

Abstract
Alzheimer's disease (AD)-linked presenilin (PS) mutations result in pronounced endoplasmic reticulum calcium disruptions that occur before detectable histopathology and cognitive deficits. More subtly, these early AD-linked calcium alterations also reset neurophysiological homeostasis, such that calcium-dependent presynaptic and postsynaptic signaling appear functionally normal yet are actually operating under aberrant calcium signaling systems. In these 3xTg-AD mouse brains, upregulated ryanodine receptor (RyR) activity is associated with a shift toward synaptic depression, likely through a reduction in presynaptic vesicle stores and increased postsynaptic outward currents through small-conductance calcium-activated potassium SK2 channels. The deviant RyR–calcium involvement in the 3xTg-AD mice also compensates for an intrinsic predisposition for hippocampal long-term depression (LTD) and reduced long-term potentiation (LTP). In this study, we detail the impact of disrupted RyR-mediated calcium stores on synaptic transmission properties, LTD, and calcium-activated membrane channels of hippocampal CA1 pyramidal neurons in presymptomatic 3xTg-AD mice. Using electrophysiological recordings in young 3xTg-AD and nontransgenic (NonTg) hippocampal slices, we show that increased RyR-evoked calcium release in 3xTg-AD mice “normalizes” an altered synaptic transmission system operating under a shifted homeostatic state that is not present in NonTg mice. In the process, we uncover compensatory signaling mechanisms recruited early in the disease process that counterbalance the disrupted RyR–calcium dynamics, namely increases in presynaptic spontaneous vesicle release, altered probability of vesicle release, and upregulated postsynaptic SK channel activity. Because AD is increasingly recognized as a “synaptic disease,” calcium-mediated signaling alterations may serve as a proximal trigger for the synaptic degradation driving the cognitive loss in AD.
Introduction
Endoplasmic reticulum (ER) calcium signaling maintains synaptic function by regulating neurotransmission, membrane excitability and synaptic plasticity (Emptage et al., 2001; Bouchard et al., 2003; Stutzmann et al., 2003; Ross et al., 2005; Raymond and Redman, 2006; Watanabe et al., 2006). Not surprisingly, ER calcium signaling impairments are implicated in many neurodegenerative diseases involving memory loss, including Alzheimer's disease (AD) (LaFerla, 2002; Stutzmann, 2007; Bezprozvanny and Mattson, 2008; Foskett, 2010). For instance, AD-linked presenilin (PS) mutations markedly increase ER calcium release, resulting in altered presynaptic and postsynaptic synaptic transmission mechanisms (Parent et al., 1999; Chakroborty et al., 2009; Zhang et al., 2009; Goussakov et al., 2010), intrinsic membrane properties (Stutzmann et al., 2004, 2006), and cytoplasmic and nuclear signaling cascades (Schapansky et al., 2007; Müller et al., 2011). Both ryanodine receptor (RyR) and IP3R are involved, with IP3R having localized effects in the soma, and RyR exerting a stronger influence within dendrites and presynaptic terminals (Smith et al., 2005; Cheung et al., 2008, 2010; Rybalchenko et al., 2008). Describing the effects of calcium dysregulation within synaptic compartments, such as presynaptic terminals and dendritic spine heads, is particularly important for understanding AD pathology, because it is the degree of synaptic dysfunction that best correlates with the devastating memory loss in AD (Selkoe, 2002; Scheff and Price, 2003; Gylys et al., 2004; Scheff et al., 2006). This relationship makes sense because synapses are the site of calcium-dependent synaptic plasticity, which serves to encode learning and memory functions (Bliss and Collingridge, 1993; Martin et al., 2000; Whitlock et al., 2006).
Although many studies have demonstrated overt impairments in synaptic plasticity coincident with amyloid deposition (Nalbantoglu et al., 1997; Chapman et al., 1999; Oddo et al., 2003; Selkoe, 2008), we and others have shown there are pronounced neuronal signaling deficits that operate “below the radar” until ER calcium stores are specifically probed (Stutzmann, 2007; Müller et al., 2011). For example, basal synaptic function and plasticity mechanisms appear similar between young nontransgenic (NonTg) and 3xTg-AD mice, yet when RyRs are manipulated, striking synaptic transmission and plasticity aberrations are revealed in the 3xTg-AD mice, whereas NonTg mice exhibit no observable effects. This is in large part attributable to an increase in calcium-induced calcium release (CICR) via RyR (Chakroborty et al., 2009; Goussakov et al., 2010, 2011). Thus, in 3xTg-AD mouse brains, compensatory mechanisms mask these early deficits and maintain a normal physiological phenotype. However, sustaining synaptic homeostasis and compensating for intracellular calcium dysregulation concurrently can likely compromise neuronal function in the long term.
In this study, we examine presynaptic and postsynaptic mechanisms that serve to initially sustain a “normal” neurophysiology phenotype in young 3xTg-AD mice but ultimately may accelerate the synaptic pathophysiology evident at later disease stages. This is manifested as increased postsynaptic small-conductance calcium-activated potassium SK2 channel function and increased presynaptic spontaneous vesicle release, both of which likely contribute to synaptic depression and increased long-term depression (LTD). Overall, we are suggesting that subtle but insidious calcium-mediated pathogenic mechanisms can exist before amyloid and tau pathology and are a proximal contributor to synaptic signaling dysfunction in AD.
Materials and Methods
Transgenic mice.
Six- to 8-week-old triple transgenic (3xTg-AD) mice and age-matched NonTg controls (males and females) were used in all experiments. The 3xTg-AD (APPswe, TauP301L, and PS1M146VKI) mice are described by Oddo et al. (2003). NonTg control mice are on the same background strain (C57BL/6/J29). We chose the 3xTg-AD mouse model for several reasons. First, the deficits in PS function and the associated calcium dysregulations can be applicable to both familial and sporadic AD (Takami et al., 1997). Second, studies comparing mutant PS, PS1/APP, APP/Tau, and 3xTg-AD mouse models indicate that the exaggerated ER calcium signaling at young ages is associated with mutant PS1, whereas mutant APP and Tau do not significantly affect ER calcium signals at these early stages. These studies also show similar synaptic phenotypes in these AD mouse models at the presymptomatic ages used here (Oddo et al., 2003; Stutzmann et al., 2006, 2007; Dewachter et al., 2008; Auffret et al., 2009; Goussakov et al., 2010).
Hippocampal slice preparation.
Transverse hippocampal slices were prepared in accordance with protocols approved by the Institutional Animal Care and Use Committee at Rosalind Franklin University. In brief, mice were deeply anesthetized with halothane and decapitated. The brains were extracted rapidly and transverse hippocampal slices (400 μm for extracellular field potential recordings and 300 μm for patch-clamp experiments) were cut with a vibrating microtome (Campden Instruments) into ice-cold oxygenated artificial CSF (aCSF) with the following composition (in mm): 125 NaCl, 2.5 KCl, 1.25 KH2PO4, 1.2 MgSO4, 2 CaCl2, 10 dextrose, and 25 NaHCO3 (Chakroborty et al., 2009). Intact slices were placed in a holding chamber containing aCSF at room temperature (27°C) and oxygenated with 95% O2/5% CO2.
Extracellular field potential recordings and drug treatment protocols.
For extracellular field potential recording, slices were transferred to an interface chamber (Harvard Apparatus), perfused with oxygenated aCSF (1.5 ml/min) at room temperature (27°C), and covered with a continuous flow of humidified gas (95% O2/5% CO2). Data were acquired at 10 kHz using pClamp 9.2 software with an AxoClamp 2B amplifier and a DigiData 1322A board for digitization (Molecular Devices). Field EPSPs (fEPSPs) were recorded in the stratum radiatum of the CA1 subfield of the hippocampus using recording microelectrodes (2–6 MΩ) filled with aCSF. Microelectrodes were pulled from glass capillaries (Harvard Apparatus) on a P-2000 pipette puller (Sutter Instruments). Synaptic responses were evoked by stimulation of the Schaffer collateral/commissural pathway with a bipolar stimulating electrode. Baseline fEPSPs were evoked at 70% of the maximum fEPSP at 0.05 Hz for 20 min before the induction of LTD. LTD was induced at baseline intensity using a low-frequency stimulation (LFS) consisting of 900 pulses at 1 Hz. Baseline fEPSPs were recorded for 1 h at 0.05 Hz after the induction of LTD. Input/output (I/O) curves were generated using stimulus intensities from 0 to 225 μA in increments of 25 μA. Paired-pulse facilitation (PPF) was assessed using interstimulus intervals of 50 ms. Ten successive response pairs were recorded at 0.05 Hz intervals. For experiments with FK506 (calcineurin blocker, 75 μm; Tocris Bioscience) and Xestospongin B (XeB) (IP3R antagonist, 5 μm, extracted and purified from the marine sponge Xestospongia exigua as described by Jaimovich et al., 2005), slices were incubated in the drug for 2–3 h before recording. For experiments with dantrolene (RyR antagonist, 10 μm), a 20 min baseline at 0.05 Hz was recorded in aCSF, after which dantrolene was perfused continuously over the slice. The same stimulus intensity (70% of maximum fEPSP) was used to record a 20 min baseline in dantrolene, after which LTD was induced, followed by a 1 h baseline in dantrolene. For experiments with (RS)-3,5-dihydroxyphenylglycine (DHPG) (mGluR1/5 agonist, 100 μm; Tocris Bioscience), a 20 min baseline at 0.05 Hz was recorded in aCSF containing 2-amino-5-phosphonopentanoic acid (APV) (NMDAR antagonist, 50 μm, Sigma-Aldrich), after which LTD was induced by washing in DHPG for 5 min, followed by a 1 h baseline at 0.05 Hz recorded in aCSF containing APV.
For analysis, responses were measured offline using pClamp 9.2 software and analyzed using Origin Pro8 and Microsoft Excel software. For LTD recordings, fEPSP slopes recorded 40–60 min after LFS were averaged and expressed as a percentage of the average slope from 20 min baseline recordings. For PPF, fEPSP amplitudes were expressed as a ratio of the second fEPSP over the first fEPSP. For I/O curves, the first 5 ms of the fEPSP slopes were measured.
Whole-cell patch-clamp recordings.
To perform whole-cell patch-clamp recordings, slices were transferred to a perfusion chamber mounted on a movable stage assembly on a fixed upright microscope (BX51; Olympus) and perfused with oxygenated aCSF (2 ml/min) at room temperature. Data were acquired in current-clamp mode (unless otherwise stated) at 10 kHz using pClamp 10.2 software with an AxoClamp 2B amplifier and a DigiData 1440 board for digitization (Molecular Devices). Hippocampal CA1 pyramidal neurons were identified visually by infrared-differential interference contrast optics and electrophysiologically by their passive membrane properties. Whole-cell patch recordings were performed from CA1 pyramidal neurons of the hippocampus using recording microelectrodes (4–6 MΩ) filled with the following solution (in mm): 135 K-methylsulfonate, 10 HEPES, 10 Na-phosphocreatine, 2 MgCl2, 4 Na-ATP, and 0.4 Na-GTP, pH adjusted to 7.3–7.4 with KOH. Microelectrodes were pulled from glass capillaries (Harvard Apparatus) on a P-87 pipette puller (Sutter Instruments). Synaptic responses were evoked by stimulation of the Schaffer collateral/commissural pathway with a monopolar stimulating electrode. Series resistance was monitored throughout the experiment and was <10 MΩ. The current–voltage relationship was assessed by measuring voltage responses to constant current pulses of 500 ms duration, varying in amplitude from −200 to +200 pA in steps of 20 pA. Spontaneous postsynaptic potentials were recorded for 1 min. To confirm that the spontaneous postsynaptic miniature potentials we recorded did not reflect action-potential-generated vesicle release, we also recorded spontaneous events with TTX (1 μm; Sigma-Aldrich) in the bath solutions. We compared amplitude and frequency of potentials with and without TTX and found no significant differences between them in either the NonTg or 3xTg-AD slices (NonTg: frequency, t(1,3) = 2.09, p = 0.13; amplitude, t(1,3) = 1.37, p = 0.26; 3xTg-AD: frequency, t(1,3) = 0.082, p = 0.94; amplitude, t(1,3) = 0.49, p = 0.67; n = 4 for both). The lack of a TTX effect likely reflects an absence, or low degree of, spontaneous action potentials at this synapse in the slice preparation. For clarity, the data shown include only non-TTX conditions. PPF was assessed using interstimulus intervals of 25, 50, 100, 200, 500, and 1000 ms. Five successive responses were recorded for each interstimulus interval at 0.05 Hz. Apamin-sensitive medium afterhyperpolarization (mAHP) current was measured in voltage-clamp mode at −55 mV using either a single depolarizing pulse for 100 ms or 10 depolarizing pulses at 70 Hz. Five successive response pairs at 0.05 Hz intervals were recorded for each interstimulus interval. Experiments were performed with dantrolene (RyR antagonist, 10 μm) or apamin (SK channel antagonist, 1 μm; Sigma-Aldrich) in the bath solution. Responses were measured using pClamp 10.2 and further analyzed using Origin Pro8 and Microsoft Excel. The amplitude of mAHP current was measured 50 ms after the offset of the depolarizing pulse for the single-pulse protocol and 50 ms after the offset of the 10th depolarizing pulse for the 10-pulse protocol. Spontaneous postsynaptic potentials were analyzed offline using MiniAnalysis software (version 6.0.9; Synaptosoft), and cumulative probability histograms were generated to compare amplitudes and frequencies.
Immunoblot analysis.
Hippocampal tissue was harvested from 6- to 8-week-old 3xTg-AD and NonTg mice (n = 6 each). Tissue was homogenized on ice in Tissue Protein Extraction Reagent (Invitrogen) containing protease inhibitors (Roche). Total hippocampal protein was quantified and separated by SDS-PAGE on 3–8% Tris-acetate NuPAGE gradient gels (Invitrogen). Protein was transferred onto polyvinylidene difluoride membranes (Hybond-P; GE Healthcare) at 30 V for 2 h under reducing conditions. Membranes were blocked with 5% nonfat milk in TBS for 1 h at room temperature. Rabbit anti-KCNN2 (29 kDa; short form) and rabbit anti-β-tubulin primary antibodies were diluted 1:1000 in 2.5% nonfat milk and applied to the respective half of the membranes for 72 h at 4°C with shaking. Rabbit anti-KCNN2 (60 kDa; long form) was diluted 1:1000 and applied for 48 h after azide quench. HRP-conjugated goat secondary antibodies were applied for 1 h at room temperature. All antibodies were obtained from Abcam. KCNN2 (short and long forms) density levels are represented relative to β-tubulin. Images were acquired using the VersaDoc Gel imaging system and quantified using NIH ImageJ software.
Data analysis and statistics.
Data are expressed as mean ± SEM and assessed for significance using Student's two-tailed t test or paired t test, one-way or two-way ANOVA with Scheffé's post hoc analysis, where n denotes the number of slices examined in extracellular field experiments and number of neurons in patch-clamp experiments. To assess for statistical significance in cumulative probability histograms for spontaneous potentials, the nonparametric Kolmogorov–Smirnov (K–S) test was performed with Origin Pro8 software.
Results
The rationale driving this study stems from previous work demonstrating abnormal and potentially disruptive aberrations in calcium-regulated synaptic transmission mechanisms. For example, our previous studies in young presymptomatic 3xTg-AD mice demonstrated a significantly reduced threshold for inducing RyR-mediated CICR during basal synaptic transmission, exaggerated RyR-evoked calcium release in dendrites and dendritic spine heads, and enhanced basal neurotransmission and altered short- and long-term plasticity with RyR block (Chakroborty et al., 2009; Goussakov et al., 2010, 2011). These data led to questions regarding possible underlying shifts toward synaptic depression resulting from aberrant RyR-mediated calcium signaling. The phenomenon of LTD is little studied in AD, and here we seek to identify alterations in synaptic depression and investigate contributing factors in an effort to define deficits in synaptic function in early AD.
Blocking RyR-mediated calcium release reveals shift to synaptic depression
Because RyR-mediated CICR is necessary for the induction of long-term plasticity (Obenaus et al., 1989; Alford et al., 1993; Harvey and Collingridge, 1993; Reyes and Stanton, 1996), we further explored the consequences of dysregulated ER calcium signaling on synaptic physiology in young 3xTg-AD mice by examining NMDAR-dependent LTD induced by a LFS (see Materials and Methods). Under control conditions LTD appeared similar between both 3xTg-AD (n = 7, 40.4 ± 1.2% decrease) and NonTg (n = 5, 40.5 ± 1.1% decrease) mice (p > 0.05; Fig. 1A), as did basal synaptic transmission as measured with I/O functions and short-term presynaptic plasticity as measured by PPF ratios (3xTg-AD: n = 19, 13, respectively; NonTg: n = 12, 9 respectively; p > 0.05; data not shown). This I/O and PPF phenotype is consistent with our previous observations (Chakroborty et al., 2009).
Figure 1.

RyR blockade enhances hippocampal LTD at the Schaffer collateral synapse in 3xTg-AD mice. A, Left, Graph shows averaged time course of LTD from NonTg and 3xTg-AD mice recorded in standard aCSF. Inset (above) shows representative traces from baseline before (1) and after (2) LFS in the NonTg (black) and 3xTg-AD (gray) mice. Right, Bar graph shows percentage change in post-LFS baseline relative to pre-LFS baseline from NonTg and 3xTg-AD mice. B, Left, Graph shows averaged time course of LTD from NonTg and 3xTg-AD mice recorded in 10 μm dantrolene. Inset (above) shows representative traces from baseline in standard aCSF (1), baseline in dantrolene (2), and after LFS (3) in the NonTg (black) and 3xTg-AD (gray) mice. Right, Bar graph shows percentage change in post-LFS baseline relative to pre-LFS dantrolene baseline from NonTg and 3xTg-AD mice. Baseline fEPSPs were recorded at 0.05 Hz for 20 min before and 60 min after induction of LTD by LFS (900 pulses at 1 Hz denoted by arrow). *p < 0.05, significantly different from NonTg.
However, when RyRs were blocked with 10 μm dantrolene, different signaling patterns emerged, suggesting that 3xTg-AD mice use alternative signaling mechanisms distinct from RyR pathways. In NonTg mice, dantrolene had no effect on I/O or PPF functions (n = 6, p > 0.05; data not shown; Chakroborty et al., 2009) but significantly increased these functions in 3xTg-AD mice (n = 5, t(1,4) = −5.3; t(1,4) = −11.4, respectively; p < 0.05; data not shown; Chakroborty et al., 2009; Tables 1, 2). We next compared LTD against both a control and a drug treatment baseline. Here, dantrolene abolished LTD in NonTg mice (n = 9, p > 0.05) but generated very different responses in 3xTg-AD mice. Dantrolene increased baseline responses by 50.8 ± 1.4% (n = 5, t(1,4) = 22.6; p < 0.05) and further enhanced LTD to 72.5 ± 1.2% below dantrolene baseline and 58.5 ± 1.2% below control aCSF baseline (Fig. 1B).
Table 1.
Effects of drug treatments on basal synaptic transmission (I/O functions) in NonTg Vs 3xTg-AD mice (compared with effect in control aCSF before drug)
| Treatment | NonTg | 3xTg-AD |
|---|---|---|
| Dantrolene | No change | Significantly increased |
| FK506 | No change | No change |
| FK506 + Dantrolene | No change | Significantly increased |
| XeB | No change | No change |
| XeB + Dantrolene | No change | Significantly increased |
Table 2.
Effects of drug treatments on presynaptic plasticity (PPF) in NonTg Vs 3xTg-AD mice (compared with effect in control aCSF before drug)
| Treatment | NonTg | 3xTg-AD |
|---|---|---|
| Dantrolene | No change | Significantly increased |
| FK506 | No change | No change |
| FK506 + Dantrolene | No change | Significantly increased |
| XeB | No change | No change |
| XeB + Dantrolene | No change | Significantly increased |
Possible compensatory mechanisms in 3xTg-AD mice
We next explored calcium-dependent signaling cascades that could be recruited as an alternative mechanism to support LTD. We first examined whether changes in calcineurin function may serve as an underlying factor in the 3xTg-AD responses. Calcineurin is a calcium-dependent protein phosphatase that contributes to NMDAR-dependent LTD by dephosphorylating AMPA receptor subunits and facilitating their internalization and removal from the synapse. To investigate whether calcineurin is serving distinct functions in the 3xTg-AD mice and thereby underlying the divergent effects on RyR antagonism, we blocked calcineurin activity with 75 μm FK506 and applied the same synaptic stimulus protocols as described in Figure 1. Blocking calcineurin did not affect I/O functions (p > 0.05; Fig. 2A) or PPF (p > 0.05; Fig. 2D) in either NonTg (n = 12, 16 respectively) or 3xTg-AD (n = 11, 14 respectively) mice. FK506 abolished LTD in both NonTg mice (n = 9), consistent with existing literature (Mulkey and Malenka, 1992; Mulkey et al., 1994), and in the 3xTg-AD mice (n = 7; Fig. 2G). Although FK506 can modulate RyR (Brillantes et al., 1994; Xiao et al., 1997; Ozawa, 2008), what effects it may have on murine neuronal RyR are unclear. In the present experiments, however, FK506 did not differentially affect synaptic transmission between the NonTg and 3xTg-AD hippocampal neurons, suggesting the FK506– RyR interactions are not different between the NonTg and 3xTg-AD strains.
Figure 2.

RyR blockade alters presynaptic and postsynaptic functions and reveals an alternative form of LFS–LTD in 3xTg-AD mice. A–C, I/O function shows changes in fEPSP slope with increasing stimulus intensity (0–225 μA) from the following: A, NonTg and 3xTg-AD mice with 75 μm FK506; B, NonTg mice in FK506 before and after 10 μm dantrolene (Dant) treatment; and C, 3xTg-AD mice in FK506 before and after dantrolene treatment. Insets A–C show representative fEPSP traces from NonTg and 3xTg-AD mice for each condition. D–F, Bar graphs show paired-pulse ratio from the following: D, NonTg and 3xTg-AD mice with FK506; E, NonTg mice with FK506 before and after treatment with 10 μm dantrolene; and F, from 3xTg-AD mice with FK506 before and after dantrolene treatment. Insets D–F show representative PPF traces from NonTg and 3xTg-AD mice for each condition. G, Left, Graph shows averaged time course of LTD (LFS, NMDA-dependent) in FK506 from NonTg and 3xTg-AD mice; insets (above) show representative fEPSP traces before LFS in FK506 (1) and after LFS in FK506 (2) from NonTg (black) and 3xTg-AD (gray) mice. Bar graphs on right show averaged percentage change in post-LFS baseline relative to pre-LFS baseline from NonTg and 3xTg-AD mice in FK506. H, Left, Graph shows averaged time course of LTD in FK506 with dantrolene from NonTg and 3xTg-AD mice. Insets (above) show representative fEPSP baseline traces in FK506 (1), baseline in FK506 and dantrolene (2), and after LFS in FK506 and dantrolene (3) from NonTg (black) and 3xTg-AD (gray) mice. Bar graphs on right show averaged percentage change in post-LFS baseline relative to pre-LFS baseline from NonTg and 3xTg-AD mice in FK506 with dantrolene. For FK506 experiments, slices were pretreated for 2 h with drug before recording. Baseline fEPSPs were recorded at 0.05 Hz for 20 min before and 60 min after induction of LTD. The arrow indicates the time of LFS. *p < 0.05, significantly different from 3xTg-AD before dantrolene treatment; **p < 0.05, significantly different from NonTg.
We next examined patterns of synaptic plasticity when both calcineurin and RyR were blocked. Coapplication of dantrolene and FK506 had little effect on basal synaptic transmission (n = 6; p > 0.05; Fig. 2B) or PPF (n = 7; p > 0.05; Fig. 2E) in NonTg mice but, consistent with RyR antagonism alone, increased both the I/O function (n = 5, t(1,4) = −6.7; p < 0.05; Fig. 2C) as well as PPF (n = 5, t(1,4) = −4.1; p < 0.05; Fig. 2F) in 3xTg-AD mice. With LTD, blocking both RyR and calcineurin in NonTg mice did not change baseline responses and abolished LTD (n = 7; p > 0.05). However, in 3xTg-AD mice, blocking both RyR and calcineurin still generated a synaptic phenotype similar to blocking RyR alone. Baseline responses increased (26.2 ± 0.8%, n = 7, t(1,6) = −19.9; p < 0.05) and modest LTD was generated (35.9 ± 0.6% relative to dantrolene/FK506 baseline and 19.2 ± 0.6% relative to aCSF baseline; Fig. 2H), although the magnitude of LTD was less than during RyR blockade alone (t(1,11) = −31.83; p < 0.05). Thus, it appears that a signaling pathway for LTD can be recruited in 3xTg-AD mice that does not require calcineurin or RyR activity. We therefore examined alternative signaling cascades, such as the following mGluR-mediated mechanism for LTD.
The role of IP3R in basal neurotransmission and plasticity is similar between NonTg and 3xTg-AD mice
Because there is some functional overlap between RyR and IP3R in plasticity mechanisms (Fitzjohn and Collingridge, 2002), we examined whether aberrant interactions exist between these ER calcium channels in 3xTg-AD mice. Blocking IP3R with 5 μm XeB had little effect on I/O or PPF functions (p > 0.05; Fig. 3A,D) in NonTg (n = 16, 19, respectively) or 3xTg-AD (n = 13, 17, respectively) mice, and there were no differences between the NonTg and 3xTg-AD responses (p > 0.05; data not shown). Administering the LFS while blocking IP3R resulted in a polarity shift from LTD to long-term potentiation (LTP) in both NonTg (n = 6, 134.19 ± 2.6% over baseline) and 3xTg-AD (n = 9, 100.4 ± 3.4% over baseline) mice (Fig. 3G), although the degree of LTP is significantly less in 3xTg-AD mice (t(1,14) = 7.8; p < 0.05). This is consistent with previous observations demonstrating that IP3R-mediated calcium release suppresses LTP (Fujii et al., 2000; Taufiq et al., 2005).
Figure 3.
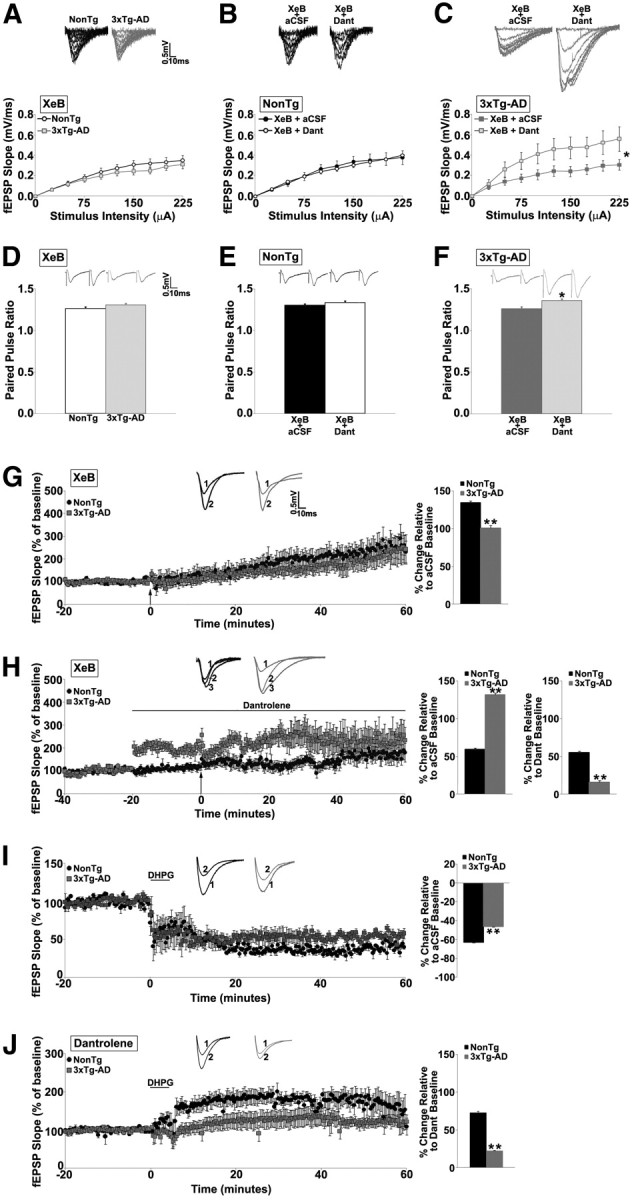
Blunted IP3R-dependent LTD expression in 3xTg-AD mice. A–C, I/O function shows changes in fEPSP slope with increasing stimulus intensity (0–225 μA) from the following: A, NonTg and 3xTg-AD mice with 5 μm XeB; B, NonTg mice in XeB before and after 10 μm dantrolene (Dant) treatment; and C, 3xTg-AD mice in XeB before and after dantrolene treatment. Insets A–C show representative fEPSP traces from NonTg and 3xTg-AD mice for each condition. D–F, Bar graphs show paired-pulse ratio from the following: D, NonTg and 3xTg-AD mice with 5 μm XeB; E, NonTg mice with XeB before and after treatment with dantrolene; and F, from 3xTg-AD mice with XeB before and after dantrolene treatment. Insets D–F show representative PPF traces from NonTg and 3xTg-AD mice for each condition. G, Left, Graph shows averaged time course of LTD (LFS) in XeB from NonTg and 3xTg-AD mice; insets (above) show representative fEPSP traces before LFS in XeB (1) and after LFS in XeB (2) from NonTg (black) and 3xTg-AD (gray) mice. Bar graphs on right show averaged percentage change in post-LFS baseline relative to pre-LFS baseline from NonTg and 3xTg-AD mice in XeB. H, Left, Graph shows averaged time course of LTD in XeB with dantrolene from NonTg and 3xTg-AD mice. Insets (above) show representative fEPSP baseline traces in XeB (1), baseline in XeB and dantrolene (2), and after LFS in XeB and dantrolene (3) from NonTg (black) and 3xTg-AD (gray) mice. Bar graphs on right show averaged percentage change in post-LFS baseline relative to pre-LFS baseline from NonTg and 3xTg-AD mice in XeB with dantrolene. I, Left, Graph shows averaged time course of LTD (chemical induction, mGluR-dependent) in 100 μm DHPG from NonTg and 3xTg-AD mice; insets (above) show representative fEPSP traces before (1) and after (2) DHPG from NonTg (black) and 3xTg-AD (gray) mice. Bar graphs on right show averaged percentage change in post-DHPG baseline relative to pre-DHPG baseline from NonTg and 3xTg-AD mice. J, Left, Graph shows averaged time course of LTD in DHPG with dantrolene from NonTg and 3xTg-AD mice. Insets (above) show representative fEPSP baseline traces in dantrolene, before (1) and after (2) DHPG from NonTg (black) and 3xTg-AD (gray) mice. Bar graphs on right show averaged percentage change in post-DHPG baseline relative to pre-DHPG baseline from NonTg and 3xTg-AD mice in dantrolene. For XeB experiments, slices were pretreated for 2 h with XeB before recording. Baseline fEPSPs were recorded for 20 min at 0.05 Hz before and for 60 min at 0.05 Hz after induction of LTD. The arrow indicates the time of LFS. For DHPG experiments, slices were treated with DHPG for 5 min after baseline fEPSPs were recorded. DHPG experiments were done in the presence of APV to block NMDAR-dependent LTD. *p < 0.05, significantly different from 3xTg-AD before dantrolene treatment; **p < 0.05, significantly different from NonTg.
When blocking both IP3R and RyR, I/O (n = 10; p > 0.05; Fig. 3B) and PPF (n = 13; p > 0.05; Fig. 3E) functions were not altered in NonTg mice. However, in 3xTg-AD mice, I/O (n = 8, t(1,7) = −6.0; p < 0.05; Fig. 3C) and PPF (n = 10, t(1,9) = −3.6; p < 0.05; Fig. 3F) increased, similar to effects observed with RyR block alone. Blocking RyR and IP3R while administering the LFS resulted in divergent effects on plasticity expression (Fig. 3H). In NonTg mice, baseline responses were not altered (n = 8; p > 0.05) and a polarity shift to LTP was still observed (69.7 ± 1.8% over aCSF baseline and 67.1 ± 1.8% over dantrolene/XeB baseline). In contrast, in 3xTg-AD mice, blocking both channels greatly increased baseline responses (98.0 ± 2.0%, t(1,8) = −39.1; p < 0.05) and blocked LTP relative to drug baseline (p > 0.05). Because evoked baseline responses are increased in the presence of dantrolene, likely because of the removal of SK2 channel activity (Chakroborty et al., 2009), this may reflect a “ceiling effect” of the postsynaptic response.
To identify RyR-independent mechanisms that could account for the enhanced LTD observed during RyR blockade in 3xTg-AD mice, we next examined mGluR-dependent LTD, which invokes Gq activation and subsequent IP3 generation (Oliet et al., 1997). We stimulated the mGlu receptors with 100 μm DHPG while simultaneously inhibiting NMDAR–LTD with APV (see Materials and Methods) (Palmer et al., 1997; Huber et al., 2001). These conditions generated a robust mGluR–LTD in both NonTg (n = 5, 62.6 ± 0.6% decrease) and 3xTg-AD mice (n = 5, 46.0 ± 0.5% decrease), with LTD magnitude being modestly but significantly lower in 3xTg-AD mice (t(1,9) = −20.0; p < 0.05; Fig. 3I). The possible role of RyR in mGluR–LTD has not been examined previously, and it is feasible that the increased CICR in the 3xTg-AD mice may result in aberrant recruitment of RyR-sensitive calcium stores in mGluR–LTD. To test this hypothesis, we stimulated the mGluR while simultaneously inhibiting NMDAR and RyR (with APV and dantrolene, respectively). Interestingly, this treatment resulted in a polarity shift from LTD to LTP in both NonTg (n = 5, 72.2 ± 2.0% over baseline) and 3xTg-AD (n = 6, 21.9 ± 0.9% over dantrolene baseline) mice, although LTP was still relatively blunted in the 3xTg-AD mice compared with NonTg (t(1,10) = 22.93; p < 0.05; Fig. 3J). These intriguing findings suggest that RyR-sensitive calcium stores make an active contribution to mGluR–LTD and serve to suppress synaptic potentiation. This observation has not been reported previously and appears analogous to the phenomenon of IP3R activation suppressing synaptic potentiation (Fujii et al., 2000; Taufiq et al., 2005). The polarity shift to LTP during RyR blockade in mGluR–LTD is similar in the NonTg and 3xTg-AD mice, albeit the magnitude continues to be lower in the AD mice, suggesting that separate signaling pathways are not recruited in the mouse strains. However, in the 3xTg-AD mice, IP3-regulated signaling pathways may have a lesser influence in mediating synaptic plasticity, perhaps in response to the upregulation of RyR function. The blunted LTD and LTP, induced by DHPG and DHPG + dantrolene, respectively, in the 3xTg-AD mice, may also reflect a partial occlusion of plasticity mechanisms and impaired synaptic scaling (see below). This hypothesis is based on studies in 3xTg-AD neurons and mutant PS-expressing cells demonstrating altered IP3R gating properties, resulting in increased Po and reduced threshold for IP3-evoked responses, such that endogenous IP3 levels can be sufficient to trigger calcium release at the single-channel level. This ER calcium leak constitutively upregulates kinases and signaling cascades (e.g., CaMKIV) important for plasticity encoding (Cheung et al., 2008, 2010; Müller et al., 2011). Because these neurons are already operating under increased calcium-regulated kinase activity in their resting state, subsequent IP3-mediated signaling cascades will not generate the same magnitude of plasticity. Similar plasticity occlusion effects have been observed in neuronal circuits in which mGluR-mediated signaling cascades are upregulated (McCutcheon et al., 2011).
Presynaptic modifications in CA3 terminals in 3xTg-AD mice
RyR-mediated CICR can facilitate spontaneous neurotransmitter vesicle release from presynaptic terminals and alter their release probability and PPF functions (Emptage et al., 1999; Llano et al., 2000; Bardo et al., 2002). Spontaneous neurotransmission is a common property of all synapses (Katz, 1969) and is involved in the maturation and stabilization of synaptic connections (McKinney et al., 1999; Verhage et al., 2000), the modulation of protein synthesis in dendrites (Sutton et al., 2004), and driving action potentials (Carter and Regehr, 2002). Spontaneous release events also have the ability to set the concentration of neurotransmitter within the synaptic cleft and thereby shape postsynaptic responses (Wasser and Kavalali, 2009). Here, we examined whether altered ER calcium release affects spontaneous vesicle release properties and calcium-dependent short-term plasticity in CA3 terminals. Using whole-cell patch-clamp recordings, we compared the amplitude and frequency of spontaneous events in control aCSF and dantrolene from NonTg (n = 13; Fig. 4A) and 3xTg-AD (n = 12; Fig. 4B) CA1 pyramidal neurons.
Figure 4.

Effects of RyR blockade on spontaneous postsynaptic potentials. A, B, Representative traces of spontaneous postsynaptic potentials recorded from NonTg (A) and 3xTg-AD (B) CA1 pyramidal neurons in aCSF (top) and 10 μm dantrolene (bottom). C, D, Cumulative probability histograms showing interevent interval (C) and amplitude of spontaneous postsynaptic potentials (D) from NonTg and 3xTg-AD neurons with 10 μm dantrolene. *p < 0.05, significantly different from 3xTg-AD before dantrolene treatment.
In control aCSF, the frequency distribution of spontaneous EPSPs in 3xTg-AD neurons was significantly different than NonTg neurons, indicative of a higher release frequency (K–S test; p < 0.05 for 3xTg-AD/aCSF vs NonTg/aCSF; Fig. 4C). RyR block normalized the frequency of spontaneous events in 3xTg-AD neurons (p < 0.05 for 3xTg-AD/aCSF vs 3xTg-AD/dantrolene), with little effect in NonTg neurons (p > 0.05). Our findings are consistent with recent observations that chronic ER stress, a common feature in 3xTg-AD and other AD mouse models (Katayama et al., 2001; Salminen et al., 2009; Quiroz-Baez et al., 2011; Stutzmann and Mattson, 2011), increases the sensitivity of presynaptic calcium sensors associated with spontaneous neurotransmitter release (Nosyreva and Kavalali, 2010). In addition, although evoked and spontaneous vesicle release involves calcium sensors, there can be distinct mechanisms and binding affinities supporting each. For example, the synaptotagmin-1 calcium-binding domains which support spontaneous vesicle release are different from the binding domains required for evoked release (Xu et al., 2009), whereas the doc2b and synaptotagmin-12 calcium sensors specifically mediate spontaneous release and are not recruited for evoked vesicle release (Maximov et al., 2007; Groffen et al., 2010). The SNARE complex assembly also appears to more stringently regulate evoked release over spontaneous release in a calcium-dependent manner (Deák et al., 2006). Any of these mechanisms may become increasingly sensitized to calcium in 3xTg-AD neurons. Thus, increased calcium sensitivity of spontaneous release machinery coupled with the aberrant CICR and ER stress in 3xTg-AD neurons can substantially increase the frequency of spontaneous events and render neurons more vulnerable to excitotoxicity and synaptic pathology (Carter and Regehr, 2002; Cavelier and Attwell, 2005; Wasser and Kavalali, 2009).
In our studies, PPF measured across a range of interstimulus intervals (25–1000 ms) suggests there are mechanisms compensating for the increased probability of vesicle release (Fig. 5). In aCSF, PPF trended lower in 3xTg-AD neurons when compared with NonTg neurons but does not achieve statistical significance (p > 0.05 for all interstimulus intervals; n = 10 for both). However, blocking the RyR increases PPF in 3xTg-AD neurons relative to NonTg, consistent with a reduction in release probability, particularly at longer interstimulus intervals (p < 0.05 for 50, 200, and 1000 ms). The amplitude of spontaneous EPSCs was also significantly increased in 3xTg-AD neurons on RyR block relative to 3xTg-AD responses in aCSF and NonTg responses in aCSF and dantrolene (K–S test; p < 0.05; Fig. 4D). This may reflect, in part, increased membrane input resistance when RyRs are blocked in 3xTg-AD neurons (see below), as well as compensatory synaptic scaling events (Pratt et al., 2011) and depletion of synaptic vesicles. Because the number of vesicles in the readily releasable pool is limited, the availability of synaptic vesicles for exocytosis becomes the limiting step during action potential firing, which can then lead to synaptic depression if vesicles are rapidly depleted (Schneggenburger et al., 2002; Fioravante and Regehr, 2011). This depletion would likely be accelerated in the 3xTg-AD neurons because of the increased RyR-calcium tone and increased spontaneous vesicle release.
Figure 5.

Increased RyR-mediated calcium tone alters PPF properties in 3xTg-AD neurons. A, B, PPF was measured at interstimulus intervals of 25, 50, 200, and 1000 ms. Bar graph shows paired-pulse ratio from NonTg (A) and 3xTg-AD (B) neurons before and after treatment with 10 μm dantrolene at 25, 50, 200, and 1000 ms. Inset, Representative PPF traces from NonTg and 3xTg-AD neurons with dantrolene at 25, 50, 200, and 1000 ms. *p < 0.05, significantly different from 3xTg-AD before dantrolene treatment.
Postsynaptic SK channel activity is increased in CA1 neurons in 3xTg-AD mice
The SK channels underlie the mAHP and modulate neuronal excitability by limiting firing frequency (Sah, 1996; Stocker et al., 1999; Bond et al., 1999; Sah and Davies, 2000). RyR-evoked calcium release can contribute to the activation of the mAHP (Sah, 1996; Pineda et al., 1999; Shah and Haylett, 2000; Stutzmann et al., 2003; van de Vrede et al., 2007), and previous studies suggest an enhanced coupling efficiency between the SK channels and RyR in AD mice (Stutzmann et al., 2006). Thus, we examined whether dysregulated RyR calcium signaling altered postsynaptic SK channel function in 3xTg-AD CA1 neurons. Under basal conditions, 3xTg-AD neurons (n = 15) had a significantly greater mAHP current amplitude than NonTg neurons (n = 12) in response to a single depolarizing voltage step (t(1,26) = −3.5; p < 0.05; Fig. 6A), as well as a train of depolarizing voltage steps (t(1,26) = −2.3; p < 0.05; Fig. 6B). RyR block normalized the mAHP amplitude in 3xTg-AD neurons to NonTg levels (F(3,26) = 6.6; p < 0.05, Scheffé's post hoc analysis, p < 0.05 for 3xTg-AD/aCSF vs 3xTg-AD/dantrolene, NonTg/aCSF, and NonTg/dantrolene). Apamin reduced the mAHP current in both NonTg (t(1,11) = 6.1; p < 0.05) and 3xTg-AD (t(1,14) = 6.4; p < 0.05) neurons, confirming involvement of the SK2 channel (Fig. 6C). Notably, there were no significant differences in hippocampal SK2 channel expression levels, for either the long form (60 kDa) or short form (29 kDa), between NonTg and 3xTg-AD as measured by Western blot analysis (p > 0.05; Fig. 6D).
Figure 6.

Increased SK channel activity in 3xTg-AD neurons. A, B, Left, Bar graph showing mAHP current amplitude in response to a single depolarizing pulse (A) and 10 depolarizing pulses (B) from NonTg and 3xTg-AD CA1 pyramidal neurons before and after treatment with 10 μm dantrolene (Dant). Right, Representative traces of mAHP current from NonTg and 3xTg-AD neurons before and after treatment with dantrolene. C, Left, Bar graph showing mAHP current amplitude in response to a single depolarizing pulse from NonTg and 3xTg-AD CA1 pyramidal neurons before and after treatment with 1 μm apamin (Apa). Right, Representative traces of mAHP current from NonTg and 3xTg-AD neurons before and after treatment with 1 μm apamin. D, Bar graphs show relative protein expression levels of short-form SK2 (SK2-S, left) and long-form SK2 (SK2-L, middle) and immunoblot images of SK2-S and SK2-L (right) from the hippocampus of NonTg and 3xTg-AD mice. Density levels are relative to β-tubulin levels. *p < 0.05, significantly different from 3xTg-AD before dantrolene treatment; **p < 0.05, significantly different from NonTg before apamin treatment; ***p < 0.05, significantly different from 3xTg-AD before apamin treatment.
To measure the impact of calcium signaling disruptions on membrane properties, we next measured peak and steady-state voltage responses to a range of injected current steps. RyR block significantly increased the amplitude of peak voltage responses to negative current injections in 3xTg-AD neurons (n = 14, t(1,13) = 3.7; p < 0.05) but had little effect on NonTg neurons (n = 15; p > 0.05; Fig. 7C). This increased voltage response in 3xTg-AD neurons could reflect the increase in membrane resistance during closure of constitutively activated SK channels, because no differences in voltage-gated calcium channel activity have been observed in presymptomatic 3xTg-AD mice in previous studies (Stutzmann et al., 2004, 2006; Goussakov et al., 2010, 2011). Steady-state voltage responses were not affected by RyR block in either NonTg (t(1,14) = 1.4; p > 0.05) or 3xTg-AD (t(1,13) = 3.2; p > 0.05) neurons (Fig. 7D). Measured at resting membrane potential (−65 mV), membrane input resistance was also significantly increased in 3xTg-AD neurons during RyR block (t(1,13) = −63.3; p < 0.05) but not in NonTg neurons (p > 0.05) (Fig. 7E).
Figure 7.

Increase in input resistance of 3xTg-AD neurons on RyR blockade. A, B, Representative voltage traces of the neuron in response to 500 ms constant-current pulses ranging from −200 to +200 pA before and after treatment with 10 μm dantrolene from NonTg and 3xTg-AD CA1 pyramidal neurons. C, Graph showing current–voltage relationship at peak response between NonTg and 3xTg-AD neurons before and after treatment with 10 μm dantrolene (Dant). D, Graph showing current–voltage relationship at steady state between NonTg and 3xTg-AD neurons before and after treatment with dantrolene. E, Population plots of membrane input resistance measured from NonTg and 3xTg-AD neurons before and after treatment with dantrolene. *p < 0.05, significantly different from 3xTg-AD before dantrolene treatment.
Discussion
AD is increasingly referred to as a “synaptic disease” because of the central role synaptic breakdown has in the devastating cognitive deficits (Selkoe, 2002; Coleman and Yao, 2003; Chong et al., 2011). Here we propose that aberrant ER calcium signaling plays an early role in AD synaptic dysfunction, before the onset of histopathology or cognitive decline. Our previous studies in presymptomatic 3xTg-AD mice demonstrate markedly reduced CICR threshold in synaptic compartments, such that basal synaptic stimulation or NMDAR activation can evoke greatly elevated ER calcium responses within presynaptic and postsynaptic compartments in which ER calcium release is typically negligible to small (Chakroborty et al., 2009; Goussakov et al., 2010, 2011). Despite this deviant calcium release under basal conditions, synaptic transmission appears normal. In this study, we uncover markedly altered synaptic transmission properties that exist below the radar that reflect a blunted or depressed hippocampal network. These early deficits may drive the synaptic breakdown associated with AD-linked cognitive impairment.
Early increases in LTD may be reversed by increased RyR levels in AD
To explore mechanisms accounting for synaptic depression in 3xTg-AD mice, we examined two prominent LTD signaling pathways: NMDAR–LTD and mGluR–LTD (Table 3). NMDAR–LTD is generated by LFS of the NMDAR and is mediated by calcineurin and RyR-evoked calcium release (Mulkey et al., 1994; Reyes and Stanton, 1996). mGluR–LTD involves mGluR1/5 activation, which generates IP3 and subsequent ER calcium release (Oliet et al., 1996). Although blocking calcineurin (also called protein phosphatase 2B) abolished LTD in both NonTg and 3xTg-AD mice, blocking both calcineurin and RyR revealed a separate LTD induction mechanism in 3xTg-AD mice that is independent of both these signaling factors. These may reflect a reduction in presynaptic vesicle stores and increased calcium-activated K+ currents, each of which are facilitated by sensitized CICR in 3xTg-AD mice. IP3R contributions to NMDAR- dependent LTD were also compared between NonTg and 3xTg-AD mice. Blocking IP3R resulted in a shift to LTP in both mouse strains, consistent with previous reports (Fujii et al., 2000; Nishiyama et al., 2000), yet the magnitude of plasticity was blunted in 3xTg-AD mice, perhaps as a result of constitutive upregulation of calcium-regulated kinases (see Results; Müller et al., 2011). Blocking both RyR and IP3R abolished LTP in 3xTg-AD mice relative to the dantrolene baseline, yet plasticity is maintained in the control mice. This may reflect an additional “ceiling effect,” because blocking RyR increases basal synaptic strength in 3xTg-AD mice by suppressing inhibitory SK2 channels (Chakroborty et al., 2009). These findings demonstrate a recalibration toward synaptic depression when RyRs are blocked in 3xTg-AD mice.
Table 3.
Effects of drug treatments on synaptic plasticity (LTD) in NonTg Vs 3xTg-AD mice
| Treatment | NonTg | 3xTg-AD |
|---|---|---|
| Dantrolene | LTD is abolished | LTD is increased |
| FK506 | LTD is abolished | LTD is abolished |
| FK506 + Dantrolene | LTD is abolished | Moderate LTD |
| XeB | LTP | LTP (less than NonTg) |
| XeB + Dantrolene | LTP | LTP (less than NonTg) |
| DHPG | LTD | LTD (less than NonTg) |
| DHPG + Dantrolene | LTP | LTP (less than NonTg) |
Increased neurotransmitter vesicle release from 3xTg-AD mice presynaptic terminals
LTD can reflect both presynaptic and postsynaptic changes in synaptic efficacy, with the former reflecting a reduction in the probability of calcium-dependent neurotransmitter release (Stanton et al., 2003; Enoki et al., 2009). Because RyR can trigger spontaneous vesicle release (Emptage et al., 1999; Llano et al., 2000; Bardo et al., 2002), the sensitized CICR responses in 3xTg-AD neurons can diminish vesicle reserves and impose long-term consequences for synaptic transmission. Under “normal” conditions, CICR mobilizes vesicles from the reserve pool to the readily releasable pool, thereby maintaining a responsive vesicle population primed for rapid release (Kuromi and Kidokoro, 2002; Zucker and Regehr, 2002); however, upregulated CICR can accelerate vesicle pool depletion and reduce release probability. Although this may be normalized initially by increasing neurotransmitter synthesis and vesicle cycling, sustaining these compensatory mechanisms can introduce metabolic stress, promoting synapse loss (Verkhratsky, 2002; Stutzmann, 2007; Mattson, 2010). Reduced vesicle reserves and cycling capacity may also underlie the homeostatic shift to LTD in 3xTg-AD mice. The effects of increased RyR calcium on facilitating vesicle release are sustained over longer intervals in 3xTg-AD mice. Residual calcium levels continue to support PPF with interstimulus intervals up to 1 s, suggesting that calcium release can expend vesicle pool contents and drive a network into a state of synaptic depression. Conversely, blocking RyR increased PPF in 3xTg-AD mice, consistent with a reduction in presynaptic calcium levels and lowered vesicle release probability.
Increased postsynaptic SK channel activity in CA1 pyramidal neurons
The calcium-activated SK2 channels underlie the mAHP in CA1 pyramidal neurons and regulate spiking frequency, membrane excitability, and plasticity (Sah, 1996; Stocker et al., 1999; Sah and Davies, 2000). Suppressing SK2 channel function facilitates memory, whereas upregulating SK2 function impairs LTP and hippocampal-dependent learning and memory (Fournier et al., 2001; Stackman et al., 2002; Hammond et al., 2006; Allen et al., 2011). Here we demonstrate that, under basal conditions, upregulated RyR function underlies increased SK2 currents in 3xTg-AD neurons, which is distinct from the activity-dependent trigger through L-type voltage-gated calcium channels. The broader implications of this phenomenon are uncovered when RyR are blocked, resulting in increased I/O functions. The recruitment of SK2 channels via RyR-mediated calcium release is further supported by apamin (an SK2-specific channel blocker), increasing the I/O function similarly to dantrolene in 3xTg-AD mice but having no effect in NonTg mice (Chakroborty et al., 2009). Although the aberrant RyR calcium release can initially serve to offset postsynaptic excitation resulting from increased neurotransmitter release, this same phenomenon can shift homeostatic set points for membrane excitability and spiking thresholds (Bear, 1995; Sah and Bekkers, 1996; Hirschberg et al., 1999). The increased SK2 currents are not attributable to increased SK2 expression; rather, because SK2 channels are expressed in CA1 dendritic spines (Allen et al., 2011), it is likely that the deviant RyR-evoked calcium is more tightly coupled to SK2 channels as a result of physical proximity.
An additional process to consider is that of homeostatic synaptic scaling, which is a calcium-dependent compensatory form of plasticity that maintains neuronal excitability within optimal ranges. Neurons adapt to sustained changes in network activity by adjusting their synaptic strengths to stabilize firing rates and neural networks coordinating complex functions, such as memory encoding. Pratt et al. (2011) uncovered that mutant PS-expressing neurons had impaired synaptic scaling responses and concluded that PS1 is required for scaling up excitatory synaptic strengths in response to a suppression of network activity but is not needed for scaling down in response to enhanced network activity or setting mEPSC amplitude. As this relates to our findings, mutant PS may impair the ability of the hippocampal network to increase responsivity and increased RyR function may be an attempt to compensate for this deficit; therefore, when RyRs are blocked, the network reverts to a depressed synaptic state. Although this idea will require additional refinement, it is consistent with our overall hypothesis that deficits in synaptic homeostasis may contribute to brain dysfunction in AD.
Implications for AD
3xTg-AD neurons, as well as other model AD systems (Stutzmann et al., 2006; Zhang et al., 2009; Cheung et al., 2010; Pratt et al., 2011), exhibit RyR-mediated calcium deficits early in AD, before β-amyloid and tau deposition and cognitive impairments. This alters synaptic transmission properties in a way that is initially difficult to detect because of the recruitment of signaling mechanisms that maintain the appearance of “normalcy” (Fig. 8). These mechanisms may initially stabilize synaptic activity, but prolonged metabolic demands and aberrant neuronal activity may ultimately stress neurotransmission and plasticity functions. For example, mutant PS upregulates RyR expression, which in turn increases calcium-dependent presynaptic neurotransmitter release as well as postsynaptic SK channel activity. Alternatively, preexisting synaptic deficits may generate depressed synaptic networks, and increased RyR expression serves as the compensatory mechanism to “stabilize” the network. As an example, RyR3 expression is upregulated in late-stage AD pathology, possibly as a neuroprotective mechanism against Aβ1–42 deposition (Supnet et al., 2010). Similar alterations in RyR expression are also observed in human brain studies. Patients with mild cognitive impairment demonstrate elevated levels of RyR2 linked with cognitive decline and synaptic loss (Kelliher et al., 1999; Bruno et al., 2011), consistent with early increases in RyR2 expression in several AD mouse models. However, regardless of fundamental cause, the early dyshomeostasis in synaptic transmission and plasticity mechanisms likely contribute to the cognitive deficits in later AD stages. For example, disruptions in LTD introduce destabilizing effects on synaptic networks. This is because LTD optimizes the flexibility and efficiency of synapses by facilitating the acquisition of new associations while rapidly suppressing old or irrelevant ones (Dayan and Willshaw, 1991; Kemp and Manahan-Vaughan, 2004). This has direct implications for episodic memory, which is greatly affected in AD patients and results from deficiencies in encoding and storing new information. In AD patients, this is manifested as a reduced capacity to learn new material and retain memories of recent events (Walker et al., 2007), and as discussed previously, this aspect of late-stage memory loss is highly correlated with earlier synaptic pathology (Selkoe, 2002; Coleman and Yao, 2003; Chong et al., 2011). Thus, although targeting late-stage AD features is an attractive goal, the increasing body of evidence demonstrating the contribution of early pathogenic mechanisms to late-stage AD symptoms warrants the consideration of new diagnostic and treatment strategies targeting more proximal disease processes (Goussakov et al., 2010; Müller et al., 2011; Pratt et al., 2011).
Figure 8.

Proposed role of ER calcium underlying early disruptions in synaptic homeostasis at the CA3–CA1 synapse in AD. At presynaptic CA3 terminals, RyR-evoked CICR can trigger spontaneous release of neurotransmitter vesicles. During high-frequency activity, CICR can be evoked by voltage-gated calcium influx to increase the residual calcium levels and release probability. In AD, increased RyR expression greatly increases CICR. Increased CICR can alter neurotransmitter vesicle cycling and result in depletion of vesicles from the readily releasable pool as well as the reserve pool. This can initially be remedied by increasing neurotransmitter synthesis and vesicle cycling. Unfortunately, such adaptive mechanisms have the potential to cause metabolic and oxidative stress that may result in apoptosis and synapse loss. At postsynaptic CA1 terminals, NMDAR-mediated calcium signal is amplified by RyR-mediated CICR in dendritic spines, and this phenomenon is required for the induction of synaptic plasticity. In a separate but parallel plasticity pathway, the mGluR–PLC pathway generates IP3, activating IP3R. Activation of IP3Rs supports regenerative calcium waves, which may also be involved in plasticity and gene expression. RyR calcium also activates SK channels, which modulate neuronal excitability by hyperpolarizing the membrane and by setting the frequency of action potential firing. In AD, increased RyR-evoked calcium release increases SK channel activity and can thus affect membrane potential and synaptic plasticity dynamics. Increased SK channel currents decrease neuronal excitability and increase the threshold for induction of synaptic plasticity and thus hinder plasticity. In the AD brains, ER calcium is serving an aberrant role in synaptic transmission and plasticity and is upregulating these presynaptic and postsynaptic calcium-dependent events that alter synaptic homeostasis. SC input, Schaffer collateral input; VGCC, voltage-gated calcium channel.
Footnotes
We thank Drs. Clark Briggs, Kuei Tseng, and Hey-Kyoung Lee for substantive comments and suggestions.
The authors declare no competing financial interests.
References
- Alford S, Frenguelli BG, Schofield JG, Collingridge GL. Characterization of Ca2+ signals induced in hippocampal CA1 neurons by the synaptic activation of NMDA receptors. J Physiol. 1993;469:693–716. doi: 10.1113/jphysiol.1993.sp019838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen D, Bond CT, Luján R, Ballesteros-Merino C, Lin MT, Wang K, Klett N, Watanabe M, Shigemoto R, Stackman RW, Jr, Maylie J, Adelman JP. The SK2-long isoform directs synaptic localization and function of SK2-containing channels. Nat Neurosci. 2011;14:744–749. doi: 10.1038/nn.2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auffret A, Gautheron V, Repici M, Kraftsik R, Mount HT, Mariani J, Rovira C. Age-dependent impairment of spine morphology and synaptic plasticity in hippocampal CA1 neurons of a presenilin 1 transgenic mouse model of Alzheimer's disease. J Neurosci. 2009;29:10144–10152. doi: 10.1523/JNEUROSCI.1856-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardo S, Robertson B, Stephens GJ. Presynaptic internal Ca2+ stores contribute to inhibitory neurotransmitter release onto mouse cerebellar Purkinje cells. Br J Pharmacol. 2002;137:529–537. doi: 10.1038/sj.bjp.0704901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear MF. Mechanism for a sliding synaptic modification threshold. Neuron. 1995;15:1–4. doi: 10.1016/0896-6273(95)90056-x. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer's disease. Trends Neurosci. 2008;31:454–463. doi: 10.1016/j.tins.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: LTP in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Bond CT, Maylie J, Adelman JP. Small-conductance calcium-activated potassium channels. Ann N Y Acad Sci. 1999;868:370–378. doi: 10.1111/j.1749-6632.1999.tb11298.x. [DOI] [PubMed] [Google Scholar]
- Bouchard R, Pattarini R, Geiger JD. Presence and functional significance of presynaptic ryanodine receptors. Prog Neurobiol. 2003;69:391–418. doi: 10.1016/s0301-0082(03)00053-4. [DOI] [PubMed] [Google Scholar]
- Brillantes AB, Ondrias K, Scott A, Kobrinsky E, Ondriasová E, Moschella MC, Jayaraman T, Landers M, Ehrlich BE, Marks AR. Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell. 1994;77:513–523. doi: 10.1016/0092-8674(94)90214-3. [DOI] [PubMed] [Google Scholar]
- Bruno AM, Huang JY, Bennett DA, Marr RA, Hastings ML, Stutzmann GE. Altered ryanodine receptor expression in mild cognitive impairment and Alzheimer's disease. Neurobiol Aging. 2011;33:1001.e1–e6. doi: 10.1016/j.neurobiolaging.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AG, Regehr WG. Quantal events shape cerebellar interneuron firing. Nat Neurosci. 2002;5:1309–1318. doi: 10.1038/nn970. [DOI] [PubMed] [Google Scholar]
- Cavelier P, Attwell D. Tonic release of glutamate by a DIDS-sensitive mechanism in rat hippocampal slices. J Physiol. 2005;564:397–410. doi: 10.1113/jphysiol.2004.082131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakroborty S, Goussakov I, Miller MB, Stutzmann GE. Deviant ryanodine receptor-mediated calcium release resets synaptic homeostasis in presymptomatic 3xTg-AD mice. J Neurosci. 2009;29:9458–9470. doi: 10.1523/JNEUROSCI.2047-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Marshall VJ, Irizarry M, Younkin L, Good MA, Bliss TV, Hyman BT, Younkin SG, Hsiao KK. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci. 1999;2:271–276. doi: 10.1038/6374. [DOI] [PubMed] [Google Scholar]
- Cheung KH, Shineman D, Müller M, Cárdenas C, Mei L, Yang J, Tomita T, Iwatsubo T, Lee VM, Foskett JK. Mechanism of Ca2+ disruption in Alzheimer's disease by presenilin regulation of InsP3 receptor channel gating. Neuron. 2008;58:871–883. doi: 10.1016/j.neuron.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung KH, Mei L, Mak DO, Hayashi I, Iwatsubo T, Kang DE, Foskett JK. Gain-of-function enhancement of IP3 receptor modal gating by familial Alzheimer's disease-linked presenilin mutants in human cells and mouse neurons. Sci Signal. 2010;3:ra22. doi: 10.1126/scisignal.2000818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong SA, Benilova I, Shaban H, De Strooper B, Devijver H, Moechars D, Eberle W, Bartic C, Van Leuven F, Callewaert G. Synaptic dysfunction in hippocampus of transgenic mouse models of Alzheimer's disease: a multi-electrode array study. Neurobiol Dis. 2011;44:284–291. doi: 10.1016/j.nbd.2011.07.006. [DOI] [PubMed] [Google Scholar]
- Coleman PD, Yao PJ. Synaptic slaughter in Alzheimer's disease. Neurobiol Aging. 2003;24:1023–1027. doi: 10.1016/j.neurobiolaging.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Dayan P, Willshaw DJ. Optimizing synaptic learning rules in linear associative memories. Biol Cybern. 1991;65:253–265. doi: 10.1007/BF00206223. [DOI] [PubMed] [Google Scholar]
- Deák F, Shin OH, Kavalali ET, Südhof TC. Structural determinants of synaptobrevin 2 function in synaptic vesicle fusion. J Neurosci. 2006;26:6668–6676. doi: 10.1523/JNEUROSCI.5272-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewachter I, Ris L, Croes S, Borghgraef P, Devijver H, Voets T, Nilius B, Godaux E, Van Leuven F. Modulation of synaptic plasticity and Tau phosphorylation by wild-type and mutant presenilin1. Neurobiol Aging. 2008;29:639–652. doi: 10.1016/j.neurobiolaging.2006.11.019. [DOI] [PubMed] [Google Scholar]
- Emptage N, Bliss TV, Fine A. Single synaptic events evoke NMDA receptor-mediated release of calcium from internal stores in hippocampal dendritic spines. Neuron. 1999;22:115–124. doi: 10.1016/s0896-6273(00)80683-2. [DOI] [PubMed] [Google Scholar]
- Emptage NJ, Reid CA, Fine A. Calcium stores in hippocampal synaptic boutons mediate short-term plasticity, store-operated Ca2+ entry, and spontaneous transmitter release. Neuron. 2001;29:197–208. doi: 10.1016/s0896-6273(01)00190-8. [DOI] [PubMed] [Google Scholar]
- Enoki R, Hu YL, Hamilton D, Fine A. Expression of long-term plasticity at individual synapses in hippocampus is graded, bidirectional, and mainly presynaptic: optical quantal analysis. Neuron. 2009;62:242–253. doi: 10.1016/j.neuron.2009.02.026. [DOI] [PubMed] [Google Scholar]
- Fioravante D, Regehr WG. Short-term forms of presynaptic plasticity. Curr Opin Neurobiol. 2011;21:269–274. doi: 10.1016/j.conb.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzjohn SM, Collingridge GL. Calcium stores and synaptic plasticity. Cell Calcium. 2002;32:405–411. doi: 10.1016/s0143416002001999. [DOI] [PubMed] [Google Scholar]
- Foskett JK. Inositol trisphosphate receptor Ca2+ release channels in neurological diseases. Pflugers Arch. 2010;460:481–494. doi: 10.1007/s00424-010-0826-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier C, Kourrich S, Soumireu-Mourat B, Mourre C. Apamin improves reference memory but not procedural memory in rats by blocking small conductance Ca2+-activated K+ channels in an olfactory discrimination task. Behav Brain Res. 2001;121:81–93. doi: 10.1016/s0166-4328(00)00387-9. [DOI] [PubMed] [Google Scholar]
- Fujii S, Matsumoto M, Igarashi K, Kato H, Mikoshiba K. Synaptic plasticity in hippocampal CA1 neurons of mice lacking type 1 inositol-1,4,5-triphosphate receptors. Learn Mem. 2000;7:312–320. doi: 10.1101/lm.34100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goussakov I, Miller MB, Stutzmann GE. NMDA-mediated Ca2+ influx drives aberrant ryanodine receptor activation in dendrites of young Alzheimer's disease mice. J Neurosci. 2010;30:12128–12137. doi: 10.1523/JNEUROSCI.2474-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goussakov I, Chakroborty S, Stutzmann GE. Generation of dendritic Ca2+ oscillations as a consequence of altered ryanodine receptor function in AD neurons. Channels (Austin) 2011;5:9–13. doi: 10.4161/chan.5.1.14124. [DOI] [PubMed] [Google Scholar]
- Groffen AJ, Martens S, Díez Arazola R, Cornelisse LN, Lozovaya N, de Jong AP, Goriounova NA, Habets RL, Takai Y, Borst JG, Brose N, McMahon HT, Verhage M. Doc2b is a high affinity Ca2+ sensor for spontaneous neurotransmitter release. Science. 2010;327:1614–1618. doi: 10.1126/science.1183765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gylys KH, Fein JA, Yang F, Wiley DJ, Miller CA, Cole GM. Synaptic changes in Alzheimer's disease: increased amyloid-beta and gliosis in surviving terminals is accompanied by decreased PSD-95 fluorescence. Am J Pathol. 2004;165:1809–1817. doi: 10.1016/s0002-9440(10)63436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond RS, Bond CT, Strassmaier T, Ngo-Anh TJ, Adelman JP, Maylie J, Stackman RW. Small-conductance Ca2+-activated K+ channel type 2 (SK2) modulates hippocampal learning, memory, and synaptic plasticity. J Neurosci. 2006;26:1844–1853. doi: 10.1523/JNEUROSCI.4106-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey J, Collingridge GL. Signal transduction pathways involved in the acute potentiation of NMDA responses by 1S, 3R-ACPD in rat hippocampal slices. Br J Pharmacol. 1993;109:1085–1090. doi: 10.1111/j.1476-5381.1993.tb13733.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschberg B, Maylie J, Adelman JP, Marrion NV. Gating properties of single SK channels in hippocampal CA1 pyramidal neurons. Biophys J. 1999;77:1905–1913. doi: 10.1016/S0006-3495(99)77032-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Roder JC, Bear MF. Chemical induction of mGluR5- and protein synthesis-dependent long-term depression in hippocampal area CA1. J Neurophysiol. 2001;86:321–325. doi: 10.1152/jn.2001.86.1.321. [DOI] [PubMed] [Google Scholar]
- Jaimovich E, Mattei C, Liberona JL, Cardenas C, Estrada M, Barbier J, Debitus C, Laurent D, Molgó J. Xestospongin B, a competitive inhibitor of IP3-mediated Ca2+ signaling in cultured rat myotubes, isolated myonuclei, and neuroblastoma (NG108–15) cells. FEBS Lett. 2005;579:2051–2057. doi: 10.1016/j.febslet.2005.02.053. [DOI] [PubMed] [Google Scholar]
- Katayama T, Imaizumi K, Honda A, Yoneda T, Kudo T, Takeda M, Mori K, Rozmahel R, Fraser P, George-Hyslop PS, Tohyama M. Disturbed activation of endoplasmic reticulum stress transducers by Familial Alzheimer's disease-linked presenilin-1 mutations. J Biol Chem. 2001;276:43446–43454. doi: 10.1074/jbc.M104096200. [DOI] [PubMed] [Google Scholar]
- Katz B. The release of neural transmitter substances. Vol 10. Liverpool, UK: Liverpool UP; 1969. [Google Scholar]
- Kelliher M, Fastbom J, Cowburn RF, Bonkale W, Ohm TG, Ravid R, Sorrentino V, O'Neill C. Alterations in ryanodine receptor calcium release channel correlate with Alzheimer's disease neurofibrillary and beta-amyloid pathologies. Neuroscience. 1999;92:499–513. doi: 10.1016/s0306-4522(99)00042-1. [DOI] [PubMed] [Google Scholar]
- Kemp A, Manahan-Vaughan D. Hippocampal long term depression and long term potentiation encode different aspects of novelty acquisition. Proc Natl Acad Sci U S A. 2004;101:8192–8197. doi: 10.1073/pnas.0402650101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuromi H, Kidokoro Y. Selective replenishment of two vesicle pools depends on the source of Ca2+ at the Drosophila synapse. Neuron. 2002;35:333–343. doi: 10.1016/s0896-6273(02)00777-8. [DOI] [PubMed] [Google Scholar]
- LaFerla FM. Calcium dyshomeostasis and intracellular signaling in Alzheimer's disease. Nat Rev Neurosci. 2002;3:862–872. doi: 10.1038/nrn960. [DOI] [PubMed] [Google Scholar]
- Llano I, González J, Caputo C, Lai FA, Blayney LM, Tan YP, Marty A. Presynaptic calcium stores underlie large-amplitude miniature IPSCs and spontaneous calcium transients. Nat Neurosci. 2000;3:1256–1265. doi: 10.1038/81781. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Grimwood PD, Morris RG. Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci. 2000;23:649–711. doi: 10.1146/annurev.neuro.23.1.649. [DOI] [PubMed] [Google Scholar]
- Mattson MP. ER calcium and Alzheimer's disease: in a state of flux. Sci Signal. 2010;3:pe10. doi: 10.1126/scisignal.3114pe10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximov A, Shin OH, Liu X, Südhof TC. Synaptotagmin-12, a synaptic vesicle phosphoprotein that modulates spontaneous neurotransmitter release. J Cell Biol. 2007;176:113–124. doi: 10.1083/jcb.200607021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCutcheon JE, Loweth JA, Ford KA, Marinelli M, Wolf ME, Tseng KY. Group I mGluR activation reverses cocaine-induced accumulation of calcium-permeable AMPA receptors in nucleus accumbens synapses via a protein kinase C-dependent mechanism. J Neurosci. 2011;31:14536–14541. doi: 10.1523/JNEUROSCI.3625-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney RA, Capogna M, Dürr R, Gähwiler BH, Thompson SM. Miniature synaptic events maintain dendritic spines via AMPA receptor activation. Nat Neurosci. 1999;2:44–49. doi: 10.1038/4548. [DOI] [PubMed] [Google Scholar]
- Mulkey RM, Malenka RC. Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus. Neuron. 1992;9:967–975. doi: 10.1016/0896-6273(92)90248-c. [DOI] [PubMed] [Google Scholar]
- Mulkey RM, Endo S, Shenolikar S, Malenka RC. Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature. 1994;369:486–488. doi: 10.1038/369486a0. [DOI] [PubMed] [Google Scholar]
- Müller M, Cárdenas C, Mei L, Cheung KH, Foskett JK. Constitutive cAMP response element binding protein (CREB) activation by Alzheimer's disease presenilin-driven inositol trisphosphate receptor (InsP3R) Ca2+ signaling. Proc Natl Acad Sci U S A. 2011;108:13293–13298. doi: 10.1073/pnas.1109297108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalbantoglu J, Tirado-Santiago G, Lahsaïni A, Poirier J, Goncalves O, Verge G, Momoli F, Welner SA, Massicotte G, Julien JP, Shapiro ML. Impaired learning and LTP in mice expressing the carboxy terminus of the Alzheimer amyloid precursor protein. Nature. 1997;387:500–505. doi: 10.1038/387500a0. [DOI] [PubMed] [Google Scholar]
- Nishiyama M, Hong K, Mikoshiba K, Poo MM, Kato K. Calcium stores regulate the polarity and input specificity of synaptic modification. Nature. 2000;408:584–588. doi: 10.1038/35046067. [DOI] [PubMed] [Google Scholar]
- Nosyreva E, Kavalali ET. Activity-dependent augmentation of spontaneous neurotransmission during endoplasmic reticulum stress. J Neurosci. 2010;30:7358–7368. doi: 10.1523/JNEUROSCI.5358-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obenaus A, Mody I, Baimbridge KG. Dantrolene-Na (Dantrium) blocks induction of long-term potentiation in hippocampal slices. Neurosci Lett. 1989;98:172–178. doi: 10.1016/0304-3940(89)90505-3. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Aβ and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Oliet SH, Malenka RC, Nicoll RA. Bidirectional control of quantal size by synaptic activity in the hippocampus. Science. 1996;271:1294–1297. doi: 10.1126/science.271.5253.1294. [DOI] [PubMed] [Google Scholar]
- Oliet SH, Malenka RC, Nicoll RA. Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neuron. 1997;18:969–982. doi: 10.1016/s0896-6273(00)80336-0. [DOI] [PubMed] [Google Scholar]
- Ozawa T. Effects of FK506 on Ca2+ release channels. Perspect Medicin Chem. 2008;2:51–55. doi: 10.4137/pmc.s382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer MJ, Irving AJ, Seabrook GR, Jane DE, Collingridge GL. The group I mGlu receptor agonist DHPG induces a novel form of LTD in the CA1 region of the hippocampus. Neuropharmacology. 1997;36:1517–1532. doi: 10.1016/s0028-3908(97)00181-0. [DOI] [PubMed] [Google Scholar]
- Parent A, Linden DJ, Sisodia SS, Borchelt DR. Synaptic transmission and hippocampal long-term potentiation in transgenic mice expressing FAD-linked presenilin 1. Neurobiol Dis. 1999;6:56–62. doi: 10.1006/nbdi.1998.0207. [DOI] [PubMed] [Google Scholar]
- Pineda JC, Galarraga E, Foehring RC. Different Ca2+ source for slow AHP in completely adapting and repetitive firing pyramidal neurons. Neuroreport. 1999;10:1951–1956. doi: 10.1097/00001756-199906230-00029. [DOI] [PubMed] [Google Scholar]
- Pratt KG, Zimmerman EC, Cook DG, Sullivan JM. Presenilin 1 regulates homeostatic synaptic scaling through Akt signaling. Nat Neurosci. 2011;14:1112–1114. doi: 10.1038/nn.2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiroz-Baez R, Ferrera P, Rosendo-Gutiérrez R, Morán J, Bermúdez-Rattoni F, Arias C. Caspase-12 activation is involved in amyloid-β protein-induced synaptic toxicity. J Alzheimers Dis. 2011;26:467–476. doi: 10.3233/JAD-2011-110326. [DOI] [PubMed] [Google Scholar]
- Raymond CR, Redman SJ. Spatial segregation of neuronal calcium signals encodes different forms of LTP in rat hippocampus. J Physiol. 2006;570:97–111. doi: 10.1113/jphysiol.2005.098947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes M, Stanton PK. Induction of hippocampal long-term depression requires release of Ca2+ from separate presynaptic and postsynaptic intracellular stores. J Neurosci. 1996;16:5951–5960. doi: 10.1523/JNEUROSCI.16-19-05951.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross WN, Nakamura T, Watanabe S, Larkum M, Lasser-Ross N. Synaptically activated Ca2+ release from internal stores in CNS neurons. Cell Mol Neurobiol. 2005;25:283–295. doi: 10.1007/s10571-005-3060-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybalchenko V, Hwang SY, Rybalchenko N, Koulen P. The cytosolic N-terminus of presenilin-1 potentiates mouse ryanodine receptor single channel activity. Int J Biochem Cell Biol. 2008;40:84–97. doi: 10.1016/j.biocel.2007.06.023. [DOI] [PubMed] [Google Scholar]
- Sah P. Ca2+-activated K+ currents in neurons: types, physiological roles and modulation. Trends Neurosci. 1996;19:150–154. doi: 10.1016/s0166-2236(96)80026-9. [DOI] [PubMed] [Google Scholar]
- Sah P, Bekkers JM. Apical dendritic location of slow afterhyperpolarization current in hippocampal pyramidal neurons: implications for the integration of long-term potentiation. J Neurosci. 1996;16:4537–4542. doi: 10.1523/JNEUROSCI.16-15-04537.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah P, Davies P. Calcium-activated potassium currents in mammalian neurons. Clin Exp Pharmacol Physiol. 2000;27:657–663. doi: 10.1046/j.1440-1681.2000.03317.x. [DOI] [PubMed] [Google Scholar]
- Salminen A, Kauppinen A, Suuronen T, Kaarniranta K, Ojala J. ER stress in Alzheimer's disease: a novel neuronal trigger for inflammation and Alzheimer's pathology. J Neuroinflammation. 2009;6:41. doi: 10.1186/1742-2094-6-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapansky J, Olson K, Van Der Ploeg R, Glazner G. NF-kappa B activated by ER calcium release inhibits Abeta-mediated expression of CHOP protein: enhancement by AD-linked mutant presenilin 1. Exp Neurol. 2007;208:169–176. doi: 10.1016/j.expneurol.2007.04.009. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Price DA. Synaptic pathology in Alzheimer's disease: a review of ultrastructural studies. Neurobiol Aging. 2003;24:1029–1046. doi: 10.1016/j.neurobiolaging.2003.08.002. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Schmitt FA, Mufson EJ. Hippocampal synaptic loss in early Alzheimer's disease and mild cognitive impairment. Neurobiol Aging. 2006;27:1372–1384. doi: 10.1016/j.neurobiolaging.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Sakaba T, Neher E. Vesicle pools and short-term synaptic depression: lessons from a large synapse. Trends Neurosci. 2002;25:206–212. doi: 10.1016/s0166-2236(02)02139-2. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res. 2008;192:106–113. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah M, Haylett DG. Ca2+ channels involved in the generation of the slow afterhyperpolarization in cultured rat hippocampal pyramidal neurons. J Neurophysiol. 2000;83:2554–2561. doi: 10.1152/jn.2000.83.5.2554. [DOI] [PubMed] [Google Scholar]
- Smith IF, Hitt B, Green KN, Oddo S, LaFerla FM. Enhanced caffeine-induced Ca2+ release in the 3xTg-AD mouse model of Alzheimer's disease. J Neurochem. 2005;94:1711–1718. doi: 10.1111/j.1471-4159.2005.03332.x. [DOI] [PubMed] [Google Scholar]
- Stackman RW, Hammond RS, Linardatos E, Gerlach A, Maylie J, Adelman JP, Tzounopoulos T. Small conductance Ca2+-activated K+ channels modulate synaptic plasticity and memory encoding. J Neurosci. 2002;22:10163–10171. doi: 10.1523/JNEUROSCI.22-23-10163.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton PK, Winterer J, Bailey CP, Kyrozis A, Raginov I, Laube G, Veh RW, Nguyen CQ, Müller W. Long-term depression of presynaptic release from the readily releasable vesicle pool induced by NMDA receptor-dependent retrograde nitric oxide. J Neurosci. 2003;23:5936–5944. doi: 10.1523/JNEUROSCI.23-13-05936.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker M, Krause M, Pedarzani P. An apamin-sensitive Ca2+-activated K+ current in hippocampal pyramidal neurons. Proc Natl Acad Sci U S A. 1999;96:4662–4667. doi: 10.1073/pnas.96.8.4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutzmann GE. The pathogenesis of Alzheimer's disease is it a lifelong “calciumopathy”? Neuroscientist. 2007;13:546–559. doi: 10.1177/1073858407299730. [DOI] [PubMed] [Google Scholar]
- Stutzmann GE, Mattson MP. Endoplasmic reticulum Ca2+ handling in excitable cells in health and disease. Pharmacol Rev. 2011;63:700–727. doi: 10.1124/pr.110.003814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutzmann GE, LaFerla FM, Parker I. Ca2+ signaling in mouse cortical neurons studied by two-photon imaging and photoreleased inositol triphosphate. J Neurosci. 2003;23:758–765. doi: 10.1523/JNEUROSCI.23-03-00758.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutzmann GE, Caccamo A, LaFerla FM, Parker I. Dysregulated IP3 signaling in cortical neurons of knock-in mice expressing an Alzheimer's-linked mutation in presenilin1 results in exaggerated Ca2+ signals and altered membrane excitability. J Neurosci. 2004;24:508–513. doi: 10.1523/JNEUROSCI.4386-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutzmann GE, Smith I, Caccamo A, Oddo S, LaFerla FM, Parker I. Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer's disease mice. J Neurosci. 2006;26:5180–5189. doi: 10.1523/JNEUROSCI.0739-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutzmann GE, Smith I, Caccamo A, Oddo S, Parker I, Laferla F. Enhanced ryanodine-mediated calcium release in mutant PS1-expressing Alzheimer's mouse models. Ann N Y Acad Sci. 2007;1097:265–277. doi: 10.1196/annals.1379.025. [DOI] [PubMed] [Google Scholar]
- Supnet C, Noonan C, Richard K, Bradley J, Mayne M. Up-regulation of the type 3 ryanodine receptor in neuroprotective in the TgCRND8 mouse model of Alzheimer's disease. J Neurochem. 2010;112:356–365. doi: 10.1111/j.1471-4159.2009.06487.x. [DOI] [PubMed] [Google Scholar]
- Sutton MA, Wall NR, Aakalu GN, Schuman EM. Regulation of dendritic protein synthesis by miniature synaptic events. Science. 2004;304:1979–1983. doi: 10.1126/science.1096202. [DOI] [PubMed] [Google Scholar]
- Takami K, Terai K, Matsuo A, Walker DG, McGeer PL. Expression of presenilin-1 and -2 mRNAs in rat and Alzheimer's disease brains. Brain Res. 1997;748:122–130. doi: 10.1016/s0006-8993(96)01274-7. [DOI] [PubMed] [Google Scholar]
- Taufiq AM, Fujii S, Yamazaki Y, Sasaki H, Kaneko K, Li J, Kato H, Mikoshiba K. Involvement of IP3 receptors in LTP and LTD induction in guinea pig hippocampal CA1 neurons. Learn Mem. 2005;12:594–600. doi: 10.1101/lm.17405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Vrede Y, Fossier P, Baux G, Joels M, Chameau P. Control of IsAHP in mouse hippocampus CA1 pyramidal neurons by RyR3-mediated calcium-induced calcium release. Pflugers Arch. 2007;455:297–308. doi: 10.1007/s00424-007-0277-4. [DOI] [PubMed] [Google Scholar]
- Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer RE, van den Berg TK, Missler M, Geuze HJ, Südhof TC. Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science. 2000;287:864–869. doi: 10.1126/science.287.5454.864. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A. The endoplasmic reticulum and neuronal calcium signaling. Cell Calcium. 2002;32:393–404. doi: 10.1016/s0143416002001896. [DOI] [PubMed] [Google Scholar]
- Walker M, Chan D, Thom M. Hippocampus and human disease. In: Andersen P, Morris R, Amaral D, Bliss T, O'Keefe J, editors. The hippocampus book. Oxford: Oxford UP; 2007. pp. 769–812. [Google Scholar]
- Wasser CR, Kavalali ET. Leaky synapses: regulation of spontaneous neurotransmission in central synapses. Neuroscience. 2009;158:177–188. doi: 10.1016/j.neuroscience.2008.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Hong M, Lasser-Ross N, Ross WN. Modulation of calcium wave propagation in the dendrites and to the soma of rat hippocampal pyramidal neurons. J Physiol. 2006;575:455–468. doi: 10.1113/jphysiol.2006.114231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock JR, Heynen AJ, Shuler MG, Bear MF. Learning induces long-term potentiation in the hippocampus. Science. 2006;313:1093–1097. doi: 10.1126/science.1128134. [DOI] [PubMed] [Google Scholar]
- Xiao RP, Valdivia HH, Bogdanov K, Valdivia C, Lakatta EG, Cheng H. The immunophilin FK506-binding protein modulates Ca2+ release channel closure in rat heart. J Physiol. 1997;500:343–354. doi: 10.1113/jphysiol.1997.sp022025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Pang ZP, Shin OH, Südhof TC. Synaptotagmin-1 functions as a Ca2+ sensor for spontaneous release. Nat Neurosci. 2009;12:759–766. doi: 10.1038/nn.2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Wu B, Beglopoulos V, Wines-Samuelson M, Zhang D, Dragatsis I, Südhof TC, Shen J. Presenilins are essential for regulating neurotransmitter release. Nature. 2009;460:632–636. doi: 10.1038/nature08177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]