

Abstract
Objectives
Identify SNPs associated with mild statin-induced side effects.
Background
Statin-induced side effects can interfere with therapy. SNPs in cytochrome P450 enzymes impair statin metabolism; the reduced function SLCO1B1*5 allele impairs statin clearance and is associated with simvastatin-induced myopathy with CK elevation.
Methods
The STRENGTH study was a pharmacogenetics study of statin efficacy and safety. Subjects (n=509) were randomized to atorvastatin 10mg, simvastatin 20mg, or pravastatin 10mg followed by 80mg, 80mg, and 40mg, respectively. We defined a composite adverse event (CAE) as discontinuation for any side effect, myalgia, or CK>3× baseline during follow-up. We sequenced CYP2D6, CYP2C8, CYP2C9, CYP3A4, and SLCO1B1 and tested seven reduced function alleles for association with the CAE.
Results
The CAE occurred in 99 subjects (54 discontinuations, 49 myalgias, and nine CK elevations). Sex was associated with CAE (percent female in CAE vs. no CAE groups, 66% vs. 50%, p<0.01). SLCO1B1*5 was associated with CAE (percent with ≥ 1 allele in CAE vs. no CAE groups, 37% vs. 25%, p=0.03) and those with CAE with no significant CK elevation (p≤ 0.03). Furthermore, there was evidence for a gene-dose effect (percent with CAE in those with 0, 1, or 2 alleles: 19%, 27%, and 50%, trend p = 0.01). Finally, the CAE risk appeared to be highest in those carriers assigned to simvastatin.
Conclusions
SLCO1B1*5 genotype and female sex were associated mild statin-induced side effects. These findings expand the results of a recent genome wide association study of statin myopathy with CK > 3 times normal to milder, statin-induced, muscle side effects.
Keywords: hydroxymethylglutaryl-CoA Reductase Inhibitors, pharmacogenetics, single nucleotide polymorphisms, muscular diseases, clinical trial, myopathy
Introduction
Statins are widely prescribed medications for the prevention of myocardial infarction and stroke, and also reduce cardiovascular mortality (1). Despite their proven efficacy, 25 – 50% of patients with CAD are nonadherent with statin medications after one year (2). Although a multifactorial problem, many believe that statin nonadherence is primarily caused by side effects (3). In numerous placebo-controlled clinical trials, statins have a well-defined safety profile with a small but real risk of predominantly musculoskeletal side effects of increasing severity: myalgia, CK elevations, and rhabdomyolysis (1). In clinical practice the incidence of mild statin-induced side effects appears to be higher than that seen in controlled trials and is estimated at 5-10% (4). This discrepancy is unexplained. However, it is the consensus that these symptoms can be caused by statins, often in the absence of CK elevations (5). This consensus is supported by observations that statin-induced side effects appear to be a class effect (5), improve with withdrawal of the drug, and recur with rechallenge of drug (6).
Although the mechanisms for these side effects are unclear, certain patient characteristics have been identified that contribute to the risk: lower body mass, female sex, and hepatic or renal dysfunction (7). Several observations have identified that these side effects are dose-dependent (8), are increased by concomitant drugs that impair statin disposition and metabolism (8), and are associated with elevated levels of statin metabolites (6). Furthermore, SNPs that impair the activity of drug metabolizing enzymes (9) have been associated with the development of musculoskeletal side effects. In particular, the *5 allele (Val174Ala, rs4149056) in the hepatic drug transporter SLCO1B1 interferes with localization of the transporter to the plasma membrane(10) and leads to higher systemic statin concentrations(11-13). The *5 allele was identified in a genome wide association study as a dominant cause of severe statin-induced myopathy (defined as CK >10× the upper limit of normal in those with symptoms and >3× in those without symptoms) in patients taking 80mg of simvastatin (14). Whether the SLCO1B1*5 and other SNPs in drug metabolizing enzymes are responsible for milder side effects caused by statins other than simvastatin is unknown.
Therefore, we sought to test the hypothesis that common genetic polymorphisms that lead to reduced function in drug metabolizing or transporting enzymes would be associated with mild statin-induced side effects, particularly those without CK elevations. We tested this hypothesis in the context of a large pharmacogenetics trial of three commercially available statins, the STRENGTH (Statin Response Examined by Genetic Haplotype Markers) study, where the main objectives were to identify genetic associations with statin safety and efficacy.
Methods
Study Population
The STRENGTH study was a pharmacogenetic study of statin efficacy and safety (15). In brief, it was a 16-week, randomized, open label study of three statins tested in 509 outpatients with hypercholesterolemia conducted between 2001 and 2002. Subjects were randomly assigned to eight weeks of 10 mg/day atorvastatin, 20 mg/day simvastatin, or 10 mg/day pravastatin followed by eight weeks of 80 mg/day atorvastatin, 80 mg/day simvastatin, and 40 mg/day pravastatin, respectively.
The following 57 subjects were excluded from all analyses due to incomplete follow up: 10 withdrew consent prior to drug administration, 21 discontinued therapy for reasons not related to symptoms, 13 were lost to follow up, seven were removed for protocol violations or noncompliance with study drug, one for pregnancy, and five for other reasons.
Follow up, laboratory testing, and definition of primary outcome
In-person visits with a research coordinator were scheduled every other week to assess for adverse events and compliance. Any symptom, physical sign, syndrome, or disease that either emerged during the study or, if present at screening, worsened during the study, regardless of the suspected cause of the event was documented as a potential adverse event. Symptoms may have been volunteered spontaneously by the subject or discovered as a result of general questioning by the study staff or by physical examination. At each visit the subject was asked, “Have you experienced any problems since your last visit?” In order to avoid vague, ambiguous, or colloquial expressions, all symptoms were recorded in standard medical terminology in addition to the subject's own words. For example, symptoms were recorded as “myalgia”, “muscle cramps”, or “elevated creatine phosphokinase” as appropriate. At both the first and final visit (Week 16 or at time of termination) a thorough physical examination was performed and any significant change that occurred during the trial recorded as a potential adverse event. Finally, in addition to these assessments, at the following intervals routine laboratory analyses for chemistries, liver function studies, and CK were measured: baseline and weeks 6, 8, 12, and 16.
For this analysis, we prospectively defined the primary outcome as a composite adverse event (CAE) of any of the following at any time point in the study: 1) premature discontinuation of study drug due to any side effect, 2) myalgia/muscle cramps (irrespective of CK values), and 3) CK elevations greater than three times the upper limit of normal (irrespective of symptoms).
Candidate gene selection, Power calculation, and SNP selection
As part of the STRENGTH protocol over 160 potential candidate genes were sequenced or genotyped as previously described (15). For the purposes of this focused analysis, we selected from this list of candidate genes, five genes (CYP2D6, CYP2C8, CYP2C9, CYP3A4, and SLCO1B1) thought to be implicated in statin pharmacokinetics based on information compiled by the PharmGKB database (www.pharmgkb.org) (16) and recent reviews of statin pharmacogenetics pathways (17). The percent missing genotypes and race stratified tests for Hardy-Weinberg Equilibrium were evaluated using Haploview software (18) as a means of quality control. The average call rate was 98%.
We prospectively defined allele frequency cutoffs based on power calculations performed using the QUANTO program (19). Based on the 99 subjects with the CAE included in our primary analysis, assuming a relative risk of 2.0 associated with the risk allele and a dominant model, we estimated that we would have 50% power with minor allele frequencies (MAF) of 0.05, 73% power with MAF of 0.10, 82% power with MAF of 0.15, and 85% power with MAF of 0.20. Based on this, we chose to include only those alleles with frequencies > 0.10 in STRENGTH Caucasians for further analysis. Finally, because we were primarily interested in those genes implicated in statin pharmacokinetics we limited our analysis to functional SNPs, i.e., those with alleles that had been previously tested in vivo or in vitro and found to reduce the enzymatic function of the protein.
Statin metabolite measurements
Simvastatin and pravastatin metabolites were measured on plasma collected from samples collected the day after the last low- or high- dose statin was taken. We randomly chose 57 subjects assigned to simvastatin and 55 assigned to pravastatin with available samples, stratified 1:1 based on SLCO1B1 genotype and sex. Metabolites were analyzed by liquid chromatography – tandem mass spectrometry system as previously described (20) with modifications. The lower limits of quantification in plasma were: 0.018, 0.018, 0.041, and 0.041 ng/mL for simvastatin acid, simvastatin lactone, pravastatin acid, and pravastatin lactone, respectively. In two simvastatin and six pravastatin samples, metabolites of both lactone and acid were undetectable (suggesting nonadherence) and were excluded. In addition, three simvastatin and one pravastatin high dose measurements were less than 5% of their respective low dose concentrations (in contrast to the remainder of the group whose high dose measurements were 4-8× fold higher than their low dose measurement), suggesting nonadherence at high dose and therefore were also excluded from further analysis.
Statistical analysis
Continuous variables were compared using Student's t-tests, categorical variables were compared using Chi square tests, and trends across ordinal variables were assessed with the Cochran-Armitage trend test. Triglycerides and CK values were log-transformed to approximate normal distributions. Multivariate models were constructed using logistic regression for categorical variables. A generalized linear model with the least square means method was used to estimate percent reduction in LDL by genotype. For changes in CK throughout the study, a repeated measures ANOVA was employed. To account for the multiple comparisons we calculated the false discovery rate (FDR) as described by Benjamini and Hochberg (21).
Statin metabolite concentrations were log transformed to approximate a normal distribution. Generalized linear models were constructed to evaluate the association between genotype (coded as 1, 2, 3) or sex and the log-transformed drug metabolite concentrations. For each model, residual plots were examined to confirm a normal distribution of errors. No influential outliers were observed.
A p-value of < 0.05 was considered significant for all analyses except for interaction tests where we used a p-value of < 0.2 to allow greater power to detect interactions as suggested by Selvin (22) and others (23). SAS Version 9.1 (SAS Institutes, Cary, NC) was employed for all analyses.
Results
At total of 452 subjects (88% of total STRENGTH participants) received study drug and were available for follow up. During the course of the study, 99 (22%) met at least one criterion for the CAE with 54 discontinuing drug for any side effect, 61 developing myalgias/muscle cramps, and nine developing CK elevations greater than 3× the upper limit of normal. The baseline characteristics of the entire cohort and those with and without the CAE are described in Table 1. Of these characteristics, only sex was significantly different between groups with or without the CAE, with a greater percentage of females in the CAE group (odds ratio (OR), [95% confidence interval (CI)], p-value: 2.0, [1.2, 3.1], 0.004). Importantly, assigned statin type was not associated with the CAE (p = 0.9). Seven genetic variants in the five candidate genes (Table 2) met the analysis criteria of having a minor allele frequency greater than 0.10 that were also known to reduce enzymatic function. One SNP (CYP3A4*1b) deviated significantly from Hardy-Weinberg equilibrium in Caucasian controls (p = 0.002), however the magnitude of the deviation was small and therefore the SNP was retained for statistical analysis. When these seven SNPs were tested in dominant models, one SNP in SLCO1B1 (SLCO1B1*5, rs4149056, V174A) was associated with the the CAE during the STRENGTH trial (χ2 = 4.2, p = 0.03, FDR = 0.24). In multivariate analyses adjusted for race, female sex and SLCO1B1*5 genotype were independently associated with the CAE (OR, 95% CI, p-value: 2.2, [1.4, 3.6], p = 0.001 and 1.7, [1.04, 2.8], p = 0.03, respectively). When we analyzed the individual components of the CAE, we observed that drug discontinuation due to a side effect was the primary determinant of the associations for SLCO1B1*5 and female sex with the CAE (p < 0.05 for both)
Table 1. Baseline characteristics of STRENGTH cohort stratified by the composite adverse event.
| Characteristic | Entire cohort (n = 452) | CAE (n = 99) | No CAE (n = 353) |
|---|---|---|---|
| Age, years (SD) | 56 (10) | 58 (10) | 56 (11) |
|
| |||
| Sex, Number (%) | |||
| Male | 209 (46) | 33 (33) | 176 (50) |
| Female | 243 (54) | 66 (67)* | 177 (50) |
|
| |||
| Body mass index, kg/m2 (SD) | 29.1 (5.2) | 29.1 (4.7) | 29.1 (5.4) |
|
| |||
| Smoking status, number (%) | |||
| Smoker | 84 (19) | 18 (18) | 66 (19) |
| Nonsmoker | 368 (81) | 81 (82) | 287 (81) |
|
| |||
| Race, Number (%) | |||
| Caucasian | 389 (86) | 87 (89) | 300 (85) |
| African American | 22 (5) | 3 (3) | 19 (5) |
| Other | 43 (9) | 9 (8) | 44 (10) |
|
| |||
| Total cholesterol mg/dl (SD) | 258 (32) | 263 (32) | 256 (32) |
|
| |||
| LDL mg/dl (SD) | 173 (25) | 176 (24) | 173 (26) |
|
| |||
| HDL mg/dl (SD) | 49 (13) | 51 (13) | 48 (12) |
|
| |||
| Triglycerides mg/dl (SD) | 177 (68) | 182 (73) | 176 (66) |
|
| |||
| Baseline creatinine kinase units/L (SD) | 121 (81) | 123 (94) | 121 (71) |
|
| |||
| Assigned statin, Number (%) | |||
| Atorvastatin | 147 (32) | 31 (31) | 116 (33) |
| Simvastatin | 162 (36) | 37 (37) | 125 (35) |
| Pravastatin | 143 (32) | 31 (31) | 112 (32) |
All comparisons are between those with and without the composite adverse event (CAE);
= 0.004;
SD = standard deviation
Table 2. Reduced function alleles of statin pharmacokinetic genes and frequency in STRENGTH.
| SNP | rsID | Protein effect | Enzyme/Transporter Effect* | Entire cohort (n = 452) | CAE (n = 99) | No CAE (n = 353) |
|---|---|---|---|---|---|---|
| CYP2D6*4 | rs3892097 | Spicing defect | Reduced due to haploinsufficiency | 142 (32%) | 34 (34%) | 108 (31%) |
| CYP2D6*10 | rs1065852 | Missense, S34P | Decreased | 161 (36%) | 40 (40%) | 121 (35%) |
| CYP2C8*3 | rs10509681 | Missense, R399K | Decreased | 94 (21%) | 15 (15%) | 79 (23%) |
| CYP2C8*4 | rs1058930 | Missense, M264I | Decreased(29) | 46 (10%) | 8 (8%) | 38 (11%) |
| CYP2C9*3 | rs1057910 | Missense, L395I | Decreased | 47 (11%) | 14 (14%) | 33 (9%) |
| CYP3A4*1b | rs2740574 | −392 A>G | Decreased | 60 (13%) | 14 (14%) | 46 (13%) |
| SLCO1B1*5 | rs4149056 | Missense, V174A | Decreased (10) | 123 (28%) | 35 (35%) | 88 (25%)† |
CAE = composite adverse event;
Unless otherwise stated, enzymatic effects taken from http://www.cypalleles.ki.se
p = 0.03
Prior studies have demonstrated that there is a gene dose effect with the SLCO1B1*5 allele in relationship to statin drug concentrations (12). Therefore, we sought to find a similar relationship with the CAE. In STRENGTH, we found that there indeed was evidence for a gene-dose effect: the proportion of individuals with the CAE increased with increasing numbers of the SLCO1B1*5 risk allele. For noncarriers (n = 325), carriers of 1 allele (n = 115), and carriers of 2 alleles (n = 8) the proportions [95% CI] with the CAE were 0.19 [0.15, 0.24], 0.27 [0.19, 0.36], and 0.50 [0.16, 0.84], respectively (one sided trend p-value = 0.01).
Sequencing SLCO1B1 in the STRENGTH study identified eight additional polymorphisms with MAF > 1% in STRENGTH Caucasians. When these additional SNPs were tested individually for their association with the CAE, one SNP in the 13th intron, rs4149080, provided a stronger association with the CAE (χ2 = 7.5, p = 0.006) compared with SLCO1B1*5, however there was significant linkage disequilibrium (LD) between rs4149080 and SLCO1B1*5 (r2 = 0.85) in STRENGTH Caucasians.
Because of the two-stage trial design, we also examined the timing of the occurrence of CAE with respect to sex and SLCO1B1 genotype. Of the 99 subjects who developed CAE, many (n = 61, 62%) developed these during the first eight weeks of the trial during the low dose phase. When restricting the analysis of the CAE to the low dose phase of the trial, female sex and SLCO1B1*5 genotype continued to associate with the CAE (p = 0.009 and p = 0.03, respectively).
Because of the randomized drug allocation, we also observed the proportions with the CAE with respect to SLCO1B1*5 carrier status and sex stratified by assigned drug treatment (Figure). Subjects who carried at least one allele of SLCO1B1*5 and were assigned to simvastatin had a higher incidence of the CAE and those assigned to atorvastatin showed a similar trend when compared with those with no alleles. Interestingly, carriers of SLCO1B1*5 had no excess occurrence of the CAE if they were assigned to pravastatin compared with noncarriers. These trends were not, however, statistically significant (gene*treatment interaction p=0.4). In contrast, the effect of sex on the CAE appeared consistent across all three statins when compared with men.
Figure. Statin stratified analyses of adverse events.
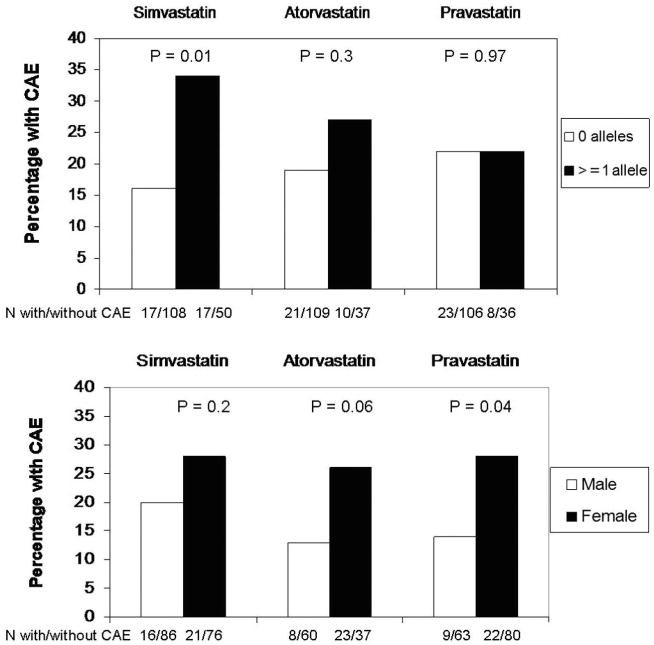
Percentage and number of those with the composite adverse event outcome (CAE) in STRENGTH stratified by SLCO1B1*5 genotype (top panel) and sex (bottom panel) for each assigned statin type is displayed. Carriers of SLCO1B1*5 appear to have no excess risk of CAE when assigned to pravastatin, whereas females appear to have an increased risk with all three statins.
Changes in LDL cholesterol and CK levels during the low- and high-dose phases of the trial were measured. We found that the percent reduction in LDLc cholesterol was similar for carriers and noncarriers of SLCO1B1*5 at both low and high dose treatment (statin adjusted percent LDL reduction ± SE in carriers vs. noncarriers at low and high dose: −31 ± 1 vs. −32 ± 1 and −41 ± 1 vs. −41 ± 1, p > 0.5 for both comparisons). Analysis of CK levels revealed that there was an association between statin dose and marginally higher CK levels: average CK (mg/dl) at baseline, during low-dose, and during high dose phases: 121 ± 81, 123 ± 90, and 126 ± 82, repeated measures ANOVA p = 0.004). In the entire STRENGTH cohort, however, SLCO1B1*5 genotype was not associated with CK levels alone, neither during the low-dose nor high-dose phases (p > 0.5).
In exploratory analyses to understand the statin specific associations of SLCO1B1*5 with the CAE, we measured simvastatin and pravastatin acid and lactone concentrations from stored plasma samples. (Table 3) Statin lactone concentrations in STRENGTH demonstrated no significant differences by SLCO1B1*5 genotype for pravastatin or simvastatin at low or high doses (p > 0.3 for all comparisons). In contrast, the concentration of simvastatin acid, was positively correlated with SLCO1B1*5 both at 20mg (p = 0.006) and 80mg (p = 0.03). This pattern, however, was not evident for pravastatin acid (p > 0.5). When we tested for a gene × statin interaction, there was a suggestion for statistical evidence with low dose statin acid concentrations (gene × treatment interaction p-value = 0.21) when using the more liberal alpha threshold as has been proposed for evaluating interactions (22,23). With respect to sex, there were no significant differences in statin acid or lactone concentrations at low dose or high doses of simvastatin or pravastatin (p > 0.5 for all comparisons).
Table 3. Simvastatin and Pravastatin Metabolite Concentrations in STRENGTH.
| Sex | SLCO1B1 genotype | ||||
|---|---|---|---|---|---|
| Metabolite | Male | Female | *1/*1 | *1/*5 | *5/*5 |
| Simvastatin Acid (20mg) | 28 1.7 [0.8 – 2.9] | 30 1.1 [0.4 – 2.1] | 28 1.2 [0.5 – 2.1] | 27 1.4 [0.7 – 2.9] | 3 11.2 [7.9 – 17.2] 0.006* |
| Simvastatin Lactone (20mg) | 28 0.5 [0.2 – 1.0] | 30 0.4 [0.3 – 0.8] | 28 0.5 [0.3 – 1.0] | 27 0.4 [0.2 – 0.9] | 3 0.4 [0.2 – 0.6] |
| Simvastatin Acid (80mg) | 27 4.1 [1.5 – 8.2] | 26 3.5 [1.6 – 5.3] | 28 3.7 [1.3 – 5.3] | 24 3.7 [1.6 – 9.2] | 1 77.5 0.03* |
| Simvastatin Lactone (80mg) | 27 1.3 [0.7 – 4.0] | 26 1.2 [0.8 – 2.4] | 28 1.1 [0.8 – 3.5] | 24 1.4 [0.7 – 3.3] | 1 3.7 |
| Pravastatin acid (10mg) | 28 0.2 [0.1 – 0.6] | 25 0.2 [0.1 – 0.5] | 25 0.2 [0.07 – 0.5] | 27 0.2 [0.1 - 0.5] | 1 0.2 |
| Pravastatin lactone (10mg) | 25 0.01 [0.01 – 0.02] | 23 0.02 [0.01 - 0.03] | 23 0.02 [0.01 – 0.03] | 24 0.01 [0.01 – 0.02] | 1 0.002 |
| Pravastatin acid (40mg) | 29 1.2 [0.6 – 3.6] | 24 0.9 [0.4 – 1.9] | 26 1.0 [0.4 – 2.1] | 27 1.0 [0.5 – 3.6] | N/A |
| Pravastatin lactone (40mg) | 29 0.06 [0.03 – 0.11] | 25 0.05 [0.02 – 1.1] | 26 0.07 [0.02-0.14] | 27 0.05 [0.03 – 0.10] | N/A |
Each cell contains the number tested, median (ng/ml), 25th and 75th percentiles (in brackets), and p-value where appropriate (based on linear regression on log transformed values);
N/A = no available sample;
p-value for comparison across genotypes;
To assess whether SLCO1B1*5 was associated mild side effects without significant CK elevations we limited the population to either the 90 cases with CK levels < 3× ULN (9 cases with CK > 3× ULN excluded) or the 71 cases with musculoskeletal side effects (remaining 18 with gastrointestinal side effects coded as controls). In these analyses the association of SLCO1B1*5 and sex with mild side effects either remained or were strengthened. (Table 4). Finally, when we included those subjects who received study drug but were originally excluded (n = 57, coded as controls), female sex and carrier status continued to be associated with the CAE (p = 0.003 and p=0.04, respectively).
Table 4. Sensitivity Analyses Using Various Endpoint Definitions.
| Factor | Drug discontinuation due to any side effect + muscle symptoms + CK > 3× ULN (n = 99) | Drug discontinuation due to any side effect + muscle symptoms (n = 90) | Drug discontinuation due to musculoskeletal side effect + muscle symptoms (n = 71) | |
|---|---|---|---|---|
| SLCO1B1*5 | p-value | 0.03 | 0.01 | 0.03 |
| χ2 statistic | 4.8 | 8.9 | 4.5 | |
|
| ||||
| Female Sex | p-value | 0.004 | 0.0002 | 0.0001 |
| χ2 statistic | 8.4 | 14.1 | 14.4 | |
ULN = upper limit of normal.
Discussion
In this study, we sought to identify common, functional genetic variants in drug metabolizing enzymes or a hepatic transporter that were associated with a composite measure of mild statin-induced side effects, in a large, prospective pharmacogenetics statin challenge study where one of the primary objectives was understanding statin safety. We defined a composite adverse event, the CAE, as discontinuation for any side effect, the development of myalgia, and CK elevation greater than three times the upper limit of normal. We found that carriers of the reduced function allele (*5) of the organic acid transporter SLCO1B1 and females were at higher risk of developing this CAE. This finding not only confirms the association with simvastatin-induced myopathy with CK elevations identified in a genome wide association study(14), but also expands this association to the most common statin-induced side effects (e.g. myalgia or muscle ache without significant CK elevations) in a population treated with various statins. Further, we found that the risk of the CAE was highest in those SLCO1B1*5 carriers assigned to simvastatin and negligible in those assigned to pravastatin and is consistent with our observation that the acid metabolite concentration of the drug was elevated in carriers who received simvastatin but not in subjects receiving pravastatin.
Although the precise mechanisms for statin-induced side effects are unknown, they likely are caused by a combination of patient and statin characteristics. Several patient-specific risk characteristics have been identified such as older age, reduced body mass, hypothyroidism, and female sex (7). Our findings are consistent with a prior association of female sex and simvastatin-induced myopathy(14). The reason for the effect of sex is unclear, but one study identified higher concentrations of pravastatin in women than in men (13). In STRENGTH, we found no association between sex and statin metabolite concentrations (Table 3). Further, the association of female sex with the CAE in this analysis appeared consistent across statin types. These observations suggest that the mechanism for the increased risk of side effects in women may not relate to altered statin pharmacokinetics or statin-specific characteristics.
Whether statin-induced side effects are class effects has been debated. Cerivastatin, according to some, has the highest risk of musculoskeletal side effects - especially rhabodomyolysis - when compared with other statins (24). In the present study, we observed that carriers of SLCO1B1*5 had no excess risk of adverse events if assigned to pravastatin, even though the CAE rates were the same for all three statins. There are two likely reasons why carriers appear to tolerate pravastatin better than simvastatin. First, we showed that after eight weeks of statin therapy, carriers accumulate higher concentrations of simvastatin acid, but not pravastatin acid. Although both statins are substrates for SLCO1B1(11), prior studies have all been performed after single dose challenge studies. We speculate that during extended statin therapy there may be alternative, compensatory routes of elimination for pravastatin (e.g. renal) such that carriers of the *5 allele are protected from drug accumulation. Second, the two statin metabolites differ markedly in their in vitro myotoxicity such that simvastatin acid is nearly ten-fold more myotoxic than pravastatin acid (25). Therefore, the higher exposure to simvastatin acid – a potentially more myotoxic metabolite compared with pravastatin acid -- may mediate, in part, the side effects seen in carriers assigned to simvastatin. These results, however, need to be evaluated in other, larger populations and if replicated, prescribing pravastatin would be a reasonable first choice for patients who carry SLCO1B1*5 to avoid statin-induced side effects. The more potent statins could then be reserved for those who do not achieve their LDL goals.
Despite the interesting findings of the present study, there are several limitations that deserve mention. First, our finding for SLCO1B1*5 in a dominant model does not survive correction for multiple comparisons using the FDR. However, our analysis was conservative in three ways: SNPs that belonged to CYP2D6 were treated as independent tests even though, due to LD in STRENGTH Caucasians they are not truly independent; the tests of SLCO1B1*5 and the other variants might logically have been 1-sided due to the known direction of their function; and the test of a gene dose effect increases the statistical significance of SLCO1B1*5′s effect. Moreover, SLCO1B1*5 was associated with simvastatin myopathy in a genome wide association study with a much stronger level of association (p = 1 × 10−9) (26) therefore our results are confirmatory of this initial report. Consistently, in STRENGTH, the association of SLCO1B1*5 with adverse events in patients assigned to simvastatin appeared to be the strongest.
Second, we had no placebo control arm as part of the study. Several placebo-controlled trials of statins demonstrate that a significant proportion of patients assigned to placebo experience adverse events at rates comparable to those assigned to statin (27,28). Therefore, in the present study, we cannot quantify the attributable risk of adverse events in carriers of SLCO1B1*5. However, we can calculate the relative risk of adverse events in carriers vs. noncarriers and our estimates are similar in magnitude to those obtained from the recently reported genome wide association study of simvastatin-induced myopathy (26).
Lastly, we used a composite endpoint that reflects mild statin-induced side effects that developed during the trial. However, when we limited the analysis to those without significant CK elevation or those with only musculoskeletal symptoms without CK elevations, the association with SLCO1B1*5 holds (Table 3).
There were several methodological strengths to our approach. First, we provided a clear biological rationale for the candidate genes and SNPs of interest based on known statin pharmacogenetics. By choosing genes implicated in statin pharmacology and SNPs that were known to be functional in prior studies we limited the number of statistical tests being performed and the risk of finding a false positive result. For example, the SLCO1B1*5 allele has been well studied with respect to leading to higher statin drug concentrations (11-13) and was identified as a major cause of simvastatin-induced myopathy with CK elevations(14). Second, our hypothesis was tested within the context of a large, prospective statin pharmacogenetics challenge study. The main benefit of such a trial is to provide clear, consistent, a priori definitions of the clinical phenotype of interest in order to minimize misclassification error. In addition, all subjects were followed, interviewed, and assessed in the identical manner therefore minimizing potential biases. Lastly, we provide in vivo data of statin metabolites that parallel the clinical associations.
Overall, statins are well tolerated medications, though a portion of patients experience adverse events that limit dose escalation, statin adherence, and ultimately achieving target LDL levels. We report here that carriers of the SLCO1B1*5 allele are at a two-fold relative risk of mild statin induced side effects – the majority of which had normal CK levels. These results could have potential implications for clinical practice since the vast majority of patients who are intolerant to statins have mild symptoms without associated CK elevations. For these patients, our findings suggest that pravastatin – instead of simvastatin – may be a reasonable first choice statin for carriers of the SLCO1B1*5 allele, whereas women may benefit from increased surveillance for symptoms. However, further studies of the role of this variant and gender in statin adherence and prognosis, and of statin-specific effects are necessary.
Acknowledgments
Funding Sources: This work was supported by institutional funds from the Duke Institute for Genome Sciences & Policy. Dr. Voora was supported by an NIH T32 training grant (T32HL007101).
Common abbreviation list
- CK
creatine kinase
- LDLc
low density lipoprotein cholesterol
- SNP
single nucleotide polymorphism
- AST
aspartate aminotransferase
- ALT
alanine aminotransferase
- CAD
coronary artery disease
Footnotes
Financial Disclosures: This trial was conducted by Genaissance Pharmaceuticals which is now a part of Clinical Data, Inc. and all analyses were performed by investigators at Duke University. Two authors (CRR and BAS) are employees and shareholders of Clinical Data, Inc. SHS received unrestricted research funding from Medtronic, Inc. SA is participating in a post-doctoral fellowship in which time is spent at Novartis Pharmaceuticals functioning as a clinical trial leader.
References
- 1.Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366:1267–78. doi: 10.1016/S0140-6736(05)67394-1. [DOI] [PubMed] [Google Scholar]
- 2.Ho PM, Magid DJ, Shetterly SM, et al. Medication nonadherence is associated with a broad range of adverse outcomes in patients with coronary artery disease. American Heart Journal. 2008;155:772–779. doi: 10.1016/j.ahj.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 3.Jacobson TA. Toward “Pain-Free” Statin Prescribing: Clinical Algorithm for Diagnosis and Management of Myalgia. Mayo Clin Proc. 2008;83:687–700. doi: 10.4065/83.6.687. [DOI] [PubMed] [Google Scholar]
- 4.Nichols GA, Koro CE. Does Statin Therapy Initiation Increase the Risk for Myopathy? An Observational Study of 32,225 Diabetic and Nondiabetic Patients. Clinical Therapeutics. 2007;29:1761–1770. doi: 10.1016/j.clinthera.2007.08.022. [DOI] [PubMed] [Google Scholar]
- 5.Bays H. Statin safety: an overview and assessment of the data--2005. Am J Cardiol. 2006;97:6C–26C. doi: 10.1016/j.amjcard.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 6.Hermann M, Bogsrud MP, Molden E, et al. Exposure of atorvastatin is unchanged but lactone and acid metabolites are increased several-fold in patients with atorvastatin-induced myopathy. Clin Pharmacol Ther. 2006;79:532–9. doi: 10.1016/j.clpt.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 7.Gotto JAM. Statins, Cardiovascular Disease, and Drug Safety. The American Journal of Cardiology. 2006;97:S3–S5. doi: 10.1016/j.amjcard.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 8.Thompson PD, Clarkson PM, Rosenson RS. An Assessment of Statin Safety by Muscle Experts. The American Journal of Cardiology. 2006;97:S69–S76. doi: 10.1016/j.amjcard.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 9.Frudakis TN, Thomas MJ, Ginjupalli SN, Handelin B, Gabriel R, Gomez HJ. CYP2D6*4 polymorphism is associated with statin-induced muscle effects. Pharmacogenet Genomics. 2007;17:695–707. doi: 10.1097/FPC.0b013e328012d0a9. [DOI] [PubMed] [Google Scholar]
- 10.Kameyama Y, Yamashita K, Kobayashi K, Hosokawa M, Chiba K. Functional characterization of SLCO1B1 (OATP-C) variants, SLCO1B1*5, SLCO1B1*15 and SLCO1B1*15+C1007G, by using transient expression systems of HeLa and HEK293 cells. Pharmacogenet Genomics. 2005;15:513–22. doi: 10.1097/01.fpc.0000170913.73780.5f. [DOI] [PubMed] [Google Scholar]
- 11.Pasanen MK, Neuvonen M, Neuvonen PJ, Niemi M. SLCO1B1 polymorphism markedly affects the pharmacokinetics of simvastatin acid. Pharmacogenet Genomics. 2006;16:873–9. doi: 10.1097/01.fpc.0000230416.82349.90. [DOI] [PubMed] [Google Scholar]
- 12.Pasanen MK, Fredrikson H, Neuvonen PJ, Niemi M. Different effects of SLCO1B1 polymorphism on the pharmacokinetics of atorvastatin and rosuvastatin. Clin Pharmacol Ther. 2007;82:726–33. doi: 10.1038/sj.clpt.6100220. [DOI] [PubMed] [Google Scholar]
- 13.Niemi M, Pasanen MK, Neuvonen PJ. SLCO1B1 polymorphism and sex affect the pharmacokinetics of pravastatin but not fluvastatin[ast] Clin Pharmacol Ther. 2006;80:356–366. doi: 10.1016/j.clpt.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 14.Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy--a genomewide study. N Engl J Med. 2008;359:789–99. doi: 10.1056/NEJMoa0801936. [DOI] [PubMed] [Google Scholar]
- 15.Voora D, Shah SH, Reed CR, et al. Pharmacogenetic predictors of statin mediated LDLc reduction and dose response. Circ Cardiovasc Genet. 2008;1 doi: 10.1161/CIRCGENETICS.108.795013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klein TE, Chang JT, Cho MK, et al. Integrating genotype and phenotype information: an overview of the PharmGKB project. Pharmacogenetics Research Network and Knowledge Base. Pharmacogenomics J. 2001;1:167–70. doi: 10.1038/sj.tpj.6500035. [DOI] [PubMed] [Google Scholar]
- 17.Mangravite LM, Thorn CF, Krauss RM. Clinical implications of pharmacogenomics of statin treatment. Pharmacogenomics J. 2006;6:360–74. doi: 10.1038/sj.tpj.6500384. [DOI] [PubMed] [Google Scholar]
- 18.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–5. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 19.Gauderman WJ, Morrison JM. QUANTO 1.1: A computer program for power and sample size calculations for genetic-epidemiology studies. 2006 http://hydra.usc.edu/gxe.
- 20.Zhao JJ, Rogers JD. Proceedings of the 47th ASMS Conference on Mass Spectrommetry; 1999; p. 1003. [Google Scholar]
- 21.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300. [Google Scholar]
- 22.Selvin S. Statistical Analysis of Epidemiologic Data. New York, NY: Oxford University Press; 1991. [Google Scholar]
- 23.Frankel DS, Meigs JB, Massaro JM, et al. Von Willebrand Factor, Type 2 Diabetes Mellitus, and Risk of Cardiovascular Disease: The Framingham Offspring Study. Circulation. 2008;118:2533–2539. doi: 10.1161/CIRCULATIONAHA.108.792986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cziraky MJ, Willey VJ, McKenney JM, et al. Statin Safety: An Assessment Using an Administrative Claims Database. The American Journal of Cardiology. 2006;97:S61–S68. doi: 10.1016/j.amjcard.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 25.Skottheim IB, Gedde-Dahl A, Hejazifar S, Hoel K, Asberg A. Statin induced myotoxicity: The lactone forms are more potent than the acid forms in human skeletal muscle cells in vitro. Eur J Pharm Sci. 2008 doi: 10.1016/j.ejps.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 26.Cardiovascular disease and steroid hormone contraception. Report of a WHO Scientific Group. World Health Organ Tech Rep Ser. 1998;877:i–vii. 1–89. [PubMed] [Google Scholar]
- 27.Cannon CP, Braunwald E, McCabe CH, et al. Intensive versus Moderate Lipid Lowering with Statins after Acute Coronary Syndromes. N Engl J Med. 2004;350:1495–1504. doi: 10.1056/NEJMoa040583. [DOI] [PubMed] [Google Scholar]
- 28.Pfeffer MA, Keech A, Sacks FM, et al. Safety and Tolerability of Pravastatin in Long-Term Clinical Trials: Prospective Pravastatin Pooling (PPP) Project. Circulation. 2002;105:2341–2346. doi: 10.1161/01.cir.0000017634.00171.24. [DOI] [PubMed] [Google Scholar]
- 29.Singh R, Ting JG, Pan Y, Teh LK, Ismail R, Ong CE. Functional role of Ile264 in CYP2C8: mutations affect haem incorporation and catalytic activity. Drug Metab Pharmacokinet. 2008;23:165–74. doi: 10.2133/dmpk.23.165. [DOI] [PubMed] [Google Scholar]