

Abstract
The evolutionary conserved kinase mTOR couples cell growth and metabolism to environmental inputs in eukaryotes. T cells depend on mTOR signalling to integrate immune signals and metabolic cues for their proper maintenance and activation. Under steady-state conditions, mTOR is actively controlled by multiple inhibitory mechanisms, and this enforces normal T cell homeostasis. Antigen recognition by naïve CD4+ and CD8+ T cells triggers mTOR activation, which in turn programs their differentiation into functionally distinct lineages. This Review focuses on the signalling mechanisms of mTOR in T cell homeostatic and functional fates and therapeutic implications of targeting mTOR in T cells.
The mammalian target of rapamycin (mTOR; now officially known as the mechanistic target of rapamycin), a conserved serine/threonine kinase, is a central regulator of cell growth and metabolism1. mTOR senses and integrates diverse environmental signals including nutrients and growth factors, many of which deliver the inputs to the PI3K–AKT pathways that ultimately activate mTOR. mTOR exists in two multiprotein complexes in metazoans. mTOR complex 1 (mTORC1) contains the scaffolding protein, regulatory associated protein of mTOR (RAPTOR), and is sensitive to the immunosuppressant rapamycin (FIG. 1a). mTOR complex 2 (mTORC2) has a distinct scaffolding protein, rapamycin-insensitive companion of TOR (RICTOR), and is relatively resistant to rapamycin except under prolonged period of treatment. Aberrantly elevated mTOR activity is frequently associated with human malignancies, and consequently mTOR has been the subject of extensive investigation in cancer biology1. However, emerging evidence highlights a critical role of mTOR signalling in both the innate and adaptive immune systems.
Figure 1. Regulation and function of mTOR signalling pathways in T cells.


(a) Components of mTOR signalling. In T cells, mammalian target of rapamycin (mTOR) can be activated by multiple signals, including the conventionally defined signals 1–3 (antigenic stimulation, co-stimulation and cytokines), growth factors and immunomodulatory factors (such as leptin and sphingosine 1-phosphate (S1P)), and nutrients. The tuberous sclerosis 1 (TSC1)–TSC2 complex integrates signals from phosphoinositide 3-kinase (PI3K)–AKT and liver kinase B1 (LKB1)–AMP-activated protein kinase (AMPK) pathways, and this is mediated by reciprocal regulation of TSC2 activity through AKT-dependent Thr1462 and AMPK-dependent Ser1387 phosphorylation. Upon antigen stimulation, TSC is inactivated by T cell receptor (TCR) signals to mediate mTORC1 activation, but TSC function is maintained in naïve T cells to keep mTOR complex 1 (mTORC1) in check. Additionally, AKT and AMPK can directly modulate mTORC1 functions independently of TSC and RAS homologue enriched in brain (RHEB), and amino acids activate mTORC1 via the RAG family of small GTPases. mTORC1 is best known for its function to promote translation initiation and protein synthesis by directly phosphorylating the substrates S6 kinases (S6Ks) and eIF4E-binding proteins (4E-BPs). Additional mTORC1 targets include the regulatory proteins in cell signalling, metabolism and autophagy. mTORC2 is important for full activation of AKT by inducing Ser473 phosphorylation (thus AKT can be both upstream of mTORC1 and downstream of mTORC2) and for phosphorylation of various protein kinase C (PKC) isoforms including the activation of PKCθ/NF-κB in T cells and serum and glucocorticoid-inducible kinase 1 (SGK1). EAA, essential amino acid; PIP2, phosphatidylinositol-4,5-bisphosphate; PIP3, phosphatidylinositol-3,4,5-triphosphate.
(b) Control of T cell homeostasis by active inhibition of mTOR. Under steady state, negative inhibitory molecules for mTOR actively maintain the homeostasis of T cells in the thymus and periphery by preventing them from engaging alternative paths. Although phosphatase and tensin homologue (PTEN), TSC1 and LKB1 have shared capacity to inactivate mTOR, they exert distinct effects to enforce T cell homeostasis.
(c) Role of mTORC1 and mTORC2 in functional differentiation of CD4+ T cells. Following antigen stimulation, mTOR signalling promotes differentiation of T helper 1 (TH1), TH2 and TH17 effector cells, and inhibits the induction of regulatory T (TReg) cells.
The outcome of an adaptive immune response depends on the sensing of antigenic and inflammatory signals by T cells. T cells have evolved to perceive these immune stimuli and further coordinate them with diverse environmental and metabolic cues through the evolutionarily conserved mTOR pathway. Thus, mTOR endows T cells with the ability to properly integrate a multitude of signals to dictate the outcome of adaptive immunity. Whereas the first major function ascribed to mTOR in T cells was the promotion of cell cycle progression, more recent studies have established mTOR signalling as a fundamental determinant of cell fate decision both under steady-state and following cognate antigen recognition. mTOR likely affects these diverse processes in T cells by serving as a signalling node to coordinately regulate immune receptor signalling pathways, metabolic programmes and migratory activity. As a number of excellent reviews have covered the immune functions of mTOR2–4, this Review mainly focuses on the most recent genetic studies that have identified new roles for mTOR signalling in T cell fate decision, and on the therapeutic implications of modulating mTOR functions in T cells. Following the hierarchy of signal transduction, first the extracellular inputs that feed into the mTOR pathway in T cells are addressed. Then the mechanisms through which negative and positive components of mTOR signalling impinge upon cell fate decision in T cells are described, with a special focus on T cell homeostasis and T helper cell differentiation (Table 1). Finally, mTOR downstream effector pathways involved in T cell metabolism, and the implications of targeting mTOR and metabolic pathways for disease therapeutics are discussed.
Table 1.
Genetic models of T cell-specific deletion of mTOR components and negative regulators.
| Target genes | Cre types | Biochemical defects | Thymic phenotypes | Peripheral phenotypes | Refs |
|---|---|---|---|---|---|
| Mtor | CD4-Cre | Abrogated mTORC1 and mTORC2 activities | None | Diminished TH1, TH2 and TH17 and spontaneous induced TReg differentiation | 6, 30 |
| Rheb | CD4-Cre | Abrogated mTORC1 activity | None | Diminished TH1 and TH17 and increased TH2 differentiation | 30 |
| Rictor | CD4-Cre; dLck-iCre | Abrogated mTORC2 activity | None | Diminished TH2 (both lines) and TH1 (in dLck-iCre) differentiation | 30, 77 |
| Pten | CD4-Cre; Lck-Cre | Increased AKT and mTOR activities | Lymphoma; minor defects in development | Autoimmune disease | 44–47, 49–51 |
| Tsc1 | CD4-Cre; Lck-Cre | Increased mTORC1 and decreased mTORC2 activities | None (CD4-Cre) or minor reduction of cell number (Lck-Cre) | Loss of T cell quiescence and survival; diminished antigen-specific response | 7–9 |
| Lkb1 | CD4-Cre; Lck-Cre | Diminished AMPK and increased mTORC1 activities | Reduced survival; defective α-selection and positive selection | Reduced survival and TCR-induced proliferation; increased metabolism and cytokine production | 57–60 |
Upstream input signals to mTOR
In T cells, mTOR senses three broad categories of instructive signals. The first is the immune activation signals transduced from dendritic cells (DCs), including antigens, co-stimulation and inflammatory cytokines, collectively known as signals 1–3, that are essential to direct proper T cell activation and differentiation5. The other two instructive signals are mediated by environmental stimuli such as growth and immunomodulatory factors, and metabolic cues derived mainly from nutrients (FIG. 1a). Whereas the immune activation signals are unique to T cells, the environmental and metabolic stimuli act on all eukaryotic cells. Many of the upstream signals activate mTORC1 through the GTP-loaded form of the small GTPase RHEB (FIG. 1a). RHEB is tightly regulated by the tuberous sclerosis 1 (TSC1)–TSC2 complex, which, through its GTPase-activating protein (GAP) activity toward RHEB, inactivates RHEB and mTORC1. The TSC complex serves as a molecular switch to integrate upstream signals, and in particular the positive and negative signals transduced from PI3K–AKT and AMP-activated protein kinase (AMPK) pathways, respectively1. RHEB deficiency in T cells blunts mTORC1 activation in response to T cell receptor (TCR) stimulation6, whereas loss of TSC1 disrupts the entire TSC complex and enhances basal and TCR-induced mTORC1 activity7–9. These results highlight a crucial role for the TSC-RHEB axis in T cell responses.
TSC-independent pathways also engage mTORC1, although the importance of these mechanisms in T cells remains to be determined. Activation of mTORC1 in response to amino acids requires the RAG family of GTPases10, 11, whereas AMPK-mediated direct phosphorylation of RAPTOR inhibits mTORC1 during energy stress12. Further, the stress-activated p38β MAP kinase targets different components of mTORC1 to either positively or negatively regulate its activity under different environmental stresses13, 14. Upstream pathways that activate mTORC2 are just beginning to be identified, with recent studies showing a role for the ribosome to link PI3K to mTORC2 activation15, 16. Moreover, another small GTPase RAC1 binds directly to mTOR to activate mTORC1 and mTORC2, which provides a means to regulate both mTOR complexes simultaneously17. The convergence of multiple signals on mTOR suggests that its basic function is a signal integrator.
Immune signals 1–3
Both mTORC1 and mTORC2 are activated within minutes after TCR stimulation. Magnitude of mTOR activation is directly correlated with the duration of T cell–DC interaction and the dose of the cognate antigen18, 19. mTOR activity is further shaped by co-stimulatory signals. CD28 co-stimulation is a classic PI3K–AKT activating signal that in turn upregulates mTOR activity induced by TCR, to facilitate productive T cell activation20–22. Another co-stimulatory receptor, OX40, a member of the tumour necrosis factor receptor (TNFR) family, assembles a signalling complex by recruiting PI3K–AKT to augment TCR-dependent AKT signalling23. By contrast, the PD1–PDL1 axis, a negative T cell co-stimulatory pathway, downregulates mTOR signalling to mediate immune tolerance24.
As compared with TCR-dependent rapid activation of mTOR, the homeostatic cytokine interleukin-7 (IL-7) induces delayed yet sustained PI3K/AKT and mTOR activation. This activation depends on the transcriptional activity of signal transducer and activator of transcription 5 (STAT5), and contributes to IL-7-mediated glucose uptake and tropic effects in T cells25, 26. However, increased mTORC1 activity as a result of TSC1 deficiency also impairs IL-7-dependent survival response in naïve T cells7, suggesting that a defined threshold of mTOR activity is important to mediate IL-7 response. In antigen-stimulated CD8+ T cells, IL-12 enhances and prolongs TCR-dependent mTOR activation, to programme functional maturation of effector cells. Similar to IL-7-stimulated naïve T cells, IL-12-induced mTOR activation in antigen-stimulated CD8+ T cells is indirect and depends on STAT signalling (in this case, STAT4)27. In T helper 2 (TH2) and TH17 cells, mTOR is activated by IL-4 and IL-1, respectively, to facilitate cell cycle progression28, 29. Furthermore, there is extensive interplay between mTOR and STAT signalling in T cells6, 30 and other cells31.
Leptin receptor
Leptin, an adipose-derived hormone regulating energy metabolism, has long been known to directly regulate T cell proliferation and cytokine production, thereby linking nutritional status and proinflammatory immune responses32. More recently, regulatory T (TReg) cells were shown to express leptin and its receptor and contain more abundant mTOR activity relative to conventional T cells33. Neutralization of leptin or deletion of leptin receptor considerably diminishes mTOR activity in TReg cells, suggesting a link between autocrine secretion of leptin and mTOR activation in TReg cells. The elevated mTOR activity that results from leptin signalling maintains the anergic state of TReg cells, because transient inhibition of mTOR or neutralization of leptin reverses the hyporesponsiveness of TReg cells to TCR stimulation, resulting in their robust proliferation33, 34. Leptin also activates mTOR in autoreactive CD4+ T cells to promote their survival and mediate autoimmune neuroinflammaton35. As the leptin level is directly correlated with nutrient status, the leptin–mTOR axis has been proposed to bridge metabolism and immunity33.
S1PR1
Sphingosine 1-phosphate receptor 1 (S1PR1), a G protein-coupled receptor for the bioactive lipid sphingosine 1-phosphate (S1P), is a crucial regulator of T cell egress out of the thymus and secondary lymphoid organs. In TReg cells, S1PR1 activates AKT and mTOR, and this delivers a cell-intrinsic negative signal to restrain TReg cell suppressive activity36. In conventional CD4+ T cells, S1PR1 is dispensable for immediate mTOR activation but is important to sustain mTOR activity during their differentiation into effector cells37. S1PR1-dependent activation of mTOR inhibits the generation of TReg cells while driving TH1 cell development in a reciprocal manner. These studies identify an S1PR1–mTOR axis that controls TReg function and T cell lineage choices36, 37.
TLRs
Ligation of Toll-like receptors (TLRs) expressed on innate immune cells induces copious amounts of proinflammatory molecules that are important for immediate immune defense. Additionally, both CD4+ and CD8+ T cells express functional TLRs, and recent results suggest a T cell-intrinsic role for TLRs in immune responses. TLR2, via signalling through the adaptor molecule MyD88, activates mTOR and promotes the expression of T-bet in effector CD8+ T cells38. Since mTOR is also downstream of TCR signalling, activation of mTOR therefore bridges TCR and TLR signals and promotes effector CD8+ T cell function38 and contributes to T cell memory formation39. However, MyD88 deficiency in T cells diminishes initial T cell expansion, but not the subsequent generation of the memory population40. Further study is required to clarify how TLR2 and MyD88 signalling affects mTOR activation in T cells.
mTOR is also activated by additional extracellular signals in T cells. Insulin induces activation of mTORC1 and promotes mTOR-dependent T-bet expression in antigen-activated CD8+ T cells27. Also, NOTCH1 activates AKT–mTOR during early thymic development and relies on AKT-dependent metabolic and trophic effects to regulate thymocyte differentiation and survival41. Finally, mTOR is intimately linked to nutrient sensing in all eukaryotic cells1, and the physiological relevance of metabolic regulation of mTOR is discussed in further details below.
mTOR in immune homeostasis
T cells develop in the thymus through a step-wise differentiation process. Once mature T cells are released into the periphery, they circulate through the blood and peripheral lymphoid organs in a quiescent state characterized by small cell size and low metabolic activity. Survival of naïve T cells relies on the engagement of TCRs by self-peptide–MHC complexes and on the availability of IL-7, and is further shaped by growth factors and nutrients for metabolic fitness42, 43. Given the central role of mTOR as an environmental sensor, it might be predicted that mTOR activity is required for the homeostasis of thymocytes and peripheral T cells. Surprisingly, deletion of the Mtor gene after initial thymocyte development (using the CD4-Cre system) had minimal effects on the homeostasis of peripheral T cells under steady state6, although the requirement of mTOR in early thymic development has yet to be determined. By contrast, several negative regulators of mTOR signalling were found to enforce normal homeostasis of T cells, as manifested by the disrupted development and maintenance in T cells lacking these inhibitory molecules (FIG. 1b).
PTEN suppresses lymphoma and autoimmunity
Phosphatase and tensin homologue (PTEN) mainly functions as a lipid phosphatase and hydrolyzes phosphatidylinositol-3,4,5-triphosphate, thereby mediating the reverse reaction of PI3K (FIG. 1a). Deletion of PTEN in T cells causes lymphoma that originates in the thymus and is largely driven by c-Myc overexpression44–49. Pten−/− thymocytes exhibit markedly elevated AKT and mTOR activities even before transformation47, 50. Importantly, tumour development is blocked by mTOR inhibition following rapamycin treatment or deletion of 3-phosphoinositide-dependent protein kinase 1 (PDK1), indicating an crucial role of AKT and mTOR signalling in this process47, 48. Somewhat unexpectedly, PTEN deficiency does not overtly disrupt homeostasis of pre-malignant thymocytes, except for some minor defects in cell size and generation of double-positive thymocytes and invariant natural killer T (iNKT) cells47, 50, 51.
Pten+/− mice develop a late-onset autoimmune disease associated with resistance to FAS-mediated apoptosis52. Consistent with this observation, complete loss of PTEN in T cells impairs central tolerance as well as peripheral tolerance44 and the induction of forkhead box P3 (FOXP3) expression53. Recent studies demonstrate that development of autoimmunity in Pten−/− mice can be exclusively mediated by peripheral T cells. Therefore, the two main defects that result from the loss of PTEN function, lymphoma and autoimmunity, are separable and mediated by T cells of distinct developmental stages49. Because of the pivotal roles for PTEN in immune homeostasis and function, molecular signals regulating PTEN have received considerable interest. Work from several independent groups has revealed that the microRNA cluster 17–92 represses the expression of PTEN to promote AKT-mTOR activity in lymphocytes, and consequently controls PTEN-dependent autoimmune and oncogenic functions54–56.
TSC1 maintains quiescence of naïve T cells
TSC1 and TSC2 function as an integral complex to stringently control mTORC1 activity (FIG. 1a). We and others have recently found that TSC1-mediated control of mTOR signalling enforces a quiescent programme in naïve T cells by controlling cell size, cell cycle entry and metabolic machinery7–9. Abrogation of quiescence predisposes TSC1-deficient T cells to apoptosis, and this results in the loss of conventional T cells and iNKT cells. The remaining Tsc1−/− T cells exist in a unique ‘semi-activated’ (CD44+CD122−) status in vivo and exhibit increased activation, cell cycle entry and cytokine expression after acute TCR stimulation. Despite this, TSC1-deficient mice fail to generate effective anti-bacterial immune responses, even when the excessive apoptosis is largely blocked, which suggests that maintenance of naïve T cell quiescence is important for a productive immune response7. The precise mechanism by which TSC1 deficiency dampens the immune response is uncertain but may involve premature activation of the cell cycle and metabolic machineries and transcriptional responses in Tsc1−/− naïve cells before they receive proper TCR signals7. These studies identify TSC1 as a bona fide factor in establishing naïve T cell quiescence to facilitate immune homeostasis and function.
Tsc1−/− peripheral T cells exhibit increased mTORC1 but diminished mTORC2 activities7–9. Treatment of Tsc1−/− mice with rapamycin partly rectifies the altered T cell homeostasis and survival, whereas loss of mTORC2 alone has no apparent effect. These results indicate a crucial contribution for mTORC1 activation to T cell homeostasis7. By contrast, in mature thymocytes lacking TSC1, mTORC1 activity is increased but cell survival is not affected7. Moreover, although PTEN-deficient T cells upregulate mTORC1 activity, they largely maintain their quiescence prior to tumour development47, 50, and this suggests a crucial but cell context-dependent effect of mTORC1 on T cell homeostasis. Collectively, these data illustrate that TSC1-dependent active control of mTORC1 is a key checkpoint to enforce quiescence of naïve T cells in the periphery7–9.
LKB1 promotes T cell development and survival
Liver kinase B1 (LKB1) phosphorylates and activates AMPK subfamily members in response to energy depletion. Deletion of LKB1 in T cells results in profound defects in multiple compartments, including extensive apoptosis of T cells, impaired thymic selection and dysregulated metabolism and proliferation57–60. Despite the well-documented role of the LKB1–AMPK axis in mTORC1 inhibition (FIG. 1a)61, signalling mechanisms downstream of LKB1 in T cells are not fully understood. The increased mTORC1 activity in Lkb1−/− T cells contributes to excessive cytokine production60, but whether it leads to reduced cell survival or other defects is unclear. Also, although AMPK activity is diminished in Lkb1−/− T cells57–60 and AMPK is activated by TCR stimulation62, loss of AMPKα1 (the predominant AMPK isoform in T cells) causes only slight disturbance of T cell homeostasis60, 63. Therefore, T cell homeostasis likely requires additional AMPK-related kinases regulated by LKB161.
Altogether, analyses of T cells deficient in the tumour suppressors PTEN, TSC1 and LKB1 have provided new insight into mechanisms of immune homeostasis. Notably, hematopoietic stem cells (HSCs) lacking these molecules exhibit some analogous defects in the control of proliferation, survival or function as mutant T cells64–70, suggesting that HSCs and naïve T cells have a common requirement for these pathways for the proper maintenance. Further, the defects in PTEN or TSC1-deficient HSCs are largely responsive to rapamycin correction64, 66, 67, but LKB1 functions independent of mTOR in stem cells68–70. Therefore, despite the shared ability to inhibit mTOR signalling, these molecules employ distinct pathways for the homeostatic control of HSCs and possibly T cells. Aside from these well characterized mTOR inhibitory molecules, recent studies have identified additional negative regulators of mTOR with important roles in cell signalling and disease regulation, such as DEPTOR71, SESTRIN72, and the mTORC1 component PRAS4073, 74. The roles of these molecules in T cell-mediated adaptive immunity have not been addressed.
Moreover, mTOR may further interact and crosstalk with transcriptional and immune signalling pathways to mediate T cell homeostasis. In particular, forkhead box O1 (FOXO1), a crucial transcription factor for naïve T cell survival and trafficking75, 76, is under the stringent control of AKT-mediated phosphorylation and nuclear exclusion and thus could serve as an important downstream target of mTORC2. Consistent with this idea, transcriptional targets of FOXO1 including IL-7 receptor α (IL-7Rα) and trafficking molecules such as CD62L are altered in T cells treated with mTOR- or AKT-inhibitors, or in PTEN-, RICTOR- and PDK1-deficient T cells75, 77–79. The functional link and physiological relevance of these interactions remain to be fully defined. Diacylglycerol kinases have also been shown to inhibit TCR-induced mTOR activity by downregulating diacylglycerol-mediated RAS signalling80, which may contribute to T cell homeostasis and function.
mTOR in T helper cell differentiation
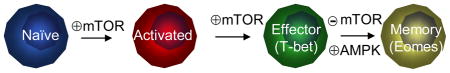
Early studies on mTOR signalling in T cell responses centered upon the anti-proliferative effect of the inhibitor rapamycin. Analysis of Mtor−/− T cells has confirmed a role for mTOR in cell cycle progression, although T cell proliferation is delayed but not abolished6. Accumulative evidence, however, highlights a central role for mTOR as a fundamental determinant of cell fate decision of antigen-activated CD4+ and CD8+ T cells. Inhibition of mTOR with rapamycin facilitates induction of anergic and regulatory CD4+ T cells, two crucial components of peripheral tolerance (Box 1), as well as differentiation of memory CD8+ T cells (Box 2). Excellent reviews discuss the role of mTOR signalling in the differentiation of regulatory, effector and memory T cells4, 81–83, so here I mainly focus on more recent genetic evidence that establishes a crucial role for mTOR in T helper cell differentiation.
Box 1. mTOR in peripheral tolerance.
Induced regulatory T (TReg) cells act in synergy with naturally occurring TReg cells to establish immune tolerance139. Inhibition of the mammalian target of rapamycin (mTOR) activity has been shown to induce de novo forkhead box P3 (FOXP3) expression140, 141, or to expand preexisting natural TReg cells113. Conversely, increasing AKT activity, through either deletion of phosphatase and tensin homologue (PTEN) or enforced expression of constitutively active AKT, leads to mTOR-dependent inhibition of induced TReg cell differentiation53, 142. Moreover, several upstream receptors, including programmed cell death ligand 1 (PDL1) and sphingosine 1-phosphate receptor 1 (S1PR1), inhibit the generation of induced TReg cells through mTOR activation24, 37. Two downstream pathways mediate the inhibitory effects of mTOR on induced TReg cell differentiation (FIG. 2a). First, SMAD3, a key transcription factor downstream of transforming growth factor β (TGF-β) signalling for FOXP3 induction, is antagonized by mTOR signalling in multiple cell types including T cells6,37,143. Second, forkhead box O1 (FOXO1) and FOXO3, which induce FOXP3 expression144–146, are inactivated by AKT-dependent phosphorylation, although how this is controlled by mTOR complex 2 (mTORC2) signalling requires further studies147. Interestingly, FOXOs have been shown to form a complex with SMAD3 to control neuroepithelial and glioblastoma cell proliferation148, and the integration of SMAD and FOXO signalling by mTOR in T cells will be an interesting topic to explore.
Aside from immune suppression mediated by TReg cells, another important mechanism of peripheral tolerance is the induction of T cell anergy. T cell anergy is usually induced by TCR (signal 1) alone, in the absence of co-stimulation (signal 2). Among the pathways strongly activated by signal 2 are AKT and mTOR21, 22. Indeed, blocking mTOR activity by rapamycin induces T cell anergyin vitro, even in the presence of both signals 1 and 2. In vivo, mTOR inhibition promotes T cell anergy under conditions that would normally induce active priming, indicating that mTOR has a central role in dictating the decision between T cell activation and anergy21, 22. Although rapamycin treatment inhibits the G1 phase of the cell cycle, blocking T cell proliferation alone does not induce anergy4. Instead, blocking metabolic pathways necessary for mTOR activation promotes T cell energy, suggesting that mTOR-dependent metabolic control is a key determinant of T cell activation and anergy21, 22.
Box 2. mTOR in memory CD8+ T cell differentiation.
CD8+ T cells constitute an important arm of adaptive immunity due to their response to pathogens by clonal expansion and differentiation into cytotoxic effector cells149. During the contraction phase that follows, the majority of effector cells die by apoptosis, while the remaining small subset of antigen-specific T cells develops into long-lived memory T cells. Recent results have revealed a crucial role of mTOR in the lineage decisions between the short-lived effector T cells and memory T cell precursors. Arakai et al. described that mTOR modulated memory CD8+ T cell formation in a kinetic and dose-dependent manner119. In the model of lymphocytic choriomeningitis virus (LCMV) infection, rapamycin treatment during the T cell expansion phase diminished apoptotic death of effector cells, leading to an increase in the quantity of memory T cells. By contrast, rapamycin treatment during the contraction phase promoted the protective capacity of memory T cells and thus the quality of T cell memory. These effects were dose-dependent as CD8+ T cell expansion was blocked by high dose of rapamycin treatment. Silencing of RAPTOR in T cells recapitulated rapamycin effect, thereby establishing a cell-intrinsic effect of mTORC1 in memory CD8+ T cell formation119. However, how mTORC1 is activated by upstream signals in CD8+ T cells is unclear, as it appears to require PI3K27 but occurs largely independent of AKT79.
Pearce et al. independently identified an inhibitory role of mTOR in memory formation and further linked this function to an upstream regulator, TRAF696. In a model ofListeria monocytogenes infection, Traf6−/− CD8+ T cells showed normal effector responses but could not develop into memory cells. This defect was associated with the failure to upregulate fatty acid oxidation (FAO), and was rectified by pharmacological activation of AMP-activated protein kinase (AMPK) or inhibition of mTOR96. More recently, FAO has been shown to promote mitochondrial respiratory capacity selectively in memory T cells as an important mechanism to promote their survival150. In another related study, Rao et al. found that rapamycin treatment diminished CD8+ effector function, but promoted memory formation and tumour immunity27. mTOR inhibition decreased T-bet levels while inducing EOMES expression as important mechanisms to regulate T cell differentiation27. However, deletion of PTEN does not strongly affect CD8+ memory formation151, and thus the underlying molecular mechanisms remain to be identified. These studies collectively support a central role for mTOR in dictating effector and memory fate of CD8+ T cells in infectious and tumour immunity.
Decision making between effector and TReg cells
Naïve CD4+ T cells respond to antigen stimulation by developing into distinct lineages, including TH1, TH2 and TH17 cells, with specialized properties and effector functions (FIG. 1c), and induced TReg cells to prevent excessive immune reaction84. Powell and colleagues revealed that mTOR promotes the differentiation of effector T cells6. Mtor−/− T cells show normal TCR-induced activation markers and IL-2 production, but fail to differentiate into TH1, TH2 and TH17 effector cells. Furthermore, they are unable to activate the selective STAT proteins or express the lineage-selective transcription factors (STAT4 and T-bet, STAT6 and GATA3, and STAT3 and RORγt for TH1, TH2 and TH17 cells, respectively). These results identify an indispensable role for mTOR in effector CD4+ T cell differentiation6. Conversely, TCR stimulation of Mtor−/− T cells results in the spontaneous induction of FOXP3, even in the absence of exogenous cytokines6. This phenotype is not observed in either RHEB or RICTOR-deficient T cells, indicating that both mTOR complexes contribute to the inhibition of TReg cell induction, possibly through distinct downstream mechanisms (FIG. 2a)6, 30, 77.
Figure 2. mTOR-dependent signalling in CD4+ T cell differentiation.


(a) Differentiation of induced regulatory T (TReg) cells. Induction of forkhead box P3 (FOXP3) expression depends on the transcription factors SMAD3, SMAD4, FOXO1 and FOXO3. mTOR inhibits induced TReg cell differentiation by antagonizing the function of SMAD3 and SMAD4 downstream of transforming growth factor β (TGF-β) signalling, and by inducing nuclear exclusion of FOXO1 and FOXO3. These two effects are likely mediated by mTOR complex 1 (mTORC1) and mTORC2, respectively.
(b) Differentiation of T helper 1 (TH1) cells. mTORC1 inhibits induction of suppressor of cytokine signalling 3 (SOCS3), a crucial negative regulator of signal transducer and activator of transcription 4 (STAT4), to promote interleukin-12 (IL-12) signalling and TH1 cell differentiation. Additionally, mTORC2 is required for activation of AKT that also contributes to interferon γ (IFN-γ) production.
(c) Differentiation of TH2 cells. mTORC2 promotes TH2 cell differentiation via two mechanisms: by preventing expression of SOCS5, a negative regulator of IL-4 and STAT6 signalling, and by activating protein kinase C-theta (PKCθ) signalling and nuclear factor-κB (NF-κB)-mediated transcription. By contrast, mTORC1 negatively constrains STAT6 signalling and TH2 cell differentiation.
TH1 and TH17 cell differentiation
The two mTOR complexes exert disparate effects on effector T cell differentiation. Deficiency of RHEB, and consequently the loss of mTORC1 activity, largely recapitulate the impaired TH1 and TH17 cell differentiation in Mtor−/− T cells, suggesting that mTORC1 mediates mTOR-dependent TH1 and TH17 cell differentiation30. Rheb−/− T cells express increased levels of suppressor of cytokine signalling 3 (SOCS3), a negative regulator of STAT signalling, and silencing of SOCS3 expression restores TH1 cell differentiation in these cells (FIG. 2b). Thus, mTORC1 signalling promotes TH1 cell differentiation by modulating cytokine signalling30. Since mTORC1 activity can also be regulated by upstream signals other than RHEB12, how such RHEB-independent pathways contribute to T cell differentiation awaits future investigation. Additionally, deficiency of RICTOR and thus loss of mTORC2 activity reduce TH1 cell differentiation through the downregulation of AKT signalling (FIG. 2b)77, although another independent study shows a dispensable role for mTORC2 in TH1 cell differentiation30. How mTOR affects TH17 cell differentiation remains to be fully established, but could partly involve the upregulation of hypoxia-inducible factor 1α (HIF1α) (see below for details)85, 86.
TH2 cell differentiation
In contrast to RHEB/mTORC1 signalling, mTORC2 is required for TH2 cell differentiation (FIG. 2c). Two groups have independently demonstrated that loss of RICTOR impairs TH2 cell differentiation in vitro and in vivo, without appreciably affecting the development of TH17 cells30, 77. Lee et al. attributed the inability of Rictor−/− T cells for TH2 cell differentiation to decreased protein kinase C-theta (PKCθ) activity and nuclear factor-κB (NF-κB)-mediated transcription, as complementation with activated PKCθ restored the TH2 defects77. Delgoffe et al. described elevated SOCS5 expression in Rictor−/− T cells, which accounted for the diminished TH2 response30. Collectively, these results establish mTORC2 as a crucial regulator of TH2 cell differentiation30, 77, and future studies will determine whether PKCθ/NF-κB and SOCS5 act in separate pathways or are components of the same signalling cascade. By contrast, mTORC1 negatively controls TH2 cell differentiation, as indicated by the increased STAT6 activation and GATA3 expression in Rheb−/− T cells (FIG. 2c)30.
In summary, mTOR dictates cell fate decision between effector and regulatory T cells, with mTORC1 and mTORC2 exerting distinct effects on immune receptor signalling. These effects are further shaped by the metabolic pathways, which will be discussed below. Moreover, loss of mTORC1 or mTORC2 activity impairs T cell proliferation, with a stronger effect observed in Rheb−/− T cells30, 77. Cell cycle progression is known to be a prerequisite for T cell differentiation, partly by allowing epigenetic remodeling of cytokine loci87. It would be informative to examine how mTORC1 and mTORC2 regulate cell cycle progression, and whether this affects epigenetic regulation or lineage differentiation.
Effector pathways and metabolic regulation
To orchestrate T cell homeostasis and differentiation, mTOR affects several downstream pathways, including those involved in immune receptor signalling, metabolic programming and T cell trafficking (Box 3). Whereas the effects of mTOR on immune receptor signalling molecules, as described above, are easy to appreciate, the relative contribution of mTOR to T cell metabolism and trafficking and how this influences cell fate decision in vivo remain under debate4, 81, 83. However, evidence is emerging that mTOR serves as a signalling node to regulate both T cell metabolism and migration and further link them to immune signalling and transcriptional networks, to ensure that the metabolic programme and migratory activity match cell fate decision of T cells. In particular, recent studies have revealed exciting new findings on how mTOR-dependent metabolism controls T cell fate, an area with notable therapeutic implications.
Box 3. mTOR in T cell trafficking.
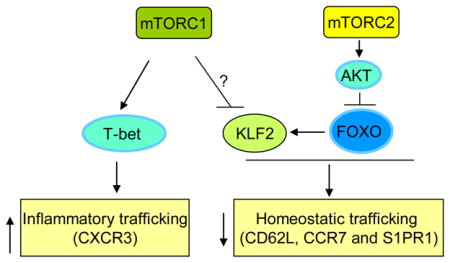
Trafficking of naïve and memory T cells requires signals transduced through chemokine receptors and trafficking molecules, such as CC chemokine receptor 7 (CCR7), CD62L and sphingosine 1-phosphate receptor 1 (S1PR1). Antigen-stimulated effector T cells downregulate these surface molecules, but upregulate proinflammatory chemokine receptors and adhesion molecules that endow these cells with increased ability to traffic to sites of inflammation instead of entering secondary lymphoid organs152. Therefore, quiescent and activated T cells are characterized by differential migratory activities, and proper regulation of this process is essential for a productive immune response. A role for mTOR in T cell migration was revealed by the observation that rapamycin treatment prevented the downregulation of CCR7, CD62L and S1PR1 in CD8+ T cells responding to TCR or cytokine signals78. Rapamycin-treated effector cells tend to migrate to lymph nodes and spleen rather than nonlymphoid tissues27, 78. Conversely, deficiency of phosphatase and tensin homologue (PTEN) or tuberous sclerosis 1 (TSC1) is sufficient to downregulate these surface receptors7, 75, 78. mTOR-induced effects may involve both mTORC2–AKT–FOXO and AKT–mTORC1 pathways, and depend upon KLF2, a critical transcription factor for CCR7, CD62L and S1PR1 expression83. This is further supported by the observations that inhibition of AKT activity or deficiency of 3-phosphoinositide-dependent protein kinase 1 (PDK1) enhances KLF2 expression and the downstream trafficking molecules79. In addition to controlling KLF2 expression, mTOR is required for the expression of T-bet27, which, among many of its downstream effects, directs the expression of proinflammatory chemokine receptors to coordinate T cell migration with effector and regulatory activities153, 154. These results highlight that regulation of T cell migration is an important mechanism by which AKT-mTOR signalling modulates immune function in vivo83.
Metabolism in T cell activation and differentiation
Naïve T cells utilize a catabolic metabolism where they generate ATP through the tricarboxylic acid (TCA) cycle and oxidative phosphorylation. T cell activation markedly elevates uptake and consumption of glucose and glutamine, with a concomitant suppression of fatty acid oxidation (FAO) (FIG. 3a)43, 88–90. Among the central regulators of this metabolic reprogramming induced by TCR stimulation are PI3K–AKT signalling and c-Myc, which regulate glucose metabolism and a global metabolic transcriptome, respectively88, 91, 92. Additional transcription factors such as estrogen-related receptor-alpha (ERRα) also contribute93, but HIF1α, which has overlapping functions with c-Myc in cancer cell metabolism94, is not required for TCR activation-induced metabolic reprogramming88. PI3K–AKT pathways signal through mTOR, which further engages an extensive crosstalk with c-Myc in many cellular contexts95. In activated T cells, rapamycin inhibits expression of c-Myc and induction of glycolysis85,88, whereas T cells lacking c-Myc fail to fully activate mTORC1 in response to TCR signals88. Consistent with a pivotal role of mTOR in T cell metabolism, Tsc2−/− T cells, with constitutive mTOR activity, are highly glycolytic after TCR stimulation93. Moreover, mTOR orchestrates the metabolic programme of naïve T cells under steady state, as gene expression programmes for glucose, nucleotide and amino acid metabolism are abnormally upregulated in Tsc1−/− naïve T cells that exhibit increased mTORC1 activity7. In summary, mTOR has a vital role in orchestrating the metabolic programmes in both naïve and activated T cells.
Figure 3. mTOR in T cell metabolism.

(a) Metabolic programmes in T cells (left) and mTOR activity in T cell subsets (right). Only the prominent metabolic programs are presented. FAO, fatty acid oxidation; TCA, tricarboxylic-acid cycle; ETC, electron-transport chain; ROS, reactive oxygen species; OXPHOS, oxidative phosphorylation.
(b) Proposed mechanisms of mTOR-mediated metabolic pathways in T cell differentiation. An important upstream signal to activate mTOR is nutrients, in particular essential amino acids (EAAs) whose levels are actively controlled by dendritic cells (DCs). Once activated, mTOR serves as a platform to engage several downstream effector pathways including immune receptor signalling, metabolic programme and migratory activity. mTOR promotes metabolism via activating a gene expression programme consisting of metabolic gene targets of transcription factors hypoxia-inducible factor 1α (HIF1α), c-Myc and SREBP, which in turn impinge upon biosynthetic and bioenergetic pathways. Three potential downstream mechanisms are proposed to explain how these metabolic pathways regulate T cell differentiation, including (1) signal crosstalk; (2) feedback control; (3) selective expansion and survival. Among metabolites with signalling activities, reactive oxygen species (ROS) oxidize the catalytic cysteine residues of phosphatases to cause their inactivation, whereas NAD+ is required for the activity of sirtuin family deacetylases.
Recent studies further demonstrate that a metabolic switch to AMPK-dependent FAO is required for the differentiation of CD8+ memory T cells and CD4+ TReg cells96, 97, both of which are less anabolic than their effector counterparts4. Defective memory formation in Traf6−/− CD8+ T cells is associated with the failure to upregulate FAO, and enhancement of FAO by rapamycin or metformin (to activate AMPK) treatment restores the memory formation96. Notably, AKT-independent metabolism has been identified in CD8+ T cell responses79, 83, although such findings do not necessarily exclude a role for mTOR in this process because of a weak effect of AKT inhibition on mTOR activity in CD8+ T cells79. In CD4+ T cells, elevated FAO and AMPK activities are associated with differentiation into TReg but not effector cells, and FAO inhibition prevents rapamycin-induced TReg cell induction97. Therefore, a common component for the differentiation of memory and regulatory T cells is the selective requirement of FAO, in a process reciprocally regulated by mTOR and AMPK.
In contrast to TReg cells and memory CD8+ T cells, effector T cell differentiation is accompanied by strong upregulation of glycolysis. Pharmacological blocking of mTOR or glycolysis reduces TH1, TH2 and TH17 cell differentiation85, 97. HIF1α is selectively expressed in T cells undergoing TH17 cell differentiation and its induction requires signalling through mTOR85, 86. HIF1α is required for mediating glycolysis during TH17 cell differentiation and contributes to the lineage choices between TH17 and induced TReg cells85. Therefore, mTOR-dependent induction of transcription factors c-Myc and HIF1α orchestrates a metabolic checkpoint in TCR-activated and TH17-polarized T cells, respectively85, 88. HIF1α has also been shown to exert direct effects to promote degradation of FOXP3 and the transcriptional activity of RORγt86. Interestingly, similar to the induction of TReg cells, TH17 cell differentiation appears to be particularly sensitive to metabolic perturbations. Impairment in TH17 cell differentiation results not only from blockade of glycolysis85, but also from depletion of selective amino acids98 and increases in lipid metabolism99. As mTORC1 is a key regulator of amino acid and lipid metabolism100, it will be interesting to examine whether these additional metabolic processes in TH17 cells are regulated by mTOR.
Sensing and propagating metabolic cues
As a central environmental sensor, mTOR links growth factor signalling and availability of nutrients especially amino acids1, 101. This ancient pathway of amino acid metabolism is acquired by the immune system as an important mechanism for immune regulation102. In response to TReg cell-mediated suppression, DCs upregulate the enzymes that consume multiple essential amino acids present in the tissue microenvironment. Consequently, T cells respond to nutrient starvation by downregulation of mTORC1 activity and induction of FOXP3103. Although other nutrient sensors such as GCN2 have been identified in T cells104, mTORC1 appears to have a dominant role in sensing amino acid availability103, 105. Consistent with this notion, inhibition of mTOR-dependent amino acid or glucose metabolism attenuates T cell activation and instead induces T cell anergy (Box 1)21, 22.
Despite these advances, the extent to which mTOR-dependent metabolic pathways regulate T cell differentiation remains a contentious issue4, 81, 83. As T cell fate is ultimately manifested as lineage-specific gene expression programme, how do the metabolic intermediates downstream of mTOR activation dictate signalling and transcriptional events? Here I would like to discuss three potential mechanisms (FIG. 3b). First, certain metabolic intermediates, such as reactive oxidative species (ROS) and nicotinamide adenine dinucleotide (NAD) whose production in the metabolic flux depends on mTOR, are now recognized to exert direct signalling activities by serving as substrates or modifiers of enzymes and other regulators, thereby functioning as metabolic checkpoints106. Given the recently identified roles of ROS and NAD+-dependent sirtuin 1 in T cell function and differentiation107, 108, mTOR-dependent metabolic flux may directly engage signalling transduction and crosstalk. Second, nutrient/energetic signals are able to mediate positive and negative feedbacks on mTOR activity itself, which may affect not only cell metabolism but also other mTOR-dependent events such as immune receptor signalling. In particular, activation of mTORC1 by amino acids and the crosstalk with c-Myc upregulate the expression of the amino acid transporter CD98, which establishes a positive loop that amplifies mTOR signalling in T cells88, 101. Considering the classic role of mTOR in protein translation, which consumes amino acids, it is perhaps appropriate that mTOR activity is regulated by such a positive loop. Consistent with this notion, inhibition of translation with cycloheximide considerably promotes mTORC1 activity, presumably by elevating the levels of intracellular amino acids109. However, excessive mTORC1 activation may deplete cellular ATP and cause energetic stress, which activates AMPK to negatively control mTOR activity110. Third, metabolism is intricately linked to cell cycle and apoptotic machineries111, 112. T cells with proper metabolism are likely to exhibit preferential survival and/or expansion, and consequently on a population level, are selected to develop into a particular lineage. In support of this notion, rapamycin differentially affects the proliferation and survival of effector and TReg cells113–116. Future efforts to identify the molecular components orchestrating these processes should provide exciting insight into the interface between metabolism and immunity.
Therapeutic targeting of mTOR
The immunosuppressive effect of rapamycin dates back to the 1970s, before the identification of its molecular target. Recent studies suggest that blocking mTOR not only mediates immunosuppression in transplant rejections and autoimmune disorders, but also boosts immunity under selective conditions and impacts other aspects of T cell homeostasis and functions.
mTOR inhibition for immunosuppression
In late 1990s, FDA approved the use of rapamycin to prevent rejection in kidney transplants. A chief mechanism of action for rapamycin is the induction and expansion of TReg cells and the inhibition of effector T cell differentiation, as described above. This is distinct from other immunosuppressants such as cyclosporine A and FK506 that mainly block Ca2+ and calcineurin activation downstream of TCRs, thereby allowing the design of combinatorial therapy of rapamycin and other immunosuppressive agents. This is important because rapamycin monotherapy exerts a rather weak effect to prevent graft rejection, despite its multiple immunomodulatory functions.
The immunosuppressive effect of rapamycin is also apparent in patients and experimental models of systemic autoimmune diseases such as systemic lupus erythematosus and rheumatoid arthritis, as well as organ-specific autoimmune disorders117, 118. Rapamycin likely establishes long-term immune tolerance in these models by expanding TReg cells and inhibiting effector T cell differentiation and function. Notably, rapamycin also exerts potent effects on DCs and other immune components2, and the effects of rapamycin in vivo might be attributed to these cells, in addition to T cells.
Rapamycin for vaccine development to boost immunity
Despite the well established immunosuppressive effects of mTOR inhibition, rapamycin and metformin promote the generation of protective T cell memory in the models of infection with lymphocytic choriomeningitis virus (LCMV) and Listeria monocytogenes. Mechanistically, this has been associated with the induction of a metabolic switch from glycolysis to FAO in the presence of rapamycin and metformin96, 119. The immunostimulatory effect of rapamycin for CD8+ T cell responses has been extended to additional infectious models120, 121, and anti-tumour immune responses122, 123. Therefore, mTOR inhibitors may serve as novel adjuvants for vaccine development against pathogens and tumours. In particular, this strategy of tumour immunotherapy is promising due to its synergy with direct suppression of tumour growth by mTOR inhibitors. Notably, the effect of mTOR inhibition depends on the dose range and kinetics of the treatment. Administration of a very high dose of rapamycin prevents CD8+ T cell expansion, whereas the duration and time points of rapamycin treatment impact the quantity and quality of memory T cell responses82, 119. To explore the basis for rapamycin-mediated immunosuppressive and immunostimulatory effects on CD8+ T cell responses, Ferrer et al. compared the effects of rapamycin on immune responses elicited by L. monocytogenes and by a skin transplant against the same antigen120. Treatment with rapamycin augmented antigen-specific T cell responses to the pathogen but not to the transplant. Thus, the environment in which an antigen is presented influences the effects of rapamycin on T cell responses120.
T cell malignancy and metabolic dysregulation
A common hematological malignancy is T cell acute lymphoblastic leukemia (T-ALL) that frequently harbors activating NOTCH1 mutations and/or loss-of-function mutations of PTEN. Interestingly, both types of mutations activate mTOR signalling, suggesting a pivotal role for mTOR in T-ALL development. Consistently with this notion, in a PTEN-null T-ALL mouse model, rapamycin was effective to halt T-ALL initiation and development. However, upon rapamycin withdrawal, the majority of PTEN-null mice rapidly became sick and died precipitously. These results indicate that leukemia stem cells and leukemia blasts have differential responses to rapamycin, which could contribute to the limited clinical efficacy of rapamycin in cancer48.
Chronic inflammation in the adipose tissue has been established as a major mechanism to cause insulin resistance and subsequent development of type 2 diabetes. Although earlier studies mainly implicated macrophages as a major inflammatory infiltrate124, recent reports identify the recruitment of effector CD8+ T cells and FOXP3+ TReg cells to the adipose tissue, where they function to exacerbate and ameliorate the inflammation, respectively125–127. Given a prominent immunomodulatory role of mTOR in T cells, whether mTOR signalling in T cells contributes to the pathogenesis of metabolic disease will be interesting to explore. This raises the notion that therapeutic targeting of mTOR in metabolic diseases may have a dual effect through the regulation of T cell metabolism and effector function as well as the direct modulation of the function of metabolic tissues (such as adipose tissue, skeletal muscle and liver), in which mTOR signalling has a central role.
Targeting mTOR: beyond rapamycin
Rapamycin inhibits mTOR through an unusual allosteric mechanism that requires binding to its intracellular partner FKBP12. Whereas rapamycin strongly inhibits the functions of S6 kinases (S6Ks) downstream of mTORC1, it exerts a surprisingly weak effect on the phosphorylation of the other major mTORC1 target, eIF4E-binding proteins (4E-BPs)128. Moreover, rapamycin treatment frequently abrogates feedback inhibition of mTORC1 on PI3K–AKT signalling, leading to paradoxical enhancement of AKT activity129. The incomplete inhibition on mTORC1 and the reversal of the feedback loop by rapamycin attenuate its therapeutic effects, as reflected by the disappointing therapeutic outcomes of rapamycin and other first-generation mTOR inhibitors in cancer patients130. Therefore, selective ATP-competitive mTOR inhibitors, including Torin1 and PP242, were recently developed that completely inhibit mTORC1, including rapamycin-resistant phosphorylation of 4E-BPs131–133. As compared with rapamycin, these second-generation inhibitors exert stronger inhibition on mTORC2 and thus are less likely to activate the feedback loop131–133. Indeed, these new inhibitors exhibit greater anti-tumour effects than rapamycin134, 135, and some of them have entered clinical trials as new cancer therapeutics130. The immunomodulatory function is anticipated to be extensively investigated in the near future.
Because of the pleiotropic effects of mTOR in T cells, targeting the upstream and downstream components of mTOR signalling, rather than mTOR itself, offers an alternative strategy for added specificity. In particular, targeting mTOR-dependent metabolic pathways is efficacious for modulating T cell-mediated diseases. A classic example is the AMPK-activators metformin and AICAR that block energy-mediated activation of mTORC1, without interfering with the PI3K–AKT-mediated growth factor signalling. Aside from modulating T cell memory as described above96, the AMPK activators exert both prophylactic and therapeutic effects on EAE by attenuating the severity of clinical disease136, 137. Similarly, blocking glycolysis protects mice from autoimmune neuroinflammation mediated by TH17 cells85. Moreover, blocking upstream activators of mTOR in T cells, such as S1PR1 and leptin receptor, shows similar effects as rapamycin in modulating T cell responses in selective models33, 37. Further studies of mTOR-dependent signalling axes will provide more opportunities to specifically targeting mTOR-associated pathways.
Concluding remarks
Cell fate decision mediated by mTOR is of particular importance to T cells because of their unique features: a constant exposure to unpredictable pathogen threats, extensive proliferation following antigenic stimulation, and continuous migration in a variety of tissues. Whereas the recent advances have established mTOR as a fundamental determinant of T cell homeostatic and functional fates, many open questions remain. Further studies are needed to elucidate the upstream signals and downstream effectors that mediate T cell homeostasis and antigen-specific immune responses. Also, we have yet to define the emerging roles of mTOR in certain cell biological processes such as autophagy, a process crucial for T cell homeostasis and function138. Much of our current knowledge on the mTOR-controlled signalling networks is derived from the use of in vitro systems and cell lines. However, identification of T cell-specific and context-dependent signals promises to provide more insight into regulation of adaptive immunity. From this perspective, the use of sophisticated genetic systems offers the ultimate molecular and cellular specificities to dissect the underlying processes. Moreover, although research in this area has benefited from the use of rapamycin over the last two decades, the development and application of more potent and selective inhibitors of mTOR are essential to advance the research and clinical application. The continued expansion of our understanding of mTOR signalling will manifest legitimate therapeutic opportunities for a number of T cell-mediated disorders.
Acknowledgments
I acknowledge the large number of researchers who have contributed to this field whose work was not cited owing to space limitations. I thank Drs. Ruoning Wang and Yanyan Wang for critical comments on the manuscript, and members of my laboratory for helpful discussions. The author’s research is supported by US National Institutes of Health (K01 AR053573 and R01 NS064599), National Multiple Sclerosis Society (RG4180-A-1), Lupus Research Institute, Cancer Research Institute, and the American Lebanese Syrian Associated Charities.
Glossary
- Metabolism
Intracellular chemical reactions that convert nutrients and endogenous molecules into energy and biomass (proteins, nuclear acids and lipids). Naïve T cells utilize a catabolic metabolism where they use glucose, fatty acids, and amino acids for ATP generation through the TCA cycle and oxidative phosphorylation. Upon antigen stimulation, the bioenergetic demands of a T cell increase dramatically over the resting state and transition into anabolic metabolism mediated by glycolysis and glutaminolysis
- AMP-activated protein kinase (AMPK)
A group of serine/threonine kinases that is activated in response to energy depletion. AMPK is an important activator of fatty acid oxidation and a potent inhibitor of mTORC1
- FOXP3+ TReg cells
A critical subset of CD4+ T cells for the maintenance of immune tolerance. Although Treg cells mainly develop in the thymus as a separate lineage of CD4+ T cells, known as naturally occurring TReg cells, a second subset of Treg cells arises de novo from conventional T cells in the periphery upon antigen stimulation in the presence of TGF-β (induced TReg). Invariant natural killer T (iNKT) cells. A subset of immune cells that shares properties with both T cells and natural killer cells
- Central tolerance
A process that eliminates self-reactive lymphocytes during their ontogeny. For T cells, this occurs mainly through clonal deletion in the thymus
- Peripheral tolerance
A process that downregulates activation of self-reactive T cells in secondary lymphoid organs (i.e., lymph nodes and spleen). Two of the most important mechanisms are the suppression by Treg cells and induction of T cell anergy
- Catabolic metabolism
The breakdown of complex substances into simpler ones, often accompanied by ATP production. Examples include the oxidation of fatty acids and amino acids
- Oxidative phosphorylation
A metabolic pathway that produces ATP from the oxidation of nutrients and transfer of electrons in a two-step process in mitochondria. The first reaction involves the conversion of intermediate molecules (pyruvate and fatty acids) to acetyl coenzyme A (acetyl-CoA) and the degradation of acetyl-CoA to carbon dioxide in the tricarboxylic-acid (TCA) cycle, yielding free electrons that are carried by NADH and FADH2. The second reaction involves the transfer of electrons from NADH and FADH2 to the electron-transport chain (ETC), resulting in the movement of protons out of the mitochondrial matrix and the generation of electrochemical potential for ATP synthesis
- Fatty acid oxidation (FAO)
An important metabolic process to derive energy by mobilization and oxidation of fatty acids, mainly in the mitochondrion matrix. FAO is positively and negatively regulated by AMPK and mTOR, respectively
- T cell anergy
A state of T cell unresponsiveness to antigen stimulation by failing to proliferate and produce IL-2
- Autophagy
A recycling process in which the cell degrades cytoplasmic organelles and proteins in lysosomes
Biography
Hongbo Chi received his Ph.D. from the University of Rochester, and his postdoctoral training from Yale University School of Medicine. In 2007, he started his independent research program at St. Jude Children’s Research Hospital, where he is currently an Associate Member in the Department of Immunology. His research focuses on the signalling mechanisms, in particular the kinase pathways mediated by mTOR and MAPK, in the immune system.
Footnotes
Competing interest statement
The author declares no competing financial interests.
References
- 1.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thomson AW, Turnquist HR, Raimondi G. Immunoregulatory functions of mTOR inhibition. Nat Rev Immunol. 2009;9:324–337. doi: 10.1038/nri2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Araki K, Ellebedy AH, Ahmed R. TOR in the immune system. Curr Opin Cell Biol. 2011 doi: 10.1016/j.ceb.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Powell JD, Delgoffe GM. The mammalian target of rapamycin: linking T cell differentiation, function, and metabolism. Immunity. 2010;33:301–311. doi: 10.1016/j.immuni.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iwasaki A, Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science. 2010;327:291–295. doi: 10.1126/science.1183021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Delgoffe GM, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30:832–844. doi: 10.1016/j.immuni.2009.04.014. Provides the first genetic evidence for mTOR in promoting CD4+ effector T cell differentiation while inhibiting induction of TReg cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang K, Neale G, Green DR, He W, Chi H. The tumor suppressor Tsc1 enforces quiescence of naive T cells to promote immune homeostasis and function. Nat Immunol. 2011;12:888–897. doi: 10.1038/ni.2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu Q, et al. The tuberous sclerosis complex-Mammalian target of rapamycin pathway maintains the quiescence and survival of naive T cells. J Immunol. 2011;187:1106–1112. doi: 10.4049/jimmunol.1003968. References 7 and 8 describe that maintenance of T cell quiescence requires TSC1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Brien TF, et al. Regulation of T-cell survival and mitochondrial homeostasis by TSC1. Eur J Immunol. 2011;41:3361–3370. doi: 10.1002/eji.201141411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sancak Y, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320:1496–1501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sancak Y, et al. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141:290–303. doi: 10.1016/j.cell.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gwinn DM, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zheng M, et al. Inactivation of Rheb by PRAK-mediated phosphorylation is essential for energy-depletion-induced suppression of mTORC1. Nat Cell Biol. 2011;13:263–272. doi: 10.1038/ncb2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu XN, et al. Phosphorylation of Raptor by p38beta participates in arsenite-induced mammalian target of rapamycin complex 1 (mTORC1) activation. J Biol Chem. 2011;286:31501–31511. doi: 10.1074/jbc.M111.233122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zinzalla V, Stracka D, Oppliger W, Hall MN. Activation of mTORC2 by association with the ribosome. Cell. 2011;144:757–768. doi: 10.1016/j.cell.2011.02.014. [DOI] [PubMed] [Google Scholar]
- 16.Oh WJ, et al. mTORC2 can associate with ribosomes to promote cotranslational phosphorylation and stability of nascent Akt polypeptide. EMBO J. 2010;29:3939–3951. doi: 10.1038/emboj.2010.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saci A, Cantley LC, Carpenter CL. Rac1 regulates the activity of mTORC1 and mTORC2 and controls cellular size. Mol Cell. 2011;42:50–61. doi: 10.1016/j.molcel.2011.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Turner MS, Kane LP, Morel PA. Dominant role of antigen dose in CD4+Foxp3+ regulatory T cell induction and expansion. J Immunol. 2009;183:4895–4903. doi: 10.4049/jimmunol.0901459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Katzman SD, et al. Duration of antigen receptor signaling determines T-cell tolerance or activation. Proc Natl Acad Sci U S A. 2010;107:18085–18090. doi: 10.1073/pnas.1010560107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Colombetti S, Basso V, Mueller DL, Mondino A. Prolonged TCR/CD28 engagement drives IL-2-independent T cell clonal expansion through signaling mediated by the mammalian target of rapamycin. J Immunol. 2006;176:2730–2738. doi: 10.4049/jimmunol.176.5.2730. [DOI] [PubMed] [Google Scholar]
- 21.Zheng Y, et al. A role for mammalian target of rapamycin in regulating T cell activation versus anergy. J Immunol. 2007;178:2163–2170. doi: 10.4049/jimmunol.178.4.2163. [DOI] [PubMed] [Google Scholar]
- 22.Zheng Y, Delgoffe GM, Meyer CF, Chan W, Powell JD. Anergic T cells are metabolically anergic. J Immunol. 2009;183:6095–6101. doi: 10.4049/jimmunol.0803510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.So T, Choi H, Croft M. OX40 complexes with phosphoinositide 3-kinase and protein kinase B (PKB) to augment TCR-dependent PKB signaling. J Immunol. 2011;186:3547–3555. doi: 10.4049/jimmunol.1003156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Francisco LM, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009;206:3015–3029. doi: 10.1084/jem.20090847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wofford JA, Wieman HL, Jacobs SR, Zhao Y, Rathmell JC. IL-7 promotes Glut1 trafficking and glucose uptake via STAT5-mediated activation of Akt to support T-cell survival. Blood. 2008;111:2101–2111. doi: 10.1182/blood-2007-06-096297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rathmell JC, Farkash EA, Gao W, Thompson CB. IL-7 enhances the survival and maintains the size of naive T cells. J Immunol. 2001;167:6869–6876. doi: 10.4049/jimmunol.167.12.6869. [DOI] [PubMed] [Google Scholar]
- 27.Rao RR, Li Q, Odunsi K, Shrikant PA. The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and Eomesodermin. Immunity. 2010;32:67–78. doi: 10.1016/j.immuni.2009.10.010. Describes mTOR-dependent regulation of T-bet and EOMES as important mechanisms for CD8+ effector and memory differentiation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stephenson LM, Park DS, Mora AL, Goenka S, Boothby M. Sequence motifs in IL-4R alpha mediating cell-cycle progression of primary lymphocytes. J Immunol. 2005;175:5178–5185. doi: 10.4049/jimmunol.175.8.5178. [DOI] [PubMed] [Google Scholar]
- 29.Gulen MF, et al. The Receptor SIGIRR Suppresses Th17 Cell Proliferation via Inhibition of the Interleukin-1 Receptor Pathway and mTOR Kinase Activation. Immunity. 2010;32:54–66. doi: 10.1016/j.immuni.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Delgoffe GM, et al. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol. 2011;12:295–303. doi: 10.1038/ni.2005. Describes the differential requirements of mTORC1 and mTORC2 in TH1, TH2 and TH17 differentiation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou J, et al. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc Natl Acad Sci U S A. 2007;104:16158–16163. doi: 10.1073/pnas.0702596104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lord GM, et al. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature. 1998;394:897–901. doi: 10.1038/29795. [DOI] [PubMed] [Google Scholar]
- 33.Procaccini C, et al. An oscillatory switch in mTOR kinase activity sets regulatory T cell responsiveness. Immunity. 2010;33:929–941. doi: 10.1016/j.immuni.2010.11.024. Describes a leptin-mTOR axis that modulates the responsiveness of TReg cells to TCR signals. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.De Rosa V, et al. A key role of leptin in the control of regulatory T cell proliferation. Immunity. 2007;26:241–255. doi: 10.1016/j.immuni.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 35.Galgani M, et al. Leptin modulates the survival of autoreactive CD4+ T cells through the nutrient/energy-sensing mammalian target of rapamycin signaling pathway. J Immunol. 2010;185:7474–7479. doi: 10.4049/jimmunol.1001674. [DOI] [PubMed] [Google Scholar]
- 36.Liu G, et al. The receptor S1P1 overrides regulatory T cell-mediated immune suppression through Akt-mTOR. Nat Immunol. 2009;10:769–777. doi: 10.1038/ni.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu G, Yang K, Burns S, Shrestha S, Chi H. The S1P(1)-mTOR axis directs the reciprocal differentiation of T(H)1 and T(reg) cells. Nat Immunol. 2010;11:1047–1056. doi: 10.1038/ni.1939. Describes an important role of S1PR1 to activate mTOR and modulate TH1 and Treg differentiation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Geng D, et al. When Toll-like receptor and T-cell receptor signals collide: a mechanism for enhanced CD8 T-cell effector function. Blood. 2010;116:3494–3504. doi: 10.1182/blood-2010-02-268169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Quigley M, Martinez J, Huang X, Yang Y. A critical role for direct TLR2-MyD88 signaling in CD8 T-cell clonal expansion and memory formation following vaccinia viral infection. Blood. 2009;113:2256–2264. doi: 10.1182/blood-2008-03-148809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rahman AH, et al. Antiviral memory CD8 T-cell differentiation, maintenance, and secondary expansion occur independently of MyD88. Blood. 2011;117:3123–3130. doi: 10.1182/blood-2010-11-318485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ciofani M, Zuniga-Pflucker JC. Notch promotes survival of pre-T cells at the beta-selection checkpoint by regulating cellular metabolism. Nat Immunol. 2005;6:881–888. doi: 10.1038/ni1234. [DOI] [PubMed] [Google Scholar]
- 42.Sprent J, Surh CD. Normal T cell homeostasis: the conversion of naive cells into memory-phenotype cells. Nat Immunol. 2011;12:478–484. doi: 10.1038/ni.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Michalek RD, Rathmell JC. The metabolic life and times of a T-cell. Immunol Rev. 2010;236:190–202. doi: 10.1111/j.1600-065X.2010.00911.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suzuki A, et al. T cell-specific loss of Pten leads to defects in central and peripheral tolerance. Immunity. 2001;14:523–534. doi: 10.1016/s1074-7613(01)00134-0. [DOI] [PubMed] [Google Scholar]
- 45.Xue L, Nolla H, Suzuki A, Mak TW, Winoto A. Normal development is an integral part of tumorigenesis in T cell-specific PTEN-deficient mice. Proc Natl Acad Sci U S A. 2008;105:2022–2027. doi: 10.1073/pnas.0712059105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hagenbeek TJ, Spits H. T-cell lymphomas in T-cell-specific Pten-deficient mice originate in the thymus. Leukemia. 2008;22:608–619. doi: 10.1038/sj.leu.2405056. [DOI] [PubMed] [Google Scholar]
- 47.Finlay DK, et al. Phosphoinositide-dependent kinase 1 controls migration and malignant transformation but not cell growth and proliferation in PTEN-null lymphocytes. J Exp Med. 2009 doi: 10.1084/jem.20090219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guo W, et al. Suppression of leukemia development caused by PTEN loss. Proc Natl Acad Sci U S A. 2011;108:1409–1414. doi: 10.1073/pnas.1006937108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu X, et al. Distinct roles for PTEN in prevention of T cell lymphoma and autoimmunity in mice. J Clin Invest. 2010;120:2497–2507. doi: 10.1172/JCI42382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hagenbeek TJ, et al. The loss of PTEN allows TCR alphabeta lineage thymocytes to bypass IL-7 and Pre-TCR-mediated signaling. J Exp Med. 2004;200:883–894. doi: 10.1084/jem.20040495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kishimoto H, et al. The Pten/PI3K pathway governs the homeostasis of Valpha14iNKT cells. Blood. 2007;109:3316–3324. doi: 10.1182/blood-2006-07-038059. [DOI] [PubMed] [Google Scholar]
- 52.Di Cristofano A, et al. Impaired Fas response and autoimmunity in Pten+/− mice. Science. 1999;285:2122–2125. doi: 10.1126/science.285.5436.2122. [DOI] [PubMed] [Google Scholar]
- 53.Sauer S, et al. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc Natl Acad Sci U S A. 2008;105:7797–7802. doi: 10.1073/pnas.0800928105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xiao C, et al. Lymphoproliferative disease and autoimmunity in mice with increased miR-17–92 expression in lymphocytes. Nat Immunol. 2008;9:405–414. doi: 10.1038/ni1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Olive V, et al. miR-19 is a key oncogenic component of mir-17–92. Genes & development. 2009;23:2839–2849. doi: 10.1101/gad.1861409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jiang S, et al. Molecular dissection of the miR-17–92 cluster’s critical dual roles in promoting Th1 responses and preventing inducible Treg differentiation. Blood. 2011;118:5487–5497. doi: 10.1182/blood-2011-05-355644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tamas P, et al. LKB1 is essential for the proliferation of T-cell progenitors and mature peripheral T cells. Eur J Immunol. 2010;40:242–253. doi: 10.1002/eji.200939677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cao Y, et al. The serine/threonine kinase LKB1 controls thymocyte survival through regulation of AMPK activation and Bcl-XL expression. Cell Res. 2010;20:99–108. doi: 10.1038/cr.2009.141. [DOI] [PubMed] [Google Scholar]
- 59.Cao Y, et al. LKB1 regulates TCR-mediated PLCgamma1 activation and thymocyte positive selection. EMBO J. 2011;30:2083–2093. doi: 10.1038/emboj.2011.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maciver NJ, et al. The Liver Kinase B1 Is a Central Regulator of T Cell Development, Activation, and Metabolism. J Immunol. 2011;187:4187–4198. doi: 10.4049/jimmunol.1100367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tamas P, et al. Regulation of the energy sensor AMP-activated protein kinase by antigen receptor and Ca2+ in T lymphocytes. J Exp Med. 2006;203:1665–1670. doi: 10.1084/jem.20052469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mayer A, Denanglaire S, Viollet B, Leo O, Andris F. AMP-activated protein kinase regulates lymphocyte responses to metabolic stress but is largely dispensable for immune cell development and function. Eur J Immunol. 2008;38:948–956. doi: 10.1002/eji.200738045. [DOI] [PubMed] [Google Scholar]
- 64.Yilmaz OH, et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 2006;441:475–482. doi: 10.1038/nature04703. [DOI] [PubMed] [Google Scholar]
- 65.Zhang J, et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature. 2006;441:518–522. doi: 10.1038/nature04747. [DOI] [PubMed] [Google Scholar]
- 66.Chen C, et al. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J Exp Med. 2008;205:2397–2408. doi: 10.1084/jem.20081297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gan B, et al. mTORC1-dependent and -independent regulation of stem cell renewal, differentiation, and mobilization. Proc Natl Acad Sci U S A. 2008;105:19384–19389. doi: 10.1073/pnas.0810584105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gan B, et al. Lkb1 regulates quiescence and metabolic homeostasis of haematopoietic stem cells. Nature. 2010;468:701–704. doi: 10.1038/nature09595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gurumurthy S, et al. The Lkb1 metabolic sensor maintains haematopoietic stem cell survival. Nature. 2010;468:659–663. doi: 10.1038/nature09572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nakada D, Saunders TL, Morrison SJ. Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells. Nature. 2010;468:653–658. doi: 10.1038/nature09571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Peterson TR, et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137:873–886. doi: 10.1016/j.cell.2009.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008;134:451–460. doi: 10.1016/j.cell.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sancak Y, et al. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007;25:903–915. doi: 10.1016/j.molcel.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 74.Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol. 2007;9:316–323. doi: 10.1038/ncb1547. [DOI] [PubMed] [Google Scholar]
- 75.Kerdiles YM, et al. Foxo1 links homing and survival of naive T cells by regulating L-selectin, CCR7 and interleukin 7 receptor. Nat Immunol. 2009;10:176–184. doi: 10.1038/ni.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ouyang W, Beckett O, Flavell RA, Li MO. An essential role of the Forkhead-box transcription factor Foxo1 in control of T cell homeostasis and tolerance. Immunity. 2009;30:358–371. doi: 10.1016/j.immuni.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lee K, et al. Mammalian target of rapamycin protein complex 2 regulates differentiation of Th1 and Th2 cell subsets via distinct signaling pathways. Immunity. 2010;32:743–753. doi: 10.1016/j.immuni.2010.06.002. Describes the roles of mTORC2 in TH1 and TH2 differentiation by affecting AKT and PKCθ signaling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sinclair LV, et al. Phosphatidylinositol-3-OH kinase and nutrient-sensing mTOR pathways control T lymphocyte trafficking. Nat Immunol. 2008;9:513–521. doi: 10.1038/ni.1603. Provides the first evidence for the regulatory role of mTOR in T cell trafficking. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Macintyre AN, et al. Protein kinase B controls transcriptional programs that direct cytotoxic T cell fate but is dispensable for T cell metabolism. Immunity. 2011;34:224–236. doi: 10.1016/j.immuni.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gorentla BK, Wan CK, Zhong XP. Negative regulation of mTOR activation by diacylglycerol kinases. Blood. 2010;117:4022–4031. doi: 10.1182/blood-2010-08-300731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Peter C, Waldmann H, Cobbold SP. mTOR signalling and metabolic regulation of T cell differentiation. Curr Opin Immunol. 2010;22:655–661. doi: 10.1016/j.coi.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 82.Araki K, Youngblood B, Ahmed R. The role of mTOR in memory CD8 T-cell differentiation. Immunol Rev. 2010;235:234–243. doi: 10.1111/j.0105-2896.2010.00898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Finlay D, Cantrell DA. Metabolism, migration and memory in cytotoxic T cells. Nat Rev Immunol. 2011;11:109–117. doi: 10.1038/nri2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations. Annu Rev Immunol. 2010;28:445–489. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shi LZ, et al. HIF1a-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. 2011;208:1367–1376. doi: 10.1084/jem.20110278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dang EV, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell. 2011;146:772–784. doi: 10.1016/j.cell.2011.07.033. References 85 and 86 describe an important role for HIF1α and metabolic pathways in TH17 and TReg differentiation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bird JJ, et al. Helper T cell differentiation is controlled by the cell cycle. Immunity. 1998;9:229–237. doi: 10.1016/s1074-7613(00)80605-6. [DOI] [PubMed] [Google Scholar]
- 88.Wang R, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35:871–882. doi: 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jones RG, Thompson CB. Revving the engine: signal transduction fuels T cell activation. Immunity. 2007;27:173–178. doi: 10.1016/j.immuni.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 90.Pearce EL. Metabolism in T cell activation and differentiation. Curr Opin Immunol. 2010 doi: 10.1016/j.coi.2010.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Frauwirth KA, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity. 2002;16:769–777. doi: 10.1016/s1074-7613(02)00323-0. [DOI] [PubMed] [Google Scholar]
- 92.Rathmell JC. T cell Myc-tabolism. Immunity. 2011;35:845–846. doi: 10.1016/j.immuni.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Michalek RD, et al. Estrogen-related receptor-alpha is a metabolic regulator of effector T-cell activation and differentiation. Proc Natl Acad Sci U S A. 2011;108:18348–18353. doi: 10.1073/pnas.1108856108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gordan JD, Thompson CB, Simon MC. HIF and c-Myc: sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell. 2007;12:108–113. doi: 10.1016/j.ccr.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 96.Pearce EL, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. 2009;460:103–107. doi: 10.1038/nature08097. References 96 and 119 are the first reports to implicate mTOR and metabolic pathways in CD8+ memory formation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Michalek RD, et al. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. 2011;186:3299–3303. doi: 10.4049/jimmunol.1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sundrud MS, et al. Halofuginone inhibits TH17 cell differentiation by activating the amino acid starvation response. Science. 2009;324:1334–1338. doi: 10.1126/science.1172638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cui G, et al. Liver X receptor (LXR) mediates negative regulation of mouse and human Th17 differentiation. J Clin Invest. 2011;121:658–670. doi: 10.1172/JCI42974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Duvel K, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010;39:171–183. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nicklin P, et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell. 2009;136:521–534. doi: 10.1016/j.cell.2008.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Grohmann U, Bronte V. Control of immune response by amino acid metabolism. Immunol Rev. 2010;236:243–264. doi: 10.1111/j.1600-065X.2010.00915.x. [DOI] [PubMed] [Google Scholar]
- 103.Cobbold SP, et al. Infectious tolerance via the consumption of essential amino acids and mTOR signaling. Proc Natl Acad Sci U S A. 2009;106:12055–12060. doi: 10.1073/pnas.0903919106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Munn DH, et al. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity. 2005;22:633–642. doi: 10.1016/j.immuni.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 105.Cobbold SP, Adams E, Nolan KF, Regateiro FS, Waldmann H. Connecting the mechanisms of T-cell regulation: dendritic cells as the missing link. Immunol Rev. 2010;236:203–218. doi: 10.1111/j.1600-065X.2010.00913.x. [DOI] [PubMed] [Google Scholar]
- 106.Locasale JW, Cantley LC. Metabolic flux and the regulation of mammalian cell growth. Cell Metab. 2011;14:443–451. doi: 10.1016/j.cmet.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lee K, Won HY, Bae MA, Hong JH, Hwang ES. Spontaneous and aging-dependent development of arthritis in NADPH oxidase 2 deficiency through altered differentiation of CD11b+ and Th/Treg cells. Proc Natl Acad Sci U S A. 2011;108:9548–9553. doi: 10.1073/pnas.1012645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhang J, et al. The type III histone deacetylase Sirt1 is essential for maintenance of T cell tolerance in mice. J Clin Invest. 2009;119:3048–3058. doi: 10.1172/JCI38902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Beugnet A, Tee AR, Taylor PM, Proud CG. Regulation of targets of mTOR (mammalian target of rapamycin) signalling by intracellular amino acid availability. Biochem J. 2003;372:555–566. doi: 10.1042/BJ20021266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Choo AY, et al. Glucose addiction of TSC null cells is caused by failed mTORC1-dependent balancing of metabolic demand with supply. Mol Cell. 2010;38:487–499. doi: 10.1016/j.molcel.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Buchakjian MR, Kornbluth S. The engine driving the ship: metabolic steering of cell proliferation and death. Nat Rev Mol Cell Biol. 2010;11:715–727. doi: 10.1038/nrm2972. [DOI] [PubMed] [Google Scholar]
- 112.Mason EF, Rathmell JC. Cell metabolism: An essential link between cell growth and apoptosis. Biochim Biophys Acta. 2011;1813:645–654. doi: 10.1016/j.bbamcr.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Battaglia M, Stabilini A, Roncarolo MG. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood. 2005;105:4743–4748. doi: 10.1182/blood-2004-10-3932. [DOI] [PubMed] [Google Scholar]
- 114.Strauss L, et al. Selective survival of naturally occurring human CD4+CD25+Foxp3+ regulatory T cells cultured with rapamycin. J Immunol. 2007;178:320–329. doi: 10.4049/jimmunol.178.1.320. [DOI] [PubMed] [Google Scholar]
- 115.Zeiser R, et al. Differential impact of mammalian target of rapamycin inhibition on CD4+CD25+Foxp3+ regulatory T cells compared with conventional CD4+ T cells. Blood. 2008;111:453–462. doi: 10.1182/blood-2007-06-094482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Strauss L, Czystowska M, Szajnik M, Mandapathil M, Whiteside TL. Differential responses of human regulatory T cells (Treg) and effector T cells to rapamycin. PLoS ONE. 2009;4:e5994. doi: 10.1371/journal.pone.0005994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Young DA, Nickerson-Nutter CL. mTOR--beyond transplantation. Curr Opin Pharmacol. 2005;5:418–423. doi: 10.1016/j.coph.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 118.Fernandez D, Bonilla E, Mirza N, Niland B, Perl A. Rapamycin reduces disease activity and normalizes T cell activation-induced calcium fluxing in patients with systemic lupus erythematosus. Arthritis Rheum. 2006;54:2983–2988. doi: 10.1002/art.22085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Araki K, et al. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460:108–112. doi: 10.1038/nature08155. References 96 and 119 are the first reports to implicate mTOR and metabolic pathways in CD8+ memory formation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ferrer IR, et al. Cutting edge: Rapamycin augments pathogen-specific but not graft-reactive CD8+ T cell responses. J Immunol. 2010;185:2004–2008. doi: 10.4049/jimmunol.1001176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Turner AP, et al. Sirolimus enhances the magnitude and quality of viral-specific CD8+ T-cell responses to vaccinia virus vaccination in rhesus macaques. Am J Transplant. 2011;11:613–618. doi: 10.1111/j.1600-6143.2010.03407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Li Q, et al. A central role for mTOR kinase in homeostatic proliferation induced CD8+ T cell memory and tumor immunity. Immunity. 2011;34:541–553. doi: 10.1016/j.immuni.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wang Y, Wang XY, Subjeck JR, Shrikant PA, Kim HL. Temsirolimus, an mTOR inhibitor, enhances anti-tumour effects of heat shock protein cancer vaccines. Br J Cancer. 2011;104:643–652. doi: 10.1038/bjc.2011.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chawla A, Nguyen KD, Goh YP. Macrophage-mediated inflammation in metabolic disease. Nat Rev Immunol. 2011;11:738–749. doi: 10.1038/nri3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Winer S, et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med. 2009;15:921–929. doi: 10.1038/nm.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Nishimura S, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med. 2009;15:914–920. doi: 10.1038/nm.1964. [DOI] [PubMed] [Google Scholar]
- 127.Feuerer M, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. 2009;15:930–939. doi: 10.1038/nm.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci U S A. 2008;105:17414–17419. doi: 10.1073/pnas.0809136105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.O’Reilly KE, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–1508. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Benjamin D, Colombi M, Moroni C, Hall MN. Rapamycin passes the torch: a new generation of mTOR inhibitors. Nat Rev Drug Discov. 2011;10:868–880. doi: 10.1038/nrd3531. [DOI] [PubMed] [Google Scholar]
- 131.Apsel B, et al. Targeted polypharmacology: discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nat Chem Biol. 2008;4:691–699. doi: 10.1038/nchembio.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Feldman ME, et al. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009;7:e38. doi: 10.1371/journal.pbio.1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Thoreen CC, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284:8023–8032. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hsieh AC, et al. Genetic dissection of the oncogenic mTOR pathway reveals druggable addiction to translational control via 4EBP-eIF4E. Cancer Cell. 2010;17:249–261. doi: 10.1016/j.ccr.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Janes MR, et al. Effective and selective targeting of leukemia cells using a TORC1/2 kinase inhibitor. Nat Med. 2010;16:205–213. doi: 10.1038/nm.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Nath N, et al. 5-aminoimidazole-4-carboxamide ribonucleoside: a novel immunomodulator with therapeutic efficacy in experimental autoimmune encephalomyelitis. J Immunol. 2005;175:566–574. doi: 10.4049/jimmunol.175.1.566. [DOI] [PubMed] [Google Scholar]
- 137.Nath N, et al. Metformin attenuated the autoimmune disease of the central nervous system in animal models of multiple sclerosis. J Immunol. 2009;182:8005–8014. doi: 10.4049/jimmunol.0803563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Dunkle A, He YW. Apoptosis and autophagy in the regulation of T lymphocyte function. Immunol Res. 2011;49:70–86. doi: 10.1007/s12026-010-8195-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Haribhai D, et al. A requisite role for induced regulatory T cells in tolerance based on expanding antigen receptor diversity. Immunity. 2011;35:109–122. doi: 10.1016/j.immuni.2011.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kang J, Huddleston SJ, Fraser JM, Khoruts A. De novo induction of antigen-specific CD4+CD25+Foxp3+ regulatory T cells in vivo following systemic antigen administration accompanied by blockade of mTOR. J Leukoc Biol. 2008;83:1230–1239. doi: 10.1189/jlb.1207851. [DOI] [PubMed] [Google Scholar]
- 141.Gao W, et al. Contrasting effects of cyclosporine and rapamycin in de novo generation of alloantigen-specific regulatory T cells. Am J Transplant. 2007;7:1722–1732. doi: 10.1111/j.1600-6143.2007.01842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Haxhinasto S, Mathis D, Benoist C. The AKT-mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. J Exp Med. 2008;205:565–574. doi: 10.1084/jem.20071477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Song K, Wang H, Krebs TL, Danielpour D. Novel roles of Akt and mTOR in suppressing TGF-beta/ALK5-mediated Smad3 activation. EMBO J. 2006;25:58–69. doi: 10.1038/sj.emboj.7600917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ouyang W, et al. Foxo proteins cooperatively control the differentiation of Foxp3+ regulatory T cells. Nat Immunol. 2010;11:618–627. doi: 10.1038/ni.1884. [DOI] [PubMed] [Google Scholar]
- 145.Harada Y, et al. Transcription factors Foxo3a and Foxo1 couple the E3 ligase Cbl-b to the induction of Foxp3 expression in induced regulatory T cells. J Exp Med. 2010;207:1381–1391. doi: 10.1084/jem.20100004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kerdiles YM, et al. Foxo transcription factors control regulatory T cell development and function. Immunity. 2010;33:890–904. doi: 10.1016/j.immuni.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Merkenschlager M, von Boehmer H. PI3 kinase signalling blocks Foxp3 expression by sequestering Foxo factors. J Exp Med. 2010;207:1347–1350. doi: 10.1084/jem.20101156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Seoane J, Le HV, Shen L, Anderson SA, Massague J. Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell. 2004;117:211–223. doi: 10.1016/s0092-8674(04)00298-3. [DOI] [PubMed] [Google Scholar]
- 149.Zhang N, Bevan MJ. CD8(+) T cells: foot soldiers of the immune system. Immunity. 2011;35:161–168. doi: 10.1016/j.immuni.2011.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.van der Windt GJ, et al. Mitochondrial respiratory capacity is a critical regulator of CD8(+) T cell memory development. Immunity. 2012;36:68–78. doi: 10.1016/j.immuni.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Hand TW, et al. Differential effects of STAT5 and PI3K/AKT signaling on effector and memory CD8 T-cell survival. Proc Natl Acad Sci U S A. 2010;107:16601–16606. doi: 10.1073/pnas.1003457107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Finlay D, Cantrell D. Phosphoinositide 3-kinase and the mammalian target of rapamycin pathways control T cell migration. Ann N Y Acad Sci. 2010;1183:149–157. doi: 10.1111/j.1749-6632.2009.05134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Koch MA, et al. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol. 2009;10:595–602. doi: 10.1038/ni.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Taqueti VR, et al. T-bet controls pathogenicity of CTLs in the heart by separable effects on migration and effector activity. J Immunol. 2006;177:5890–5901. doi: 10.4049/jimmunol.177.9.5890. [DOI] [PubMed] [Google Scholar]