hours after TAT treatment, animals were inoculated intranasally with varying doses (103, 105, and 106) of colony-forming units (CFUs) of S. pneumoniae serotype 3 wild-type strain (American Type Culture Collection, number 6303) in PBS (50 μl). Morphine pellets were a generous gift from the National Institute on Drug Abuse. To determine opioid specificity, animals were implanted with a naltrexone pellet 4 h before morphine pellet implantation.
Primary microglial cultures.
Primary microgial cultures were prepared as follows. Microglia was prepared from postnatal days 1–3 mouse brains. Microglial cell cultures prepared in this manner are >99% pure, as determined by Mac-1 antibody staining (Roche Applied Science). Cortices were isolated and trypsinized. Three brains were triturated and plated onto tissue culture T-75 flasks in DMEM/F12 with l-glutamine (Invitrogen) containing 20% heat-inactivated FBS. After 24 h, all media and tissue were removed and replaced with fresh media. After 7 d, one-half of the media was replaced and cells were maintained at a mixed glia culture until day 14. At 14 d in vitro, microglia was removed from the mixed glial culture via a rotating shaker at 200 rpm for 45 min and the total number of microglia isolated per flask was calculated. The typical yield of microglial cells was ∼250,000 cells/brain.
Primary neuronal cultures.
Primary neuronal cultures were prepared as follows. Brain tissues were harvested from fetal (E17–E18) mice. Briefly, dissociated cortical cells were plated in poly-l-lysine-coated six-well plates using a plating medium of MEM-Eagle's salts (supplied glutamine-free and lacking progesterone or other hormones) supplemented with 5% heat-inactivated horse serum, 5% fetal bovine serum, glutamine (2 mm), and glucose (final concentration: 21 mm). After plating, cells were incubated at 37°C in a 5% CO2 humidified atmosphere and incubated for 11 d without media replacement. On day 11, the plating media was replaced with fresh feeding medium and typically ready for use. Primary neuronal cells with final density of 12,000 cells/well in 96-well plates used for caspase assays.
Ex vivo culture of lungs and brain.
Mice were anesthetized using isoflurane and cardially perfused with cold PBS (0.1 m) solution. After perfusion, mice were killed by CO2 asphyxiation and brain and lungs were aseptically removed and homogenized in cold PBS (2 ml). Serial dilutions of the tissue homogenates were plated on blood agar plates. Plates were incubated overnight at 37°C, and S. pneumoniae colonies (CFU) were counted.
Immunohistochemistry.
The mouse brains from all groups were snap frozen and cryostat sections (5 μm) were stained to detect apoptosis using a terminal dexoynucleotidyl transferase-mediated digoxigenin-dUTP nick end-labeling (TUNEL) assay (according to the manufacturer's instructions; Roche Applied Science). To detect apoptosis in specific cells, cells were immunolabeled with primary antibodies such as mouse anti-NeuN antibody (Millipore) to identify neuronal cells and rabbit ionized calcium binding adaptor molecule (IBA-1) (Serotec) antibody for microglial cells. Subsequently, cells were stained with secondary antibodies (Dylight549 rabbit anti-mouse IgG and rhodamine red goat anti-rabbit IgG respectively; Jackson ImmunoResearch Laboratories) after washing with 0.01% PBS-Tween. For identifying astrocytes, GFAP (Alexa Fluor 555; Cell Signaling Technology) monoclonal antibody was used. The slides were washed and mounted using Prolong Gold antifade reagent with DAPI (nuclei marker; Invitrogen). Immunofluorescence images were obtained on a Nikon Eclipse Ti confocal microscope using a 60× oil-immersion objective. Sectioning was performed on a minimum of five random sections from each of four individual mice per experiment. The TUNEL (green)/nuclei (blue, DAPI) images were quantified using the ImageJ software.
RNA preparation, semiquantitative reverse transcription-PCR, and quantitative real-time PCR.
Total RNA was isolated from primary microglia cells using the RNeasy minikit (Qiagen). cDNA was synthesized using 1 μg of total RNA, oligo d(T) primers, and RNase MMLV reverse transcriptase, according to the manufacture's specification (Promega). Primer sequences used in the amplification of TLR genes are TLR4 (accession no. NM_021297), TLR2 (accession no. NM_011905), and TLR9 (accession no. NM_031178) (all from Origene Techonologies). Tested transcript levels were normalized to 18S rRNA transcript levels from the same preparations of cDNA. Our previous study showed that morphine treatment alone does not change 18S rRNA transcript levels. PCR products were electrophoresed through 1.5% agarose gel and ImageJ software was used to quantify the band intensity. Real-time PCR was performed using SyBR-Green master mix (Applied Biosystems) on the ABI Prism 7500 sequence detection system (Applied Biosystems). The relative levels of TLR expression were quantified using the ΔCt-ΔCt method and were expressed as percentage-fold change.
Flow cytometry.
Cell surface TLRs expression on primary microglial cells was analyzed by flow cytometry. Nonspecific binding to the Fcγ receptor was reduced by incubating cells with purified rat anti-mouse CD16/CD32 (BD Biosciences) for 10 min at 4°C. Cells were subsequently stained for TLR2 (Alexa Fluor 488; Serotec), TLR4 (PE; BD Biosciences), and TLR9 (FITC; BD Biosciences) for 30 min at 4°C. Appropriate isotype controls for each of the anti-TLRs were included. After washing with 0.1% PBS–0.1% sodium azide (PBS-NaN3), cells (10,000 events) were acquired by FACS Canto II cytometer and data were analyzed using Diva software (BD Bioscience).
Transfection of HEK-293 cells.
HEK-293 cells stably transfected with either TLR2, 4, or 9 genes were purchased from Invivogen and transiently transfected with the mu-opioid receptor (MOR) plasmid and the NFKB-luciferase reporter system as previously described (Wang et al., 2011).
Human microglia cell culture.
Human microglia (CHME-5) cells or primary human microglia (ScienCell) were cultured in a humidified chamber with a 5% CO2-95% air mixture at 37°C and maintained in DMEM supplemented with 10% FBS and 1% penicillin–streptomycin (Invitrogen), and all the assays (proinflammatory cytokine levels and TLR surface expression) were conducted within 10–15 cell passages after thawing the cells from the liquid nitrogen (to keep the best morphological and biological characteristics). For detecting cytokine (TNF-α, IL-6, and MCP-1) levels, human ELISA kit (R&D Systems) was used according to manufacturer's protocol. To detect the surface TLR expression on CHME-5, anti-TLR2 (Alexa Fluor 488), anti-TLR4 (Biotin), and anti-TLR9 (PE) (all from eBioscience) human antibodies were used. Streptavidin PerCP (BD Bioscience) was used as secondary antibody to detect TLR4 expression. Control isotope antibodies with appropriate fluorochrome combination were used to assess the nonspecific anitibody binding. After staining, cells were washed and resuspended in 0.1% PBS-NaN3 and 10,000 events were acquired by FACS Canto II cytometer and data were analyzed using Diva software (BD Bioscience).
Preparation of brain homogenates.
Mouse brain homogenates were prepared by rapidly washing the brain following harvest in ice-cold 10 mm Tris–HCl, pH 7.5 (1/10, w/v). The cerebral (whole-brain) tissue was rapidly dissected, put on ice, and weighed. Around 500 mg of tissue was immediately homogenized in cold 10 mm Tris–HCl, pH 7.5 (1/10, w/v). The homogenate was centrifuged for 10 min at 4000 × g and the supernatant used for reactive oxygen species (ROS) and nitrite determination.
Nitric oxide production.
Nitric oxide (NO) levels were estimated by measuring the concentrations of nitrites (NO2−), which are the resulting NO metabolites. Briefly, supernatants free from cellular debris were mixed with Griess reagent [1 part 1% (w/v) sulfanilamide in 5% H3PO4, 1 part 0.1% (w/v) N-1-naphthylethylenediamine (v/v)] in 96-well tissue culture plates for 10 min at room temperature in the dark. The absorbance at 540 nm was determined using a microplate reader (Bio-Rad Laboratories).
Determination of ROS production.
ROS generation was monitored by flow cytometry using the peroxide-sensitive fluorescent marker 2′,7′-dichlorofluorescein diacetate (DCFH-DA; Invitrogen). In brief, either cells or brain homogenates were incubated with DCFH-DA (20 μm) at 37°C. After incubation with the dye, cells or brain homogenates were suspended in ice-cold PBS and placed on ice in a dark environment. DCFH levels as a marker for ROS production were determined at 488 nm excitation and 525 nm emission wavelengths using a fluorescence plate reader (BMG Labtech).
Measurement of caspase-3 activity.
Primary neuronal cells were seeded and exposed to 500 μl of microglial spent medium treated with vehicle, S. pneumoniae lysate (1 × 103), morphine (1 μm), TAT (100 ng/ml), or S. pneumoniae lysate + morphine + TAT. After 24 h incubation, the cells were lysed and caspase-3 activity was determined using the CaspACE fluorometric activity assay (Promega) according to the manufacturer's instruction. Protein levels in the cell lysate samples were determined using the bicinchoninic acid protein assay kit with an absorption band of 570 nm (Pierce) to normalize the cell numbers between control and different treated groups. Caspase 3 activity in brain homogenates were measured.
Statistical analysis.
Data were collected from three independent experiments and expressed as mean ± SEM. Significances were determined by Student's t test and two-way ANOVA analysis. Individual group comparisons were made by the two-tailed Student's t test. Statistical significance was accepted at p < 0.05. Survival was evaluated for differences using a log-rank test.
Results
Synergistic increase in mortality in animals chronically treated with morphine and TAT and infected with S. pneumoniae
We have previously shown, in an opioid-dependent model, that chronic morphine treatment significantly increases dissemination of S. pneumoniae, resulting in high mortality in a mouse model of S. pneumoniae infection (Wang et al., 2005, 2008). To determine whether the threshold for infection and mortality are lowered when animals are coadministered HIV-1 TAT protein, experiments were initially performed to determine the dose (CFU) of bacteria that resulted in minimal or no mortality in morphine-treated animals. In these experiments, S. pneumoniae was administered intranasally (to simulate the natural entry route) with varying doses (CFU) of bacteria in 50 μl of saline. It was determined that a dose of 1 × 103 CFU/50 μl resulted in minimal mortality in mice that were morphine-treated (Fig. 1A). However, when this concentration of bacteria was administered to mice that had been treated with morphine (25 and 75 mg pellet) and injected intravenously every other day with recombinant full-length HIV-1 TAT (82 aa, 10 μg/kg; Immunodiagnostics), there was a synergistic and dose-dependent increase in mortality, with almost 50% and 100% mortality observed after 48 h in animals that received the 25 and 75 mg morphine pellet, respectively (Fig. 1B). The dose for TAT injection was selected based on previously published data (Westendorp et al., 1995; Xiao et al., 2000; Toborek et al., 2005; Rumbaugh et al., 2006; Eugenin et al., 2007). Intravenous injection of TAT was chosen to simulate systemic infection conditions. These doses are consistent with the concentrations used in other studies to determine physiological roles of TAT in tissues, where the localized TAT concentration is expected to be slightly greater than that detected in serum from HIV-infected individuals (2 to 40 ng/ml). To determine the role of the mu-opioid receptor in morphine's effect, MORKO mice were treated with morphine (25 and 75 mg) pellets and treated with TAT (10 μg/kg) and infected with S. pneumoniae (1 × 103 CFU/50 μl). The synergistic increase in mortality was abolished in the MORKO mice (Fig. 1C), establishing the role of MOR in morphine's effects.
Figure 1.

Kaplan–Meier plots showing dose-dependent increase in mortality with increasing dose of bacterial load and synergistic increase in mortality in animals chronically treated with morphine and TAT and infected with S. pneumoniae. A, In these experiments, S. pneumoniae was administered intranasally (to simulate the natural entry route) with varying doses (CFU) of bacteria in saline (50 μl). It was determined that a dose of 1 × 103 CFU/50 μl resulted in minimal mortality in mice that were morphine-treated. B, Animals were divided into four groups (6 animals per group) and implanted with either a placebo pellet or morphine pellets (75 or 25 mg) and then injected intravenously with TAT (10 μg/kg). Twenty-four hours later, animals were inoculated intranasally with 1 × 103 CFU S. pneumoniae (serotype 3). C, WT and MORKO mice were treated as described in A and B and survived for 5 d.
Synergistic increase in bacterial dissemination into the CNS and lung in animals chronically treated with morphine and TAT and infected with S. pneumoniae
To determine bacterial dissemination at different time points, animals were killed and bacterial counts measured using serially diluted brain and lung homogenates in a blood agar plate (Fig. 2A). Significant bacterial dissemination into the lung as well as in the CNS was observed in animals that were treated with morphine (25 mg) plus TAT (10 μg/kg) and infected with S. pneumoniae (1 × 103 CFU) compared with all other groups. Results shown are from animals that were treated with morphine + TAT for 48 h postinfection. To determine whether the effects observed with morphine was mediated through the mu-opioid receptors, WT and MORKO mice were treated with morphine (25 mg) plus TAT (10 μg/kg, i.v.) and inoculated with S. pneumoniae (1 × 103 CFU). The synergistic increase in bacterial dissemination was significantly attenuated in the MORKO mice (Fig. 2B), implicating the mu-opioid receptor in modulating morphine's effect.
Figure 2.

Synergistic increase in bacterial dissemination into the CNS and lung in animals chronically treated with morphine and TAT and infected with S. pneumoniae (S.p). A, Bacterial dissemination into lung and brain in wild-type mice treated with morphine + TAT. Animals were divided into four groups (6 per group) as described in Figure 1. Tissue was harvested at 48 h following infection and bacterial load in the tissue was measured by homogenizing the tissue in medium. The homogenate was serially diluted and plated on blood agar plates; colonies were counted after 24 h incubation at 37°C (**p < 0.01). B, To determine the role of the mu-opioid receptor in morphine-mediated effects, wild-type and MORKO mice (six per group) was treated as in Figure 1 and bacterial dissemination into the CNS measured as described in A. The data shown in A and B represent bacterial counts in tissues at 48 h following infection in animals that received morphine pellet (25 mg). Error bars are mean ± SEM. **p < 0.001.
Morphine increases bacterial dissemination in a TATtg mouse model
To determine whether the synergistic effect of morphine and intravenous TAT injection can be recapitulated in a TATtg mouse model, a well characterized constitutive TAT expression model was used. In this model, TAT driven by a Simian Virus-40-promoter is expressed constitutively in all tissues. Several papers published with this model (Prakash et al., 1997, 1998a,b, 2000, 2001; Avraham et al., 2004) show TAT expression levels and functionality comparable to that observed in HIV-infected patients, including decreased immune function and disruption of blood–brain barrier. To determine the effect of morphine treatment in this model, WT and TATtg mice were implanted with either a 75 or 25 mg morphine pellet. Twenty-four hours after pellet implantation, animals were injected intranasally with 1 × 103 CFU S. pneumoniae (serotype 3). Significant mortality was observed within 24 h of infection when mice were implanted with a 75 mg morphine pellet. One hundred percent mortality was observed within 36 h (Fig. 3A) in the 75 mg morphine-pellet-implanted group. Mortality observed when implanted with the 25 mg morphine pellet was similar to intravenous injection with TAT (10 μg/kg), validating our results that intravenous injection of TAT is an acceptable model. Bacterial dissemination, when measured 48 h postinfection, was also significantly greater in morphine (25 mg pellet)-treated TATtg mice when compared with placebo control and wild-type mice treated with 25 mg morphine pellet (Fig. 3B). Since most animals died within 36 h of infection, bacterial dissemination could not be determined in the TATtg mice implanted with the 75 mg morphine pellet group. These results also support our hypothesis that TAT synergizes with morphine to potentiate bacterial dissemination.
Figure 3.

Chronic morphine treatment accelerates mortality and increases dissemination in a TATtg mouse model. Wild-type and TATtg mice (6 per group) were implanted subcutaneously with either placebo pellet or morphine pellet (75 or 25 mg). Twenty-four hours after pellet implantation, animals were inoculated with S. pneumonia (S.p.; 1 × 103 CFU). A, B, Survival (A) and bacterial dissemination (B) were followed for 5 d. Bacterial dissemination was determined as described in the tex. Data presented shows dissemination after 48 h following infection. Each data points represent at least six animals. Error bars are mean ± SEM. *p < 0.001, **p < 0.0001.
Morphine treatment in the presence of TAT and S. pneumoniae markedly increases CNS apoptosis
To determine whether the increased CNS dissemination of S. pneumoniae in morphine (25 mg pellet) + Tat (10 μg/kg)-treated group resulted in a synergistic increase in neuronal apoptosis, cryostat section of brains harvested from animals treated with morphine + TAT + S. pneumoniae were evaluated for TUNEL staining (Fig. 4A) and TUNEL staining quantified using the metamorph software (Fig. 4B). Significant apoptosis was observed only in the morphine + TAT + S. pneumoniae group (Fig. 4A,B). In contrast, very little apoptosis was observed in animals that received either morphine or TAT alone, although these animals received the same dose of S. pneumoniae. Similar synergistic increase in apoptotic cells were observed when TATtg mice were treated with morphine (25 mg pellet) and inoculated with 1 × 103 CFU S. pneumoniae when compared with animals that were treated with either morphine or TAT alone and inoculated with 1 × 103 CFU (Fig. 4C). These studies further support our hypothesis that chronic morphine treatment in the presence of TAT in the context of infection with S. pneumoniae synergistically induces CNS apoptosis.
Figure 4.

Confocal microscopic images and quantification of apoptotic bodies in the brain tissue. Morphine treatment in the presence of TAT and S. pneumoniae (S.p.) markedly increases CNS apoptosis. A, To determine apoptosis, animals (6 per group) were treated as described in Figure 1 and killed at 72 h. Brains were removed and snap frozen in liquid nitrogen. Cryostat sections (5 μm) were used to evaluate apoptosis using TUNEL staining (Intergen) according to the manufacturer's instruction. Anatomical location from where sections were taken is indicated (top). DAPI staining shows the nuclei of cells. Scale bars, 10 μm. B, The number of apoptotic cells were also determined using metamorph software and expressed as percentage of total cells. C, The graph represents quantification of apoptotic cells in TATtg mouse brain treated with morphine/placebo pellet and S. pneumoniae. The data are presented as mean ± SD of three independent experiments. *p < 0.01.
Morphine + TAT + S. pneumoniae induces apoptosis predominantly in the neuronal cell population
To determine the cell population that was undergoing apoptosis, sections derived from animals treated as Figure 4A were subjected to immunocytochemical staining as described in Materials and Methods. TUNEL staining was colocalized with the neuronal marker, NeuN (Fig. 5), with very little apoptotic cells colocalized with the microglial marker, IBA-1 or the astrocyte marker, GFAP (Fig. 5). However, marked activation of both astrocytes and microglia were observed, as evidenced by the intense GFAP and IBA-1 staining.
Figure 5.
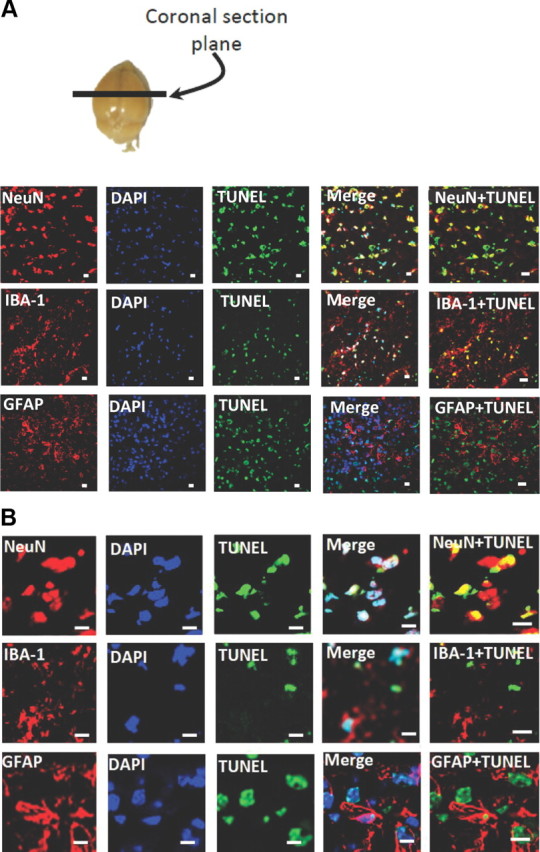
Morphine + TAT + S. pneumoniae induces apoptosis predominantly in the neuronal cell population. Animals were treated as described in the text and cryostat sections were processed for TUNEL staining and incubated with anti-NeuN (neuronal nuclei marker), anti-GFAP (astrocyte marker), and anti-IBA-1 (microglial marker). Anatomical location from where sections were taken is indicated (A, top). A, B, Representative confocal images of NeuN, IBA-1, and GFAP staining with TUNEL and DAPI are shown. Scale bars, 10 μm.
Morphine treatment in the presence of TAT and S. pneumoniae markedly increases TNF α, IL-6, and MCP-1 synthesis in CNS
To determine whether S. pneumoniae infection results in proinflammatory cytokine synthesis in the CNS of animals chronically treated with morphine and TAT, brain homogenates were analyzed for TNF-α, IL-6, and MCP-1 protein synthesis using ELISA (R&D System). We determined the time course of the inflammatory response. As shown in Figure 6A, the time course of the inflammatory response revealed a significant but transient increase in TNF-α levels that peaked at ∼36 h and decreased over the next 48 h. In contrast, there was a delay in the induction but a persistent increase in the expression levels of both IL-6 and MCP-1. This increase was sustained even at 96 h. Brain homogenates from animals treated with morphine + TAT + S. pneumoniae (48 h after infection with 1 × 103 CFU) were also subjected to mouse inflammation antibody array (Raybiotec) (Fig. 6B). From an average of four separate experiments, the data show marked increase in TNF-α, IL-6, and MCP-1 secretion, confirming our ELISA data in Figure 6A. In addition, an increased intensity in arrays spotted with RANTES antibodies was also observed, suggesting a synergistic increase in the induction of proinflammatory cytokines and chemokines when all three insults (morphine, TAT, and S. pneumoniae) were present at the same time.
Figure 6.

Morphine treatment in the presence of TAT and S. pneumoniae markedly increases TNF-α, IL-6, and MCP-1 synthesis in CNS. Animals were treated as described in Figure 1. Animals were treated with morphine pellet (25 mg) and TAT (10 μg/kg). A, The data represents cytokine levels in brain homogenate at various time points (0–96 h) following infection. Animals were killed at the end of every time point and brains were isolated and homogenized. Brain homogenates were centrifuged at 12,000 RPM and the supernatant analyzed for protein determination using an ELISA kit (R&D Systems) or subjected to mouse inflammation antibody array according to the manufacturer's instructions. Data represents mean of three independent experiments. *p < 0.05. B, Antibody array (Raybiotec) determinations after 48 h following infection. 1, IL-6; 2, TNF-α; 3, MCP-1; 4, RANTES.
Morphine synergizes with S. pneumoniae and TAT and increases TNF-α, IL-6, and MCP-1 synthesis in primary microglial cultures
To determine whether the in vivo effects can be recapitulated in vitro, primary microglial cultures were purified from postnatal day 1–3 mice, as described in Materials and Methods, above, and treated with morphine (1 μm) for 24 h. Cells were then treated with TAT (100 ng/ml) and activated with S. pneumoniae (1 × 103 CFU) lysate (100 μl). TNF-α, IL-6, and MCP-1 were measured using ELISA (Quantikine kit; R&D). Only cells that were treated with morphine + TAT + S. pneumoniae lysate showed significant and marked increases in proinflammatory cytokines synthesis. Cells treated with morphine alone or TAT alone in the presence of S. pneumoniae lysates showed marginal increase when compared with cells treated with a combination of morphine (1 μm) and TAT (100 ng/ml) (Fig. 7A). The effect of morphine was dose-dependent and naltrexone pretreatment of microglial cells significantly reduced the synergistic increase in proinflammatory cytokine synthesis (Fig. 7B).
Figure 7.

Morphine synergizes with S. pneumoniae and TAT and increases TNF-α, IL-6, and MCP-1 synthesis in primary microglial cultures. A, Primary microglial cells were purified as described in Materials and Methods and treated with morphine (1 μm) for 24 h before the addition of TAT (100 ng/ml). Cells were then treated with either vehicle or S. pneumoniae (S.p.) bacterial lysates (1 × 103) for 48 h. B, Primary microglial cells were purified as described in Materials and Methods and treated with varying doses of morphine (10 nm–1 μm) in the presence and absence of Naltrexone (10 μm) for 24 h before the addition of TAT (100 ng/ml). Cells were then treated with either vehicle or S. pneumoniae bacterial lysates (1 × 103) for 48 h. Proinflammatory cytokine synthesis was measured using ELISA as described in Materials and Methods. Data represents mean ± SEM of three independent experiments. *p < 0.05.
Morphine treatment results in upregulation of TLR 2, 4, and 9 expression levels in CNS and in primary microglial cells
To determine whether TLRs were involved in the mechanism by which morphine and TAT synergistically increase the synthesis of proinflammatory cytokine synthesis, WT and MORKO mice were treated with either placebo or morphine pellet, in the presence and absence of Naltrexone, for 24 h and TLR 2, 4, and 9 mRNA expression determined in total RNA isolated from brain homogenates. Morphine treatment resulted in a significant increase in TLR 2, 4, and 9 expressions (Fig. 8A). Morphine-induced expression was completely abolished in the MORKO mice and significantly attenuated in animals that were treated with both morphine and Naltrexone pellets (Fig. 8A). Since TLRs are predominantly expressed on microglial cells, we next determine whether morphine treatment of primary microglial cells can lead to TLR induction. Primary microglial cells were purified as described in the Materials and Methods, above, and treated with morphine (1 μm) for 24 h. To determine the opioid receptor type that was mediating the effects, microglial cells were pretreated overnight with Naltrexone (10 μm), CTOP (10 μm), Naltrindole (10 μm), or norBNI (10 μm) before the addition of morphine (1 μm) for 24 h. Morphine treatment resulted in a threefold to fourfold induction of TLR2, 4, and 9 mRNA. Morphine-induced increase in TLR expression was antagonized by Naltrexone and CTOP but not when cells were treated with Naltrindole or norBNI, implicating the mu-opioid receptor in morphine-induced modulation of TLR expression (Fig. 8B). To determine whether S. pneumoniae or TAT can modulate morphine-induced TLR expression, primary microglia cells were treated with either morphine, TAT, or S. pneumoniae lysates, alone or in combination. Only morphine treatment resulted in an increase in TLR 2, 4, and 9 mRNA expressions (Fig. 8C). Interestingly, treatment with either TAT or S. pneumoniae lysate alone did not lead to an increase in TLR expression. TLR expression levels in cells that were treated with a combination of morphine, TAT, and S. pneumoniae lysate did not show any further increase when compared with morphine treatment alone, suggesting that the increase in TLR expression observed in the cells that were treated with a combination of morphine + TAT + S. penumoniae lysates were predominantly mediated by morphine (Fig. 8C). When protein expression levels of TLR 2, 3, and 9 were quantified using flow cytometry (Fig. 8D), our data showed significant increases in TLR 2, 4, and 9 protein expression levels in cells treated with morphine (1 μm). The data suggest the role of TLR expression as a potential mechanism for the synergistic increase observed with morphine treatment.
Figure 8.

Morphine treatment increases TLR 2, 4, and 9 mRNA and protein expression levels in CNS and mouse primary microglial cells. A, WT and MORKO mice were treated with either placebo or morphine pellet. To determine opioid specificity, WT mice were treated with Naltrexone pellet (30 mg) 4 h before morphine (25 mg) pellet implantation. Total RNA was isolated following 24 h of morphine pellet implantation and subjected to real-time PCR analysis with primers specific for TLR 2, 4, and 9. Data represent expression normalized to 18S RNA. Each experiment had six animals per group. Data represent mean of three independent experiments. *p < 0.05. B, Primary microglial cells were prepared as described in Materials and Methods and treated with either morphine (1 μm) or saline in the presence of Naltrexone (10 μm), CTOP (10 μm), Naltrindole (10 μm), or norBNI (10 μm) for 24 h. Cells were then harvested for mRNA and subjected to real-time PCR analysis with primers specific for TLR 2, 4, and 9 and normalized to 18S RNA. Data represent the mean of three independent experiments. *p < 0.05. C, Primary microglial cells treated with either morphine (1 μm) or saline for 24 h and then treated with TAT (100 ng/ml) and/or S. pneumoniae (S.p.) lysates (1 × 103). Messenger RNA were isolated from the microglial cells and subjected to real-time PCR for TLR 2, 4, and 9 mRNA expression. Data represents mean of three independent real-time PCR experiments. Error bars are SEM. *p < 0.05. D, Microglial cells were treated with saline, morphine (1 μm), Naltrexone (10 μm), or morphine (1 μm) + Naltrexone (10 μm). TLR expression on the surface of microgial cells was analyzed by flow cytometry. A total of 10,000 events were acquired in all the experiments using FACS cantocytometer and analyzed with BD DIVA software (BD Biosciences). Histograms show the mean fluorescence intensity of TLR2, 4, and 9 expressions on CD11b+ cells.
S. pneumoniae activates TLR 2-, 4-, and 9-induced NF-κB activation and TAT and morphine treatment synergizes with S. pneumoniae-induced activation
Since TAT and S. pneumoniae lysates did not modulate TLR expression, we sought to investigate whether the underlying mechanism for the synergistic increase may be due to an increase in TLR activation by TAT and S. pneumoniae. To test the hypothesis, stably transfected TLR 2, 4, and 9 HEK-293 cells were transiently transfected with MOR and NF-κB reporter plasmid and treated with morphine (1 μm), TAT (100 ng/ml), or S. pneumoniae (1 × 103) lysates, either alone or in combination. Treatment with S. pneumoniae lysate alone resulted in a significant increase in NF-κB activation in cells stably transfected with TLR 2, 4, and 9 (Fig. 9). No significant activation was seen in cells that were stably transfected with the empty vector (Fig. 9, top). Interestingly, we show for the first time that TAT can synergistically increase S. pneumoniae-induced TLR 2 and 9 NF-κB activation (Fig. 9). While morphine treatment alone did not result in any significant activation of NF-κB, in the presence of S. pneumoniae lysates, morphine treatment resulted in significant activation of NF-κB in TLR 2, 4, and 9 stably transfected cells. NF-κB activation was significantly amplified when cells were treated with a combination of morphine, TAT, and S. pneumoniae lysate (Fig. 9).
Figure 9.
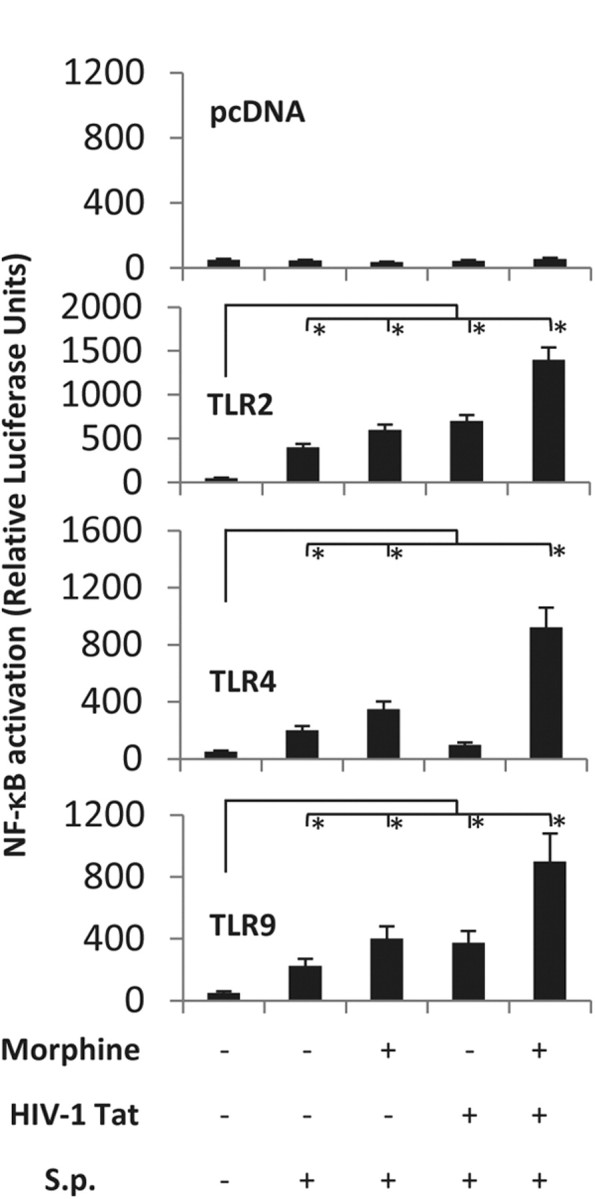
S. pneumoniae (S.p.) lysate activates TLR 2-, 4-, and 9-induced NF-κB activation and TAT and morphine treatment synergizes with S. pneumoniae-induced activation in HEK293 cells. Cells (100,000 cells/ml) were stably transfected with TLR 2, 4, and 9 and transiently transfected with hemagglutin-tagged MOR and NF-κB-luciferase reporter plasmid and treated with TAT (100 ng/ml) for 6 h and then activated with S. pneumoniae lysates (1 × 103) for 6 h. Cells were cotransfected with Renilla luciferase to determine transfection efficiency. Data are expressed as ratio of Firefly luciferase to Renilla luciferase and normalized to hemagglutin-tagged MOR expression. Heat-denatured TAT showed no activation (data not shown). Data represents mean ± SEM of three independent experiments. *p < 0.05.
Morphine + TAT + S. pneumoniae infection induced synergistic increase in bacterial dissemination and proinflammatory cytokines are significantly attenuated in TLR 2, 4 knock-outs and TLR2/4 double knock-out mice
To further determine whether morphine's synergistic effects were mediated via Toll-like receptors, we treated TLR2KO, TLR4KO, and wild-type mice with morphine (25 mg) for 24 h, followed by intravenous treatment with HIV-1 Tat protein (10 μg/kg) and then inoculated intranasally with luciferase-tagged S. pneumoniae (1 × 103, Xen10; Xenogen). After 48 h, mice were anesthetized with isoflurane (Halocarbon Products) and cardially perfused with 0.1 m PBS. Brain was aseptically removed and imaged using the IVIS50 CCD camera system (Xenogen). Total photon emission was quantitated by deriving a gate around each brain using IgorPro 4.09A software. Data showed a significant reduction in S. pneumoniae dissemination in the CNS of TLR 2 and 4 knock-out mice in the presence morphine compared with the wild-type (Fig. 10A,B). Further, the TUNEL assay reveals no apoptotic nuclear bodies in the CNS of TLR 2 and 4 knock-out mice (Fig. 10C). In addition, the synergistic increase in proinflammatory cytokines (TNF-α, IL-6, and the chemokine MCP-1) were significantly reduced in brain homogenates harvested from TLR 2 and 4 knock-out mice and dramatically reduced in the TLR2/4 double knock-out mice (Fig. 10D), confirming our hypothesis that TLRs are involved in the synergistic increase in morphine + TAT induced proinflammatory cytokine release. To further determine whether decreased bacterial dissemination and proinflammatory response correlated with increased survival, TLR2KO, TLR4KO, and TLR2/4 double knock-out mice were treated with morphine and TAT and infected with S. pneumoniae and survival observed over 5 d. Significant mortality was observed with the WT mice; however, the same treatment resulted in no mortality in the TLR2/4 double KO mice and 10% and 20% mortality in the TLR2KO and TLR4KO mice, respectively (Fig. 10E).
Figure 10.
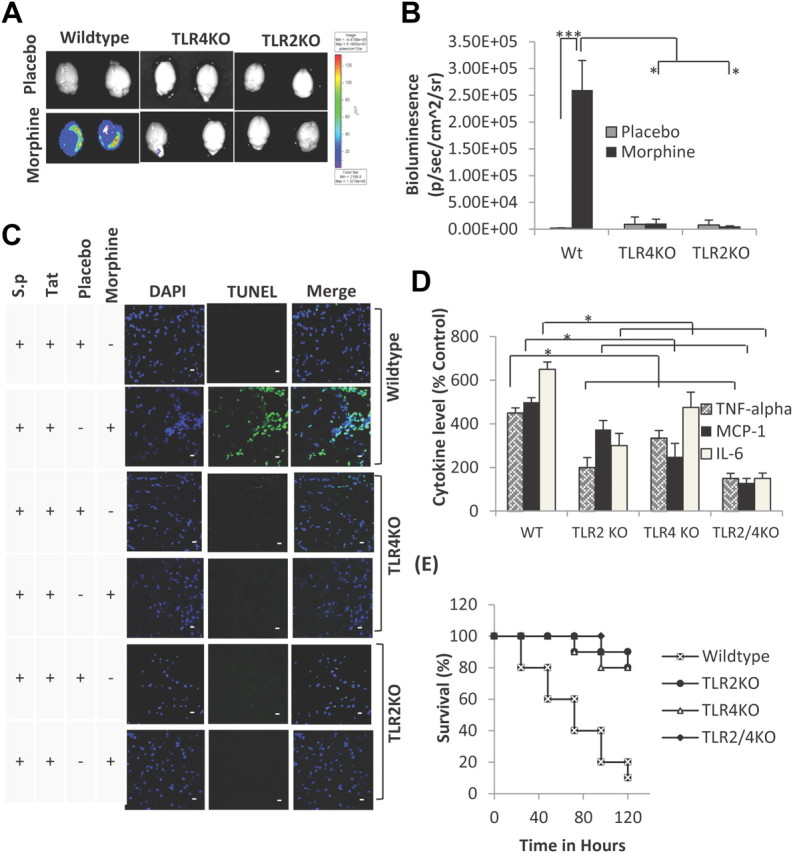
Morphine + TAT + S. pneumoniae (luciferase tagged) induced synergistic increase in bacterial dissemination and proinflammatory cytokines are significantly attenuated in TLR 2, 4 knock-outs and TLR2/4 double knock-out mice. A, A significant decrease in bacterial dissemination into the CNS of TLR 2, 4 knock-outs and TLR2/4 double knock-out mice following morphine, TAT, and S. pneumoniae lysate treatment. B, Quantification of bacterial dissemination in the brain tissue of WT, TLR2KO, and TLR4KO mice. Data represents mean ± SEM of three independent experiments. *p < 0.01, ***p < 0.001. C, To determine apoptosis, animals (6 per group) were treated as described in Figure 4A and killed at 72 h. Brains were removed and snap frozen in liquid nitrogen. Cryostat sections (5 μm) were used to evaluate apoptosis using TUNEL staining (Intergen) according to the manufacturer's instruction. DAPI staining shows the nuclei of cells. Apoptosis is significantly lower in the TLR knock-out animals compared with wild-type following treatment with morphine, TAT, and S. pneumoniae (S.p.) lysate. Scale bars, 10 μm. D, Synergistic increase in proinflammatory cytokines in WT mice following morphine, TAT, and S. pneumoniae lysate treatment is significantly attenuated in brain homogenate harvested from TLR 2, 4 knock-outs and TLR2/4 double knock-out mice. Data represents mean ± SEM of three independent experiments. *p < 0.01. E, Survival curve of WT and TLR 2, 4 knock-outs and TLR2/4 double knock-out mice treated with morphine + TAT + S. pneumoniae. Animals were treated as described in Figure 4A and survival followed for 5 d. Data represents mean ± SEM of three independent experiments.
Morphine treatment increases TLR expression on CHME-5 and synergizes with TAT and S. pneumoniae to increase proinflammatory cytokines in primary human microglial cells and in CHME-5
To determine whether human microglial cells would respond similarly to murine glial cells, a well characterized human microglial cell line (CHME-5) was treated with morphine in the presence of S. pneumoniae lysate and TAT. RT-PCR analysis reveals that morphine significantly increased TLR 2, 4, and 9 mRNA expressions (Fig. 11A) and flow cytometry analysis reveals increase in TLR protein expression (Fig. 11B) in human microglia cells line similar to the effects observed with mouse microglia. We next investigated whether proinflammatory cytokines levels are modulated in the human microglial cell line following morphine treatment in the presence or absence of TAT and S. pneumoniae lysates. As was observed in the murine model in vivo and in vitro treatment, morphine treatment in the presence of TAT and S. pneumoniae lysates showed the greatest expression of IL-6, TNF-α, and MCP-1 in CHME-5 cells (Fig. 11C). Morphine-induced activation was significantly attenuated when cells were pretreated with Naltrexone. Similar responses were seen with primary human microglial cells (ScienCell Research Laboratories) (Fig. 11D). These data confirm that treatment in human cells recapitulates the observation demonstrated in the murine model, thus validating the murine model as an appropriate model to investigate consequences of drug abuse in the context of opportunistic infection.
Figure 11.

Morphine treatment increases TLR expression on human microglia cell line (CHME-5) and synergizes with TAT and S. pneumoniae to increase proinflammatory cytokines in primary human microglial cells and in CHME-5. A, CHME-5 microglial cells were treated with either morphine (1 μm) or saline for 24 h and then treated with TAT (100 ng/ml) and/or S. pneumoniae (S.p.) lysates (1 × 103). mRNA were isolated from the microglial cells and subjected to real-time PCR for TLR 2, 4, and 9 mRNA expression. Data represents mean ± SEM of three independent real-time PCR experiments. *p < 0.05. B, CHME-5 microglial cells were treated with either morphine (MS, 1 μm) or saline for 24 h and then treated with TAT (100 ng/ml) and/or S. pneumoniae lysates (1 × 103). The surface TLR expression was analyzed through flow cytometry. A total of 10,000 events were acquired in all the experiments using FACS cantocytometer and analyzed with BD DIVA software (BD Biosciences). Histograms show the mean fluorescence intensity of TLR 2, 4, and 9 expressions on CD11b+ cells. C, CHME-5 microglial cells were treated with morphine (MS; 1 μm) for 24 h before the addition of TAT (100 ng/ml). Cells were then treated with S. pneumoniae bacterial lysates (1 × 103) for 48 h. Proinflammatory cytokine were measured using ELISA as described in Materials and Methods. Data represents mean ± SEM of three independent experiments. **p < 0.01. D, Primary human microglial cells were treated as described above and IL-6 levels were measured using ELISA as described in Materials and Methods. Data represent mean ± SEM of three independent experiments.*p < 0.05.
Treatment of microglial with TLR ligands increase ROS and NO production
Chronic activation of microglia can induce neuronal damage through the release of potentially cytotoxic molecules such as ROS and reactive nitrogen intermediates (NO). To determine whether activation of TLR cognate ligands in combination with morphine and TAT result in the induction and release of reactive oxygen species, primary microglial cells were pretreated with morphine (1 μm) and TAT (100 ng/ml) and then activated with either S. pneumoniae lysates (1 × 103) or TLR cognate ligands, TLR2-Pam3CSK4 (100 ng), TLR4-LPS (100 ng), and TLR-9-CpG (1 μg) (all from Invivogen). Although treatment with morphine and TAT alone resulted in no significant increase in ROS (Fig. 12A) and NO (Fig. 12B) production, treatment in the presence of S. penumoniae lysates and the cognate TLR ligands resulted in a robust and significant increase in ROS and NO production.
Figure 12.

Morphine and TAT treatment in combination with TLR ligands increase ROS and NO production in primary microglial cells. Primary microglial cells were purified from WT mice as described in Materials and Methods and treated with morphine (1 μm) for 24 h before the addition of TAT (100 ng/ml). Cells were then treated with S. pneumonia (S.p.) lysates or TLR 2, 4, and 9 cognate ligands (Pam3CSK4, LPS, and CpG respectively). A, ROS generation was monitored by flow cytometry using the peroxide-sensitive fluorescent probe 2′,7′-dichlorofluorescein diacetate. Data are represented as percentage increase over vehicle control. DCFH levels as a marker for ROS production were measured by flow cytometer (FACScalibur; Becton Dickinson), which emitted a fluorescent signal at 525 nm. Each group was acquired with >10,000 individual cells and compared with vehicle-treated group. B, NO levels were estimated by measuring the concentrations of nitrites (NO2−), which are the resulting NO metabolites. Supernatants free from cellular debris were mixed with Griess reagent [1 part 1% (w/v) sulfanilamide in 5% H3PO4, 1 part 0.1% (w/v) N-1-naphthylethylenediamine (v/v)] in 96-well tissue culture plates for 10 min at room temperature in the dark. The absorbance at 540 nm was determined using a microplate reader. Each group was acquired with >10,000 individual cells and compared with vehicle-treated group. Error bars are mean ± SEM. *p < 0.01.
Morphine + TAT + S. pneumoniae-treated microglial culture supernatant increases neuronal apoptosis
To determine whether microglial-treated culture supernatant will induce neuronal apoptosis, microglial cells were pretreated as described above with morphine and TAT and then treated with S. penumoniae lysates for 24 h and the spent culture supernatant was added to primary neuronal cells in culture. Cell-free culture medium incubated with morphine and TAT and S. pneumoniae lysates was used as control. Treatment with morphine and TAT resulted in no significant increase in neuronal apoptosis, as quantified using caspase 3 fluorometric assay kit. Treatment of coculture with a combination with morphine, TAT, and S. pneumoniae lysate resulted in a significant and robust increase in caspase 3 induction (Fig. 13). To further evaluate if the neuronal apoptosis observed was mediated through the production of ROS or NO, microglial cells were pretreated with either the iNOS inhibitor, 1400w, or diphenylene-iodonium, an inhibitor of NADPH oxidase, the major producer of ROS in microglia, and then treated with morphine and TAT and S. pneumoniae lysates. Treatment with the inhibitors significantly decreased neuronal apoptosis when compared with the samples that were not treated with the inhibitors (Fig. 13), suggesting the role of NO and ROS in neuronal apoptosis. Direct treatment of neuronal cells with S. pneumoniae lysate + TAT + morphine alone in the absence of microglial supernatant (cell-free supernatant) showed no significant increase in caspase 3 activity when compared with treatment with S. pneumoniae alone. We therefore conclude that the apoptosis observed is not a direct effect of morphine + TAT + S. pneumoniae lysate but an indirect effect due to the release of ROS and reactive nitrogen species from microglial cells.
Figure 13.

Morphine + TAT + S. pneumoniae (S.p.)-treated microglial culture supernatant increases neuronal apoptosis. Microglial cells were treated with vehicle, morphine (1 μm), TAT (100 ng/ml), or S. pneumoniae lysate for 48 h. Microglial spent media (500 μl) were placed on seeded Neuro-2a cells (1 × 106). In parallel, Neuro-2a cells were exposed to cell free medium containing S. pneumoniae lysate, morphine (1 μm), and TAT (100 ng/ml) and served as control. Caspase-3 activity was determined using the CaspACE fluorometric activity assay according to the manufacturer's instruction. Protein levels of cell lysate samples were determined using the bicinchoninic acid protein assay kit with an absorption band of 570 nm to normalize the cell numbers between control and the different treated groups. To further evaluate if the neuronal apoptosis observed was mediated through the production of ROS or NO, microglial cells were pretreated with the iNOS inhibitor, 1400w, or the inhibitor of NADPH oxidase diphenylene-iodonium (DPI), and then treated with morphine, TAT, and S. pneumoniae lysates. Each group was acquired with 10,000 individual cells. Error bars are mean ± SEM. *p < 0.01.
ROS, NO, and caspase 3 activity are reduced in TLR2, 4 and TLR2/4 knock-out mice
To further establish that TLR expression and activation are responsible for the production of ROS and NO leading to neuronal apoptosis, WT and TLR 2, 4, and 2/4 knock-out mice were treated with either a placebo or morphine (25 mg) pellet for 24 h and then injected with TAT (10 μg/kg) and infected with S. pneumoniae (1 × 103 CFU) for an additional 48 h. Animals were killed and brain homogenates analyzed for ROS, NO, and caspase 3 activity as described in Materials and Methods. As observed with in vitro morphine treatment, in vivo morphine treatment in the context of TAT and S. pneumoniae resulted in a significant increase in ROS, NO, and caspase 3 activation (Table 1). The effects of morphine + TAT + S. pneumoniae were significantly reduced in TLR 2 and 4 knock-out mice and dramatically reduced in the TLR2/4 double knock-out mice. These data support a significant role of TLR 2 and 4 expression and activation in morphine + TAT + S. pneumonia infection-mediated neuronal apoptosis.
Table 1.
Morphine and TAT treatment in the context of S. pneumoniae infection in TLR 2, 4, and TLR2/4 knock-out mice
| Groups | Treatment | ROS (% control) | Nitrite (mol/mg) | Caspase 3 (MFI) |
|---|---|---|---|---|
| Wild type | Placebo | 175 ± 27 | 0.25 ± 0.03 | 121 ± 12 |
| Morphine | 500 ± 35* | 0.78 ± 0.07* | 350 ± 35* | |
| TLR2KO | Placebo | 125 ± 18 | 0.18 ± 00.02 | 93 ± 10 |
| Morphine | 196 ± 23 | 0.21 ± 0.019 | 143 ± 14 | |
| TLR4KO | Placebo | 135 ± 31 | 0.16 ± 0.009 | 88 ± 9 |
| Morphine | 182 ± 21 | 0.19 ± 0.02 | 132 ± 14 | |
| TLR2/4KO | Placebo | 112 ± 29 | 0.12 ± 0.005 | 56 ± 6 |
| Morphine | 105 ± 25 | 0.14 ± 0.006 | 52 ± 8 |
WT and TLR 2, 4 knock-out and TLR2/4 double knock-out mice were treated with either placebo or morphine pellet as described in Materials and Methods and infected with S. pneumoniae intranasally. Animals were killed for ROS and reactive nitrogen species measurement 24 h after infection and for caspase 3 activity 48 h after infection. ROS generation was measured using the peroxide-sensitive fluorescent probe 2′,7′-dichlorofluorescein diacetate. Data is represented as percentage increase over vehicle control. NO levels were estimated by measuring the concentrations of nitrites. Supernatants free from cellular debris were mixed with Griess reagent [1 part 1% (w/v) sulfanilamide in 5% H3PO4, 1 part 0.1% (w/v) N-1-naphthylethylenediamine (v/v)] in 96-well tissue culture plates for 10 min at room temperature in the dark. The absorbance at 540 nm was determined using a microplate reader. Caspase 3 activity was determined using the CaspACE fluorometric activity assay according to the manufacturer's instruction. Protein levels of brain homogenates samples were determined using the bicinchoninic acid protein assay kit with an absorption band of 570 nm to normalize the protein levels between control and different treated groups. Sample size in each group was six animals. Data represents mean ± SD of three independent experiments.
*p < 0.01.
Discussion
From the onset of the HIV/AIDS epidemic, the impact of illicit drug use on HIV disease progression has been a focus of many investigations. Both laboratory-based and epidemiological studies strongly indicate that drug dependence may exacerbate HIV disease progression and increase mortality and morbidity in these patients (Concha et al., 1997; Bell et al., 1998, 2006; Bouwman et al., 1998; Buscemi et al., 2007; Anthony et al., 2008; Walkup et al., 2008). Mounting evidence suggests that drug abusers have accelerated and more severe neurocognitive dysfunction compared with non-drug-abusing HIV-infected populations. It is postulated that since receptors for drugs of abuse are predominantly in the CNS, it is predictable that drugs of abuse such as morphine may synergize with neurotoxic substances released during the course of HIV infection and contribute to neuropathogenesis. However, in this report we demonstrate that administration of morphine (opioid-dependent model) or HIV-1 TAT either alone or in combination does not result in any significant neuronal apoptosis. However, when morphine and TAT are administered in the context of infection with S. pneumoniae, a synergistic increase in neuronal apoptosis is observed. Our studies corroborate outcomes in several neurodegenerative disease models, where there is evidence to support that episodes of systemic infections through induction of proinflammatory mediators result in neurocognitive deficits and are implicated to be contributory factors in disease progression (Kreutzberg, 1996; Cunningham et al., 2005). Interestingly, proinflammatory mediators play a critical role in the pathophysiology of NeuroAIDS and clinical studies reveal that proinflammatory cytokines and chemokines better correlate with dementia than viral load (Rock and Peterson, 2006).
We previously demonstrated that opiate abuse results in decreased bacterial clearance and increased susceptibility to opportunistic infection (Wang et al., 2005). Herein, we show that the threshold for infection and mortality is significantly lowered when opiates are administered in the presence of HIV-1 TAT with a log order shift in the dose–response curve for morphine in the presence of HIV-1 TAT (10 μg/kg) in a murine model of S. pneumonia. Furthermore, morphine treatment in the presence of TAT results in a synergistic decrease in bacterial clearance and an increase in bacterial dissemination. Bacterial dissemination into the CNS was only observed when animals were treated with both morphine and TAT, but cleared efficiently in placebo-treated animals that were injected with HIV-1 TAT. Furthermore, we demonstrate that increased dissemination of bacteria into the CNS resulted in a synergistic increase in proinflammatory cytokines and chemokine synthesis in animals that were treated with morphine and TAT and coinfected with S. pneumoniae. However, bacterial dissemination alone is not sufficient to drive the inflammatory process, since in vitro treatment of primary microglial cells with S. pneumoniae lysate alone resulted in a marginal increase in proinflammatory cytokines but a more robust and synergistic increase was observed when both TAT and morphine were superimposed upon S. pneumoniae infection.
Although TAT treatment alone has been shown to activate proinflammatory cytokine synthesis in several models, activation is seen only with high doses of TAT (Theodore et al., 2006). In in vivo models, significant but marginal increase in TNF-α was observed only when TAT (20 μg) was directly injected into the striatum (El-Hage et al., 2006). Furthermore, TAT has been shown to induce proinflammatory cytokine expression only in astrocytes with very little activation of proinflammatory cytokines in microglial cells (El-Hage et al., 2006). Consistent with these studies, the current study demonstrates that both in vivo and in vitro treatment of wild-type animals or primary microglial cultures with TAT (100 ng/ml) alone does not result in any significant increase in either proinflammatory cytokines or MCP-1 synthesis. Morphine treatment in addition to TAT treatment results in only a marginal increase. However when microglial cells are cotreated with S. pneumoniae bacterial lysate, or when animals are infected with S. pneumoniae, a synergistic increase in proinflammatory cytokines and MCP-1 are observed in groups that are morphine- and TAT-treated. These results suggest that morphine and TAT treatment result in activation of pathways that are distinct but are only unmasked when coinfected with S. pneumoniae to synergistically increase proinflammatory cytokine expression and synthesis.
To delineate the mechanism(s) underlying the observed synergistic increase, we investigated Toll-like receptor expression and signaling as potential mechanisms. In recent years, important progress has been made in understanding how specific receptors of the immune system recognize pathogen-associated molecular patterns to induce immune response (Kawai and Akira, 2011). A highly relevant class of pattern recognition receptors is the family of TLRs, of which 12 have been found in mammals (Bsibsi et al., 2002). In the case of Gram-positive bacteria, components of the bacterial cell wall (lipotechoic acid) interact with TLR 2 and 4. Very little is known regarding the expression pattern of TLR 2, 4, and 9 in the CNS following S. pneumoniae infection. In mouse microglial cells, nine of the TLRs are expressed at basal levels (Laflamme and Rivest, 2001). Murine astrocytes, however, display lower constitutive expression levels with a more restricted range. Ligation of distinct TLRs by different pathogen-associated molecules has the capacity to engage specific downstream intracellular signaling cascades and thus tailor the innate response to the activation stimulus. Classical TLR signaling involves recruitment of MyD88 intracellular adapter protein, which, through a cascade of events, ubiquitinizes IkB and targets it for proteosomal degradation. This results in the translocation of the active form of NF-κB (p65/p50) into the nucleus to activate promoters of proinflammatory cytokines (Kawai and Akira, 2011). To date, and to the best of our knowledge, there are no studies published implicating the Toll-like receptors in the neuropathogenesis associated with NeuroAIDS. We show for the first time that morphine treatment results in a significant increase in both mRNA and protein expression of TLR 2, 4, and 9 in murine and human microglial cells. Our unpublished data further establish direct activation of TLR 2 and 4 promoter activity following morphine treatment. Although TLR expression levels are not significantly modulated by either TAT or S. pneumoniae lysates, TLR activation was seen to be significantly amplified in the presence of TAT and S. pneumoniae lysates in HEK293 cells that are stably transfected with TLRs. From these results, we conclude that the synergistic increase in proinflammatory cytokines observed in the presence of morphine + TAT and S. pneumoniae lysates is mediated through a two-step process that includes a morphine-mediated increase in TLR expression and TAT- and S. pneumoniae-induced activation of TLR signaling. This conclusion is further supported by the observation that morphine + TAT and S. pneumoniae-induced proinflammatory cytokine synthesis was significantly attenuated in TLR 2, 4, and TKR2/4 double knock-out mice.
TLRs are similarly upregulated in human microglial cells in the presence of morphine treatment, and the synergistic increase in proinflammatory cytokines is observed in the context of TAT and S. pneumoniae infection, providing human relevance for the current study. Although TLR activation resulting in NF-κB activation and proinflammatory cytokines are well established, the mechanism by which TAT modulates and potentiates TLR activation still remains to be delineated.
Activation of TLR resulting in the induction of proinflammatory cytokines has been associated with an increase in both ROS and NO (Jomova et al., 2010; Gross et al., 2011). Excessive production and accumulation of ROS and NO leading to oxidative damage of neurons has been implicated in aging and various neurodegenerative conditions, such as stroke, Alzheimer's disease, and amyotrophic lateral sclerosis (Barcia et al., 2009; Jomova et al., 2010; Gross et al., 2011). We show in both in vitro and in vivo experiments significant induction in ROS and NO (in culture supernatant of microglial cells and in brain homogenates of animals) only following treatment with a combination of morphine + TAT + S. pneumoniae. Addition of treated microglial culture supernatant to primary neurons resulted in significant caspase 3 activation. Morphine + TAT + S. pneumoniae induced proinflammatory cytokines; ROS, NO, and neuronal apoptosis were significantly attenuated in TLR 2 and 4 knock-out mice. These results strongly support the hypothesis that expression and activation of TLRs play a dominant role in the synergistic increase in neuronal apoptosis observed in morphine + TAT + S. pneumonia-treated animals.
In summary, our studies show for the first time that morphine treatment increases TLR expression levels and TAT and S. pneumoniae synergistically increases TLR activation, resulting in a threefold synergistic increase in proinflammatory cytokines (IL-6, TNF-α) levels with concurrent increase in microglial, ROS, and NO production. These results are recapitulated when TLR 2, 4, and 9 are activated with their cognate ligands (Pam3cy, LPS, and CpG) in the presence of morphine and TAT and significantly attenuated in TRL 2, 4, and TLR2/4 double knock-out mice. Addition of culture supernatants from microglial cells treated with S. pneumoniae lysates + morphine + TAT to neuronal cells increased neuronal apoptosis, which was significantly reversed in the presence of NADPH oxidase inhibitor and NO inhibitor. Therefor, our findings clearly suggest for the first time that activation of TLRs on microglia cells by morphine, TAT, and S. pneumoniae, leading to an increase in proinflammatory cytokines, ROS, and NO, may contribute to the increased prevalence of neuropathogenesis that is commonly observed in HIV-infected opiate drug abusers (Fig. 14). These studies implicate the Toll-like receptors in the neuropathogenesis observed in HIV patients coinfected with opportunistic pathogens and pave the way for future studies targeting Toll-like receptors as potential therapeutic approach to ameliorate neuroinflammation associated with microgial activation.
Figure 14.

Schematic outlining potential synergism between morphine and HIV-1 TAT protein in the context of infection leading to neuronal damage and progression to NeuroAIDS.
Footnotes
This work was supported by NIH Grants R01 DA12104, R01 DA022935, KO2DA015349, R01 DA031202, and P50 DA11806 (S.R.) This work was also supported by funds from the Minneapolis Veterans Affairs Medical Center (to R.A.B.).
The authors declare no competing financial interests.
References
- Anthony IC, Arango JC, Stephens B, Simmonds P, Bell JE. The effects of illicit drugs on the HIV infected brain. Front Biosci. 2008;13:1294–1307. doi: 10.2741/2762. [DOI] [PubMed] [Google Scholar]
- Avraham HK, Jiang S, Lee TH, Prakash O, Avraham S. HIV-1 TAT-mediated effects on focal adhesion assembly and permeability in brain microvascular endothelial cells. J Immunol. 2004;173:6228–6233. doi: 10.4049/jimmunol.173.10.6228. [DOI] [PubMed] [Google Scholar]
- Ayuso-Mateos JL, Pereda M, Gómez Del Barrio A, Echevarria S, Fariñas MC, García-Palomo D. Slowed reaction time in HIV-1-seropositive intravenous drug users without AIDS. Eur Neurol. 2000;44:72–78. doi: 10.1159/000008200. [DOI] [PubMed] [Google Scholar]
- Bailey SL, Carpentier PA, McMahon EJ, Begolka WS, Miller SD. Innate and adaptive immune responses of the central nervous system. Crit Rev Immunol. 2006;26:149–188. doi: 10.1615/critrevimmunol.v26.i2.40. [DOI] [PubMed] [Google Scholar]
- Barcia C, Ros F, Carrillo MA, Aguado-Llera D, Ros CM, Gómez A, Nombela C, de Pablos V, Fernández-Villalba E, Herrero MT. Inflammatory response in parkinsonism. J Neural Transm Suppl. 2009;73:245–252. doi: 10.1007/978-3-211-92660-4_19. [DOI] [PubMed] [Google Scholar]
- Bell JE, Brettle RP, Chiswick A, Simmonds P. HIV encephalitis, proviral load and dementia in drug users and homosexuals with AIDS: effect of neocortical involvement. Brain. 1998;121:2043–2052. doi: 10.1093/brain/121.11.2043. [DOI] [PubMed] [Google Scholar]
- Bell JE, Arango JC, Anthony IC. Neurobiology of multiple insults: HIV-1-associated brain disorders in those who use illicit drugs. J Neuroimmune Pharmacol. 2006;1:182–191. doi: 10.1007/s11481-006-9018-2. [DOI] [PubMed] [Google Scholar]
- Bouwman FH, Skolasky RL, Hes D, Selnes OA, Glass JD, Nance-Sproson TE, Royal W, Dal Pan GJ, McArthur JC. Variable progression of HIV-associated dementia. Neurology. 1998;50:1814–1820. doi: 10.1212/wnl.50.6.1814. [DOI] [PubMed] [Google Scholar]
- Bsibsi M, Ravid R, Gveric D, van Noort JM. Broad expression of toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol. 2002;61:1013–1021. doi: 10.1093/jnen/61.11.1013. [DOI] [PubMed] [Google Scholar]
- Buscemi L, Ramonet D, Geiger JD. Human immunodeficiency virus type-1 protein tat induces tumor necrosis factor-alpha-mediated neurotoxicity. Neurobiol Dis. 2007;26:661–670. doi: 10.1016/j.nbd.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caro-Murillo AM, Castilla J, Pérez-Hoyos S, Miró JM, Podzamczer D, Rubio R, Riera M, Viciana P, López Aldeguer J, Iribarren JA, de los Santos-Gil I, Gómez-Sirvent JL, Berenguer J, Gutiérrez F, Saumoy M, Segura F, Soriano V, Peña A, Pulido F, Oteo JA, et al. Spanish cohort of naive HIV-infected patients (CoRIS): rationale, organization and initial results. Enferm Infecc Microbiol Clin. 2007;25:23–31. doi: 10.1157/13096749. [DOI] [PubMed] [Google Scholar]
- Clatts MC, Giang le M, Goldsamt LA, Yi H. Novel heroin injection practices: implications for transmission of HIV and other bloodborne pathogens. Am J Prev Med. 2007;32:S226–S233. doi: 10.1016/j.amepre.2007.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Concha M, Selnes OA, Vlahov D, Nance-Sproson T, Updike M, Royal W, Palenicek J, McArthur JC. Comparison of neuropsychological performance between AIDS-free injecting drug users and homosexual men. Neuroepidemiology. 1997;16:78–85. doi: 10.1159/000109674. [DOI] [PubMed] [Google Scholar]
- Cunningham C, Wilcockson DC, Campion S, Lunnon K, Perry VH. Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J Neurosci. 2005;25:9275–9284. doi: 10.1523/JNEUROSCI.2614-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hage N, Wu G, Wang J, Ambati J, Knapp PE, Reed JL, Bruce-Keller AJ, Hauser KF. HIV-1 tat and opiate-induced changes in astrocytes promote chemotaxis of microglia through the expression of MCP-1 and alternative chemokines. Glia. 2006;53:132–146. doi: 10.1002/glia.20262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eugenin EA, King JE, Nath A, Calderon TM, Zukin RS, Bennett MV, Berman JW. HIV-tat induces formation of an LRP-PSD-95-NMDAR-nNOS complex that promotes apoptosis in neurons and astrocytes. Proc Natl Acad Sci U S A. 2007;104:3438–3443. doi: 10.1073/pnas.0611699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebo KA, Fleishman JA, Moore RD. Hospitalizations for metabolic conditions, opportunistic infections, and injection drug use among HIV patients: trends between 1996 and 2000 in 12 states. J Acquir Immune Defic Syndr. 2005;40:609–616. doi: 10.1097/01.qai.0000171727.55553.78. [DOI] [PubMed] [Google Scholar]
- Gordon SB, Walsh AL, Chaponda M, Gordon MA, Soko D, Mbwvinji M, Molyneux ME, Read RC. Bacterial meningitis in malawian adults: pneumococcal disease is common, severe, and seasonal. Clin Infect Dis. 2000;31:53–57. doi: 10.1086/313910. [DOI] [PubMed] [Google Scholar]
- Gross O, Thomas CJ, Guarda G, Tschopp J. The inflammasome: an integrated view. Immunol Rev. 2011;243:136–151. doi: 10.1111/j.1600-065X.2011.01046.x. [DOI] [PubMed] [Google Scholar]
- Hauser KF, El-Hage N, Buch S, Nath A, Tyor WR, Bruce-Keller AJ, Knapp PE. Impact of opiate-HIV-1 interactions on neurotoxic signaling. J Neuroimmune Pharmacol. 2006;1:98–105. doi: 10.1007/s11481-005-9000-4. [DOI] [PubMed] [Google Scholar]
- Jomova K, Vondrakova D, Lawson M, Valko M. Metals, oxidative stress and neurodegenerative disorders. Mol Cell Biochem. 2010;345:91–104. doi: 10.1007/s11010-010-0563-x. [DOI] [PubMed] [Google Scholar]
- Kaul M, Lipton SA. Mechanisms of neuronal injury and death in HIV-1 associated dementia. Curr HIV Res. 2006;4:307–318. doi: 10.2174/157016206777709384. [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- Klugman KP, Madhi SA, Feldman C. HIV and pneumococcal disease. Curr Opin Infect Dis. 2007;20:11–15. doi: 10.1097/QCO.0b013e328012c5f1. [DOI] [PubMed] [Google Scholar]
- Kopnisky KL, Bao J, Lin YW. Neurobiology of HIV, psychiatric and substance abuse comorbidity research: workshop report. Brain Behav Immun. 2007;21:428–441. doi: 10.1016/j.bbi.2007.01.011. [DOI] [PubMed] [Google Scholar]
- Kreutzberg GW. Microglia: A sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- Laflamme N, Rivest S. Toll-like receptor 4: the missing link of the cerebral innate immune response triggered by circulating gram-negative bacterial cell wall components. FASEB J. 2001;15:155–163. doi: 10.1096/fj.00-0339com. [DOI] [PubMed] [Google Scholar]
- APROCO Study Group. Le Moing V, Rabaud C, Journot V, Duval X, Cuzin L, Cassuto JP, Al Kaied F, Dellamonica P, Chêne G, Raffi F. Incidence and risk factors of bacterial pneumonia requiring hospitalization in HIV-infected patients started on a protease inhibitor-containing regimen. HIV Med. 2006;7:261–267. doi: 10.1111/j.1468-1293.2006.00370.x. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Haughey NJ, Nath A. Cell death in HIV dementia. Cell Death Differ. 2005;12(Suppl 1):893–904. doi: 10.1038/sj.cdd.4401577. [DOI] [PubMed] [Google Scholar]
- McKimmie CS, Fazakerley JK. In response to pathogens, glial cells dynamically and differentially regulate toll-like receptor gene expression. J Neuroimmunol. 2005;169:116–125. doi: 10.1016/j.jneuroim.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Nath A. Human immunodeficiency virus (HIV) proteins in neuropathogenesis of HIV dementia. J Infect Dis. 2002;186(Suppl 2):S193–S198. doi: 10.1086/344528. [DOI] [PubMed] [Google Scholar]
- Nuorti JP, Butler JC, Gelling L, Kool JL, Reingold AL, Vugia DJ. Epidemiologic relation between HIV and invasive pneumococcal disease in san francisco county, california. Ann Intern Med. 2000;132:182–190. doi: 10.7326/0003-4819-132-3-200002010-00003. [DOI] [PubMed] [Google Scholar]
- Payeras A, Martinez P, Milà J, Riera M, Pareja A, Casal J, Matamoros N. Risk factors in HIV-1-infected patients developing repetitive bacterial infections: toxicological, clinical, specific antibody class responses, opsonophagocytosis and fc(gamma) RIIa polymorphism characteristics. Clin Exp Immunol. 2002;130:271–278. doi: 10.1046/j.1365-2249.2002.01986.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash O, Teng S, Ali M, Zhu X, Coleman R, Dabdoub RA, Chambers R, Aw TY, Flores SC, Joshi BH. The human immunodeficiency virus type 1 tat protein potentiates zidovudine-induced cellular toxicity in transgenic mice. Arch Biochem Biophys. 1997;343:173–180. doi: 10.1006/abbi.1997.0168. [DOI] [PubMed] [Google Scholar]
- Prakash O, Joshi BH, Zhang P, Aw TY, Teng S, Ali M, Shellito JE, Nelson S. Transgenic mouse model of ethanol as a cofactor in HIV disease. Alcohol Clin Exp Res. 1998a;22:266S–268S. doi: 10.1111/j.1530-0277.1998.tb04015.x. [DOI] [PubMed] [Google Scholar]
- Prakash O, Zhang P, Xie M, Ali M, Zhou P, Coleman R, Stoltz DA, Bagby GJ, Shellito JE, Nelson S. The human immunodeficiency virus type I tat protein potentiates ethanol-induced neutrophil functional impairment in transgenic mice. Alcohol Clin Exp Res. 1998b;22:2043–2049. [PubMed] [Google Scholar]
- Prakash O, Tang ZY, He YE, Ali MS, Coleman R, Gill J, Farr G, Samaniego F. Human kaposi's sarcoma cell-mediated tumorigenesis in human immunodeficiency type 1 tat-expressing transgenic mice. J Natl Cancer Inst. 2000;92:721–728. doi: 10.1093/jnci/92.9.721. [DOI] [PubMed] [Google Scholar]
- Prakash O, Rodriguez VE, Tang ZY, Zhou P, Coleman R, Dhillon G, Shellito JE, Nelson S. Inhibition of hematopoietic progenitor cell proliferation by ethanol in human immunodeficiency virus type 1 tat-expressing transgenic mice. Alcohol Clin Exp Res. 2001;25:450–456. [PubMed] [Google Scholar]
- Randhawa AK, Hawn TR. Toll-like receptors: their roles in bacterial recognition and respiratory infections. Expert Rev Anti Infect Ther. 2008;6:479–495. doi: 10.1586/14787210.6.4.479. [DOI] [PubMed] [Google Scholar]
- Rock RB, Peterson PK. Microglia as a pharmacological target in infectious and inflammatory diseases of the brain. J Neuroimmune Pharmacol. 2006;1:117–126. doi: 10.1007/s11481-006-9012-8. [DOI] [PubMed] [Google Scholar]
- Royal W, 3rd, Vlahov D, Lyles C, Gajewski CD. Retinoids and drugs of abuse: implications for neurological disease risk in human immunodeficiency virus type 1 infection. Clin Infect Dis. 2003;37(Suppl 5):S427–S432. doi: 10.1086/377554. [DOI] [PubMed] [Google Scholar]
- Rumbaugh J, Turchan-Cholewo J, Galey D, St Hillaire C, Anderson C, Conant K, Nath A. Interaction of HIV tat and matrix metalloproteinase in HIV neuropathogenesis: a new host defense mechanism. FASEB J. 2006;20:1736–1738. doi: 10.1096/fj.05-5619fje. [DOI] [PubMed] [Google Scholar]
- Shen JM, Blank A, Selwyn PA. Predictors of mortality for patients with advanced disease in an HIV palliative care program. J Acquir Immune Defic Syndr. 2005;40:445–447. doi: 10.1097/01.qai.0000185139.68848.97. [DOI] [PubMed] [Google Scholar]
- Shor-Posner G. Cognitive function in HIV-1-infected drug users. J Acquir Immune Defic Syndr. 2000;25(Suppl 1):S70–S73. doi: 10.1097/00042560-200010001-00011. [DOI] [PubMed] [Google Scholar]
- Theodore S, Stolberg S, Cass WA, Maragos WF. Human immunodeficiency virus-1 protein tat and methamphetamine interactions. Ann N Y Acad Sci. 2006;1074:178–190. doi: 10.1196/annals.1369.018. [DOI] [PubMed] [Google Scholar]
- Toborek M, Lee YW, Flora G, Pu H, András IE, Wylegala E, Hennig B, Nath A. Mechanisms of the blood–brain barrier disruption in HIV-1 infection. Cell Mol Neurobiol. 2005;25:181–199. doi: 10.1007/s10571-004-1383-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkup J, Blank MB, Gonzalez JS, Safren S, Schwartz R, Brown L, Wilson I, Knowlton A, Lombard F, Grossman C, Lyda K, Schumacher JE. The impact of mental health and substance abuse factors on HIV prevention and treatment. J Acquir Immune Defic Syndr. 2008;47(Suppl 1):S15–S19. doi: 10.1097/QAI.0b013e3181605b26. [DOI] [PubMed] [Google Scholar]
- Wang J, Barke RA, Charboneau R, Roy S. Morphine impairs host innate immune response and increases susceptibility to streptococcus pneumoniae lung infection. J Immunol. 2005;174:426–434. doi: 10.4049/jimmunol.174.1.426. [DOI] [PubMed] [Google Scholar]
- Wang J, Barke RA, Charboneau R, Schwendener R, Roy S. Morphine induces defects in early response of alveolar macrophages to streptococcus pneumoniae by modulating TLR9-NF-kappa B signaling. J Immunol. 2008;180:3594–3600. doi: 10.4049/jimmunol.180.5.3594. [DOI] [PubMed] [Google Scholar]
- Wang J, Ma J, Charboneau R, Barke R, Roy S. Morphine inhibits murine dendritic cell IL-23 production by modulating toll-like receptor 2 and Nod2 signaling. J Biol Chem. 2011;286:10225–10232. doi: 10.1074/jbc.M110.188680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westendorp MO, Frank R, Ochsenbauer C, Stricker K, Dhein J, Walczak H, Debatin KM, Krammer PH. Sensitization of T cells to CD95-mediated apoptosis by HIV-1 tat and gp120. Nature. 1995;375:497–500. doi: 10.1038/375497a0. [DOI] [PubMed] [Google Scholar]
- Xiao H, Neuveut C, Tiffany HL, Benkirane M, Rich EA, Murphy PM, Jeang KT. Selective CXCR4 antagonism by tat: implications for in vivo expansion of coreceptor use by HIV-1. Proc Natl Acad Sci U S A. 2000;97:11466–11471. doi: 10.1073/pnas.97.21.11466. [DOI] [PMC free article] [PubMed] [Google Scholar]