

Abstract
Organ of Zuckerkandl paragangliomas (PGLs) are rare neuroendocrine tumors that are derived from chromaffin cells located around the origin of the inferior mesenteric artery extending to the level of the aortic bifurcation. Mutations in the genes encoding succinate dehydrogenase subunits (SDH) B, C, and D (SDHx) have been associated with PGLs, but their contribution to PGLs of the organ of Zuckerkandl PGLs is not known. We aimed to describe the clinical presentation of patients with PGLs of the organ of Zuckerkandl and investigate the prevalence of SDHx mutations and other genetic defects among them. The clinical characteristics of 14 patients with PGL of the organ of Zuckerkandl were analyzed retrospectively; their DNA was tested for SDHx mutations and deletions. Eleven out of 14 (79%) of patients with PGLs of the organ of Zuckerkandl were found to have mutations of the SDHB (9), or SDHD (2) genes; one patient was found to have the Carney-Stratakis syndrome (CSS) and his PGL was discovered during surgery for gastrointestinal stromal tumor (GIST). Our results show that SDHx mutations are prevalent in pediatric and adult PGLs of the organ of Zuckerkandl. Patients with PGLs of the organ of Zuckerkandl should be screened for SDHx mutations and the CSS; in addition asymptomatic carriers of an SDHx mutation among the relatives of affected patients may benefit from tumor screening for early PGL detection.
Keywords: pheochromocytoma, paraganglioma, genetic testing
Introduction
The organ of Zuckerkandl consists of extra-adrenal chromaffin cells located just below the origin of the inferior mesenteric artery and above the bifurcation of the aorta. This collection of paraganglia was first described by Hungarian anatomist and surgeon, Emil Zuckerkandl, who initially named them Nebenorgane (auxiliary organs) (Ober 1983). The chromaffin cells that comprise the organ of Zuckerkandl originate from the same neural crest progenitor cells that give rise to the adrenal medulla. While these structures normally degenerate and involute by adolescence, small clusters of chromaffin cells remain that can serve as sites of future tumor development. At least 135 cases of paragangliomas (PGLs) of the organ of Zuckerkandl have been reported in the literature, but their association with specific mutations and/or genetic syndromes has not been described (Subramanian and Maker 2006).
Germline mutations in the genes encoding subunits B, C, and D of succinate dehydrogenase (SDH), the mitochondrial complex II, have been associated with the development of PGLs (Astuti, et al. 2003; Astuti, et al. 2001; Baysal, et al. 2000; Brouwers, et al. 2006; Ghayee, et al. 2009; Gimenez-Roqueplo, et al. 2003). The SDHB, SDHC, and SDHD genes encode mitochondrial proteins which serve as tumor suppressors, and loss-of-function mutations in these genes are linked to formation of PGLs (Gottlieb and Tomlinson 2005). The SDH enzyme is involved in cellular energy metabolism through the tricarboxylic acid cycle, oxidative phosphorylation, and the electron transport chain (Gottlieb and Tomlinson 2005). Although the mechanism that explains the link between mitochondrial dysfunction and tumor formation remains unknown, it is thought to be linked to an increase in reactive oxygen species and/or the activation of the hypoxia pathway or apoptosis (Benn and Robinson 2006; Eng, et al. 2003; Favier, et al. 2009; Gottlieb and Tomlinson 2005). The development of metastatic disease may be related to up-regulation of angiogenesis, however the exact mechanism remains to be elucidated (Favier, et al. 2002).
While an association with mutations in SDHB and SDHD genes has been found in patients with mediastinal PGLs, a specific analysis of SDH mutations in patients with Zuckerkandl organ PGLs has not been performed to date. SDHD gene mutations are specifically associated with head-and-neck PGLs and much less frequently with malignant PGLs (Baysal et al. 2000; Benn, et al. 2006; Havekes, et al. 2007; Timmers, et al. 2008). Mutations in the SDHB gene are associated with a high rate of malignancy and aggressive disease; among patients with metastatic PGLs, the frequency of SDHB mutations is between 30 and 83%. (Amar, et al. 2005; Brouwers et al. 2006; Gimenez-Roqueplo et al. 2003; Neumann, et al. 2004). SDHB mutations have also been associated with shorter survival (Amar, et al. 2007). Tumors classified as extra adrenal abdominal PGLs, (including a grouping of paraaortic/pericaval, bladder, remnants of the organ of Zuckerkandl, perirenal, retroperitoneal and periadrenal) are associated with mutations in both SDHB and SDHD (Benn et al. 2006). Anecdotal reports have described six patients with tumors of the organ of Zuckerkandl and SDHD (2) or SDHB (4) mutations; however, to our knowledge this is the first series with a specific focus on tumors of this anatomic site (Donahue, et al. 2008; Gimenez-Roqueplo et al. 2003).
From an initial observation of two patients with Zuckerkandl organ tumors who had SDHx mutations, we searched our clinical database for other patients with tumors involving this organ; we then tested the DNA of these patients retrospectively for mutations in SDHx. Three of the patients had mutations in SDHx identified prior to their evaluation at our institution. In the present study we report in detail the clinical characteristics, biochemical phenotype, and clinical course of 14 patients with Zuckerkandl organ tumors seen at the NIH and the University of Texas M.D. Anderson Cancer Center over the past 20 years; in these patients we examined the frequency of SDHx mutations.
Materials and methods
Subjects of Protocol
Fourteen patients with primary PGLs of the organ of Zuckerkandl with a median age at diagnosis of 22.5 yrs (range 9–71), seen at the National Institutes of Health, University of Texas M.D. Anderson Cancer Center and Dana-Farber Cancer Institute between 1989 and 2009 are presented in this study. A retrospective chart analysis was performed in order to identify patients with Zuckerkandl organ PGLs via review of abdominal CT scans and analysis of operative and pathology reports. We reviewed radiologic scans from all cases and included only those tumors defined by the radiologist to be consistent with the specific anatomic localization of the Zuckerkandl organ. The patients were followed up for an average of 8 years. Initial patient symptoms, biochemical phenotype, details of primary tumor, and patient outcome were recorded. The presence of disease in sites where chromaffin cells are not normally present, i.e. disease in an extra-paraganglionic site (lymph nodes, bone, liver, lung), was used to define metastatic disease as differentiated from multiple sites of disease (Gimenez-Roqueplo, et al. 2008). Consent was obtained from each patient after full explanation of the purpose and nature of all procedures used. The investigation was approved by the NICHD IRB and the M. D. Anderson Cancer Center IRB.
Genetic Studies
Three patients were known to harbor SDHx mutations prior to their evaluation at the NIH. For those patients without prior studies, genetic testing of germ-line DNA for mutations in SDHB, C, and D was performed. Blood samples for SDHx mutation analysis were collected prospectively for purposes of genetic testing. Genomic DNA was extracted from whole blood as previously described. The four exons of SDHD, eight exons of SDHB, and six exons of SDHC were amplified and sequenced by PCR based bidirectional sequencing by Mayo Medical Laboratories, Rochester, MN or by the Division of Molecular Diagnostics at the University of Pittsburgh Medical Center, as previously described (Brouwers et al. 2006). Deletion testing was performed using a combination of multiplex ligation-dependant probe amplification (MLPA) and Luminex® FlexMap Technologies (Monico, et al. 2007).
Hormone Assays
Plasma catecholamines and metanephrines and urinary catecholamines and metanephrines were measured using standard high-Pressure Liquid Chromatography (HPLC) assays, as described previously, at the NIH clinical center and Mayo Medical Laboratories, Rochester, MN (Eisenhofer, et al. 1986; Lenders, et al. 1993).
Results
Patient characteristics are presented in Table 1. The median age at diagnosis was 22.5 yrs (range 9–71), 9 males and 5 females. Eleven out of 14 (79%) of patients with organ of Zuckerkandl PGLs were found to have mutations of the SDHB (9), or SDHD (2) genes. Four of these represent nonsense mutations, two missense mutations, two deletions, two frame-shift and one splice site mutation. Three have no mutations found. Details of the mutations are presented in Table 1; two of the mutations represent novel mutations, SDHB p.Arg38ProfsX40 and SDHB Tyr61X, the remaining six are previously reported (Baysal et al. 2000; Brouwers et al. 2006; Pasini, et al. 2008; Solis, et al. 2009; Timmers, et al. 2007). Three of the 14 patients had a first degree relative with a history of pheochromocytoma or PGL. None of the 14 patients were related to one another.
TABLE 1.
| Patient | Mutation | Biochemistry | Size of tumor | Metastatic at dx? | Patient Outcome |
|---|---|---|---|---|---|
| M,49 | SDHB p.Arg38ProfsX40* | U:NE,DA,NMN | 10 cm | Yes | AWD |
| M,20 | Negative | not done | 3.7 cm | No | NED |
| F,33 | Negative | P: NE,CGA, U:NE,NMN | 3.1 cm | Yes | DOD |
| M,9 | SDHB p.Arg46X | P:NMN | 14.5 cm | No | AWD |
| F,9 | SDHB first exon deletion | P:NMN,NE,DA | 3 cm | Yes | AWD |
| M,15 | SDHB p.Arg46X | U:NMN,NE,DA; P:NMN,NE | 3.5 cm | No | AWD |
| M,34 | SDHB first exon deletion | P:NE,NMN | 2 4cm | Yes | AWD |
| F,18 | Negative | P:NMN,CGA | 1.4 cm | Yes | AWD |
| F,26 | SDHD p.Arg38X | P:NE | 6 cm | No | AWD |
| M,24 | SDHB Tyr61X* | P:NE U:NE,NMN | 2.5 cm | No | AWD |
| M,58 | SDHB p.Trp200Cys | Not done | 6 cm | Yes | AWD |
| F,71 | SDHB IVS1 + 1G>T | P:NMN,MN,NE,EPI | 10 cm | Yes | DOD |
| M,21 | SDHD p.Leu20CysfsX66 | P,U:NE,NMN | 2 cm | Yes | AWD |
| M,11 | SDHB p.Val140Phe | P:NE,NMN U:NE,NMN |
5.5 cm | No | AWD |
= novel mutation
P= plasma U= urine, NE= norepinephrine EPI= Epinephrine NMN= normetanephrine, CGA= chromogranin A, DA= dopamine AWD= alive with disese, DOD= dead of disease, NED= no evidence of disese
Eight out of 14 (57%) of patients had signs and symptoms related to catecholamine excess at the time of diagnosis. Imaging studies used to detect tumor are presented in Table 1. In all 14 patients, CT scans were positive for visualization of Zuckerkandl organ tumor. 123I-metaiodobenzylguanidine (123I-MIBG) scans were able to detect tumors in 8 out of 9 cases, and T2 MRI was positive in all 8 patients in whom it was used. Four out of 14 (30 %) patients presented with the organ of Zuckerkandl PGL as their only primary tumor. While 8 patients presented with metastatic disease, not all of these had the organ of Zuckerkandl PGL at the time of presentation. Seven out of 14 (50%) patients who had organ of Zuckerkandl tumors as part of their initial presentation also had multiple primary tumors or metastatic disease at initial diagnosis. Two out of 14 (14%) patients were identified to have the organ of Zuckerkandl PGL as an additional site of disease on long-term follow up. One patient was diagnosed with the Carney-Stratakis syndrome (CSS) and was found to have the organ of Zuckerkandl PGL incidentally during surgery for gastrointestinal stromal tumor (GIST). The size of the organ of Zuckerkandl PGLs was a mean of 5.3 ± 3.8 cm. Analysis of the biochemical data reveals that 9 patients had a noradrenergic phenotype, and 3 had both noradrenergic and dopaminergic phenotypes (Table 1). Four patients developed metastatic disease during follow-up, the time to metastases ranged from 2–19 years (6 ± 8.7). Sites of metastatic disease and multiple sites of disease are presented in Figure 1. Surgical resection of the primary tumor was performed in 11/14 (79%) of the patients, while four patients received therapeutic 131I-MIBG, two received Octreotide therapy, four received conventional radiation therapy, three patients with aggressive and metastatic disease additionally received chemotherapy.
Figure 1.
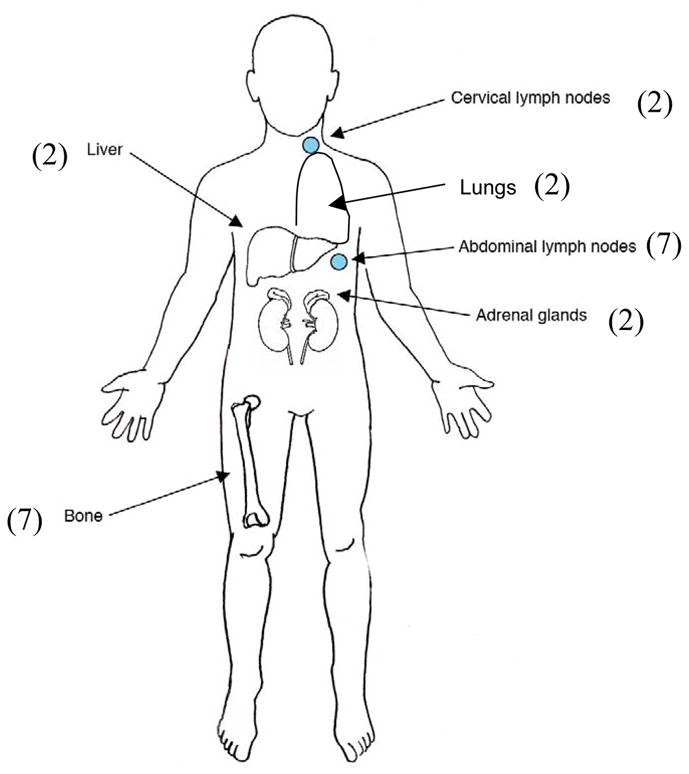
Sites of metastatic disease, n= 14 patients
Imaging studies and surgical images demonstrating the organ of Zuckerkandl PGLs are shown in Figures 2 and 3. Pathology of a representative patient’s organ of Zuckerkandl PGL that is consistent with neuroendocrine tumor is presented in Figure 4.
Figure 2. Diagnostic imaging from patient F,18.

(A)123I-metaiodobenzylguanidine (123I-MIBG) anterior maximum intensity projection showing right adrenal mass (dashed arrow) and nodule anterior to vertebrae L5 representing the organ of Zuckerkandl tumor (closed arrow). There is a focus in the mid-portion of the left kidney (open arrow) thought to be due to activity retained in the collecting system.
(B) Hybrid single photon emission computed axial tomography (SPECT) and CT (SPECT/CT) 123I-MIBG with contrast of the abdomen showing a 2.6 cm heterogeneously enhancing partially necrotic right adrenal mass (open arrow) and a 1.6 cm enhancing mass anterior to the spine and just inferior to the aortic bifurcation (closed arrow)
(C) Surgical photo, arrow indicated the site of tumor below aortic bifurcation
Figure 3. Diagnostic imaging from patient M,34.

(A) [18F]-2-fluoro-deoxy-D-glucose(FDG PET/CT) demonstrates an enhancing retroperitoneal mass (closed arrow)
(B) Magnetic resonance imaging (MRI) anterior demonstrates two retroperitoneal masses, 4 × 3 and 4 × 2 cm (closed arrows), one at the level of the right renal hilum and the other a periaortic mass between the abdominal aorta and the inferior vena cava
(C) Surgical photo demonstrating anatomical site of tumor (closed arrow) and the common iliac artery (open arrow)
Figure 4.

Low 4 × (A), medium 10 × (B), and high 40 × (C) power magnification of PGL cells in a representative patient. Immunohistochemistry for synaptophysin showing strong positivity of tumor cells (D)
Discussion
The present study including 14 patients with PGLs of the organ of Zuckerkandl establishes for the first time an association of these tumors with SDHB and SDHD mutations. Our data also shows that PGLs of the organ of Zuckerkandl are strongly associated with a noradrenergic phenotype and have an aggressive behavior, likely related to the SDHB mutational status. The identification of SDHx mutations in patients with the organ of Zuckerkandl PGLs has important implications for patient care and genetic screening of family members.
PGLs arise from sympathetic chromaffin tissue in both adrenal as well as outside of the adrenal gland in the abdomen or thorax, or alternatively originate from parasympathetic tissues in the head and neck (Benn et al. 2006). A particular grouping of PGLs, termed organ of Zuckerkandl PGLs, refer to a group of extra-adrenal chromaffin tissue situated between the origin of the inferior mesenteric artery and the bifurcation of the aorta (Ober 1983). These PGLs are remnants of the primitive sympathetic nervous system. The function of the organ of Zuckerkandl is unknown in humans, however it is thought to act as a chemoreceptor in other species (Hollinshead 1940). Mutations in genes encoding the SDH subunits B, C, and D of mitochondrial complex II have been associated with PGL development (Astuti et al. 2003; Astuti et al. 2001; Baysal et al. 2000; Brouwers et al. 2006; Ghayee et al. 2009; Gimenez-Roqueplo et al. 2003). While most head and neck PGLs are associated with SDHD gene mutations, PGLs derived from the sympathetic nervous system are often related to SDHB gene mutations. This is especially true for those deriving from extra-adrenal intra-abdominal lesions, which are frequently metastatic (Brouwers et al. 2006; Burnichon, et al. 2009). Recently, mediastinal PGLs were also found to be strongly associated with mutations of SDHB (Ghayee et al. 2009).
While the majority of our patients had mutations of the SDHx genes, there are two important exceptions. One patient with primary adrenal pheochromocytoma as well as organ of Zuckerkandl PGL who did not have a mutation in SDHx also had congenital polycythemia. She was also tested (data not shown) for a mutation of the gene encoding the prolyl hydroxylase domain 2 (PHD2) protein, but she was negative (Ladroue, et al. 2008). Another patient who presented with a GIST and was incidentally found to have an organ of Zuckerkandl PGL at the time of surgery was also not found to have SDHx mutation or deletion. Although most patients with CSS have SDHx mutations, up to 10% may harbor deletions that are not detectable by current screening methods (Carney and Stratakis 2002; Pasini et al. 2008).
Testing for SDH subunits is now recommended for all cases of pheochromocytomas and PGLs, and testing should be offered to first-degree relatives for cancer surveillance and early detection (Amar et al. 2005; Benn and Robinson 2006; Jimenez, et al. 2006; Neumann, et al. 2002; Prodanov, et al. 2009). In addition, young patients with extra-adrenal pheochromocytomas should be tested for germline mutations of the von Hippel–Lindau (VHL) gene (Boedeker, et al. 2009; Pacak, et al. 2007). Germ-line mutations in the SDHD and SDHB genes should be considered a risk factor for PGLs of the organ of Zuckerkandl and radiographic analysis of this area should be performed. However, exact surveillance recommendations for asymptomatic carriers of SDHx mutations may vary between institutions (Benn and Robinson 2006).
Clinical presentation of pheochromocytomas and PGLs is variable depending on functionality of tumor; 70–80% of PGLs of the organ of Zuckerkandl are clinically functional (Subramanian and Maker 2006). Most extra-adrenal paraganglia as well as paraganglia in the setting of a SDHB mutation primarily secrete norepinephrine or norepinephrine and dopamine, as they lack phenylethanolamine N-methyltransferase (PNMT), the enzyme responsible for the conversion of norepinephrine to epinephrine (Timmers et al. 2007). Our patient group is consistent with this finding in the predominance of noradrenergic phenotype. Interestingly, three of our patients had tumors with mixed secretion of both norepinephrine and dopamine, all of whom had SDHB mutations, consistent with previous studies linking this specific biochemical phenotype to the presence of SDHB mutations (Timmers et al. 2007). Surgical resection is the preferred treatment for organ of Zuckerkandl PGLs; 11/14 of our patients underwent resection, however a subset of patients presented with metastatic disease that was not amenable to resection. Similar to what is known about other extra-adrenal pheochromocytomas, organ of Zuckerkandl PGLs have a higher rate of malignancy than adrenal pheochromocytomas (Altergott, et al. 1985). A review of 135 cases of organ of Zuckerkandl PGLs found that 41% of these tumors were malignant based on the presence of metastases and local invasion, with the most common sites of metastatic disease including bone, liver, and lungs (Subramanian and Maker 2006). This finding is well supported by our data that shows that 86% of patients with organ of Zuckerkandl tumors had multiple sites of disease and/or metastatic disease.
We conclude that SDHB and SDHD mutations are indicators of possible organ of Zuckerkandl tumors and should lead to appropriate clinical studies and investigation of family members. This is also supported by our and previous findings that noradrenergic and dopaminergic phenotype in the same patient strongly suggest the presence of SDHB gene mutation that should be addressed in planned genetic testing. As organ of Zuckerkandl PGLs are linked to SDHx associated disease; having a PGL in the organ of Zuckerkandl impacts the probability of an SDHB or D mutation and warrants mutation testing.
Acknowledgments
Supported by intramural research funding of the Eunice Kennedy Shriver National Institute of Child Health and Human Development of the National Institutes of Health.
Funding
This work was supported by the Intramural Research Program of the National Institute of Child Health and Human Development/National Institutes of Health.
Footnotes
Disclosure statement: The authors have nothing to disclose.
Declarations of Interest
There is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
References
- Altergott R, Barbato A, Lawrence A, Paloyan E, Freeark RJ, Prinz RA. Spectrum of catecholamine-secreting tumors of the organ of Zuckerkandl. Surgery. 1985;98:1121–1126. [PubMed] [Google Scholar]
- Amar L, Baudin E, Burnichon N, Peyrard S, Silvera S, Bertherat J, Bertagna X, Schlumberger M, Jeunemaitre X, Gimenez-Roqueplo AP, et al. Succinate dehydrogenase B gene mutations predict survival in patients with malignant pheochromocytomas or paragangliomas. J Clin Endocrinol Metab. 2007;92:3822–3828. doi: 10.1210/jc.2007-0709. [DOI] [PubMed] [Google Scholar]
- Amar L, Bertherat J, Baudin E, Ajzenberg C, Bressac-de Paillerets B, Chabre O, Chamontin B, Delemer B, Giraud S, Murat A, et al. Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol. 2005;23:8812–8818. doi: 10.1200/JCO.2005.03.1484. [DOI] [PubMed] [Google Scholar]
- Astuti D, Hart-Holden N, Latif F, Lalloo F, Black GC, Lim C, Moran A, Grossman AB, Hodgson SV, Freemont A, et al. Genetic analysis of mitochondrial complex II subunits SDHD, SDHB and SDHC in paraganglioma and phaeochromocytoma susceptibility. Clin Endocrinol (Oxf) 2003;59:728–733. doi: 10.1046/j.1365-2265.2003.01914.x. [DOI] [PubMed] [Google Scholar]
- Astuti D, Latif F, Dallol A, Dahia PL, Douglas F, George E, Skoldberg F, Husebye ES, Eng C, Maher ER. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet. 2001;69:49–54. doi: 10.1086/321282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, van der Mey A, Taschner PE, Rubinstein WS, Myers EN, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287:848–851. doi: 10.1126/science.287.5454.848. [DOI] [PubMed] [Google Scholar]
- Benn DE, Gimenez-Roqueplo AP, Reilly JR, Bertherat J, Burgess J, Byth K, Croxson M, Dahia PL, Elston M, Gimm O, et al. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J Clin Endocrinol Metab. 2006;91:827–836. doi: 10.1210/jc.2005-1862. [DOI] [PubMed] [Google Scholar]
- Benn DE, Robinson BG. Genetic basis of phaeochromocytoma and paraganglioma. Best Pract Res Clin Endocrinol Metab. 2006;20:435–450. doi: 10.1016/j.beem.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Boedeker CC, Erlic Z, Richard S, Kontny U, Gimenez-Roqueplo AP, Cascon A, Robledo M, de Campos JM, van Nederveen FH, de Krijger RR, et al. Head and neck paragangliomas in von Hippel-Lindau disease and multiple endocrine neoplasia type 2. J Clin Endocrinol Metab. 2009;94:1938–1944. doi: 10.1210/jc.2009-0354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwers FM, Eisenhofer G, Tao JJ, Kant JA, Adams KT, Linehan WM, Pacak K. High frequency of SDHB germline mutations in patients with malignant catecholamine-producing paragangliomas: implications for genetic testing. J Clin Endocrinol Metab. 2006;91:4505–4509. doi: 10.1210/jc.2006-0423. [DOI] [PubMed] [Google Scholar]
- Burnichon N, Rohmer V, Amar L, Herman P, Leboulleux S, Darrouzet V, Niccoli P, Gaillard D, Chabrier G, Chabolle F, et al. The succinate dehydrogenase genetic testing in a large prospective series of patients with paragangliomas. J Clin Endocrinol Metab. 2009;94:2817–2827. doi: 10.1210/jc.2008-2504. [DOI] [PubMed] [Google Scholar]
- Carney JA, Stratakis CA. Familial paraganglioma and gastric stromal sarcoma: a new syndrome distinct from the Carney triad. Am J Med Genet. 2002;108:132–139. doi: 10.1002/ajmg.10235. [DOI] [PubMed] [Google Scholar]
- Donahue J, Sahani D, Tso L, Cusack JC., Jr Extra-adrenal pheochromocytoma involving the organ of Zuckerkandl. Surgery. 2008;143:830–832. doi: 10.1016/j.surg.2007.11.012. [DOI] [PubMed] [Google Scholar]
- Eisenhofer G, Goldstein DS, Stull R, Keiser HR, Sunderland T, Murphy DL, Kopin IJ. Simultaneous liquid-chromatographic determination of 3,4-dihydroxyphenylglycol, catecholamines, and 3,4-dihydroxyphenylalanine in plasma, and their responses to inhibition of monoamine oxidase. Clin Chem. 1986;32:2030–2033. [PubMed] [Google Scholar]
- Eng C, Kiuru M, Fernandez MJ, Aaltonen LA. A role for mitochondrial enzymes in inherited neoplasia and beyond. Nat Rev Cancer. 2003;3:193–202. doi: 10.1038/nrc1013. [DOI] [PubMed] [Google Scholar]
- Favier J, Briere JJ, Burnichon N, Riviere J, Vescovo L, Benit P, Giscos-Douriez I, De Reynies A, Bertherat J, Badoual C, et al. The warburg effect is genetically determined in inherited pheochromocytomas. PLoS One. 2009;4:e7094. doi: 10.1371/journal.pone.0007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favier J, Plouin PF, Corvol P, Gasc JM. Angiogenesis and vascular architecture in pheochromocytomas: distinctive traits in malignant tumors. Am J Pathol. 2002;161:1235–1246. doi: 10.1016/S0002-9440(10)64400-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghayee HK, Havekes B, Corssmit EP, Eisenhofer G, Hammes SR, Ahmad Z, Tessnow A, Lazurova I, Adams KT, Fojo AT, et al. Mediastinal paragangliomas: association with mutations in the succinate dehydrogenase genes and aggressive behavior. Endocr Relat Cancer. 2009;16:291–299. doi: 10.1677/ERC-08-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimenez-Roqueplo AP, Burnichon N, Amar L, Favier J, Jeunemaitre X, Plouin PF. Recent advances in the genetics of phaeochromocytoma and functional paraganglioma. Clin Exp Pharmacol Physiol. 2008;35:376–379. doi: 10.1111/j.1440-1681.2008.04881.x. [DOI] [PubMed] [Google Scholar]
- Gimenez-Roqueplo AP, Favier J, Rustin P, Rieubland C, Crespin M, Nau V, Khau Van Kien P, Corvol P, Plouin PF, Jeunemaitre X. Mutations in the SDHB gene are associated with extra-adrenal and/or malignant phaeochromocytomas. Cancer Res. 2003;63:5615–5621. [PubMed] [Google Scholar]
- Gottlieb E, Tomlinson IP. Mitochondrial tumour suppressors: a genetic and biochemical update. Nat Rev Cancer. 2005;5:857–866. doi: 10.1038/nrc1737. [DOI] [PubMed] [Google Scholar]
- Havekes B, Corssmit EP, Jansen JC, van der Mey AG, Vriends AH, Romijn JA. Malignant paragangliomas associated with mutations in the succinate dehydrogenase D gene. J Clin Endocrinol Metab. 2007;92:1245–1248. doi: 10.1210/jc.2006-1993. [DOI] [PubMed] [Google Scholar]
- Hollinshead WH. Chromaffin Tissue and Paraganglia. The Quarterly Review of Biology. 1940;15:156–171. [Google Scholar]
- Jimenez C, Cote G, Arnold A, Gagel RF. Review: Should patients with apparently sporadic pheochromocytomas or paragangliomas be screened for hereditary syndromes? J Clin Endocrinol Metab. 2006;91:2851–2858. doi: 10.1210/jc.2005-2178. [DOI] [PubMed] [Google Scholar]
- Ladroue C, Carcenac R, Leporrier M, Gad S, Le Hello C, Galateau-Salle F, Feunteun J, Pouyssegur J, Richard S, Gardie B. PHD2 mutation and congenital erythrocytosis with paraganglioma. N Engl J Med. 2008;359:2685–2692. doi: 10.1056/NEJMoa0806277. [DOI] [PubMed] [Google Scholar]
- Lenders JW, Eisenhofer G, Armando I, Keiser HR, Goldstein DS, Kopin IJ. Determination of metanephrines in plasma by liquid chromatography with electrochemical detection. Clin Chem. 1993;39:97–103. [PubMed] [Google Scholar]
- Monico CG, Rossetti S, Schwanz HA, Olson JB, Lundquist PA, Dawson DB, Harris PC, Milliner DS. Comprehensive mutation screening in 55 probands with type 1 primary hyperoxaluria shows feasibility of a gene-based diagnosis. J Am Soc Nephrol. 2007;18:1905–1914. doi: 10.1681/ASN.2006111230. [DOI] [PubMed] [Google Scholar]
- Neumann HP, Bausch B, McWhinney SR, Bender BU, Gimm O, Franke G, Schipper J, Klisch J, Altehoefer C, Zerres K, et al. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med. 2002;346:1459–1466. doi: 10.1056/NEJMoa020152. [DOI] [PubMed] [Google Scholar]
- Neumann HP, Pawlu C, Peczkowska M, Bausch B, McWhinney SR, Muresan M, Buchta M, Franke G, Klisch J, Bley TA, et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. Jama. 2004;292:943–951. doi: 10.1001/jama.292.8.943. [DOI] [PubMed] [Google Scholar]
- Ober WB. Emil Zuckerkandl and his delightful little organ. Pathol Annu. 1983;18(Pt 1):103–119. [PubMed] [Google Scholar]
- Pacak K, Eisenhofer G, Ahlman H, Bornstein SR, Gimenez-Roqueplo AP, Grossman AB, Kimura N, Mannelli M, McNicol AM, Tischler AS. Nat Clin Pract Endocrinol Metab; Pheochromocytoma: recommendations for clinical practice from the First International Symposium; October. 2005; 2007. pp. 92–102. [DOI] [PubMed] [Google Scholar]
- Pasini B, McWhinney SR, Bei T, Matyakhina L, Stergiopoulos S, Muchow M, Boikos SA, Ferrando B, Pacak K, Assie G, et al. Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur J Hum Genet. 2008;16:79–88. doi: 10.1038/sj.ejhg.5201904. [DOI] [PubMed] [Google Scholar]
- Prodanov T, Havekes B, Nathanson KL, Adams KT, Pacak K. Malignant paraganglioma associated with succinate dehydrogenase subunit B in an 8-year-old child: the age of first screening? Pediatr Nephrol. 2009;24:1239–1242. doi: 10.1007/s00467-008-1111-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solis DC, Burnichon N, Timmers HJ, Raygada MJ, Kozupa A, Merino MJ, Makey D, Adams KT, Venisse A, Gimenez-Roqueplo AP, et al. Penetrance and clinical consequences of a gross SDHB deletion in a large family. Clin Genet. 2009;75:354–363. doi: 10.1111/j.1399-0004.2009.01157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Maker VK. Organs of Zuckerkandl: their surgical significance and a review of a century of literature. Am J Surg. 2006;192:224–234. doi: 10.1016/j.amjsurg.2006.02.018. [DOI] [PubMed] [Google Scholar]
- Timmers HJ, Kozupa A, Eisenhofer G, Raygada M, Adams KT, Solis D, Lenders JW, Pacak K. Clinical presentations, biochemical phenotypes, and genotype-phenotype correlations in patients with succinate dehydrogenase subunit B-associated pheochromocytomas and paragangliomas. J Clin Endocrinol Metab. 2007;92:779–786. doi: 10.1210/jc.2006-2315. [DOI] [PubMed] [Google Scholar]
- Timmers HJ, Pacak K, Bertherat J, Lenders JW, Duet M, Eisenhofer G, Stratakis CA, Niccoli-Sire P, Tran BH, Burnichon N, et al. Mutations associated with succinate dehydrogenase D-related malignant paragangliomas. Clin Endocrinol (Oxf) 2008;68:561–566. doi: 10.1111/j.1365-2265.2007.03086.x. [DOI] [PubMed] [Google Scholar]