

Abstract
Paroxysmal nocturnal hemoglobinuria (PNH) is an acquired genetic disorder of the bone marrow that produces intravascular hemolysis, proclivity to venous thrombosis, and hematopoietic failure. Mutation in the PIG-A gene of a hematopoietic stem cell abrogates synthesis of glycosylphosphoinositol (GPI) anchors and expression of all GPI-anchored proteins on the surface of progeny erythrocytes, leukocytes, and platelets. Urokinase plasminogen activator receptor (uPAR), a GPI-linked protein expressed on neutrophils, mediates endogenous thrombolysis through a urokinase-dependent mechanism. Here we show that membrane GPI-anchored uPAR is decreased or absent on granulocytes and platelets of patients with PNH, while soluble uPAR (suPAR) levels are increased in patients’ plasma. Serum suPAR concentrations correlated with the number of GPI-negative neutrophils and were highest in patients who later develop thrombosis. In vitro, suPAR is released from PNH hematopoietic cells and from platelets upon activation, suggesting that these cells are the probable source of plasma suPAR in the absence of GPI anchor synthesis and trafficking of uPAR to the cell membrane. In vitro, the addition of recombinant suPAR results in a dose-dependent decrease in the activity of single-chain urokinase. We hypothesized that suPAR, prevents the interaction of urokinase with membrane-anchored uPAR on residual normal cells.
Introduction
Paroxysmal nocturnal hemoglobinuria (PNH) is a clonal bone marrow disorder resulting from an acquired, somatic mutation of the X chromosome PIG-A gene in an hematopoietic stem cell1–3. Affected cells cannot synthesize the GPI-moiety which anchors many different types of proteins to the cell membrane4. Red cells lacking the GPI-anchored protein CD59, a potent inhibitor of the late components of complement and reactive lysis5–7, are more susceptible to complement. Although intravascular red cell hemolysis is the hallmark of PNH, thrombosis is the most frequent complication leading to death with predilection for the hepatic and cerebral veins. In one study, 50 percent of patients with PNH died of complications of thrombosis, which appears to be more common in purely hemolytic PNH than when the disorder accompanies bone marrow aplasia8.
The pathophysiology of PNH thrombosis is not entirely understood. Studies of PNH platelets8 and procoagulants have not demonstrated consistent abnormalities. A number of stimuli have been hypothesized to contribute to thrombophilia in PNH: episodic hemolysis9 with attendant increase circulating platelet- and endothelial-derived procoagulant microparticles 10;11, complement-mediated platelet activation, and decreased fibrinolytic activity12. Urokinase plasminogen activator receptor (uPAR) is a GPI-anchored protein expressed on activated monocytes and granulocytes13–15. uPAR is synthesized as a single polypeptide chain of 313 amino acids with post-translational deletion of 30 carboxyterminal residues and transamidation linkage of the GPI-anchor. uPAR acts as a receptor for pro-urokinase, thus facilitating the generation of activated plasmin16. uPAR which is expressed on the membranes of activated normal leukocytes and is believed to play a role in fibrinolysis by promoting the conversion of urokinase to its active form. Increased levels of suPAR lacking the GPI-anchor have been described in plasma and serum from patients with PNH17;18 as well as in patients with certain types of malignancy19.
The absence of membrane uPAR on monocytes, granulocytes, and platelets might contribute to thrombosis in PNH. While some studies report suPAR to be effective in promoting fibrinolysis under certain conditions in vitro20;21, in media containing physiological concentrations of albumin, fibrinolysis is inhibited22;23. Membrane uPAR may also support fibinolysis mechanically by bringing plasminogen and urokinase into close proximity on the membrane24, 25.
In this study, we examined the levels of plasma suPAR in patients with PNH. We find that GPI-negative granulocytes and platelets release suPAR, and that the size of the PNH clone correlated with increased plasma levels in affected patients. Our studies confirm previous data that suPAR inhibits urokinase activity in vitro and we show that both LDH (a surrogate for hemolysis) and suPAR independently correlate with thrombosis in a cohort of 87 PNH patients followed for a mean of 4.1 years.
Methods and Materials
Patient selection and analysis
After informed consent, using protocols approved by the Institutional Review Board of the National Heart, Lung, and Blood Institute, serum and bone marrow samples were obtained from 87 patients with PNH sera were prepared at time of entry of the patient into study and frozen for later analysis. The diagnosis of PNH was based on flow cytometry of cell surface GPI-anchored proteins, for erythrocytes, CD59 and CD55 and, for granulocytes, CD16 and CD66 with CD15 for gating26. Patients receiving G-CSF for neutropenia were eliminated because of the drug’s influence on suPAR27. Charts were reviewed for history of thrombosis retrospectively with reviewers blinded to the suPAR value. Thrombosis of the lower extremities was documented by ultrasound, and pulmonary embolus by spiral computed tomography (CT) or high probability ventilation-perfusion scan. Head computed tomography (CT) or magnetic resonance imaging (MRI) was used to demonstrate thrombosis of the venous sinuses and ultrasound for Budd Chiari syndrome. Twenty healthy donors and ten patients with aplastic anemia without PNH were also studied.
suPAR ELISA
Soluble uPAR were measured with the kinetic ELISA (American Diagnostic, Stanford, Ct); all serum samples were analyzed in duplicate and many in triplicate.
Protease activity assays
Plasminogen activation by urokinase was quantified by adaptation of published methods28, Recombinant human single chain urokinase (scuPA) and the plasmin substrate Spectrozyme PL were purchased from American Diagnostica (Stamford, CT). Chromogenic assays were performed in 96 well microtiter plates using a total reaction volume of 200 μl in the presence of 0.5%BSA-PBS. Spectrozyme PL (0.5mM, final concentration) was mixed with variable concentrations of scuPA, and the suPAR and incubated at 37°C for 30 minutes. Following incubation, plasminogen (1μM, final concentration) was added, and the absorbance readings at 405 nm were measured and plotted against time for varying concentrations of suPAR.
Bone marrow cell separation and culture
Bone marrow was obtained by aspiration from the posterior iliac crest into syringes containing media supplemented 1:10 with heparin (O’Neill and Feldman, St Louis, MO). Mononuclear cells were isolated by density gradient centrifugation with the use of lymphocyte separation medium (Organon, Durham, NC). BMMNCs were cultured in growth factor media for four days as previously described.29 Cells were centrifuged and ELISA for suPAR was performed.
Flow cytometry
Intracellular staining for suPAR expression was performed by means of the Pharmingen Intracellular Staining Kit.16 Double-color surface staining was first performed with phycoerythrin (PE)-conjugated anti-CD13 and anti-CD42a to distinguish platelets and granulocytes. CD62-FITC was used when appropriate to identify activated platelets. Cells were permeabilized by means of a saponin-based method (Pharmingen) and stained with fluorescein isothiocyanate (FITC)-anti-suPAR (Pharmingen and BioSource, Camarillo, CA). Specificity of the antibody was confirmed by showing elimination of staining with a suPAR. Samples were analyzed by means of the Coulter (Hialeah, FL) EPICS V flow cytometer.
ELISA
We assessed the reproducibility of ELISA in clinical samples. Serum from twenty normal donors and ten PNH patients were performed in triplicate and with a high degree of reproducibility i.e. (Pearson r = 0.97). Duplicate tests performed on blood samples from 10 patients assayed during periods of comparable disease activity (GPI-anchored protein neutrophils ±10%) also showed good reproducibility; linear regression analysis demonstrated a p value of <0.0001
Immunoblotting
Fresh serum was used for immunoprecipitation which was performed as previously described30 with a polyclonal antibody directed to suPAR (American Diagnostica). Proteins (10μg/lane) were resolved in 12% Tris-glycine SDS gels (Invitrogen, Carlsbad, CA) electrophoresis at 125V and transferred to polyvinylidene difluoride (PVDF) membranes (Invitrogen, Carlsbad, CA). The membrane was blocked for 2 h with 5% BSA in PBS and 0.05% Tween-20 followed by incubation in primary suPAR mAb. Subsequently, membranes were incubated in horse radish peroxidase (HRP)-conjugated secondary anti-mouse IgG antibody and detection of the bands of interest was performed using the chemiluminescence kit with luminol (ECL plus system, Amersham Pharmacia Biotech, Buckinghamshire, UK). Membranes were stripped after the first blotting in ImmunoPure IgG elution buffer (Pierce, Rockford, IL) re-blocked and re-blotted with another antibody (Ab). To evaluate equal loading of the lanes membranes were re-blotted with anti-actin polyclonal Ab; each was probed with suPAR mAb (American Diagnostica).
Statistical methods
Basic characteristics of PNH patients and controls were described using summary statistics, such as medians, means, standard deviations, ranges and proportions. In the absence of justifiable patterns by the missing mechanism, absent data were assumed to be missing completely at random, and were deleted in the analysis. Among the PNH patients, sample means of baseline suPAR (ng/mL) along with their corresponding standard errors and 95% confidence intervals were calculated for patients grouped by sex, LDH (≤400, >400), GpI-negative neutrophils (≤500, >500), GpI percent (≤60%, >60%), ANC (≤500, >500), Mono absolute (≤300, >300), and the sum of ANC and Mono absolute (≤800, >800). P-values based on two-sample T-tests were calculated for testing equal means of baseline suPAR between patient groups. For statistical analysis using continuous covariates, natural log-transformations were used for LDH, GpI-negative neutrophils, GpI percent, ANC and Mono absolute. These transformations reduced the skewness of the covariate distributions. Linear regression models were used to evaluate the correlations between baseline suPAR and age, sex, and the log-transformations of LDH, GpI-negative neutrophils, GpI percent, ANC and Mono absolute. Univariate and multivariate Cox Proportional Hazard models were used to evaluate the effects of age, sex, baseline suPAR and the log-transformations of LDH, GpI-negative neutrophils, GpI percent, ANC and Mono absolute on the time-to-first-clot distributions. P-values based on log-rank and t statistics were used for testing the null hypothesis that the corresponding covariate had no effect on the time-to-first-clot distribution. All numerical results were computed using the Splus 7 Statistical Software Package (Insightful Inc., Seattle, WA).
Results
Soluble uPAR is increased in PNH
Soluble uPAR was measured by ELISA in the sera of 80 patients with PNH at their initial visit, and in twenty normal controls and ten patients with AA but without PNH. Soluble uPAR was increased in patients with PNH compared to AA patients without PNH and healthy controls (Table 1, Fig. 1a). Increased serum suPAR was confirmed by immunoblotting in 15 patients (example seen in Fig. 1b). Linear regression analysis demonstrated a statistically significant relationship between the absolute number of GPI-negative neutrophils and suPAR levels measured by ELISA (p<0.001).
Table 1a.
Mean, SD and 95% CI for suPAR at baseline under risk categories.
| Risk Factor | N | Mean (SD) | 95% CI for Mean | P-value |
|---|---|---|---|---|
| Sex | ||||
| Female | 33 | 2.236 (0.255) | (1.716, 2.756) | ----- |
| Male | 33 | 2.347 (0.226) | (1.887, 2.806) | 0.746 |
| LDH | ||||
| ≤400 | 33 | 1.333 (0.175) | (0.977, 1.690) | ----- |
| >400 | 32 | 3.258 (0.173) | (2.904, 3.612) | <0.001 |
| Absolute # of GPI-neg neutrophils | ||||
| ≤500 | 24 | 1.192 (0.180) | (0.818, 1.565) | ----- |
| >500 | 42 | 2.920 (0.186) | (2.545, 3.294) | <0.001 |
| % GPI-neg neutrophils percent | ||||
| ≤60% | 32 | 1.447 (0.194) | (1.052, 1.842) | ----- |
| >60% | 34 | 3.086 (0.192) | (2.695, 3.476) | <0.001 |
| ANC | ||||
| ≤500 | 15 | 1.202 (0.258) | (0.648, 1.755) | ----- |
| >500 | 50 | 2.656 (0.181) | (2.292, 3.021) | <0.001 |
| Mono Absolute | ||||
| ≤300 | 30 | 2.334 (0.230) | (1.863, 2.805) | ----- |
| >300 | 34 | 2.211 (0.248) | (1.707, 2.716) | 0.719 |
| ANC+Mono Abs. | ||||
| ≤800 | 15 | 1.092 (0.257) | (0.541, 1.644) | ----- |
| >800 | 48 | 2.676 (0.175) | (2.324, 3.028) | <0.001 |
Note: P-values are two sample T-tests.
Fig. 1. Serum level of suPAR are increased in patients with PNH and correlates with the absolute number of GPI-negative neutrophils.
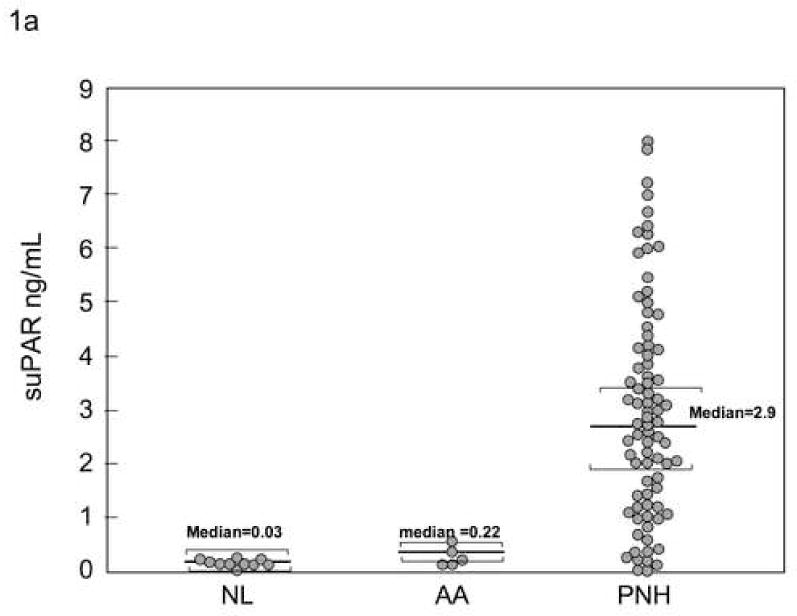
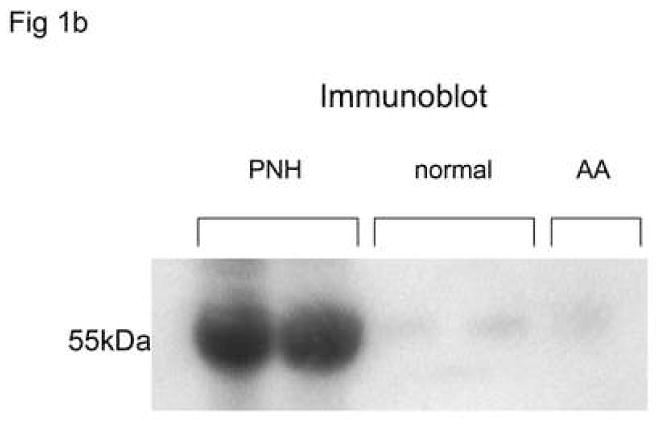
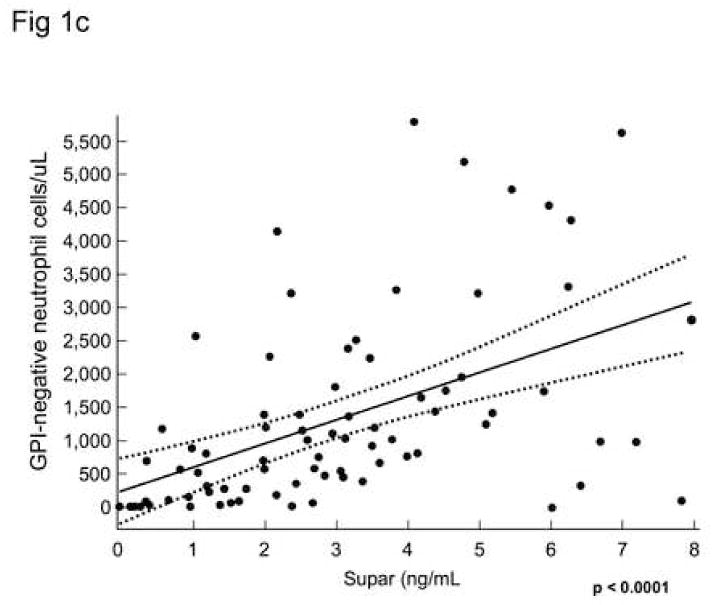
Serum sUPAR levels were measured by ELISA in patients with PNH, AA without PNH, and in healthy donors. Levels measured from patients with PNH were substantially increased compared to AA patients (without PNH) or healthy donors 1A.. Increased levels were confirmed by immunoblot showing a band at 55kDa matching the apparent molecular weight of suPAR in 15 PNH patients but not in 10 normal controls or five AA patients without PNH(1B). Linear regression analysis demonstrated a statistically significant relationship between the absolute number of GPI-negative neutrophils and suPAR levels measured by ELISA (p<0.001) (IC).
Soluble uPAR is present in GPI-negative neutrophils lacking membrane uPAR
Previously, others have demonstrated that suPAR is secreted from PNH leukocytes of patients with PNH31 and could be a potential source of suPAR in the plasma. Here we reproduced these findings but also demonstrated release of suPAR by BMMNC in culture and by activated platelets. When peripheral blood granulocytes were surface-stained with CD15-APC, CD59-PE, and anti-uPAR-FITC, a proportion of normal CD59-positive granulocytes (presumably those which are activated) but not PNH, CD59- granulocytes stained with anti-suPAR (example seen in Fig. 2a). At the same time, when these samples were intracellularly stained with anti-uPAR-FITC, most of the population of both CD59-postive and -negative granulocytes stained with anti-uPAR-FITC suggesting that uPAR is synthesized but is not surface-expressed without the GPI-anchor. Stained granulocyte populations are seen in Fig. 2b. When BMMNCs from individuals with >90% GPI-negative neutrophils and AA patients without PNH were placed in culture media for four days, centrifuged to remove the cells, and supernatants tested by ELISA, there were significant increases in supernantant levels measured by ELISA for the PNH samples only and this could not be accounted for by differences cell death (Fig. 2c). In the three patients tested immediately following thrombosis the suPAR levels declined slightly, presumably related to increased levels of uPA.
Figure 2. GPI-negative neutrophils demonstrated intracellular suPAR but not surface uPAR.
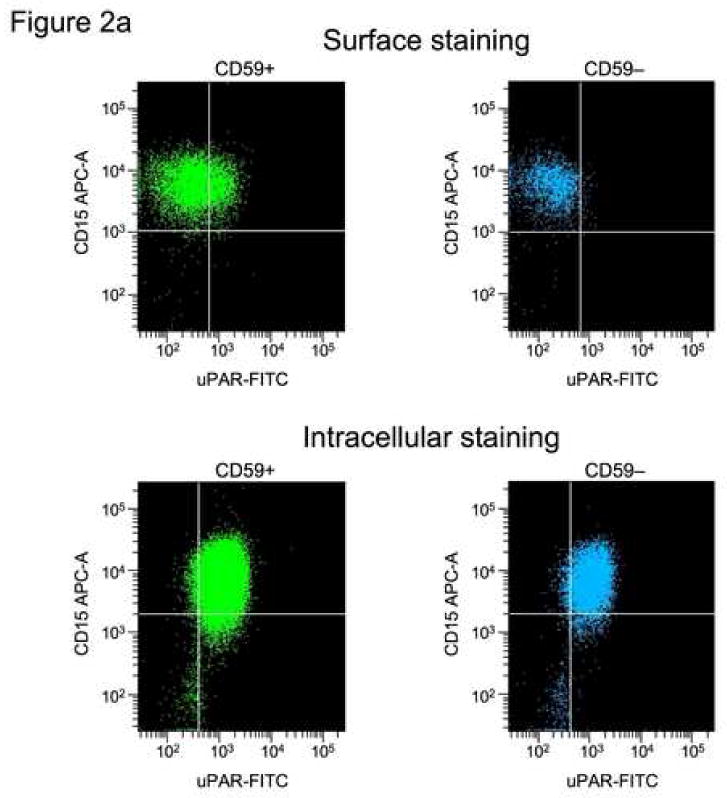
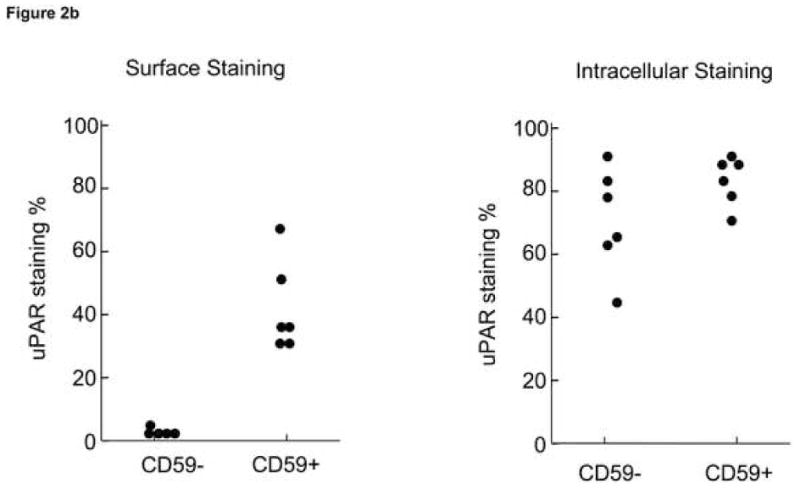
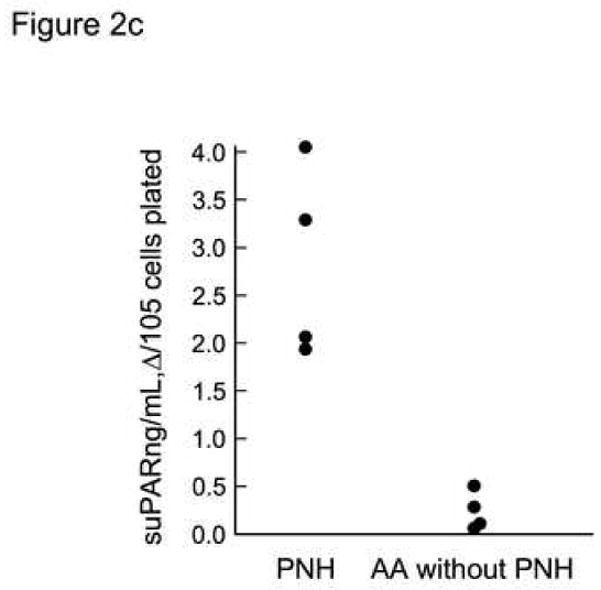
Samples of PB from PNH patients were surface-stained with CD59-PE mAb with and intracellular stained with suPAR-FITC mAb. The granulocytes were gated according to side-scatter forward scatter characteristics. CD59-positive and −negative samples were each gated and the uPAR-FITC mAb-staining cells were quantitated for each group. Consistent with previous observations that activated granulocytes express surface uPAR13, some CD59-positive granulocytes, but no CD59-negative neutrophils showed surface staining with suPAR antibody. However both CD59-positive and CD59 negative granulocytes show significant intracellular staining with uPAR (Example of staining is seen in A, results of all experiments are seen in (B)). When BMMNCs from patients with >90% GPI-negative neutrophils were placed in culture media for four days and then centrifuged, the supernatants tested by ELISA for suPAR showed significantly increased levels of sUPAR compared to AA patients (C).
GPI-anchored protein negative activated platelets release suPAR
Platelets obtained from patients PNH were activated by addition of adenosine diphosphate (ADP) (1μM) to platelet-rich plasma obtained from four PNH patients with >90% GPI-anchor protein negative platelets and four healthy donors. Platelets were identified by anti-CD42-PECy5 (anit-GPIIb,IIIa); these were gated and activated platelets were distinguished by CD62-PE staining and the percent of these cells staining with anti-uPAR-FITC were measured. Only normal, activated platelets and not platelets from PNH patients demonstrated membrane uPAR (Fig. 3a). When these platelets were centrifuged, PNH platelet poor plasma (PPP) but not normal PPP demonstrated significant increases in suPAR compared to non-activated platelets by ELISA, suggesting that PNH platelets secrete suPAR upon activation, while normal platelets express the protein on their surface. (Fig. 3b).
Figure 3. PNH platelets may represent a source of plasma suPAR.
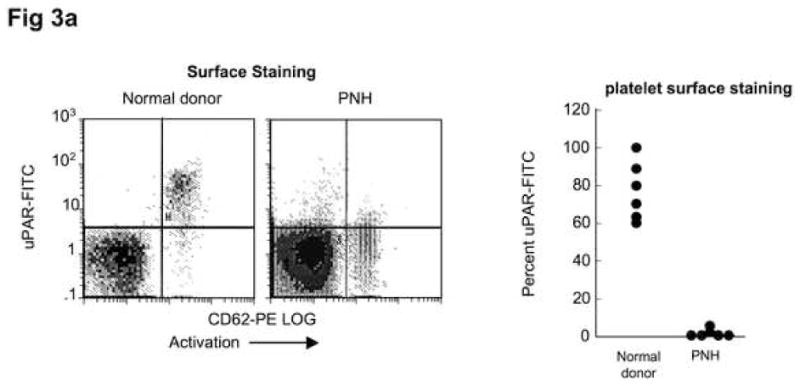
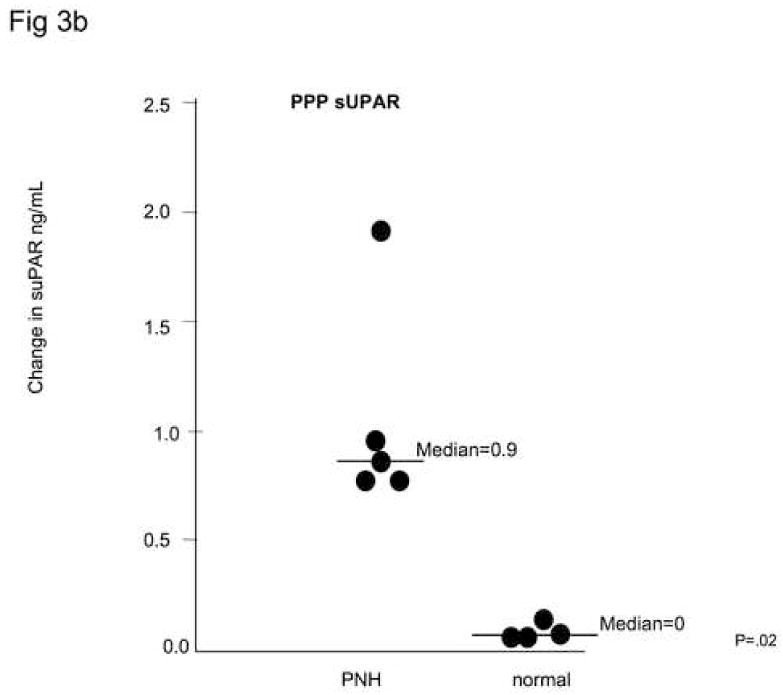
Samples of platelets from healthy donors and from PNH patients whose platelets were primarily CD59-negative were activated by adding 1μM ADP while stirring. Samples were then fixed and stained with CD62-PE, CD42-PE-Cy5, and uPAR-FITC. Platelets identified on the basis of CD42 staining were gated and activated (CD62-staining) platelets were examined for surface anti-uPAR staining. PNH platelets (N=6) demonstrated negligible uPAR surface staining, while platelets from healthy donors (N=6) demonstrated a substantial population staining for surface-uPAR (A). When the activated platelets were centrifuged and suPAR ELISA performed on the platelet poor plasma (PPP), there were measurable increases for the PNH platelets, but little suPAR was detected in the PPP of healthy platelets (B).
suPAR decreases plasmin generation
Plasmin generation rate was measured at without and with varying concentrations of suPAR by a time-dependent change in the optical density at 405nm. There was a dose-dependent decrease in the activity of single-chain urokinase following the addition of recombinant suPAR (Fig. 4).
Figure 4. Soluble uPAR decreases plasmin generation.
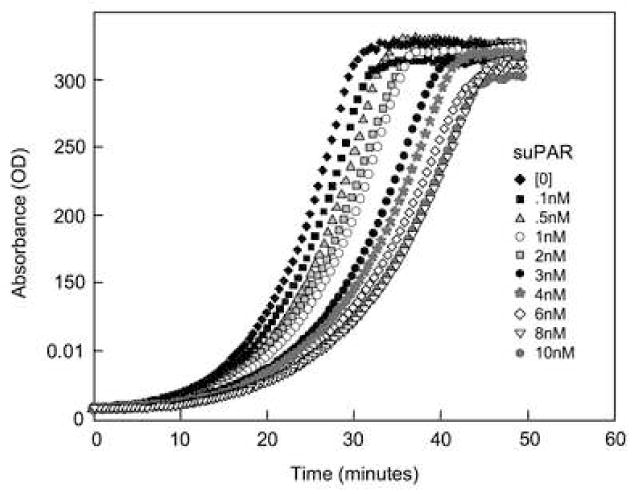
Plasminogen activation by single-chain recombinant tPA at 37°C was quantitated using a kinetic microtiter plate reader as described in Methods and Materials. Chromogenic assays were performed in 96 well microtiter plates using a total reaction volume of 200μl. The reaction occurred in the presence of 0.5%BSA-PBS. The Spectrozyme PL (0.5mM), scuPA (2nM), and the suPAR at various concentrations including controls without sUPAR were mixed and incubated at 37°C for 30 minutes. Following the incubation, plasminogen (1μM) was added and the absorbance readings at 405nm were measured and plotted against time for control and sample with varying concentrations of suPAR. Inhibition of plasmin generation by suPAR was dependent on the concentration of suPAR.
Thrombosis in PNH patients
Eighty-seven patients were stratified according to a history of thrombosis; suPAR and lactate dehydrogenase (LDH) levels were analyzed (suPAR was not available for seven patients). Univariate analysis showed that ANC, absolute number of GPI-negative neutrophils, percent GPI-negative neutrophils, and LDH correlated with suPAR levels (Table 1). For a mean follow-up of 4.1 years, there were 29 venous thrombotic events, one myocardial infarction (patient age 56 with no risk factors), and two cerebral vascular events (both under age 50, no risk factors). Thrombosis-free survival was significantly shorter in patients with LDH≥ 1000 units/mL or suPAR ≥ 2 ng/mL (Fig. 5). Multivariate analysis showed that male sex, suPAR, and LDH were independently associated with risk of thrombosis.
Figure 5. Thrombosis was increased in those patients with significant hemolysis and those with increases suPAR.
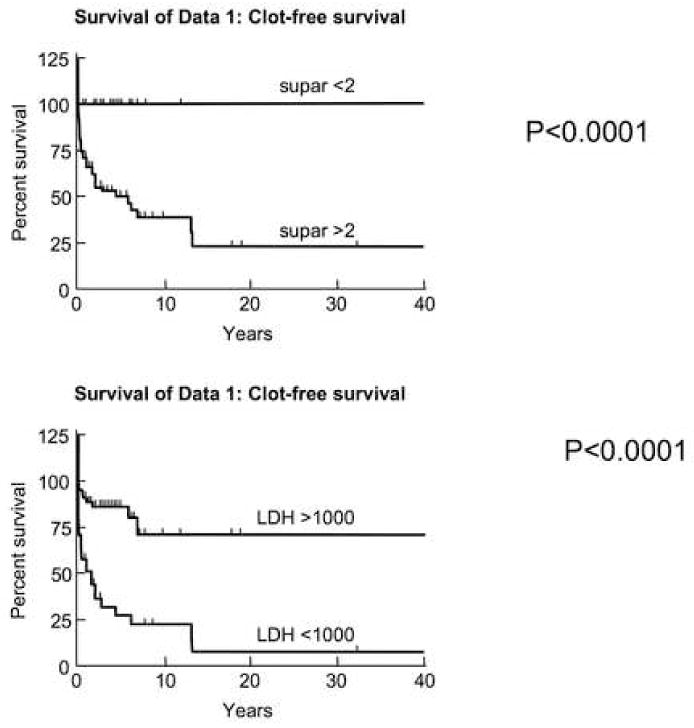
The charts of eighty-seven patients were analyzed retrospectively for history of thrombosis following entry into the study. ELISA was used to measure suPAR on frozen serum obtained on first encounter with the patients. Patients with LDH≥1000Units/L and patients with suPAR ≥ 2.0 had significantly shorter clot-free survival compared to those with lower values. Reviewer was blinded to the results of suPAR. Patients were censured after their first clot and if they were placed on warfarin prophylactically (N=1).
Discussion
Thromboembolism is frequent in PNH 32;33 where it is the most common cause of death. In this study we demonstrate increased suPAR in the sera of PNH patients who eventually developed thrombosis. Multivariate analysis of the factors associated with thrombosis in our cohort of 87 PNH patients show that LDH, male sex, and suPAR levels were independently associated with time to first thrombosis. Factors correlating with elevated suPAR levels included the absolute number and percent of GPI negative neutrolphils, and ANC. These clinical findings are consistent with our in vitro observations that PNH BMMNCs and mature activated platelets and neutrophils34 release suPAR and could be a source of this protein in PNH serum. We confirmed the results of others’35 that suPAR impedes plasmin generation from plasminogen in vitro. Defects in urokinase activation related to suPAR as well as a relative deficiency of platelets and leukocytes with GPI-linked-uPAR may contribute to the alterations in fibrinolysis12. It is likely that suPAR inhibits the generation of plasmin by competitively inhibitiing uPA-binding to GPI-anchored uPAR. A factor potentially accounting for differences in the biological behavior of soluble and GPI-linked uPAR may be related to changes in the molecular conformation of uPAR when this protein is not attached to the membrane. These changes confer an ability to bind to entirely different epitopes compared to its GPI-anchored form36;37. Some evidence suggests that uPAR possesses little intrinsic zymogenic activity, but serves the mechanical function of bringing uPA and plasminogen into juxtaposition on the cell membrane38-a role that can only be played by the GPI-anchored form.
Fibrinolytic defects such as plasminogen deficiency are not generally associated with thrombosis in the absence of additional risk factors39. In PNH, hemolysis is a likely contributor to the pro-thrombotic state. During hemolysis, plasma-free hemoglobin is released binding nitric oxide resulting in lower levels near the vessel endothelium 13. Plasma red cell and platelet microvesicles with procoagulant activity40 are formed. It is likely that among other factors, free hemoglobin released during hemolysis increases tissue factor production from the endothelium22 and complement activation of platelets results in up-regulation of platelet α-granular protein, P-selectin, and release of high molecular weight von Willebrand factor from the vascular endothelium41. Recent observations that patients on elizumab42 and anecdotal evidence suggesting that thrombi form during hemolytic episodes further support a role for hemolysis in PNH thrombophilia.
The combination of hemolysis and suPAR-related impairment in fibrinolysis in PNH (mechanism detailed in Fig. 6) may explain the greater frequency of thrombosis in patients whose disease is primarily marked by hemolysis rather than bone marrow failure8. In this circumstance more autologous PNH red cell production results in more potential for hemolysis and improved neutrophil and platelet production result in higher suPAR levels. The fact that both the ANC as well as the absolute number of GPI-negative neutrophils correlates with the suPAR levels supports this hypothesis. The relative contribution of both hemolysis and the fibrinolytic defect introduced by excess suPAR needs to be addressed, particularly in patients taking eclizumab who may be reluctant to continue their anticoagulation after their hemolysis is controlled.
Figure 6. Hypothetical mechanism for increased thrombosis in patients with PNH.
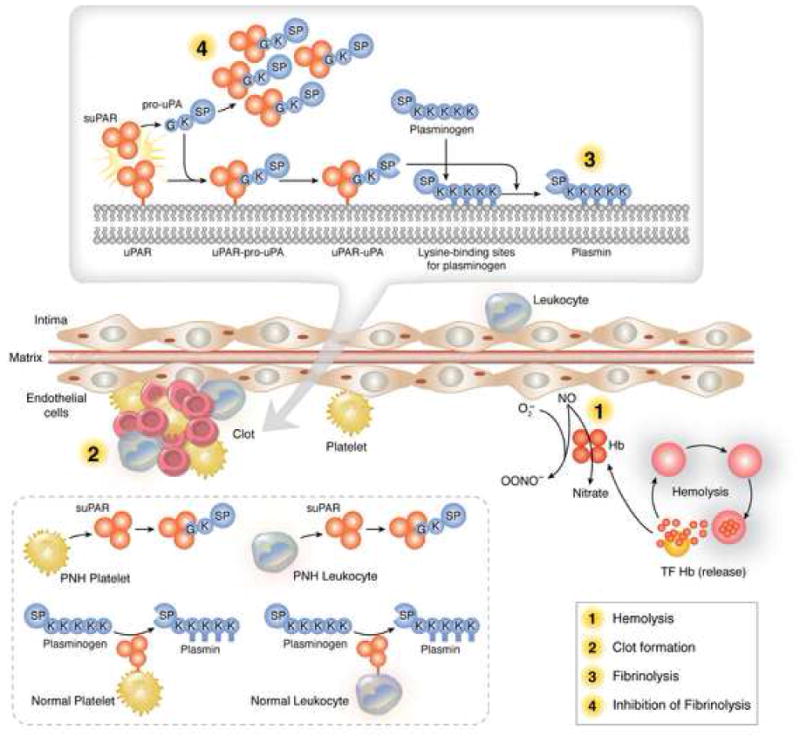
Clot forms because of the prothombotic state created by hemolysis with release of free hemoglobin (diminishing nitric oxide levels near the endothelium), red cell and platelet microvesicles (1). Fibrinolysis is impaired because of competitive binding of pro-uPA by fibrinolytically inactive suPAR (4) and the absence of fibrinolytically active membrane-uPAR normally present on platelets and leukocytes within the clot.
Table 2.
Regression coefficients and covariate adjusted relative risks obtained from the univariate and multivariate Cox Proportional Hazard Models of time to first clot versus risk factors. Large coefficient estimate (β) or relative risk (exp(β)) suggest the association of the corresponding risk factor with the worse time to first clot outcome. Natural log-transformation was used for Absolute GPI-neg neutrophils, % GPI-neg neutrophils, ANC, Mono Absolute and the sum of ANC and Mono Absolute for the purpose of reducing the skewness of the distributions.
| Risk Factor | Univariate Cox Regression | Multivariate Cox Regression | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Coefficient | Relative Risk | P (β=0) | Coefficient | Relative Risk | P (β=0) | |||||
| β | SD | RR | 95% CI | β | SD | RR | 95% CI | |||
| Age (years) | 0.011 | 0.011 | 1.01 | (0.99, 1.03) | 0.320 | -0.004 | 0.015 | 0.996 | (0.97, 1.03) | 0.800 |
| Sex (male) | 0.568 | 0.358 | 1.76 | (0.88, 3.56) | 0.108 | 0.862 | 0.404 | 2.37 | (1.07, 5.23) | 0.033 |
| LDH (units) | 0.842 | 0.169 | 2.32 | (1.66, 3.24) | <0.001 | 0.587 | 0.213 | 1.80 | (1.19, 2.37) | 0.006 |
| Abs. GPI-neg. Neu. | 0.856 | 0.219 | 2.35 | (1.53, 3.62) | <0.001 | ----- | ----- | ----- | (----, ----) | ----- |
| %GPI-neg. Neu. | 0.711 | 0.259 | 2.04 | (1.22, 3.38) | 0.004 | ----- | ----- | ----- | (----, ----) | ----- |
| ANC | 0.695 | 0.217 | 2.00 | (1.31, 3.07) | 0.001 | ----- | ----- | ----- | (----, ----) | ----- |
| Mono | 0.552 | 0.304 | 1.74 | (0.96, 3.15) | 0.072 | ----- | ----- | ----- | (----, ----) | ----- |
| ANC+Mono | 0.925 | 0.276 | 2.52 | (1.47, 4.34) | 0.001 | ----- | ----- | ----- | (----, ----) | ----- |
| suPAR at baseline | 0.399 | 0.076 | 1.49 | (1.28, 1.73) | <0.001 | 0.366 | 0.098 | 1.44 | (1.19, 1.75) | <0.001 |
Note: P-values are for log-rank tests in univariate models and t-tests for multivariate models.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Young NS. Paroxysmal nocturnal hemoglobinuria: current issues in pathophysiology and treatment. Curr Hematol Rep. 2005;4:103–109. [PubMed] [Google Scholar]
- 2.Parker C, Omine M, Richards S, Nishimura J, Bessler M, Ware R, Hillmen P, Luzzatto L, Young N, Kinoshita T, Rosse W, Socie G. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood. 2005;106:3699–3709. doi: 10.1182/blood-2005-04-1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Araten DJ, Luzzatto L. The mutation rate in PIG-A is normal in patients with paroxysmal nocturnal hemoglobinuria (PNH) Blood. 2006;108:734–736. doi: 10.1182/blood-2006-01-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nishimura J, Kanakura Y, Ware RE, Shichishima T, Nakakuma H, Ninomiya H, Decastro CM, Hall S, Kanamaru A, Sullivan KM, Mizoguchi H, Omine M, Kinoshita T, Rosse WF. Clinical course and flow cytometric analysis of paroxysmal nocturnal hemoglobinuria in the United States and Japan. Medicine (Baltimore) 2004;83:193–207. doi: 10.1097/01.md.0000126763.68170.46. [DOI] [PubMed] [Google Scholar]
- 5.Babiker AA, Ronquist G, Nilsson UR, Nilsson B. Transfer of prostasomal CD59 to CD59-deficient red blood cells results in protection against complement-mediated hemolysis. Am J Reprod Immunol. 2002;47:183–192. doi: 10.1034/j.1600-0897.2002.1o023.x. [DOI] [PubMed] [Google Scholar]
- 6.Brooimans RA, van Wieringen PAM, Van Es LA, Daha MR. Relative roles of decay-accelerating factor membrane cofactor protein and CD59 in the protection of human endothelial cells against complement-mediated lysis. Eur J Immunol. 1992;22:3135–3140. doi: 10.1002/eji.1830221216. [DOI] [PubMed] [Google Scholar]
- 7.Meri S, Morgan BP, Davies A, Daniels RH, Olavesen MG, Waldmann H, Lachmann PJ. Human protectin (CD59), an 18,000-2000 MW complement lysis restricting factor, inhibits C5b-8 catalysed insertion of C9 into lipid bilayers. Immunology. 1990;71:1–9. [PMC free article] [PubMed] [Google Scholar]
- 8.Sloand EM, Young NS. Thrombotic Complications in PNH. In: Young NS, Moss J, editors. PNH and the GPI-linked Proteins. Academic Press; San Diego, CA: 2000. pp. 101–112. [Google Scholar]
- 9.Poskitt TR, Fortwengler HP, Jr, Lunskis BJ. Activation of the alternate complement pathway by autologous red cel stroma. J Exp Med. 1973;138:715–722. doi: 10.1084/jem.138.3.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simak J, Holada K, Risitano AM, Zivny JH, Young NS, Vostal JG. Elevated circulating endothelial membrane microparticles in paroxysmal nocturnal haemoglobinuria. Br J Haematol. 2004;125:804–813. doi: 10.1111/j.1365-2141.2004.04974.x. [DOI] [PubMed] [Google Scholar]
- 11.Hugel B, Socie G, Vu T, Toti F, Gluckman E, Freyssinet JM, Scrobohaci ML. Elevated levels of circulating procoagulant microparticles in patients with paroxysmal nocturnal hemoglobinuria and aplastic anemia. Blood. 1999;93:3451–3456. [PubMed] [Google Scholar]
- 12.Grunewald M, Siegemund A, Grunewald A, Schmid A, Koksch M, Schopflin C, Schauer S, Griesshammer M. Plasmatic coagulation and fibrinolytic system alterations in PNH: relation to clone size. Blood Coagul Fibrinolysis. 2003;14:685–695. doi: 10.1097/00001721-200310000-00011. [DOI] [PubMed] [Google Scholar]
- 13.Plesner T, Behrendt N, Ploug M. Structure, function and expression on blood and bone marrow cells of the urokinase-type plasminogen activator receptor, uPAR. Stem Cells. 1997;15:398–408. doi: 10.1002/stem.150398. [DOI] [PubMed] [Google Scholar]
- 14.Ploug M, Eriksen J, Plesner T, Hansen NE, Dano K. A soluble form of the glycolipid-anchored receptor for urokinase-type plasminogen activator is secreted from peripheral blood leukocytes from patients with paroxysmal nocturnal hemoglobinuria. Eur J Biochem. 1992;208:397–404. doi: 10.1111/j.1432-1033.1992.tb17200.x. [DOI] [PubMed] [Google Scholar]
- 15.Plesner T, Ploug M, Ellis V, Ronne E, Hoyer-Hansen G, Wittrup M, Pedersen TL, Tscherning T, Dano K, Hansen NE. The receptor for urokinase-type plasminogen activator and urokinase is translocated from two distinct intracellular compartments to the plasma membrane on stimulation of human neutrophils. Blood. 1994;83:808–815. [PubMed] [Google Scholar]
- 16.Moller LB. Structure and function of the urokinase receptor. Blood Coagul Fibrinolysis. 1993;4:293–303. [PubMed] [Google Scholar]
- 17.Ronne E, Pappot H, Grondahl-Hansen J, Hoyer-Hansen G, Plesner T, Hansen NE, Dano K. The receptor for urokinase plasminogen activator is present in plasma from healthy donors and elevated in patients with paroxysmal nocturnal haemoglobinuria. Br J Haematol. 1995;89:576–581. doi: 10.1111/j.1365-2141.1995.tb08366.x. [DOI] [PubMed] [Google Scholar]
- 18.Ninomiya H, Hasegawa Y, Nagasawa T, Abe T. Excess soluble urokinase-type plasminogen activator receptor in the plasma of patients with paroxysmal nocturnal hemoglobinuria inhibits cell-associated fibrinolytic activity. Int J Hematol. 1997;65:285–291. doi: 10.1016/s0925-5710(96)00559-2. [DOI] [PubMed] [Google Scholar]
- 19.Jo M, Thomas KS, Wu L, Gonias SL. Soluble urokinase-type plasminogen activator receptor inhibits cancer cell growth and invasion by direct urokinase-independent effects on cell signaling. J Biol Chem. 2003;278:46692–46698. doi: 10.1074/jbc.M308808200. [DOI] [PubMed] [Google Scholar]
- 20.Bugge TH, Flick MJ, Danton MJ, Daugherty CC, Romer J, Dano K, Carmeliet P, Collen D, Degen JL. Urokinase-type plasminogen activator is effective in fibrin clearance in the absence of its receptor or tissue-type plasminogen activator. Proc Natl Acad Sci U S A. 1996;93:5899–5904. doi: 10.1073/pnas.93.12.5899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Higazi AA, Bdeir K, Hiss E, Arad S, Kuo A, Barghouti I, Cines DB. Lysis of plasma clots by urokinase-soluble urokinase receptor complexes. Blood. 1998;92:2075–2083. [PubMed] [Google Scholar]
- 22.Liebman HA, Feinstein DI. Thrombosis in patients with paroxysmal noctural hemoglobinuria is associated with markedly elevated plasma levels of leukocyte-derived tissue factor. Thromb Res. 2003;111:235–238. doi: 10.1016/j.thromres.2003.09.018. [DOI] [PubMed] [Google Scholar]
- 23.Ninomiya H, Hasegawa Y, Nagasawa T, Abe T. Excess soluble urokinase-type plasminogen activator receptor in the plasma of patients with paroxysmal nocturnal hemoglobinuria inhibits cell-associated fibrinolytic activity. Int J Hematol. 1997;65:285–291. doi: 10.1016/s0925-5710(96)00559-2. [DOI] [PubMed] [Google Scholar]
- 24.Ellis V. Functional Analysis of the Cellular Receptor for Urokinase in Plasminogen Activation. RECEPTOR BINDING HAS NO INFLUENCE ON THE ZYMOGENIC NATURE OF PRO-UROKINASE. J Biol Chem. 1996;271:14779–14784. doi: 10.1074/jbc.271.25.14779. [DOI] [PubMed] [Google Scholar]
- 25.Ellis V, Scully MF, Kakkar VV. Plasminogen activation initiated by single-chain urokinase-type plasminogen activator. Potentiation by U937 monocytes. J Biol Chem. 1989;264:2185–2188. [PubMed] [Google Scholar]
- 26.Maciejewski JP, Rivera C, Kook H, Dunn D, Young NS. Relationship between bone marrow failure syndromes and the presence of glycophosphatidyl inositol-anchored protein-deficient clones. Br J Haematol. 2001;115:1015–1022. doi: 10.1046/j.1365-2141.2001.03191.x. [DOI] [PubMed] [Google Scholar]
- 27.Selleri C, Montuori N, Ricci P, Visconte V, Carriero MV, Sidenius N, Serio B, Blasi F, Rotoli B, Rossi G, Ragno P. Involvement of the urokinase-type plasminogen activator receptor in hematopoietic stem cell mobilization. Blood. 2005;105:2198–2205. doi: 10.1182/blood-2004-06-2424. [DOI] [PubMed] [Google Scholar]
- 28.Horne MK, III, Goad JL, Merryman PK, Cullinane AM. Comparison of the Effect of Histidine-Rich Glycoprotein and 6-Aminohexanoic Acid on Plasmin Production and Fibrinolysis in Vitro. Thrombosis Research. 2000;99:179–186. doi: 10.1016/s0049-3848(00)00231-0. [DOI] [PubMed] [Google Scholar]
- 29.Sloand EM, Young NS, Sato T, Kumar P, Kim S, Weichold FF, Maciejewski JP. Secondary colony formation after long-term bone marrow culture using peripheral blood and bone marrow of HIV-infected patients. AIDS. 1997;11:1547–1553. doi: 10.1097/00002030-199713000-00002. [DOI] [PubMed] [Google Scholar]
- 30.Sloand E, Young NS, Sato T, Kim S, Maciejewski JP. Inhibition of interleukin-1β-converting enzyme in human hematopoietic progenitor cells results in blockade of cytokine-mediated apoptosis and expansion of their proliferative potential. Exp Hematol. 1998;26:1093–1099. [PubMed] [Google Scholar]
- 31.Ploug M, Eriksen J, Plesner T, Hansen NE, Dano K. A soluble form of the glycolipid-anchored receptor for urokinase-type plasminogen activator is secreted from peripheral blood leukocytes from patients with paroxysmal nocturnal hemoglobinuria. Eur J Biochem. 1992;208:397–404. doi: 10.1111/j.1432-1033.1992.tb17200.x. [DOI] [PubMed] [Google Scholar]
- 32.Hillmen P, Lewis SM, Bessler M, Luzzatto L, Dacie JV. Natural history of paroxysmal nocturnal hemoglobinuria. N Engl J Med. 1995;333:1253–1258. doi: 10.1056/NEJM199511093331904. [DOI] [PubMed] [Google Scholar]
- 33.Socie G, Mary JY, de GA, Rio B, Leporrier M, Rose C, Heudier P, Rochant H, Cahn JY, Gluckman E. Paroxysmal nocturnal haemoglobinuria: long-term follow-up and prognostic factors. French Society of Haematology. Lancet. 1996;348:573–577. doi: 10.1016/s0140-6736(95)12360-1. [DOI] [PubMed] [Google Scholar]
- 34.Ploug M, Eriksen J, Plesner T, Hansen NE, Dano K. A soluble form of the glycolipid-anchored receptor for urokinase-type plasminogen activator is secreted from peripheral blood leukocytes from patients with paroxysmal nocturnal hemoglobinuria. Eur J Biochem. 1992;208:397–404. doi: 10.1111/j.1432-1033.1992.tb17200.x. [DOI] [PubMed] [Google Scholar]
- 35.Behrendt N, Dano K. Effect of purified, soluble urokinase receptor on the plasminogen-prourokinase activation system. FEBS Lett. 1996;393:31–36. doi: 10.1016/0014-5793(96)00849-6. [DOI] [PubMed] [Google Scholar]
- 36.Hoyer-Hansen G, Pessara U, Holm A, Pass J, Weidle U, Dano K, Behrendt N. Urokinase-catalysed cleavage of the urokinase receptor requires an intact glycolipid anchor. Biochem J. 2001;358:673–679. doi: 10.1042/0264-6021:3580673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Higazi AA, Upson RH, Cohen RL, Manuppello J, Bognacki J, Henkin J, McCrae KR, Kounnas MZ, Strickland DK, Preissner KT, Lawler J, Cines DB. Interaction of single-chain urokinase with its receptor induces the appearance and disappearance of binding epitopes within the resultant complex for other cell surface proteins. Blood. 1996;88:542–551. [PubMed] [Google Scholar]
- 38.Ellis V. Functional analysis of the cellular receptor for urokinase in plasminogen activation. Receptor binding has no influence on the zymogenic nature of pro-urokinase. J Biol Chem. 1996;271:14779–14784. doi: 10.1074/jbc.271.25.14779. [DOI] [PubMed] [Google Scholar]
- 39.Tefs K, Gueorguieva M, Klammt J, Allen CM, Aktas D, Anlar FY, Aydogdu SD, Brown D, Ciftci E, Contarini P, Dempfle CE, Dostalek M, Eisert S, Gokbuget A, Gunhan O, Hidayat AA, Hugle B, Isikoglu M, Irkec M, Joss SK, Klebe S, Kneppo C, Kurtulus I, Mehta RP, Ornek K, Schneppenheim R, Seregard S, Sweeney E, Turtschi S, Veres G, Zeitler P, Ziegler M, Schuster V. Molecular and clinical spectrum of type I plasminogen deficiency: a series of 50 patients. Blood. 2006;108:3021–3026. doi: 10.1182/blood-2006-04-017350. [DOI] [PubMed] [Google Scholar]
- 40.Hall C, Richards S, Hillmen P. Primary prophylaxis with warfarin prevents thrombosis in paroxysmal nocturnal hemoglobinuria (PNH) Blood. 2003;102:3587–3591. doi: 10.1182/blood-2003-01-0009. [DOI] [PubMed] [Google Scholar]
- 41.Devine DV. The effects of complement activation on platelets. Curr Top Microbiol Immunol. 1992;178:101–113. doi: 10.1007/978-3-642-77014-2_7. [DOI] [PubMed] [Google Scholar]
- 42.Hillmen P, Muus P, Duhrsen U, Risitano A, Schubert J, Young NS, Schrezenmeier H, Szer J, Brodsky RA, Hill A, Socie G, Rollins S, Rother R, Bell L, Luzzatto L. The terminal complement inhibitor eculizumab reduces thrombosis in patients with paroxysmal nocturnal hemoglobinuria. Blood. 2007;108:23 (Abstr). doi: 10.1182/blood-2007-06-095646. [DOI] [PubMed] [Google Scholar]