

Abstract
Histone deacetylase inhibitors (HDACi) are a new class of compounds that induce acetylation of histone lysine tails in chromatin and modify gene expression. The FDA approved HDACi, Vorinostat or suberoylanilide hydroxamic acid (SAHA), has been shown to inhibit tumor cell growth and the production of pro-inflammatory cytokines. In preclinical allogeneic transplantation models, SAHA induces graft-versus-host disease (GVHD) amelioration in treated mice without impairing graft-versus-leukemia (GVL). LBH589 (Panobinostat), a structurally novel cinnamic hydroxamic acid class, is an HDACi more potent than SAHA. In the current work, we tested the hypothesis that LBH589 would be highly effective in the prevention of GVHD. Using mouse model of allogeneic bone marrow transplantation (BMT), we unexpectedly found that treatment with LBH589 accelerated GVHD, in contrast to the treatment with SAHA that alleviated GVHD. Accelerated GVHD in the recipients treated with LBH589 was associated with elevated Th1 cytokines in recipient serum, enhanced CXCR3 expression on donor T cells, and T-cell infiltration in the liver. The current study highlights the distinct effects of pan HDACi on allogeneic BMT, and alerts that LBH589 (Panobinostat) could have adverse effect on GVHD, and possibly on other inflammatory diseases.
Introduction
Histone deacetylase (HDAC) are enzymes that modulate chromatin structure and gene expression by removing acetyl groups on histone and other proteins. According to their structure, HDACs are classified into 4 groups. Class I (HDAC1, HDAC2, HDAC3, HDAC8), Class IIa (HDAC4, HDAC5, HDAC7, HDAC9) and IIb (HDAC6, HDAC10), class III (SIRT1–7) and Class IV (HDAC11) (1, 2). Inhibiting HDAC activity by pan-HDAC inhibitors (HDACi) has been shown to cause growth arrest and apoptosis of tumor cells. Therefore, initially, pan-HDACi were applied for cancer therapy. Recent findings showed that pan-HDACi could also prevent or alleviate inflammation in mouse models for various diseases as colitis, lupus, arthritis, and neural stroke (3-6). Pavan Reddy and others (5, 7-9) showed that SAHA, a pan-HDACi approved for the therapy of cutaneous T cell lymphoma, could alleviate GVHD after allogeneic bone marrow transplantation (BMT) in mice in indoleamine 2, 3-dioxygenase (IDO) dependent manner. LBH589 is a hydroxamic acid–based HDACi with a similar structure with SAHA. Compared with SAHA, LBH589 has much higher potency in inhibiting each of HDAC family members (10). In the current study, we evaluated the effect of LBH589 on the prevention of GVHD after allogeneic BMT, and unexpectedly we found that LBH589 worsened GVHD. The accelerated GVHD was related to higher levels of pro-inflammatory cytokines in serum, and increased CXCR3 expression on donor T cells, and T cell infiltration in the liver.
Material and Methods
Mice
C57BL/6 (B6, H-2b), BALB/c (B/c, H-2d), were purchased from NCI/NIH. All animals were housed in an American Association for Laboratory Animal Care–accredited Animal Resource Center at Moffitt Cancer Center. Experiments were carried out under protocols approved by the Institutional Animal Care and Use Committee.
Chemicals and Reagents
LBH589 powder provided by Novartis AG was dissolved in 5% dextrose (Sigma-Aldrich) and sonicated in PBS before use. SAHA was purchased from ChemieTek (Indianapolis, USA). SAHA was first dissolved in DMSO and further diluted in PBS before use. LBH589 and SAHA were administrated via intraperitoneal (i.p.) injection. CFSE 5-(and-6)-carboxyfluorescein diacetate, succinimidyl ester (CFSE) was purchased from Invitrogen.
Isolation of donor T and Bone Marrow (BM) cells
Donor mice T cells were purified by negative selection from pooled spleen and lymph node cells to remove the non-T cells using biotinylated antibodies and the MACS system (anti-biotin microbeads and LS column, Miltenyi Biotech, Auburn, CA) according to the manufacture’s instruction. The antibodies for T cell purification (anti-mouse TER-119, anti-mouse CD49b, anti-mouse CD11b and anti-human/mouse CD45R) were purchased from eBioscience. The purity of T cells was usually over 95%. BM was harvested from tibia and femurs, and T cells were depleted by incubation with anti-Thy1.2 antibody (clone 30H12, BioXCell, West Lebanon, USA), and rabbit complement (GTI Diagnostics, USA).
Bone marrow transplantation (BMT)
Female B/c mice at 7-8 weeks old were lethally irradiated (800 cGy, single dose) using a Shepherd Mark I Cesium Irradiator (J.L. Shepherd and Associates) one day prior BMT. On the day of transplantation (Day 0), 5 × 106 T cell depleted (TCD) B6 BM cells were transferred to recipients via tail vein with or without 1 × 106 B6 T cells. Mice were housed in sterilized micro isolator cages and received normal chow and autoclaved hyperchlorinated water for the first 3 weeks after BMT and autoclaved water thereafter. The clinical signs of GVHD (weight loss, ruffled fur, hunched back, and skin lesions) were monitored twice a week.
Antibodies and Flow cytometry
The following antibodies were used for cell surface staining: anti-CD4 FITC, or APC (L3T4), anti-CD8α FITC, APC, APC-Cy7 or Alexa Fluor 700 (Ly-2), anti-H-2Kb FITC, PE, or biotin (AF6), anti-mouse CD11b PE-Cy7, anti-mouse CD11c PE, anti-mouse Ly6G(Gr-1) biotin and anti-mouse CXCR3 biotin were purchased from eBioscience; anti-mouse CCR6 APC were purchased from BioLegend; anti-CD4 Pacific blue (RM4-5) and anti-mouse α4β7 PE was purchased from BD Biosciences. Detection of biotinylated antibodies was performed using APC-Cy7 or APC conjugated to streptavidin (BD Biosciences). Flow data were acquired on FACSCalibur, or LSR II (BD Biosciences) and analyzed using FlowJo (TreeStar Inc).
Measurement of T and B cell function
Splenic cells were cultured in RPMI 1640 media (Invitrogen, USA) with 10% fetal bovine serum (Atlanta Biologicals, GA, USA), penicillin (100 units/ml, Invitrogen) and streptomycin (100μg/ml, Invitrogen) in 96 well round bottom cell culture plate with or without 1μg/ml anti-CD3 antibody (BioXCell, West Lebanon, USA), or 5μg/ml lipopolysaccharide (LPS, Sigma-Aldrich, USA) for 3 days. Six hours before the end of cell culture, 1μCi [3H]-thymidine was added to each well. The proliferation of T cell or B cell was measured by a scintillation counter.
Histological analysis
Representative samples of liver, colon and small intestines were obtained from transplanted recipients and fixed in 10% neutral-buffered formalin. Samples were then embedded in paraffin, cut into 5-μm-thick sections, and stained with hematoxylin and eosin. A semi-quantitative scoring system was used to account for histologic changes consistent with the GVHD signs in the colon and liver as previously described (11). Data was presented as individual GVHD target organ scores as well as a composite score from all the tissues. All slides for GVHD analysis were coded and read in a blinded fashion. Images were visualized with an Olympus BX45 microscope. Image acquisition was performed with an Olympus DP70 digital camera (× 400) and software package.
Mouse serum cytokine analysis and intracellular cytokine staining
Mouse serum cytokine was analyzed following the protocol of BD Cytometric Bead Array (CBA) Mouse Th1/Th2/Th17 Cytokine Kit (BD Biosciences).
Statistics
The recipient survival among groups in the GVHD experiments was compared by log rank test. Student’s t-test was used for all the other experiments.
Results
Daily (QD) treatment with LBH589 accelerated GVHD after allogeneic BMT
Reduction of GVHD after treatment with pan-HDACi SAHA indicates that HDAC can alleviate the pathogenesis of GVHD. LBH589 is also a pan HDACi but with much higher potency (10). Therefore, we originally hypothesized that LBH589 would have a higher efficacy in the prevention of GVHD when compared with SAHA.
Before evaluating the efficacy of LBH589 in GVHD, we first tested its toxicity in recipients after myeloablative conditioning and BM reconstitution to determine the maximum tolerated dose. B/c mice were lethally irradiated one day before adoptive transfer of TCD-BM from B6 donors. Recipients were treated QD with LBH589 from day 0 to 4 at 2.5, 5, 10 or 20 mg/kg body weight (BW). By monitoring recipient survival, we found that treatment with 10 mg/kg LBH589 or higher dose caused significant mortality when compared with vehicle control (Fig. 1A). Treatment with 5 mg/kg LBH589 or lower dose doesn’t show significant toxicity although some mice died. To determine whether LBH589 affects donor BM reconstitution, we compared donor B cell, T cell, dendritic cell (DC) and neutrophil reconstitution in recipient spleen at day 28 after BMT. As shown in figure 1B, QD treatment with 2.5mg/kg BW LBH589 did not compromise donor lymphocyte reconstitution. In addition, the proliferative function of splenic B cell and T cell was intact (Fig. 1C).
Fig. 1. Toxicity of LBH589 with QD treatment.
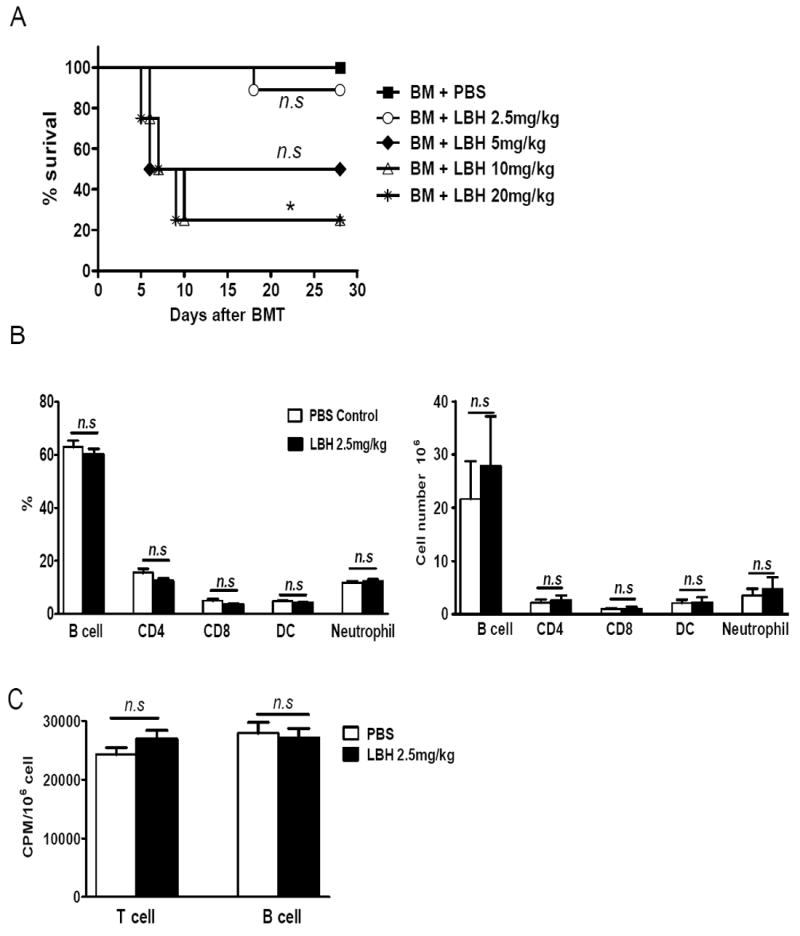
B/c mice (n = 4-5) were lethally irradiated one day before BMT. In the day of transplantation 5 × 106 TCD B6 BM cells were transferred to B/c recipients via tail vein. Recipients were treated with PBS vehicle or different dose of LBH589 daily from day 0 to 4 via i.p injection. (A) Mice survival. (B) On day 28, recipients (n=5) treated with vehicle control or LBH589 at 2.5mg/kg BW were sacrificed, and the percentages of donor B cells (B220) T cells (CD4/CD8), DC (CD11c) and neutrophils (CD11b/Gr-1) in spleen were analyzed by flow cytometry. (C) 4×105 splenic cells from recipient mice (n=5) were stimulated with 1μg/ml anti-CD3 or 5μg/ml LPS for 3 days. The proliferation of T cells or B cells was compared by [3H]-Thymidine incorporation. CPM: count per minute. This experiment was repeated 3 times and similar phenomenon was observed. The representative data from one experiment was shown. n.s, no significant difference. *p<0.05.
Since LBH589 was toxic to allogeneic BMT recipients when given a dose of 10 mg/kg or higher, we decided to evaluate the efficacy of LBH589 in the prevention of GVHD using 1.25 or 2.5 mg/kg doses QD from day 0 to 4. Recipient survival and BW changes were monitored beyond 100 days after BMT. Daily treatment with LBH589 at 2.5 mg/kg did not cause mortality or additional weight loss as compared with vehicle control on recipients transplanted with TCD-BM alone (Fig. 2). Unexpectedly, when T cells were added, the treatment with LBH589 at either dose significantly worsened GVHD, because treated recipients had accelerated death and dramatic weight loss when compared with GVHD controls (BM+T cell+PBS group) (Fig. 2 A and B).
Fig. 2. Effect of LBH589 with QD treatment on GVHD.
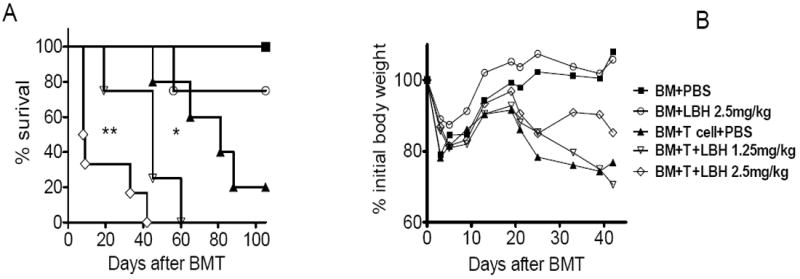
B/c mice (n = 5-6) were lethally irradiated and the next day transplanted with 5 × 106 TCD B6 BM cells with or without 1 × 106 T cells via tail vein. Recipients were treated with PBS vehicle or different dose of LBH589 daily from day 0 to 4 via i.p injection. Mice survival (A) and body weight (B) are shown. This experiment was repeated 3 times and similar results were obtained. One representative experiment was shown here. *p<0.05, **p<0.01.
Treatment with LBH589 every other day (QOD) had lower toxicity but still accelerated GVHD
Accelerated GVHD associated with QD treatment could have resulted from LBH589 toxicity or enhanced T cell activation. To lower the potential toxicity of LBH589, we decided to administer it QOD for 2 weeks. No mortality was observed on this treatment schedule when LBH589 was given at 2.5 – 7.5 mg/kg (Fig. 3A). Although treatment at 7.5 mg/kg cause noticeably more weight loss than that with vehicle control, all recipients treated with LBH589 shortly recovered their BW to the levels of those treated with vehicle control (Fig. 3B). We conclude that the QOD treatment causes lower toxicity compared to QD treatment.
Fig. 3. Toxicity of LBH589 with QOD treatment.
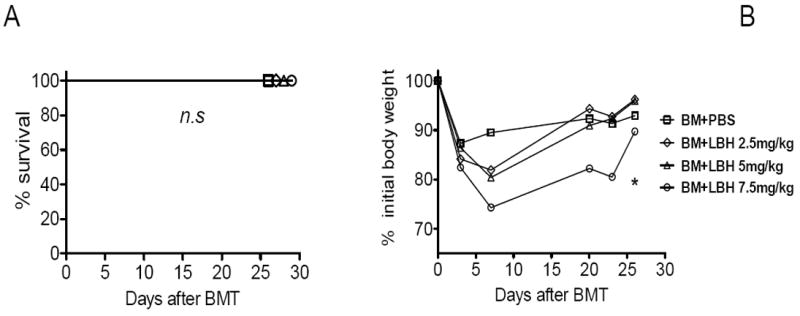
B/c mice (n = 4) were lethally irradiated and transplanted with 5 × 106 TCD B6 BM cells via tail vein. Recipients were treated with PBS vehicle or different doses of LBH589 QOD from day 0 to 13 via i.p injection. Mice survival (A) and body weight (B) are shown. The experiment was repeated 3 times and similar results were obtained. One representative experiment was shown. n.s, no significant difference. *p<0.05.
To assess the effect of LBH589 in the prevention of GVHD, we treated the recipients with LBH589 QOD at 2.5 mg/kg for 2 weeks, and found that the treatment with such dose and schedule had no appreciable toxicity in the recipients with TCD-BM alone nor on GVHD in the recipients with TCD-BM plus allogeneic T cells (Fig. 4, A and B). Further, we evaluated the LBH589 efficacy in the QOD schedule by increasing the dose to 5 mg/kg. While the QOD treatment with LBH589 at 5 mg/kg for 2 weeks did not cause noticeable toxicity on the TCD-BM group, such treatment significantly accelerated GVHD in the recipients of T cells (Fig. 4, C and D).
Fig. 4. Effect of LBH589 with QOD treatment on GVHD.
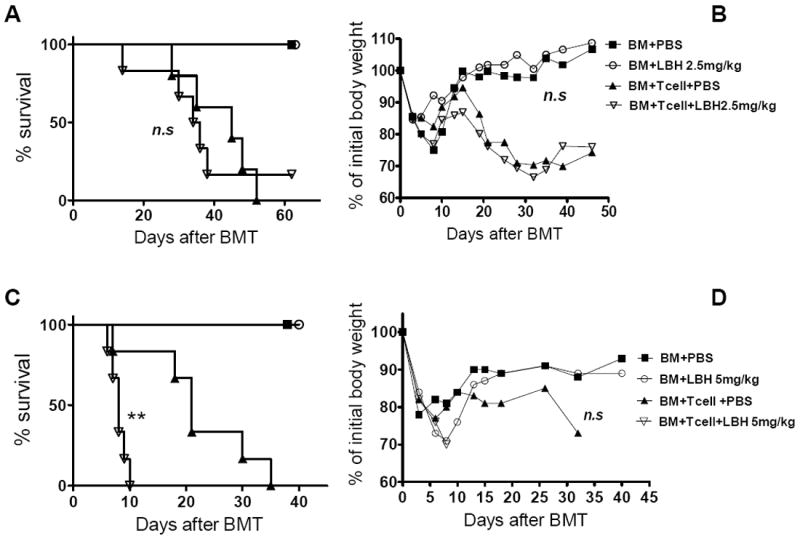
B/c mice (n = 5-6) were lethally irradiated and transplanted with 5 × 106 TCD B6 BM cells with either 0.25 × 106 or 1 × 106 B6 CD25- T cells. Recipients were treated for 12 days QOD via i.p injection with PBS vehicle or LBH589 (2.5mg/kg BW for 0.25 × 106 T cell recipients and 5mg/kg BW for 1 × 106 T cell recipients). Mice survival (A, C) and BW (B, D) are shown here. The experiment was repeated 3 times and similar results were observed. One representative experiment was shown. n.s, no significant difference; **p<0.01.
LBH589 and SAHA had an opposite effect on GVHD
LBH589 is a pan HDACi with much higher potency than SAHA. Given that independent groups showed that SAHA alleviates GVHD after allogeneic BMT (7, 9), it was highly unexpected to observe the accelerated GVHD with LBH589 treatment (Fig. 2 and 4). To exclude the possibility that the experimental systems we chose might contribute to our unexpected results, we directly compared the therapeutic effects of SAHA and LBH589 on GVHD prevention. We used the same dose and schedule of SAHA treatment shown to be effective in previously published reports by others(7, 8), and confirmed that SAHA significantly alleviated GVHD in sharp contrast with LBH589, that significantly accelerated GVHD (Fig. 5 A and B. All comparisons were made between PBS vehicle and SAHA or LBH589 treatment group). The distinct outcomes on GVHD resulted from the treatment with LBH589 or SAHA was further supported by pathologic evidence. Fig. 5C shows significantly increased damage (p<0.01) in the liver from the LBH589 treated recipients when compared with the ones that received vehicle alone. In contrast, the treatment with SAHA produced lower liver damage than observed in the control group. We also examined pathologic changes in colon and small intestine, and found that the pathologic scores were higher in the recipients treated with LBH589 than those with vehicle control although the differences were not statistically significant (data not shown). In conclusion, LBH589 and SAHA had an opposite effect on GVHD development.
Fig. 5. Effect of LBH589 and SAHA on GVHD.
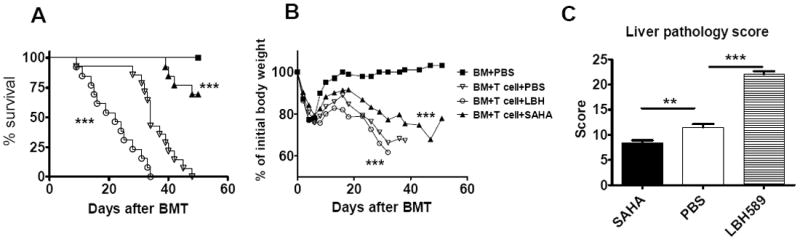
B/c mice were lethally irradiated and transplanted with 5 × 106 TCD B6 BM cells with or without 1 × 106 T cells. Recipients were treated with PBS vehicle, SAHA (25mg/kg BW, daily treatment from day 0 to 7) or LBH589 (1.25mg/kg BW, every other day, from day 0 to 12) via i.p injection. (A) Mice survival was the combined data from 3 independent experiments. (n = 12-13) (B) Mice body weight (n = 4-5). Similar results were found in the repeated experiments. (C) GVHD pathological score of mice livers from the combination of 2 independent experiment (n = 8) **p<0.01; ***p<0.001.
In vivo T cell activation was decreased by SAHA, but increased by LBH589
To understand the underlying mechanisms by which LBH589 and SAHA had opposite effects on GVHD development, we evaluated T-cell activation in response to alloantigens in vivo by measuring TNF-α and IFN-γ in recipient serum because either inflammatory cytokine plays a critical role in GVHD development. As shown in Fig. 6 A and B, the levels of TNF-α and IFN-γ were significantly lower in the recipients treated with SAHA than those treated with vehicle control, consistent with published reports by others that SAHA reduces the inflammatory cytokines (9). In contrast, the levels of IFN-γ and TNF-α were significantly higher in the recipients treated with LBH589 than those treated with vehicle control, suggesting that LBH589 enhanced T cell activation in vivo. We also compared other Th1/Th2/Th17 cytokines like IL-2, IL-4, IL-6, IL-10 and IL-17, and observed similar levels among all experimental groups (Data not shown).
Fig. 6. Effect of LBH589 and SAHA on cytokine production.
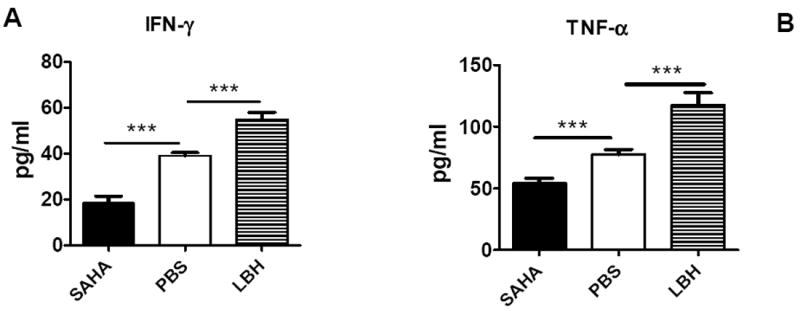
B/c mice were lethally irradiated and transplanted with 5 × 106 TCD B6 BM cells with or without 1 × 106 T cells. Recipients were treated with PBS vehicle, SAHA (25mg/kg BW, daily treatment from day 0 to 7) or LBH589 (1.25mg/kg BW, QOD, from day 0 to 12) via i.p injection. Mice were sacrificed on day 13, and serum IFN-γ (A) and TNF-α (B) were analyzed by flow cytometric bead array. The data shown here are pooled from 2 replicate experiments (n = 8) with the same setting. ***p<0.001.
Donor T cell expansion and subsequent migration into target organs are essential for the development of GVHD. We thus measured donor T cells in recipients’ spleen on day 14 after BMT, and found that treatment with LBH589 reduced the number of donor T cells in spleen whereas SAHA had no effect (Fig. 7A). Because migration of activated T cells into GVHD target organs primarily relies on chemokine receptors expression, we compared the expression of chemokine receptors on donor T cells in the recipients treated with LBH589, SAHA or vehicle control. Among the receptors (CXCR3, α4β7 and CCR6) tested, we found that donor CD4+ and CD8+ T cells expressed significantly higher levels of CXCR3 after LBH589 treatment (Fig. 7B). Given that CXCR3 is preferentially expressed on Th1 cells, these data are consistent with elevated Th1 cytokines (IFN-γ and TNF-α) in the recipients treated with LBH589 (Fig. 5). As a consequence, significantly higher numbers of donor CD4+ and CD8+ T cells were found in the liver of the recipients treated with LBH589 compared to those with vehicle control (Fig. 7C). Taken together, treatment with LBH589 increased T cell activation and migration to GVHD target organ liver in particular, which likely resulted in accelerated GVHD after allogeneic BMT. To exclude the possibility that LBH589 treatment led to bone marrow failure, we compared the percentage of donor bone marrow derived cells in the spleen at day 13. As seen in Figure7D and E, the percentage of H-2kb+ cell population (Donor bone marrow derived) were very similar among the 3 experimental groups.
Fig. 7. Effect of LBH589 and SAHA on donor T cell expansion and migration in vivo.
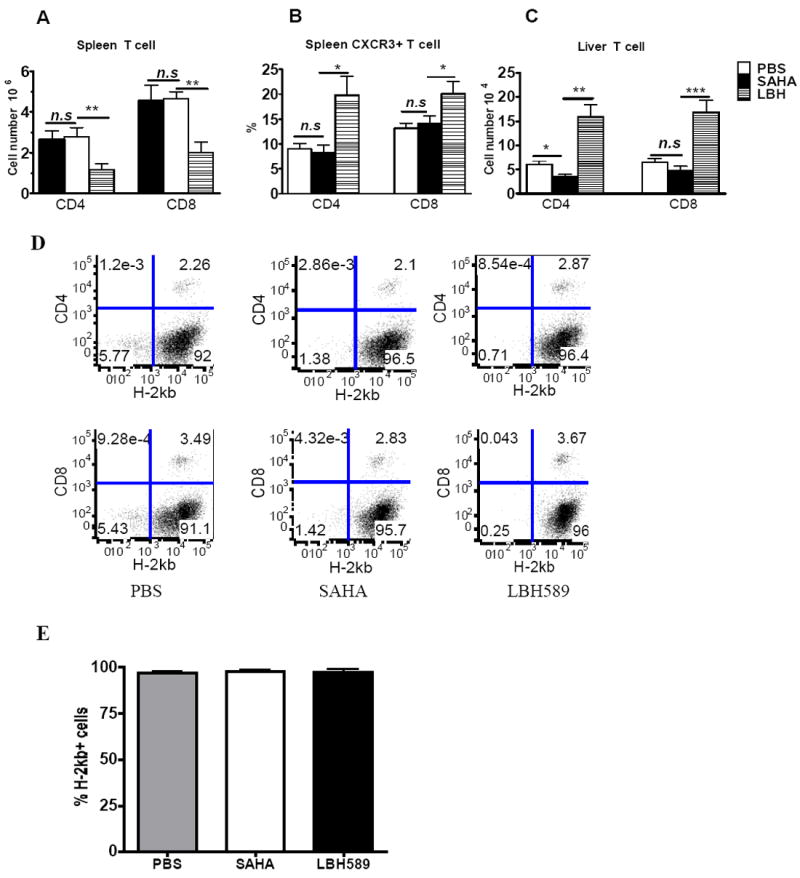
B/c mice (n = 4) were lethally irradiated and transplanted with 5 × 106 T cell depleted B6 BM cells with or without 1 × 106 T cells. Recipients were treated with PBS vehicle, SAHA (25mg/kg BW, daily treatment from day 0 to 7) or LBH589 (1.25mg/kg BW, QOD from day 0 to 12) via i.p injection. Mice were sacrificed on day 13. Splenic T cell number (A), CXCR3 expression on splenic donor T cells (B), and liver T cell infiltration (C) were analyzed by flow cytometry. The data shown are pooled from 2 replicate experiments (n = 8). (D) The flow cytometry data of H-2kb+CD4-CD8- cells in the spleen of each group. The data of one mouse from each group was shown here. Only the cells in live gate were studied. (E) The average percentage of H-2kb+ cells in spleen. Only the cells in live gate were studied. The data shown was pooled from 2 replicate experiments (n = 8). n.s, no significant difference; *p<0.05; **p<0.01; ***p<0.001.
Discussion
A pan-HDACi, SAHA, was shown to alleviate GVHD after allogeneic BMT in various mouse models (5, 7-9). Here we tested another highly potent pan-HDACi, LBH589, in the prevention of GVHD after allogeneic BMT with the expectation that LBH589 would diminish GVHD with a high efficacy. Contrary to our expectation, LBH589 accelerated rather than alleviated GVHD. The accelerated GVHD was associated with increased secretion of Th1 inflammatory cytokines. Pathological T cell infiltration in the liver was also increased by LBH589, likely through up-regulation of CXCR3 expression on donor T cells.
It is known that Th1 cytokines, including IFN-γ and TNF-α, are the critical effectors for acute GVHD, although recent reports showed that IFN-γ has dual effects in the immune regulation (12-14). IFN-γ is secreted by activated donor T cells, and increased serum levels of IFN-γ were related to acute GVHD (15, 16). TNF-α was normally secreted by host antigen presenting cells (APC) and donor T cells (17). Higher TNF-α level indicated higher host APC activity, which was correlated with increased severity of GVHD. Here, we show that LBH589 treatment significantly increased IFN-γ and TNF-α level in serum from recipients treated with LBH589. These elevated inflammatory cytokines likely contributed to accelerated GVHD. In addition, our in vitro experiments showed that at certain doses, LBH589 significantly increased IFN-γ secretion, proliferation of T cells and the antigen presentation ability of DC (Sup Fig. 1 and 2), consistent with our observation in vivo (Fig. 6). Therefore, the LBH589 doses used in vivo may fall in the concentration range that promoted T cell activation. Since LBH589 was toxic at high doses in vivo, we could not demonstrate the protective effect of LBH589 in GVHD. In accordance with our observation regarding the effect of LBH589 on cytokine production, Wang et al. recently reported that LAQ824, another pan HDACi, augmented inflammatory responses in macrophages through transcriptional regulation of IL-10 (18). SAHA does not always down-regulate the inflammatory response; it was reported to suppress the LPS-induced mRNA expression of the proinflammatory mediators Edn1, CCL7/MCP-3, and Il-12p40, but amplify the expression of the proatherogenic factors Cox-2 and Pai-1/serpine1 in primary mouse BM derived macrophages (19). Thus, the effect of pan HDACi on inflammation may be drug and disease specific.
When comparing T cell expansion in spleen and T cell infiltration in GVHD target organs at day 14, we found that there were much less donor T cells in the recipient spleen after the LBH589 treatment (Fig. 7.), but more donor T cells infiltrated the liver. Consistent with this finding, pathological analysis showed higher GVHD scores in the livers from the LBH589 treated group (Fig. 5C). Less splenic T cells might reflect less T cell expansion, or more extensive T cell migration from the spleen to the GVHD target organs upon LBH589 treatment. In fact, we found that after the LBH589 treatment, significantly more donor T cells expressed CXCR3, a chemokine receptor that mediates T cell migration to the liver and the intestine (20). We did not observe more severe GVHD in colon and small intestine in LBH589 treated recipients. It was likely due to the difference in the spatiotemporal expression of cytokine and chemokine gradients. After total body irradiation, the increase of proinflammatory cytokines and the forming of chemokine gradients (CXCL9-11) may occur earlier in liver than in gut. In addition, the expression of T-cell gut homing-receptor, α4β7, was not altered by LBH589 treatment (data not shown). Therefore, we surmised that LBH589 aggravated GVHD by increasing T-cell activation, Th1 cytokine production, and T-cell migration to recipient liver through up-regulating CXCR3 expression.
Previous studies by others have shown that pan-HDACi have therapeutic effect on many inflammatory disease models. Glauben et al. showed that oral administration of either Valproic Acid (another HDACi) or SAHA reduced the disease severity in dextran sulfate sodium-induced colitis in mice (3). The reduction of disease severity was associated with a marked suppression of colonic proinflammatory cytokines and apoptosis of lamina propria lymphocytes. Other three groups showed that SAHA alleviated GVHD after allogeneic BMT (5, 7-9). It appears that our data are contradictory to those published; however we believe that the distinct outcomes with SAHA and LBH589 can be readily reconciled.
We all used pan-HDACi without considering the functional difference of individual HDAC members. Actually, HDAC members have similar or opposite effects on the regulation of immune response. Conditional deletion of HDAC1 in T cells leads to enhanced airway inflammation and increased Th2 cytokine production (21). HDAC2 activity is indispensible to control chronic obstructive pulmonary disease by corticosteroid (22, 23). A recent report showed that HDAC6-deficiency increases the suppressive potency of regulatory T cells (24, 25) whereas our unpublished data indicated that HDAC11-deficiency resulted in T cell hyperactivation leading to increased severity of GVHD in mice. When a pan-HDACi was tested, what we observed is a compound effect on all 11 HDAC members. Considering the wide range of potency of individual HDACi on different HDAC members, and that each HDAC member plays a distinct role, it is plausible that 2 inhibitors would likely have distinct effects on a certain disease. Specifically, SAHA and LBH589 have very different potency in inhibiting each member of HDAC family, e.g. LBH589 inhibits HDAC11 40-fold more potently than SAHA (10). Given that HDAC11 may negatively regulate T cell function as supported by our unpublished data, effective blockade of HDAC11 by LBH589, but not by SAHA could contribute to the different GVHD outcome after treatment with LBH589 versus SAHA.
In addition, the substrate of HDACs are not limited to histones (26). There are more than 1,750 non-histone substrates that make the effect of HDACi even more complex (27). Given the paucity of knowledge on the functions of non-histone substrates, it is currently impossible to precisely predict the effect of HDAC inhibitors at cellular level until the role of individual HDAC is uncovered, and isoform-specific HDACi are developed. Finally, LAQ824, another pan-HDACi derived from hydroxamic acid, enhanced antitumor activity of tumor antigen-specific lymphocytes both in vitro and in vivo (28). This is consistent with our finding that LBH589 enhanced allogeneic T cell activation in vivo. LBH589’s structure is closer to LAQ824 than SAHA and accordingly, LBH589’s IC50 to HDAC members is quite similar to LAQ824. This may be the reason why both drugs improved T cell activation in vivo.
In summary, we evaluated the effect of LBH589 on the prevention of GVHD, and found that LBH589 accelerated rather than alleviated GVHD after allogeneic BMT in mice. The accelerated GVHD was associated with increased systemic Th1 cytokines and donor T cell infiltration in the recipient liver. This study highlights distinct effects of pan HDACi on allogeneic BMT, and demonstrates that LBH589 (Panobinostat) could have adverse effect on GVHD or other inflammatory diseases if used in a treatment scheme to replace other HDACi. Thus, a caution must be taken in consideration not to assume that pan-HDAC inhibitors would lead to similar therapeutic effects. Instead, the information learned from this study strongly encourages investigations on the function of individual HDAC member, and the development of isoform-specific HDACi for higher efficacy and better selectivity.
Supplementary Material
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yang XJ, Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol. 2008;9:206–218. doi: 10.1038/nrm2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Glauben R, Batra A, Fedke I, et al. Histone hyperacetylation is associated with amelioration of experimental colitis in mice. J Immunol. 2006;176:5015–5022. doi: 10.4049/jimmunol.176.8.5015. [DOI] [PubMed] [Google Scholar]
- 4.Kim HJ, Rowe M, Ren M, Hong JS, Chen PS, Chuang DM. Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: multiple mechanisms of action. J Pharmacol Exp Ther. 2007;321:892–901. doi: 10.1124/jpet.107.120188. [DOI] [PubMed] [Google Scholar]
- 5.Li N, Zhao D, Kirschbaum M, et al. HDAC inhibitor reduces cytokine storm and facilitates induction of chimerism that reverses lupus in anti-CD3 conditioning regimen. Proc Natl Acad Sci U S A. 2008;105:4796–4801. doi: 10.1073/pnas.0712051105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nasu Y, Nishida K, Miyazawa S, et al. Trichostatin A, a histone deacetylase inhibitor, suppresses synovial inflammation and subsequent cartilage destruction in a collagen antibody-induced arthritis mouse model. Osteoarthritis Cartilage. 2008;16:723–732. doi: 10.1016/j.joca.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 7.Reddy P, Maeda Y, Hotary K, et al. Histone deacetylase inhibitor suberoylanilide hydroxamic acid reduces acute graft-versus-host disease and preserves graft-versus-leukemia effect. Proc Natl Acad Sci U S A. 2004;101:3921–3926. doi: 10.1073/pnas.0400380101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reddy P, Sun Y, Toubai T, et al. Histone deacetylase inhibition modulates indoleamine 2,3-dioxygenase-dependent DC functions and regulates experimental graft-versus-host disease in mice. J Clin Invest. 2008;118:2562–2573. doi: 10.1172/JCI34712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leng C, Gries M, Ziegler J, et al. Reduction of graft-versus-host disease by histone deacetylase inhibitor suberonylanilide hydroxamic acid is associated with modulation of inflammatory cytokine milieu and involves inhibition of STAT1. Exp Hematol. 2006;34:776–787. doi: 10.1016/j.exphem.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 10.Wenlin Shao, Joseph Growney, Yun Feng, et al. HISTONE DEACETYLASE INHIBITORS AND CELL CYCLE INHIBITORS: POSTER PRESENTATIONS - PROFFERED ABSTRACTS:. 2008. AACR Meeting Abstracts; Apr 2008; 2008. p. 735. [Google Scholar]
- 11.Liang Y, Liu C, Djeu JY, et al. Beta2 integrins separate graft-versus-host disease and graft-versus-leukemia effects. Blood. 2008;111:954–962. doi: 10.1182/blood-2007-05-089573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zaidi MR, Merlino G. The Two Faces of Interferon-{gamma} in Cancer. Clin Cancer Res. 2011;17:6118–6124. doi: 10.1158/1078-0432.CCR-11-0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yi T, Chen Y, Wang L, et al. Reciprocal differentiation and tissue-specific pathogenesis of Th1, Th2, and Th17 cells in graft-versus-host disease. Blood. 2009;114:3101–3112. doi: 10.1182/blood-2009-05-219402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murphy WJ, Welniak LA, Taub DD, et al. Differential effects of the absence of interferon-gamma and IL-4 in acute graft-versus-host disease after allogeneic bone marrow transplantation in mice. J Clin Invest. 1998;102:1742–1748. doi: 10.1172/JCI3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Allen RD, Staley TA, Sidman CL. Differential cytokine expression in acute and chronic murine graft-versus-host-disease. Eur J Immunol. 1993;23:333–337. doi: 10.1002/eji.1830230205. [DOI] [PubMed] [Google Scholar]
- 16.Ferrara JL, Cooke KR, Pan L, Krenger W. The immunopathophysiology of acute graft-versus-host-disease. Stem Cells. 1996;14:473–489. doi: 10.1002/stem.140473. [DOI] [PubMed] [Google Scholar]
- 17.Piguet PF, Grau GE, Allet B, Vassalli P. Tumor necrosis factor/cachectin is an effector of skin and gut lesions of the acute phase of graft-vs.-host disease. J Exp Med. 1987;166:1280–1289. doi: 10.1084/jem.166.5.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang H, Cheng F, Woan K, et al. Histone deacetylase inhibitor LAQ824 augments inflammatory responses in macrophages through transcriptional regulation of IL-10. J Immunol. 2011;186:3986–3996. doi: 10.4049/jimmunol.1001101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Halili MA, Andrews MR, Labzin LI, et al. Differential effects of selective HDAC inhibitors on macrophage inflammatory responses to the Toll-like receptor 4 agonist LPS. J Leukoc Biol. 2010;87:1103–1114. doi: 10.1189/jlb.0509363. [DOI] [PubMed] [Google Scholar]
- 20.Duffner U, Lu B, Hildebrandt GC, et al. Role of CXCR3-induced donor T-cell migration in acute GVHD. Exp Hematol. 2003;31:897–902. doi: 10.1016/s0301-472x(03)00198-x. [DOI] [PubMed] [Google Scholar]
- 21.Grausenburger R, Bilic I, Boucheron N, et al. Conditional deletion of histone deacetylase 1 in T cells leads to enhanced airway inflammation and increased Th2 cytokine production. J Immunol. 2010;185:3489–3497. doi: 10.4049/jimmunol.0903610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barnes PJ. Targeting histone deacetylase 2 in chronic obstructive pulmonary disease treatment. Expert Opin Ther Targets. 2005;9:1111–1121. doi: 10.1517/14728222.9.6.1111. [DOI] [PubMed] [Google Scholar]
- 23.Ito K, Yamamura S, Essilfie-Quaye S, et al. Histone deacetylase 2-mediated deacetylation of the glucocorticoid receptor enables NF-kappaB suppression. J Exp Med. 2006;203:7–13. doi: 10.1084/jem.20050466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Zoeten EF, Wang L, Butler K, et al. Histone deacetylase 6 and heat shock protein 90 control the functions of Foxp3(+) T-regulatory cells. Mol Cell Biol. 2011;31:2066–2078. doi: 10.1128/MCB.05155-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beier UH, Akimova T, Liu Y, Wang L, Hancock WW. Histone/protein deacetylases control Foxp3 expression and the heat shock response of T-regulatory cells. Curr Opin Immunol. 2011 doi: 10.1016/j.coi.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spange S, Wagner T, Heinzel T, Krämer OH. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int J Biochem Cell Biol. 2009;41:185–198. doi: 10.1016/j.biocel.2008.08.027. [DOI] [PubMed] [Google Scholar]
- 27.Choudhary C, Kumar C, Gnad F, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 28.Vo DD, Prins RM, Begley JL, et al. Enhanced antitumor activity induced by adoptive T-cell transfer and adjunctive use of the histone deacetylase inhibitor LAQ824. Cancer Res. 2009;69:8693–8699. doi: 10.1158/0008-5472.CAN-09-1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.