

Abstract
To establish the druggability of a target, genetic validation needs to be supplemented with pharmacological validation. Pharmacological studies, especially in the kinase field, are hampered by the fact that many reference inhibitors are not fully selective for one target. Fortunately, the initial trickle of selective inhibitors released in the public domain has steadily swelled into a stream. However, rationally picking the most selective tool compound out of the increasing amounts of available inhibitors has become progressively difficult due to the lack of accurate quantitative descriptors of drug selectivity. A recently published approach, termed ‘selectivity entropy’, is an improved way of expressing selectivity as a single-value parameter and enables rank ordering of inhibitors. We provide a guide to select the best tool compounds for pharmacological validation experiments of candidate drug targets using selectivity entropy. In addition, we recommend which inhibitors to use for studying the biology of the 20 most investigated kinases that are clinically relevant: Abl (ABL1), AKT1, ALK, Aurora A/B, CDKs, MET, CSF1R (FMS), EGFR, FLT3, ERBB2 (HER2), IKBKB (IKK2), JAK2/3, JNK1/2/3 (MAPK8/9/10), MEK1/2, PLK1, PI3Ks, p38α (MAPK14), BRAF, SRC and VEGFR2 (KDR).
Keywords: kinase, inhibitor, selectivity, quantitative, drug target, cross-screen, profiling, tool, entropy, promiscuity
The importance of selective kinase tool compounds
In 2002, Sir Philip Cohen predicted that protein kinases would become ‘the drug targets of the 21st century’ (Cohen, 2002). So far, kinases have lived up to this expectation. In the past 10 years, 15 small molecule kinase inhibitors and five anti-kinase antibodies have been approved by the U.S. Food and Drug Administration (Table 1). These successes (and the projects that failed along the way) have yielded a wealth of reference compounds in the public domain that are useful for investigating the role of specific protein kinases in cellular processes (for reviews, see Shokat and Velleca, 2002; Carlson and White, 2011; Hodgson and Schröder, 2011).
Table 1.
Kinase inhibitors approved for therapeutic use
| Compound | Trade name | Company | Target | Cancer | Year of approval | |
|---|---|---|---|---|---|---|
| 1 | Imatinib | Gleevec/Glivec (EU) | Novartis | BCR-ABL | CML | 2001 |
| KIT | GIST | 2008 | ||||
| 2 | Gefitinib | Iressa | Astra-Zeneca | EGFR | NSCLC | 2003 |
| 3 | Sorafinib | Nexavar | Bayer/Onyx | Multiple (VEGFR, | Renal cell carcinoma | 2005 |
| PDGFR, RAF) | Liver cancer | 2007 | ||||
| 4 | Erlotinib | Tarceva | Genentech/Roche | EGFR | NSCLC | 2005 |
| Pancreatic cancer | 2005 | |||||
| 5 | Dasatinib | Sprycel | BMS | Multiple (BCR-ABL, SRC) | CML | 2006 |
| 6 | Sunitinib | Sutent | Pfizer | Multiple (PDGFR, VEGFR, KIT) | Renal cell carcinoma | 2006 |
| Imatinib-resistant GIST | 2006 | |||||
| 7 | Temsirolimus | Torisel | Wyeth | mTOR1 | Renal cell carcinoma | 2007 |
| 8 | Nilotinib | Tasigna | Novartis | BCR-ABL | Imatinib-resistant CML | 2007 |
| 9 | Lapatinib | Tykerb/Tyverb (EU) | GSK | EGFR, ERBB2 | HER2+ breast | 2007 |
| triple+ breast | 2010 | |||||
| 10 | Everolimus | Zortress/Certican (EU)/Afinitor | Novartis | mTOR1 | Advanced kidney cancer | 2009 |
| 11 | Pazopanib | Votrient | GSK | Multiple (PDGFR, VEGFR, KIT) | Renal cell carcinoma | 2009 |
| 12 | Vandetanib | Caprelsa | Astra-Zeneca | RET | Medullary thyroid cancer | 2011 |
| 13 | Ruxolitinib | Jakafi | Incyte | JAK1/2 | Myelofibrosis | 2011 |
| 14 | Vemurafenib | Zelboraf | Roche | BRAF (V600E mutant) | Metastatic melanoma | 2011 |
| 15 | Crizotinib | Xalkori | Pfizer | ALK | NSCLC | 2011 |
These compounds inhibit the TORC1 cofactor of mTOR and are therefore not kinase inhibitors in a strict sense.
Kinase inhibitors are powerful tools for pharmacological validation because their effects give direct information on the effect of therapeutic targeting of the protein. However, many of them inhibit multiple kinases, in part because they target the highly conserved ATP-binding pocket. There are many instances where inhibition of an off-target kinase contributes to, or even is solely responsible for, the observed biological effects. A recent example comes from work implicating the kinase p38α in Wnt/β-catenin signalling (Verkaar et al., 2011). p38α is a stress-activated serine/threonine kinase that mediates production of inflammatory cytokines. Multiple p38α inhibitors have been clinically evaluated for diseases of the immune system. Several researchers noted that administration of such p38α drugs to cell lines inhibits signalling through the Wnt/β-catenin pathway (Bikkavilli et al., 2008; Cervenka et al., 2010), an evolutionarily conserved signalling cascade critical for embryonic development and adult stem cell maintenance (Logan and Nusse, 2004; MacDonald et al., 2009). However, recently released cross-screening data revealed that several widely used tool compounds for p38α (for instance, SB203580) also inhibit casein kinase Iδ and CKIε (Bain et al., 2007; Verkaar et al., 2011). Both kinases are well known to be activators of Wnt/β-catenin signalling (Peters et al., 1999; Sakanaka et al., 1999). Importantly, this cross-reactivity cannot be explained by sequence similarity, as p38α and CKIs are quite distant in the phylogenetic tree. Their pharmacological similarity could only be demonstrated by profiling inhibitors in biochemical assays. Another instance where compound promiscuity confounds scientific analysis is when the same compound is used as a tool inhibitor for more than one kinase. The spectrum selective inhibitor dasatinib was used as a ‘typical’ SRC inhibitor by Gnoni et al., (2011), while An et al. (2010) used dasatinib as a ‘typical’ Abl inhibitor. Thus, it is important to thoroughly understand the selectivity of pharmacological tools in the kinase field, and to make sure that targets are validated with the most selective inhibitors (Davies et al., 2000; Knight and Shokat, 2005; Bain et al., 2007).
Whereas in the early days of kinase research, inhibitors were often named ‘selective’ on the basis of anecdotal evidence, the recent wealth of selectivity profiling data has greatly advanced the rational understanding of inhibitor promiscuity. In selectivity profiling, the activity/affinity of kinase inhibitors on a multitude of non-target kinases is tested in parallel. Here we give an overview of sources of profiling data, and illustrate how to interpret those data through new methods for quantifying selectivity. With this method, we have pinpointed the most selective inhibitors for the 20 most intensely investigated protein kinases. This review serves as a guide to picking the most selective tool compounds, thereby minimizing the chance that cross-reactivities will compromise target validation.
Technologies and study approaches in cross-screening
The most-used method to study kinase inhibitor selectivity is profiling in multiple parallel biochemical assays. Biochemical assays are preferred because the readout can be coupled with very high confidence to a particular target. Specialist profiling labs have emerged that offer selectivity profiling (Table 2). Typically, these labs run 100–400 kinase assays in parallel, using different assay formats. The largest panel even comprises 394 out of the 514 genes predicted to encode kinases in the human genome (Manning et al., 2002; Table 2).
Table 2.
Providers of kinase panel profiling services
| Provider | Location of profiling lab | Technology | Kinase panel size (wild-type kinases) | Website |
|---|---|---|---|---|
| Carna Biosciences | Kobe, Japan | Substrate mobility shift assay | 305 (272) | http://www.carnabio.com |
| DiscoverX (Ambit) | San Diego, CA, USA | Phage display competitive binding | 451 (394) | http://www.kinomescan.com |
| Millipore | Dundee, UK | Radioactive filter binding | 299 (261) | http://www.millipore.com |
| Life Technologies | Madison, WI, USA and Paisley, UK | Z'-Lyte™ and Adapta® binding assay | 317 (293) | http://www.invitrogen.com |
| ProQinase | Freiburg, Germany | Radioactive filter binding | 340 (298) | http://www.proqinase.com |
| Reaction Biology | Malvern, PA, USA | Radioactive filter binding | 385 (348) | http://www.reactionbiology.com |
| MRC Centre for Kinase Profiling | Dundee, UK | Radioactive filter binding | 126 (122) | http://www.kinase-screen.mrc.ac.uk |
Kinase panel sizes as reported on November 2011. The order of suppliers is alphabetical. The authors have had compounds profiled with most of the listed labs, and obtained robust data from all of them. See provider's websites for more information on technologies and protocols used.
The classic, ‘gold standard’ format is the radioactive filter binding assay, which combines sensitivity with a generic readout of direct kinase function (i.e. transfer of a radioactive phosphate group), and is used by more than half of the commercial kinase profiling labs (Table 2). Non-radioactive alternatives include microfluidic detection of the mobility shifts of phosphorylated substrates (often referred to as the ‘Caliper’ format), or detecting substrate phosphorylation by its protease-protective effect on fluorescent energy transfer in a probe peptide (Z'-lyte™). Another often-employed format is a competitive binding assay in which kinases expressed on bacteriophages are prevented from binding to an immobilized probe ligand by a competing inhibitor of interest. The amount of bound kinase-phage is quantified by amplification of the phage DNA with the PCR (often called the ‘Ambit’ format, after the company that developed the technology, Fabian et al., 2005).
The differences in technologies used are potentially exacerbated by differences in construct sequences and expression systems that are used. Some laboratories express kinases in Escherichia coli, others in insect cells, which results in differences in the kinase phosphorylation status. Additionally, assay conditions may vary across labs, such as buffers and incubation temperatures, concentrations of ATP and, importantly, the nature of the peptide or protein substrate. Additionally, when compounds are slow-binding allosteric inhibitors, incubation times before the readout are critical because a read out has to take place in binding equilibrium.
For all these reasons, the IC50s of reference inhibitors, as published on the websites of profiling labs, show the expected variation for IC50s measured in different labs. However, encouragingly the available data indicate that most labs find similar selectivities for similar compounds. Results from 20 compounds in enzyme activity assays and ligand binding assays were found to be comparable (Fabian et al., 2005). This was later confirmed in a massive effort where the potencies of ∼10 000 inhibitors on 40 different targets were compared (Posy et al., 2011). In a study where 16 compounds were profiled in either activity or binding assays, a single-value selectivity metric (see below) produced comparable values and similar selectivity rank ordering (Uitdehaag and Zaman, 2011).
Foregoing differences in technology and conditions used, profiling studies fall in one of the three following categories:
A dose-response binding experiment that gives a Kd for each target. This result is determined in the absence of ATP (Fabian et al., 2005).
A dose-response activity assay that determines an IC50 for each target kinase. This result is dependent on the ATP concentration in the assay. If assays are performed at an ATP concentration equal to the kinase's KM-ATP, then for a competitive inhibitor the Cheng–Prusoff relation states that IC50= 2Ki (Knight and Shokat, 2005). This Ki is the ATP-independent inhibition constant, and can be compared with the Kd.
A measurement at a single concentration of inhibitor. This results in a %-inhibition effect (or a %-remaining activity). Because fewer data points are needed, this experiment is easier to perform. However, %-inhibition data are more prone to variation than dose-response data (the difference between 20% and 80% inhibition can originate from a mere fivefold difference in IC50). They are also less informative: an inhibitor of two kinases with IC50s of 0.1 nM and 100 nM, a 1000-fold difference, would inhibit both at a similar ∼100% in a 10 µM fixed concentration screen.
In our experience, the most efficient and cost-effective strategy is to determine the selectivity of a compound in two tiers: First, the compound is tested at a single concentration (in duplicate) to determine the target kinases. Subsequently, IC50s are determined for all targets that are inhibited more than, for example, 70%. For IC50 determination, a 10-point dose-response curve is preferred, although a 5-point dose-response curve can already yield reliable data. If a compound is relatively selective, the follow-up is only a small study that makes the entire profiling study equivalent to a full-scale IC50-based profiling.
Overview of published profiling studies
Selectivity profiles are increasingly found in publications where new inhibitors are presented (see below for examples), and this practice can only be encouraged. In addition, many very interesting studies exist in which entire inhibitor sets are selectivity profiled, allowing direct comparison of the selectivities of existing inhibitors. The first of these studies (Davies et al., 2000) showed the selectivity profiles (single concentration data) of many common kinase reference compounds at the time. This was later extended with more reference inhibitors (again single concentration data, Bain et al., 2007). Both studies gave clear guidelines on which inhibitors to use when investigating the biological actions of certain kinases.
Another milestone was the study by Fabian et al. who studied the selectivity of 20 kinase inhibitors that have been investigated in clinical trials in a dose-response binding assay on 119 kinases (Fabian et al., 2005). This study demonstrated the promiscuity of some kinase drugs and drug candidates. It was followed by a larger dose-response studies of 38 clinically advanced kinase inhibitors on 317 kinases (Karaman et al., 2008) and 72 inhibitors on 442 kinases (Davis et al., 2011), which included the proposal of new tools for the quantification of selectivity. Both studies, published by Ambit, remain exquisite sources for the selectivity of known inhibitors.
Other profiling studies include the cross-reactivity of 156 commercially available protein kinase inhibitors on 60 human Ser/Thr kinases, using a single-concentration thermal shift assay (Fedorov et al., 2007). Though thermal shifts are not necessarily IC50s (Fedorov et al., 2007), and the kinase panel is a particular subset of the kinome, this study provides selectivity data on many readily available and frequently used inhibitors. Even more such compounds were recently tested at single concentration in an activity assay (178 inhibitors on 300 kinases, Anastassiadis et al., 2011). Both of these datasets provide an essential reference for interpreting old and new literature. Recently, even larger studies were released, such as the single point activity data of 577 inhibitors on 203 kinases, combined with IC50 profiles of 18 reference inhibitors (Bamborough et al., 2008), and the massive single concentration profiling study of 21851 inhibitors on 317–402 kinases (Posy et al., 2011). These studies generated interesting statistics on the pharmacological similarity of kinases. However, only few chemical structures were released. Nevertheless, they showed convincingly that, aided by X-ray structures, selectivity can be achieved on most kinase targets, starting from many different scaffolds (Johnson, 2009; Posy et al., 2011).
A great contribution to the field was the recent large-scale publication of the binding Kds of 3800 compounds against 172 kinases (Metz et al., 2011). The data and structures of 1500 compounds were uploaded to the Pubchem database (http://pubchem.ncbi.nlm.nih.gov). The value of these data for tool compound discovery is hard to overstate. For virtually any of the kinases studied, new inhibitors were identified that are more selective than the known reference inhibitors, although the stability and cellular activity of these new tool compounds still needs to be established.
A new trend is the use of cross-screening in drug discovery (Goldstein et al., 2008), as demonstrated by the recent profiling studies of 936 fragments on 30 kinases (Bamborough et al., 2011), and 118 new compounds against 353 kinases (Miduturu et al., 2011). The latter resulted in new tool compounds for previously untargeted kinases. Kinase cross-screening is an eminent method in drug discovery, because it generates potency and selectivity data in one study. Furthermore, and in contrast to high-throughput screening, it enables the picking of multi-targeted lead structures (Vieth et al., 2004; Hopkins, 2008).
Finally, it must be stressed that kinase profiling for assessing selectivity is limited in two ways. First, it uses a fixed number of assays, while there are many more biological targets in a cell. To address this, various labs have developed proteomics-based methods to capture protein targets directly from cell lysates (Bantscheff et al., 2009; Rix and Superti-Furga, 2009; Patricelli et al., 2011). In addition, other profiling panels have been set up, for example, of GPCRs and targets implicated in drug safety (Hamon et al., 2009; Heilker et al., 2009). These approaches have shown that kinase inhibitors, as any drugs, can also bind to non-kinase targets and this should be kept in mind when interpreting kinase profiles.
Second, though kinase profiling provides a good view of an inhibitor's activity on individual targets, the situation inside a living cell is more complex. Efflux pump activity, ATP levels, viscosity, protein concentration, scaffolding, target location, etc., will all affect an inhibitor's ability to bind to a target, and these factors can change with a cell's metabolic and differentiated status. Therefore, biochemical selectivity must be seen as a firm base from which to explore cellular selectivity and cellular signalling as a whole.
Displaying and quantifying selectivity profiling data
The result from any selectivity profiling is a table that contains 200–400 IC50s for each compound. The next step is then to represent those data in a meaningful way. Graphic methods to display selectivity data include heat maps (Fedorov et al., 2007; Bamborough et al., 2008), sorted activity on radar plots (Mihara et al., 2008; Verkaar et al., 2011) and dotting the phylogenetic tree of all human kinases (Manning et al., 2002) with circles, where the diameter of each circle represents compound activity (Fabian et al., 2005; Karaman et al., 2008, Figure 1). Graphic representation has the advantage that the information on individual kinases is retained. However, many selectivity questions cannot be answered by qualitative diagrams, for example: ‘what is the most selective inhibitor?’ Such quantitative questions are very relevant when choosing tool compounds.
Figure 1.
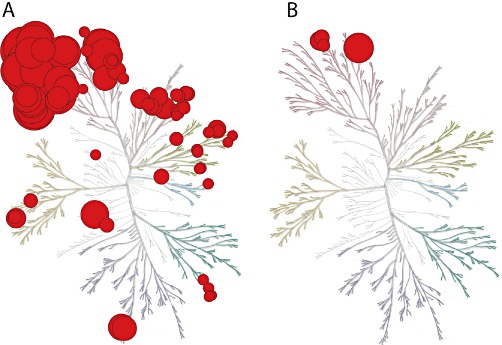
Graphical representation of inhibitor selectivity, based on the phylogenetic tree of human kinases (Manning et al., 2002). (A) Dasatinib is an example of a promiscuous inhibitor. (B) GW2580 is an example of a selective inhibitor. The larger the diameter of the red circles, the more potent the binding affinity. The kinome tree was first introduced by Manning et al. (2002). Reproduced with permission from the paper by Karaman et al. (2008). The kinome tree was reproduced courtesy of Cell Signalling Technology, Inc. (http://www.cellsignal.com).
Quantitative questions can be solved by the emerging science of deriving a single selectivity value from profiling tables. Conceptually, such a value can be compared with an IC50 or logP to indicate potency or lipophilicity, but instead indicating the general selectivity of a compound. Selectivity values can be used to rank compounds, greatly facilitating the identification of suitable tool compounds.
The first proposed methods to quantify selectivity were threshold methods such as the promiscuity score (Fedorov et al., 2007) and the selectivity score (Karaman et al., 2008). The selectivity score divides the number of kinases hit below a certain threshold (usually 3 µM) by the number of kinases tested. The method has the advantage that it is simple to calculate, but has the drawback that it is arbitrary. For instance, the score is equally high if an off-target kinase is inhibited at 1 µM or is inhibited at 1 nM, whereas the first IC50 is highly preferred in a selective inhibitor. In this way, the use of a cut-off can lead to the erroneous conclusion that more potent kinase inhibitors are less selective (Posy et al., 2011).
A method that avoids thresholds is the Gini score, which quantifies the curvature of a graph of sorted %-inhibition values (Graczyk, 2007). The higher the curvature, the more selective the inhibitor. The score is named after Corrado Gini, who used it to quantify income distribution disparities. The Gini score, thus, is a rare example of a quantitative method that has crossed over from the social sciences to the natural sciences. The Gini score has no physical–chemical meaning, and works with %-inhibition data.
A method that does have physical meaning, and which uses Kd and IC50 data, is the partition coefficient (Cheng et al., 2010). The partition coefficient determines the theoretical distribution of inhibitor molecules in a hypothetical kinase mixture, and calculates the fraction of inhibitor molecules bound to an assigned reference kinase. A similar partition coefficient is used to quantify selectivity in analytical chemistry (Peters et al., 2009). The assignment of a reference kinase makes the partition coefficient a biased approach. Depending on the project it is used for, an inhibitor can have different selectivity scores. The method also assigns similar values to an inhibitor that potently inhibits one off-target kinase and an inhibitor that less potently inhibits a plethora of off-target kinases (Cheng et al., 2010; Uitdehaag and Zaman, 2011).
The selectivity entropy
To remedy the drawbacks of the said methods, we recently proposed a selectivity entropy value. In many fields, such as bio-informatics and physical chemistry, entropy values are used to quantify the specificity of an effect by the broadness of the frequency distribution of all possible effects (Godden et al., 2000). The selectivity entropy uses this background to quantify the theoretical binding distribution of inhibitor molecules over all kinases in the selectivity panel (Uitdehaag and Zaman, 2011). It can also be regarded as a weighted summation of all possible partition coefficients in the kinase panel (Uitdehaag and Zaman, 2011).
High selectivity entropy indicates a promiscuous compound, whereas low selectivity entropy specifies selective compounds. The pan-kinase inhibitor staurosporine has an entropy score of 2.9, whereas compounds that only hit one kinase in the entire profile have an entropy score of 0 (Table 3). The selectivity entropy does not require assignment of a reference kinase, uses Kd and IC50 values, and has thermodynamic meaning. Selectivity entropy can be easily calculated in Excel (for instructions, see Uitdehaag and Zaman, 2011). In addition, we have built a website (accessible via http://www.entropy.99k.org) where a table of IC50 values can automatically be converted into a set of selectivity entropies.
Table 3.
Recommended tool compounds for important kinase targets
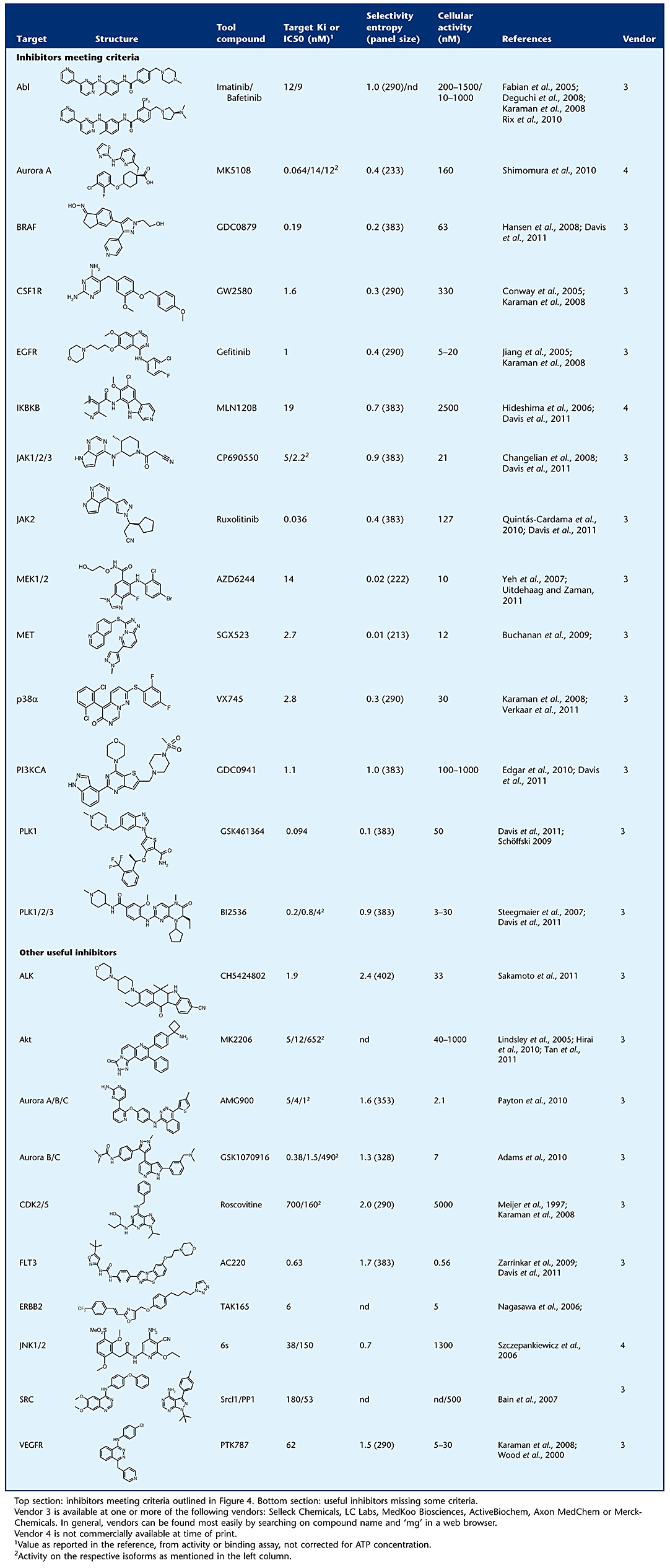 |
Recently, all methods for selectivity quantification were compared (Uitdehaag and Zaman, 2011). In general, the more advanced methods (Gini score, partition coefficient, selectivity entropy) give comparable rankings of compounds. However, in a test where 16 compounds were profiled in two different labs, the entropy method gave most consistent values, indicating that the entropy score is preferred when comparing selectivity profiles from separate labs.
Statistics of the selectivity entropy
The universality of selectivity entropy is demonstrated by large profiling datasets all showing similar average entropies and entropy distributions (Figure 2). The binding data of Karaman et al. (2008) show an average entropy of 1.8 and a median of 1.9. (2.0 and 2.1, respectively, in the follow-up work by Davis et al., 2011). A large activity-based dataset has values of 1.9 and 2.0 respectively (Uitdehaag, 2010, Figure 2). The data from Metz et al. (2011) show average and median entropies of 2.2 (Figure 2). The similarity of all these values confirms that the average selectivity entropy in a panel of about 200 kinases is 2.0 (Uitdehaag and Zaman, 2011). Compounds with an entropy score lower than 2.0 are therefore ‘more selective than average’. However, compounds with entropies below 1.0 are frequently found in cross-screens (Figure 2). Such highly selective tool compounds are much more useful for validating kinase targets than those ‘more selective than average’.
Figure 2.
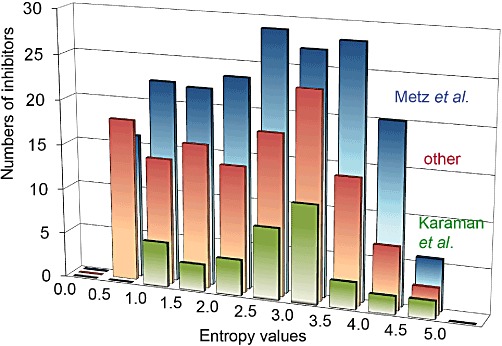
Distribution of the selectivity entropies of inhibitors in profiling datasets. The bars indicate the numbers of inhibitors in the indicated datasets with entropies between values at the x-axis. The distribution indicated ‘other’ originates from an activity-based dataset of 127 cpds profiled on 222 kinases (Uitdehaag, 2010). For the Metz et al. (2011), dataset, numbers on the y-axis need to be multiplied by 20. All three independent datasets have a similar entropy distribution.
Another crucial value is the SD on a single selectivity entropy determination for a particular compound. This is a measure of the statistical relevance of entropy differences. Errors in entropy values can arise from assay differences, different panel sizes and non-overlapping panel composition. In an experiment where 16 compounds were profiled in two different labs, all three error sources played a role resulting in an SD of 0.3 (Uitdehaag and Zaman, 2011).
When looking at the individual sources of variation, panel size is important because the entropy value tends to increase when more kinases are tested (Figure 3) (Brandt et al., 2009). When values originating from widely different panel sizes are compared, ideally a logarithmic panel size correction needs to be applied (Uitdehaag and Zaman, 2011, Figure 3). In reporting entropy values, panel sizes should therefore be given (Table 3). Secondly, variation due to panel composition can be assessed by recalculating entropies from random sample panels (Figure 3). For the archetypal promiscuous inhibitor dasatinib (Figure 1), determining an entropy score in different panels of 200 kinases results in an SD of 0.10 (Figure 3). For the selective inhibitor GW2580 (Figure 1), this is 0.19 (Figure 3). Finally, variation due to assay reproducibility was modelled by multiplying assay data (Karaman et al., 2008) with a random factor between 0.1 and 10, and recalculating entropies. Repeating this 50 times with different random factors, results in an SD of 0.20 for dasatinib, and 0.32 for GW2580. Assay reproducibility thus seems the major source of error in entropy determination, and for the selectivity entropy of any compound, an SD of about 0.3 should be taken into account.
Figure 3.
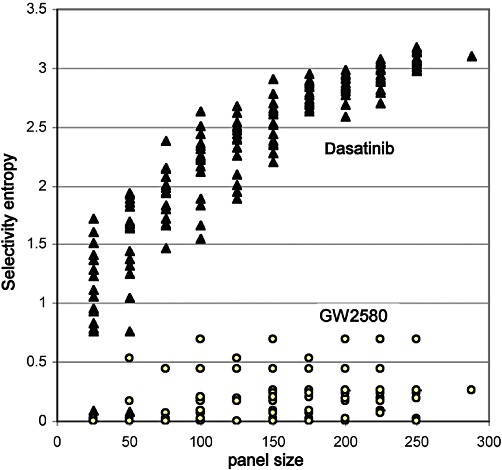
Dependency of the selectivity entropy on panel size. The Karaman et al. (2008) dataset was divided into 20 random subpanels with 11 different sizes. The selectivity entropies for the promiscuous dasatinib and the selective GW2580 were calculated for each panel. For dasatinib, a reasonable entropy guess can be made from a 200-kinase panel. For GW-2580, which inhibits only two kinases, variation is in practice independent of panel size. For every panel size, SDs were calculated for GW2580 and dasatinib. For a panel size of 200, the SD for dasatinib is 0.10, and the SD for GW2580 is 0.19. If all SDs from all panel sizes are averaged, the final average SD is 0.2.
General approach for selecting tool compounds
The advent of single-value selectivity scores allows more rational picking of selective tool compounds (Figure 4). To start such a process, the selectivity entropy scores of publicly available inhibitors need to be calculated and rank ordered. In general, the most selective of these should be chosen. If the selectivity entropies are derived from kinase panels with largely differing sizes, the inhibitors that are profiled over many kinases (>100) are preferred. The best tool inhibitors have an entropy smaller than one, but if no such exquisitely selective compounds exist, multiple compounds can be selected for use in parallel (Figure 4). However, if no compounds with an entropy score smaller than two are available, cross-reactivity becomes an unacceptable risk even when multiple inhibitors are used, and one should rather start developing a new tool compound than planning any biological experiment with one of the available inhibitors.
Figure 4.
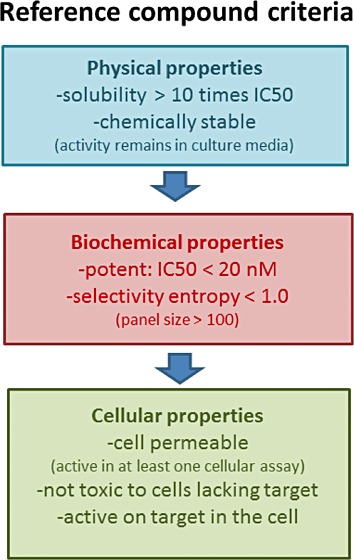
Flowchart for the selection of a good kinase tool inhibitor. Physical, biochemical and cellular properties should be assessed before using an inhibitor as reference. These criteria were used for the inhibitor selection of Table 3. Most in vivo or clinically tested inhibitors will have suitable solubility, stability and activity characteristics, leaving only the selectivity to be assessed.
The parallel use of multiple compounds is only meaningful if these compounds are structurally dissimilar and do not exhibit comparable cross-reactivity profiles. The latter could potentially be excluded by calculating correlation coefficients of both inhibitor kinase profiles, but visual inspection of the cross-screening data is often more practical.
It is very important that selected compounds have proven activity and stability in a cell, disqualifying unproven screening hits as pharmacological tools. As a guideline, Figure 4 lists properties that are essential for any tool compound for use in vitro. For in vivo applications, compounds additionally need to be stable enough to monitor the biological effect after application via the desired route and lack acute toxicities. As informal guideline, compounds that have been clinically evaluated and meet the selectivity criteria will almost certainly qualify as good tool compounds.
As not all tool compounds are equally selective, we recommend that in all cases where kinase tool compounds are used in biological studies, selectivity values are mentioned, with reference to the profiles in the literature. However, study of the individual profiles remains very important, particularly if biological validation focuses on particular off-target kinases, for instance isoforms. Only with access to all information, a pharmacological validation with kinase inhibitors can be properly assessed.
Tool compounds for clinically relevant kinases
To illustrate our guidelines for picking selective tool compound from cross-screening data, we have selected the most suitable tool inhibitors for several frequently investigated kinase drug targets, using the criteria of Figure 4 (Table 3). Targets were selected on the basis of the number of articles in Pubmed and the progression of compounds hitting these targets in the clinic. Throughout the text, the word spectrum-selective is used to indicate an inhibitor that predominantly inhibits members of a particular kinase subfamily. The suffix pan- (as in pan-Aurora) is used to indicate inhibitory activity on all isoforms of that target. Hugo Gene Nomenclature Committee-approved names for kinases are listed in the section titles (in brackets where a kinase has a dominant trivial name). For entropy calculations, all activities on non-human and mutant kinases were discarded, leading to reduced panel sizes compared with the literature (e.g. 290 kinases were included from Karaman et al., 2008 and 383 kinases from Davis et al., 2011).
Abl (ABL1)
Chronic myeloid leukaemia (CML) is caused by a chimeric BCR-ABL gene that is a driver of malignant transformation (Hantschel and Superti-Furga, 2004). The clinical success of the Abl inhibitor imatinib in treating CML heralded the emergence of kinase inhibitors as a drug target class (Druker et al., 2001). The initial euphoria brought about by the efficacy of imatinib was followed, however, by the emergence of resistant tumours (Hantschel and Superti-Furga, 2004). Resistance is caused by point mutations in Abl that render imatinib incapable of binding. Since then, second-generation Abl inhibitors that target mutant Abl and display improved potency were generated (Manley et al., 2010). Broadening the spectrum of Abl mutants being hit and losing selectivity may have gone hand in hand, because these follow-up molecules are generally less selective than imatinib. For instance, the entropy scores for nilotinib (1.7), DCC2036 (2.9), dasatinib (3.2) and PD173955 (3.3) are considerably higher than that of imatinib (0.8) (Karaman et al., 2008; Manley et al., 2010; Chan et al., 2011; Davis et al., 2011). For other Abl inhibitors, only limited selectivity information has been published, though it can be inferred that they are not exquisitely selective. For example, the dual Src/Abl inhibitor saracatinib inhibited 11 kinases with IC50s below 100 nM out of a panel of 23 kinases tested (Green et al., 2009), and bosutinib inhibited 63 kinases for more than 80% at 1 µM out of a panel of 272 kinases tested (Remsing-Rix et al., 2009; Rix and Superti-Furga, 2009). Probably, the most selective out of these second-generation Abl inhibitors is INNO406/bafetinib, which inhibited 23 kinases for more than 80% at 1 µM out of 272 kinases tested (Rix et al., 2010). We advise the use of imatinib in parallel with bafetinib to pharmacologically investigate the role of Abl or Bcr-Abl in physiologic processes.
ALK
Alk is a tyrosine kinase of which translocations drive anaplastic lymphoma, and a significant percentage of non-small cell lung cancers (Lovely et al., 2011). A clinical refocus of the dual MET/ALK inhibitor PF02341066/crizotinib has led to marked patient responses, opening the ALK field as an area of intense research. In one publication (Lovely et al., 2011), %-inhibition data are given on 96 kinases at 1 µM crizotinib, indicating activity (>80% inhibition) against in at least 20 kinases, including Abl, Axl, CSF1R, MET and ALK (Zou et al., 2007). Small profiles were released for NVP-TAE684 (Galkin et al., 2006), and more extensive profiles for X376 (Lovely et al., 2011) and AP261113 (Katayama et al., 2011). The spectrum selectivity of crizotinib and NVP-TAE684 was revealed recently by Davis et al. (2011), where they showed entropies of 3.0 and 3.7 respectively. Another thoroughly profiled and selective ALK reference compound in the public domain is CH5424802. In a 402 kinase profile at two concentrations, it inhibits ALK, LTK and GAK at single-digit nanomolar concentration, and 27 others at 10–1000 nM IC50 (Sakamoto et al., 2011). On the basis of these data, the selectivity entropy of CH5424802 is 2.4. Two compounds that only have been characterized biochemically (Metz et al., 2011), PSID (Pubchem substance ID) 103904390 and 103904391, are more selective than CH5424802. Both only hit ALK in a 172 kinase profile, and have a selectivity entropy of 0.01. Awaiting further characterization of PSID 103904390 and 103904391, CH5424802 is the most suitable tool compound for ALK.
Akt (AKT1)
Akt (or PKB) was initially identified as the gene driving oncogenic transformation through the retrovirus Akt-8 (Bellacosa et al., 1991). Consequently, Akt was found to be up-regulated in a plethora of cancers and to be instrumental for tumour growth (Mitsiades et al., 2004; Cheng et al., 2005). The allosteric Akt inhibitor MK2206 has been suggested to be a highly selective inhibitor (Lindsley et al., 2005), but no cross-screening data have been disclosed. The only reference to its selectivity over other kinases than Akt isoforms is the reiterated statement that MK2206 is selective over PKA, PKC and SGK (Lindsley et al., 2005; Hartnett et al., 2008; Wu et al., 2008; Zhao et al., 2008). Also for perifosine, which is currently under clinical evaluation for the treatment of neuroblastoma, no selectivity data are available in the public domain. The Akt inhibitors for which selectivity data have been published are not uniquely selective for Akt. For instance, GSK690693 has a calculated selectivity score of 2.7, based on a panel of 94 kinases (Rhodes et al., 2008), and A674563 has a selectivity entropy of 2.0, calculated from a profiling effort encompassing 383 kinases (Davis et al., 2011). Other Akt inhibitors include CCT128930, which inhibited seven kinases >80% at 10 µM over 47 kinases tested (Yap et al., 2011), and AT7867, which inhibited 5 out of 19 kinases tested with IC50s below 100 nM (Grimshaw et al., 2010).
Due to its allosteric binding mode, MK2206 is likely to be very selective for Akt in comparison with other Akt inhibitors. Nevertheless, the data demonstrating its selectivity should be published (Table 3).
Aurora kinases A (AURKA) and B (AURKB)
Because of the importance of Aurora kinases in mitosis, these serine/threonine kinases were among the first kinases pursued for oncology indications (Katayama and Sen, 2010). However, no Aurora inhibitors have reached the market so far. Of the three Aurora isoforms (A, B and C), both Aurora A and Aurora B have been identified as targets, and as a result, pan-Aurora inhibitors as well as those with specificity towards Aurora A or Aurora B have been developed (Yan et al., 2011).
Many Aurora inhibitor profiles have been published. A single-concentration profile of SU6668 (Bain et al., 2007), a 35-kinase profile of danusertib/PHA739358 (Carpinelli et al., 2007) and a full IC50 profile of VX680/MK0457/tozasertib (entropy 3.1, Karaman et al., 2008) have been published. These studies revealed all three inhibitors as promiscuous. Of ZM447439, which is mentioned to be ‘a more specific inhibitor’, unfortunately only a 16-kinase profile has been published (Ditchfield et al., 2003).
Full profiling showed better selectivity for the Aurora B/C selective AZD1152HQPA (entropy 1.9) and the Aurora A selective MLN8054 (entropy 1.9) (Karaman et al., 2008; Davis et al., 2011). Furthermore, recent efforts identified even more selective inhibitors, such as the pan-Aurora inhibitors SNS314 (entropy 1.4 in a 219 kinase panel, Arbitrario et al., 2010) and AMG900 (entropy 1.6 in a 353 kinase profile, Payton et al., 2010). AMG900 is about equally active on all Auroras. GSK1070916 inhibits Aurora B and Aurora C about 1000-fold more potent than Aurora A and has an entropy of 1.3 in a 328 kinase panel (Adams et al., 2010). The Aurora A inhibitor MK5108 is selective over B and C and has an entropy of 0.44 in a 233 kinase panel (Shimomura et al., 2010). All these inhibitors have single-digit nanomolar biochemical and cellular potencies (Table 3). This makes the Aurora kinase field well equipped with good tool compounds.
BRAF
The BRAF and RAF1 (CRAF) isoforms play an essential role in cell proliferation. The BRAF mutant V600E is a driving mutation in the majority of melanomas and some other cancers (Flaherty et al., 2010). Many pharmaceutical companies have developed BRAF inhibitors, culminating in astounding clinical success (Solit and Sawyers, 2010). RAF-selective tool compounds could help to contribute to our understanding of wild-type RAF signalling. A large IC50-based profile of the RAF inhibitor sorafenib revealed that it has below average selectivity (entropy 2.2, Karaman et al., 2008). For GW5074 and ZM336372, single concentration testing indicates substantial off-target activities (Bain et al., 2007). For SB590885 and SB-699393, only partial profiles have been published (Takle et al., 2008). In another publication (Hansen et al., 2008), a profile on 212 kinases is mentioned, unfortunately without disclosing the underlying data. More selective is PLX4720 that only substantially inhibits BRAF, RAF1 and Brk in a 65-kinase profile (Tsai et al., 2008), but in a 383 kinase profile surprisingly shows more potent activity on MEK5 than on BRAF (Davis et al., 2011). The best BRAF reference inhibitor is therefore GDC-0879, which only inhibits BRAF and RAF1 in panels of 140 kinases (Hoeflich et al., 2009) and383 kinases (entropy 0.2, Davis et al., 2011). (Table 3).
CDKs
Cyclin-dependent kinases (CDKs) are a highly conserved subfamily of 13 kinases, involved in regulating the cell cycle and transcription. CDKs are highly pursued potential cancer drug targets (Malumbres and Barbacid, 2009). Early drug discovery efforts resulted in the identification of several ‘classic’ CDK inhibitors, such as roscovitine/seliciclib for which the profiling in a small panel was already published in 1997 (Meijer et al., 1997). Subsequently, more extensive profiling (Fabian et al., 2005; Bain et al., 2007; Karaman et al., 2008; Davis et al., 2011) confirmed that roscovitine/seliciclib is averagely selective (entropy 2.0). Less selective are flavopiridol/alvocidib (entropy 2.5) and SNS032/BMS387032 (entropy 2.4), and the dual Aurora/CDK inhibitor JNJ7706621 (Emanuel et al., 2005), which is highly promiscuous (entropy 3.7).
Unfortunately, more recently developed compounds were only profiled in smaller panels, such as PD0332991 (Fry et al., 2004), PHA793887 (Brasca et al., 2010), P276-000 (Joshi et al., 2007), AZD5438 (Byth et al., 2009) and BS181 (Ali et al., 2009). Where newer compounds have been profiled more extensively, they have not shown high selectivity, such as R547 (entropy 2.2), AT7519 (entropy 2.4), EXEL2880 (entropy 3.2) (Squires et al., 2009; Davis et al., 2011). Nevertheless, the data suggest that it is possible to design more selective inhibitors. For instance, PHA793887 is a pan-CDK inhibitor that in a panel of 44 off-target kinases only hits GSK3β (Brasca et al., 2010). Selectivity within the CDK family is also attainable: for instance, the CDK7 selective BS181 has 40–2000 times specificity over other family members and only hits two other kinases in a 69-kinase panel (Ali et al., 2009). For proper comparison, the selectivity of these compounds needs to be quantified in larger panels. Until then, roscovitine remains one of the best CDK tool compounds (Table 3).
CSF1R
CSF1R (or Fms) is a tyrosine kinase that plays an important role in macrophage development and differentiation. CSF1R inhibitors target macrophages in inflammation and oncology (Hamilton, 2008). Many well-known tyrosine kinase inhibitors, such as imatinib, sorafenib and dasatinib, have nanomolar CSF1R activity, but profiling shows these are all spectrum-selective inhibitors (Karaman et al., 2008). An exception is GW2580 (Conway et al., 2005), which only hits CSF1R and Trk kinases in a panel of 290 kinases, and has a selectivity entropy of 0.3 (Karaman et al., 2008; Uitdehaag and Zaman, 2011). In a recent cross-comparison of CSF1R inhibitors in a panel of different assays, we confirmed that GW2580 is a very selective inhibitor (Uitdehaag et al., 2011).
EGFR and ERBB2
EGFR (Her1) and ERBB2 (Her2) are closely related receptor tyrosine kinases that are aberrantly expressed and/or activated in a plethora of cancers, most notably breast and lung cancer (Zhang et al., 2007). EGFR and ERBB2-targeting therapeutics include the antibodies cetuximab, panitumumab (both EGFR-specific) and trastuzumab (ERBB2), and the small molecule kinase inhibitors erlotinib, gefitinib, vandetanib and lapatinib (Zhang et al., 2007).
Fabian et al. (2005) and Karaman et al. (2008) showed that gefitinib and erlotinib are both very selective inhibitors of EGFR, with entropy scores of 0.4 and 0.9 respectively (Table 3). They do not possess cross-reactivity towards ERBB2 or other ERBB family members, and only significantly inhibit GAK (Karaman et al., 2008). Several other compounds, such as CI1033/canertinib (entropy score of 0.2), BIBW2992/afatinib (0.4), GW-2016 (0.6), lapatinib (0.7) and EKB569/pelitinib (1.4) have excellent selectivity but do not distinguish between EGFR and ERBB2 and are therefore not suitable as tool compounds (Fabian et al., 2005; Karaman et al., 2008; Davis et al., 2011). The marketed VEGFR/EGFR inhibitor vandetanib/ZD6474 is a promiscuous EGFR inhibitor, having an entropy score of 2.6, derived from a 119-kinase profile (Fabian et al., 2005).
Compounds that inhibit ERBB2 but not EGFR have been described (Barbacci et al., 2003; Lippa et al., 2007; Moasser, 2007). TAK165/mubritinib is the most selective over EGFR (Nagasawa et al., 2006). However, extensive selectivity data on other kinases is lacking for this and other ERBB2-specific inhibitors. The only presumed ERBB2-specific compound that has undergone rigorous selectivity testing is CP724714 (entropy score of 1.1), which was initially described to display 600-fold selectivity for ERBB2 over EGFR (Jani et al., 2007), but in binding assays appears equipotent on both kinases (Karaman et al., (2008). An enticing ERBB2-selective alternative is a compound from the Metz database with Pubchem Substance ID (PSID) 103905568). This inhibits ERBB2 with an IC50 of 1.6 nM and only cross-reacts with ERBB4 (IC50: 25 nM), yielding a selectivity entropy of 0.4 in a panel of 172 kinases.
FLT3
A translocation of the FLT3 gene (FLT3-ITD) is the driver mutation in certain types of leukaemia (Zarrinkar et al., 2009). Despite the fact that many tyrosine kinase inhibitors have FLT3 activity (Karaman et al., 2008), very few are selective for FLT3. When profiles of clinically used FLT3 inhibitors were compared (Zarrinkar et al., 2009), it appeared that MLN518/tandutinib and AC220/quizartinib are the most selective, with entropies of 1.6 and 1.8 respectively. A second profiling study showed both entropies to be 1.7 (Davis et al., 2011), which is consistent. Both compounds hit CSF1R, KIT, the PDGFRs and other receptor tyrosine kinases. AC220 is preferred as an FLT3 tool compound, because of its higher cellular potency compared with MLN518. In addition, it would be worthwhile to characterize the cellular activity of the 0.8 nM FLT3 inhibitor with PSID 103904858 (Metz et al., 2011), which hits only KIT, CSF1R, KDR and FLT1/4, and has a selectivity entropy of 1.0.
IKBKB
IKBKB (inhibitor of NF-κB kinase β, also known as IKK2 or IKKβ) is a crucial mediator of activation of NFκB signalling, which is centrally important in inflammation and cancer (Karin and Greten, 2005). For this reason, many labs have sought to develop IKBKB inhibitors, with or without specificity for its oligomeric partner IKK1. Profiles have been determined of first-generation IKBKB inhibitors such as TPCA-1 (Bamborough et al., 2008), PS1145 (Wen et al., 2006), as well as Calbiochem IKK inhibitor VII and Calbiochem inhibitor 401483 (Fedorov et al., 2007). In all cases, these studies revealed great promiscuity.
Recently, more specific inhibitors were published. For S1627, only a 12-kinase profile was published (Tegeder et al., 2004), but it was revealed that S0100230, from the same chemical class, only hits IKBKB in a 200 kinase profile (Ritzeler, 2011). Another related compound, SAR113945, is currently in clinical trials. PHA408 shows excellent specificity (350-fold) over IKK1 and only hits Pim-1 in a 30-kinase panel (Mbalaviele et al., 2009). Isoquinoline inhibitors, such as compound 21 in Christopher et al. (2009), only hit ROCK1 in a 59-kinase panel and have 25-fold selectivity over IKK1. Selectivity data for IMD1041, IMD0254 and BMS345541 are not available, unfortunately. A better characterized inhibitor is BI5700, a 9 nM IKBKB inhibitor that essentially only weakly hits off-target IKK1 and FLT3 in a 59-kinase profile, and has a cellular IC50 of 290 nM for IκBα phosphorylation (Huber et al., 2010). However, the best IKBKB inhibitor is MLN120B, which is related to PS1145 and was recently profiled (Davis et al., 2011), revealing 50-fold selectivity over IKK1 and a very low selectivity entropy of 0.7 (Table 3).
JAK2 and JAK3
Mutations in JAK2 have been implicated in polycythemia vera, whereas JAK3-deficient humans are severely immunodeficient, identifying these JAKs as targets in oncology and immunology respectively. The best known JAK inhibitor is tasocitinib/CP690550 (Changelian et al., 2008), which binds to JAK1, 2 and3 and TYK2 in a panel of 383 kinases (Davis et al., 2011), resulting in a selectivity entropy of 0.9. Tasocitinib is therefore an excellent pan-JAK reference inhibitor.
Selectivity for one specific JAK isoform seems most easily achieved for JAK2. The JAK2 specific inhibitor R723 was profiled in a 200-kinase panel, hitting 13 kinases (including JAK3) at 100 nM, and only JAK2 at 20 nM (Shide et al., 2011). SB1518 is 50-fold selective for JAK2 over JAK3, but also inhibits TYK2 and FLT3 in a 58-kinase panel (Hart et al., 2011). AZ-690 is even more selective for JAK2. When the compound was profiled at three concentrations on 83 kinases, it inhibited 11 significantly (>50%) at 100 nM, but none of the JAKs except JAK2 (Gozgit et al., 2008). The related AZD-1480 has comparable selectivity (Hedvat et al., 2009). The recommended JAK2 specific inhibitor, however, is INCB018424/ruxolitinib, a 36 pM (!) inhibitor of JAK2, which in a 383-kinase panel only significantly inhibits Tyk2 as off-target (entropy 0.4, Davis et al., 2011).
For JAK1, the dual JAK1/JAK2 reference inhibitor CYT387 has no JAK3 activity in a 141-kinase panel, profiled at two concentrations, although it potently inhibits six other kinases (Pardanani et al., 2009, estimated entropy 2.8). A fully selective JAK3 inhibitor has not yet been described.
JNK1/2/3 (MAPK8/910)
The JNK kinase family (JNK1, 2 and 3) consists of three independent genes that are activated upon a large range of cellular stressors, including cytokines, mitogens and osmotic stress (Manning and Davis, 2003). JNK2, and to a lesser extent JNK1, have been implicated in chronic inflammatory diseases, such as rheumatoid arthritis and asthma, and evidence supports a role for JNK3 in neurodegenerative disorders (Manning and Davis, 2003). The first clinically evaluated JNK inhibitor is SP600125 (Bennett et al., 2001). SP600125 has been used in over 800 articles to implicate JNK in cellular processes (Bogoyevitch and Arthur, 2008), despite the fact that profiling efforts suggested that the compound is not selective (Fabian et al., 2005; Bain et al., 2007). Indeed, the selectivity entropy score of SP600125 is 2.5, ranking it as a below-average selective inhibitor (Fabian et al., 2005). Although several inhibitors for JNK have since been described, most of these have only been termed selective without disclosure of the actual data (Scapin et al., 2003; Swahn et al., 2006; Ma et al., 2007). For instance, Kamenecka and colleagues describe a JNK inhibitor (compound 9l) which anecdotally inhibited 11 out of 400 kinases when tested at a concentration of 3 µM (Kamenecka et al., 2010). The most selective JNK inhibitor for which selectivity data have been published is compound 6s (Szczepankiewicz et al., 2006), which only inhibited JNK1, -2, -3 and ERK2 out of a panel of 74 kinases, with a selectivity entropy score of 0.7. Whereas all the said inhibitors exhibit only limited selectivity over JNK isoforms, a research compound (compound 5) developed by GSK (Angell et al., 2007) only inhibited JNK-3 >80% in a panel of 214 kinases which included JNK-1 and -2 (Bamborough et al., 2008). However, its cellular activity has not been demonstrated yet (Angell et al., 2007). In conclusion, we advise the use of compound 6s for general JNK inhibition.
MEK1/2 (MAP2K1/MAP2K2)
MEK1 and MEK2 are functionally overlapping MAP kinase kinases that act downstream of RAF. Many MEK1/2 inhibitors are currently undergoing clinical testing (Trujillo, 2011). Early on, allosteric and very selective, so-called type III inhibitors were discovered (Dudley et al., 1995; Ohren et al., 2004). All reported MEK inhibitors are dual MEK1/2 inhibitors, including UO126, PD184352, AZD6244, PD0325901, CH498765, TAK733, XL518, RDEA119 and GSK1120212, and all belong to the same chemical class. The single concentration profiles of UO126, PD184352 and PD0325901 revealed that they are very selective (Bain et al., 2007). More recently, the 222 kinase IC50 profiles of AZD6244 and PD0325901 confirmed their exquisite selectivity, with respective entropies of 0.02 and 0.55 in a panel of 222 kinases (Uitdehaag and Zaman, 2011). Of these, AZD6244/selumetinib is the most selective, essentially inhibiting no other kinases but MEK1/2 in the entire profile and exhibiting potent cellular and in vivo activity (Yeh et al., 2007).
MET
MET (or HGFR) is a tyrosine kinase of which activating mutations cause hereditary papillary renal carcinoma, and which has been implicated in many other malignancies (Munshi et al., 2010). Well-known MET inhibitors are SU11274, PHA665752 and MGCD265. However, broad kinome profiles of these inhibitors have not been published. SU11274 was profiled in a Ser/Thr kinase panel, where it inhibits at least seven kinases, most potently LOK (Fedorov et al., 2007). PF02341066/crizotinib, the dual ALK/MET inhibitor, is also not selective (see above). Recently, two highly selective inhibitors were published. ARQ197 is a non-ATP competitive inhibitor with a Ki of 355 nM, that at 10 µM only inhibits four other kinases out of a panel of 230 (Munshi et al., 2010). Even more potent and selective is SGX523, an ATP-competitive inhibitor (Ki= 2.7 nM) that at 1 µM only inhibits MET from a panel of 213 kinases (entropy 0.0, Buchanan et al., 2009), which was confirmed in a panel of 383 kinases (entropy 0.0, Davis et al., 2011). SGX523 is therefore the preferred MET reference compound.
p38α (MAPK14)
p38α is a highly pursued target for inflammatory diseases, such as rheumatoid arthritis, Crohn's disease, psoriasis and chronic obstructive pulmonary disease (Genovese, 2009; Lindstrom and Robinson, 2010). Clinical development of the earliest compounds was stopped due to liver, brain or skin toxicity (Genovese, 2009; Lindstrom and Robinson, 2010). Subsequently developed p38α inhibitors are increasingly selective. For instance, the entropy score of the clinically tested p38α inhibitor VX745 is 0.3, ranking it among the most selective tool inhibitors (Karaman et al., 2008; Uitdehaag and Zaman, 2011). Other highly selective p38α inhibitors include SCIO469 and ORG48762-0, both of which appear to have virtually no cross-reactivity towards kinases other than p38α and p38β, although they were only characterized on a limited set of kinases (Mihara et al., 2008; Verkaar et al., 2011). Of note, these kinase inhibitors are more specific than the most frequently used p38α tool inhibitors: SB203580 (entropy 2.3) and BIRB796 (entropy 1.2), the latter, for instance, being a potent inhibitor of JNK (Bain et al., 2007; Karaman et al., 2008). The in vitro potency of VX745 and SCIO469 is comparable with that of SB203580 (Verkaar et al., 2011).
PI3K family (PI3KCA/B/C/D/G and mTOR)
The PI3K family consists of 15 kinases that have pleiotropic roles in cellular signalling, such as cell growth, survival and differentiation (Katso et al., 2001). Of these kinases, mTOR and p110α, -β, -γ and –δ have been pursued for several indications. For instance, mutations in p110α are common in solid tumours (Samuels et al., 2004) and p110β is a target for the treatment of thrombosis (Jackson et al., 2005). Drug development for other PI3K isoforms (p110γ and δ) centres around inflammatory and auto-immune diseases (Rommel et al., 2007).
The most commonly used inhibitors to target PI3Ks are wortmannin, a fungal metabolite, and LY294002, a quercetin derivative. Both compounds selectively inhibit p110s and closely related kinases, such as mTOR and DNA-PK (Davies et al., 2000; Vanhaesebroeck et al., 2001; Raynaud et al., 2007), but display virtually no cross-reactivity towards 400 non-lipid kinases tested at 10 µM (Anastassiadis et al., 2011). The best-characterized pan-PI3K inhibitors are PI103 and GDC0941. Both compounds inhibit all p110 isoforms, mTOR1 and -2 with high potency (Knight et al., 2006; Davis et al., 2011). The selectivity entropy of PI103, as derived from the Karaman dataset (in which lipid kinases are relatively under-represented), is 0.05 (Karaman et al., 2008; Uitdehaag and Zaman, 2011). The entropy of PI103 calculated from a recent profiling effort that incorporated a larger panel of PI3K family members was 1.5, in which GDC0941 had an entropy of 1.0 (Davis et al., 2011). It should be noted that both compounds display virtually no cross-reactivity outside the PI3K family and are therefore excellent pan-PI3K-selective inhibitors (Davis et al., 2011). PI103 and GDC0941 are preferred over other pan-PI3K inhibitors like PP242 and TG-100–115, which have entropy scores of 3.3 and 2.1 respectively (Davis et al., 2011).
More recent drug optimization projects have yielded inhibitors that display isoform specificity. For instance, the p110γ-specific inhibitor AS-605240 displays about 10-fold selectivity over other PI3Ks (Camps et al., 2005).
Several p110δ-specific inhibitors have been described. IC287114 is approximately 10- to 20-fold selective for p110δ over other PI3Ks and does not inhibit any kinase out of 110 kinases, when tested at 10 µM (Knight et al., 2006; Jamieson et al., 2011). The IC287114 derivatives PIK293 and PIK294 inhibit p110δ with somewhat improved potency (10 nM vs. 130 nM for PIK294 and IC287114, respectively), while retaining 10- to 20-fold selectivity over other PI3Ks (Knight et al., 2006; Jamieson et al., 2011). Unfortunately, no selectivity data for PIK293 and PIK294 towards other kinases have been disclosed (Knight et al., 2006). A slightly more selective (at least 40-fold) p110δ inhibitor is CAL101, which has anecdotally been described to inhibit no kinase other than PI3K isoforms out of a panel of 402 kinases (Lannutti et al., 2011). Although these data should be disclosed or verified, CAL101 is probably the best p110δ-selective inhibitor.
PIK75 was previously described to be a p110α/γ-specific inhibitor (Knight et al., 2006), but a recent screening effort showed that apart from PI3K isoforms, PIK75 inhibited 68 out of 110 kinases at 10 µM (Jamieson et al., 2011). A more selective p110α-selective inhibitor is A66, which is 40-fold selective over other PI3Ks and only inhibited CLK4 and PI4Kβ out of 318 kinases tested at 10 µM (Jamieson et al., 2011).
Probing of p110β function is best performed with TGX221, which is 10-fold selective over other PI3Ks and inactive on 110 kinases when tested at 10 µM (Jackson et al., 2005; Jamieson et al., 2011).
Finally, the individual roles of mTORs can be dissected with rapamycin, a highly selective allosteric inhibitor of mTOR (Bain et al., 2007).
PLK1
PLK1 (polo-like kinase 1) is centrally important in mitosis and therefore an interesting drug target in oncology. Initial clinical tests in a variety of cancers showed great promise, especially in combination therapy (Schöffski, 2009). To further study the role of PLK1 in a non-therapeutic setting, inhibitors such as ON01910, HMN214 and LFM-A13 are less suited because of their activities on other kinases, including cell cycle kinases such as CDKs (Steegmaier et al., 2007; Uckun et al., 2007; Schöffski, 2009). More selective inhibitors are BI2536, BI6727 and GSK461364. The profiling data of two compounds related to GSK461364 show off-target activity (within a 100-fold window) on NEK2 and PDGFR1β (but not on Plk2) in a 57 kinase profile (Emmitte et al., 2009). For BI6727/volasertib, a 50-kinase profile was mentioned, but not published (Rudolph et al., 2009). For BI2536, a 63 kinase profile was published, showing essentially no off-target activity, except on Plk2 and Plk3 (Steegmaier et al., 2007). Recently, the first full profiling studies of BI2536 and GSK461364 were published, demonstrating their exquisite selectivity (entropies 0.9 and 0.1, respectively Davis et al., 2011). Both compound have a potent nanomolar cellular activity (Steegmaier et al., 2007; Schöffski, 2009). However, whereas BI2536 is a PLK1/2/3 inhibitor, GSK461364 is exclusively selective for PLK1, making it the most suitable tool compound.
SRC
SRC belongs to a closely related family of non-receptor tyrosine kinases, comprising nine members. SRC has been assigned critical roles in cellular survival and proliferation, and elevated SRC activity and/or expression has been observed in a variety of cancers (Belsches-Jablonski et al., 2005). Many SRC inhibitors have been reported, but most are not selective. For example, saracatinib, bosutinib and dasatinib all inhibit SRC, but also many other kinases (Karaman et al., 2008; Green et al., 2009; Remsing-Rix et al., 2009). More selective SRC inhibitors are SU-6656 (Blake et al., 2000), SRC-I1 (Tian et al., 2001), PP1 and PP2 (Hanke et al., 1996). These inhibitors have been profiled at a single concentration against 73 kinases by Bain and colleagues (Bain et al., 2007). Their analysis revealed that SU-6656 inhibited nine kinases more than 80% at 1 µM, whereas the other compounds were slightly more selective (SRC-I1, PP1 and PP2 inhibited 5, 4 and 4 kinases at 1 µM respectively). Several other inhibitors have been suggested to be selective for SRC, such as A-419259 (Wilson et al., 2002), AZM475271 (Yezhelyev et al., 2004), AP23846 (Summy et al., 2005), MNS (Wang et al., 2007) and PH006 (Ma et al., 2010), but these have not been profiled extensively. The biochemical analysis of Metz and colleagues revealed an interesting compound (PSID 103905316) that inhibits SRC (IC50 of 3.2 nM), and significantly cross-reacts with Fyn and EGFR only (IC50s of 1 and 32 nM, respectively) and has an entropy score of 1.1.
As PP1 and PP2 are structurally similar compounds with overlapping cross-reactivities, we recommend using either of these inhibitors in parallel with SRC-I1 to probe SRC function, as previously suggested by Bain and colleagues (Bain et al., 2007).
VEGFRs (FLT1/KDR/FLT4)
Members of the VEGFR family of receptor tyrosine kinases (VEGFR1/FLT1, VEGFR2/KDR/FLK1 and VEGFR3/FLT4) are important mediators of pro-angiogenic signals, and VEGFR signalling is imperative for tumour angiogenesis (Rini, 2007; Bhargava and Robinson, 2011). Three compounds that inhibit VEGFRs have reached the market in the last 5 years: sorafenib, sunitinib and pazopanib. Treatment with any of these compounds is associated with adverse events, and subsequent drug development activities have centred on improving selectivity (Bhargava and Robinson, 2011). Indeed, sunitinib and sorafenib are not selective VEGFR inhibitors, with entropy scores of 2.0 and 2.2 (Uitdehaag and Zaman, 2011). Likewise, pazopanib has an entropy score of 2.0 when tested over a panel of 61 kinases (Kumar et al., 2009). Other VEGFR inhibitors that were profiled by Karaman et al. (2008) include ZD-6474/vandetanib (entropy score of 2.9), CHIR265 (2.5), AMG706 (2.4), GW786034 (2.1), ABT869 (1.9) and PTK787/vatalanib (1.5). Vatalanib inhibits VEGFR1-3 and significantly cross-reacts with several related kinases, such as KIT, CSF1R and PDGFRs. Other VEGFR inhibitors, such as KRN951/tivozanib (Nakamura et al., 2006) and AZD2171/cediranib (Wedge et al., 2005), are also not selective over these closely related kinases. A possible exception is brivanib, which is inactive on PDGFRs, but which has only been profiled on nine kinases (Bhide et al., 2006). Vatalanib is therefore recommended as a pan-VEGFR tool compound.
Some VEGFR2-specific compounds have been described. Of these, ZM323881 is probably the most selective over VEGFR1 (Whittles et al., 2002; Spencer et al., 2009). However, its activity towards other kinases was not adequately covered by a selectivity panel, as this panel consisted of only five kinases (Whittles et al., 2002).
Conclusion
Cross-reactivity is an inherent property of most kinase inhibitors. Nonetheless, it is still common practice to describe compounds as ‘selective’ based purely on anecdotal evidence. Fortunately, it is becoming increasingly customary to accompany the description of a new kinase inhibitor with kinase panel screening data. These data are supplemented with large-scale cross-screening initiatives, which expand our knowledge of the selectivity of established reference inhibitors. Such activities will not only fill our compound tool box, but will also provide information on which tools to use for pharmacological experiments.
We have provided a guideline for picking the most selective tool compound for a large set of clinically relevant and intensively pursued kinase drug targets. Tool compounds for many other kinases can be found in the compound literature and cross-screening studies. For the targets discussed here, the recommended inhibitors provide a snapshot of the publicly available inhibitors and their cross-reactivities known at this moment in time. Updates will follow in due course.
Acknowledgments
FV was funded by the Dutch Top Institute Pharma initiative (Project D1-105). The authors thank Anne Watts for critically reading the manuscript, and Patrick Zarrinkar for providing high-resolution files for Figure 1.
Glossary
- PSID
Pubchem Substance Identification no
Conflict of interest
The authors declare no conflicts of interest for this paper.
References
- Adams ND, Adams JL, Burgess JL, Chaudhari AM, Copeland RA, Donatelli CA, et al. Discovery of GSK1070916, a potent and selective inhibitor of Aurora B/C kinase. J Med Chem. 2010;53:3973–4001. doi: 10.1021/jm901870q. [DOI] [PubMed] [Google Scholar]
- Ali S, Heathcote DA, Kroll SHB, Jogalekar AS, Scheiper B, Patel H, et al. The development of a selective cyclin-dependent kinase inhibitor that shows antitumor activity. Cancer Res. 2009;69:6208–6215. doi: 10.1158/0008-5472.CAN-09-0301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An X, Tiwari AK, Sun Y, Ding PR, Ashby CR, Jr, Chen ZS. BCR-Abl tyrosine kinase inhibitors in the treatment of Philadelphia chromosome positive chronic myeloid leukemia: a review. Leuk Res. 2010;34:1255–1268. doi: 10.1016/j.leukres.2010.04.016. [DOI] [PubMed] [Google Scholar]
- Anastassiadis T, Deacon SW, Devarajan K, Ma H, Peterson JR. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1039–1045. doi: 10.1038/nbt.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angell RM, Atkinson FL, Brown MJ, Chuang TT, Christopher JA, Cichy-Knight M, et al. N-(3-cyano-4,5,6,7-tetrahydro-1-benzothien-2-yl)amides as potent, selective, inhibitors of JNK2 and JNK3. BioOrg Med Chem Lett. 2007;17:1296–1301. doi: 10.1016/j.bmcl.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Arbitrario JP, Belmont BJ, Evanchik MJ, Flanagan WM, Fucini RV, Hansen SK, et al. SNS-314, a pan-aurora kinase inhibitor, shows potent anti-tumor activity and dosing flexibility in vivo. Cancer Chemother Pharmacol. 2010;65:707–717. doi: 10.1007/s00280-009-1076-8. [DOI] [PubMed] [Google Scholar]
- Bain J, Plater L, Elliott M, Shpiro N, Hastie J, McLaughlan H, et al. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamborough P, Drewry D, Harper G, Smith GK, Schneider K. Assessment of chemical coverage of kinome space and its implications for kinase drug discovery. J Med Chem. 2008;51:7898–7914. doi: 10.1021/jm8011036. [DOI] [PubMed] [Google Scholar]
- Bamborough P, Brown MJ, Christopher JA, Chung CW, Mellor GW. Selectivity of kinase inhibitor fragments. J Med Chem. 2011;54:5131–5143. doi: 10.1021/jm200349b. [DOI] [PubMed] [Google Scholar]
- Bantscheff M, Scholten A, Heck AJ. Revealing promiscuous drug-target interactions by chemical proteomics. Drug Discov Today. 2009;14:1021–1029. doi: 10.1016/j.drudis.2009.07.001. [DOI] [PubMed] [Google Scholar]
- Barbacci EG, Pustilnik LR, Rossi AM, Emerson E, Miller PE, Boscoe BP, et al. The biological and biochemical effects of CP-654577, a selective erbB2 kinase inhibitor, on human breast cancer cells. Cancer Res. 2003;63:4450–4459. [PubMed] [Google Scholar]
- Bellacosa A, Testa JR, Staal SP, Tsichlis PN. A retroviral oncogene, akt, encoding a serine-threonine kinase containing an SH2-like region. Science. 1991;254:274–277. doi: 10.1126/science.254.5029.274. [DOI] [PubMed] [Google Scholar]
- Belsches-Jablonski AP, Demory ML, Parsons JT, Parsons SJ. The Src pathway as a therapeutic strategy. Drug Discov Today. 2005;2:313–321. [Google Scholar]
- Bennett BL, Sasaki DT, Murray BW, O'Leary EC, Sakata ST, Xu W, et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci U S A. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhargava P, Robinson MO. Development of second-generation VEGFR tyrosine kinase inhibitors: current status. Curr Oncol Rep. 2011;13:103–111. doi: 10.1007/s11912-011-0154-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhide RS, Cai ZW, Zhang YZ, Qian L, Wei D, Barbosa S, et al. Discovery and preclinical studies of (R)-1-(4-(4-fluoro-2-methyl-1H-indol-5-yloxy)-5-methylpyrrolo[2,1-f][1,2,4]triazin-6-yloxy)propan-2-ol (BMS-540215), an in vivo active potent VEGFR-2 inhibitor. J Med Chem. 2006;49:2143–2146. doi: 10.1021/jm051106d. [DOI] [PubMed] [Google Scholar]
- Bikkavilli RK, Feigin ME, Malbon CC. p38 mitogen-activated protein kinase regulates canonical Wnt-beta-catenin signalling by inactivation of GSK3β. J Cell Sci. 2008;121:3598–3607. doi: 10.1242/jcs.032854. [DOI] [PubMed] [Google Scholar]
- Blake RA, Broome MA, Liu X, Wu J, Gishizky M, Sun L, et al. SU6656, a selective src family kinase inhibitor, used to probe growth factor signaling. Mol Cell Biol. 2000;20:9018–9027. doi: 10.1128/mcb.20.23.9018-9027.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogoyevitch MA, Arthur PG. Inhibitors of c-Jun N-terminal kinases- JuNK no more? Biochim Biophys Acta. 2008;1784:76–93. doi: 10.1016/j.bbapap.2007.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt P, Jensen AJ, Nilsson J. Small kinase assay panels can provide a measure of selectivity. Bioorg Med Chem Lett. 2009;19:5861–5863. doi: 10.1016/j.bmcl.2009.08.083. [DOI] [PubMed] [Google Scholar]
- Brasca MG, Albanese C, Alzani R, Amici R, Avanzi N, Ballinari D, et al. Optimization of 6,6-dimethyl pyrrolo[3,4-c]pyrazoles: identification of PHA-793887, a potent CDK inhibitor suitable for intravenous dosing. Bioorg Med Chem Lett. 2010;18:1844–1853. doi: 10.1016/j.bmc.2010.01.042. [DOI] [PubMed] [Google Scholar]
- Buchanan SG, Hendle J, Lee PS, Smith CR, Bounaud PY, Jessen KA. SGX523 is an exquisitely selective, ATP competitive inhibitor of the MET receptor tyrosine kinase with antitumor activity in vitro. Mol Cancer Ther. 2009;8:3181–3190. doi: 10.1158/1535-7163.MCT-09-0477. [DOI] [PubMed] [Google Scholar]
- Byth KF, Thomas A, Hughes G, Forder C, McGregor A, Geh C, et al. AZD5438, a potent oral inhibitor of cyclin-dependent kinases 1, 2 and 9, leads to pharmacodynamic changes and potent antitumor effects in human tumor xenografts. Mol Cancer Ther. 2009;8:1856–1866. doi: 10.1158/1535-7163.MCT-08-0836. [DOI] [PubMed] [Google Scholar]
- Camps M, Rückle T, Ji H, Ardisonne V, Rintelen F, Shaw J, et al. Blockade of PI3Kgamma suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nature Med. 2005;11:936–943. doi: 10.1038/nm1284. [DOI] [PubMed] [Google Scholar]
- Carlson SM, White FM. Using small molecules and chemical genetics to interrogate signaling networks. ACS Chem Biol. 2011;6:75–85. doi: 10.1021/cb1002834. [DOI] [PubMed] [Google Scholar]
- Carpinelli P, Ceruti R, Giorgini ML, Cappella P, Gianellini L, Croci V, et al. PHA-739358, a potent inhibitor of Aurora kinases with a selective target inhibition profile relevant to cancer. Mol Cancer Ther. 2007;6:3158–3168. doi: 10.1158/1535-7163.MCT-07-0444. [DOI] [PubMed] [Google Scholar]
- Cervenka I, Wolf J, Masek J, Krejci P, Wilcox WR, Kozubik A, et al. Mitogen activated protein kinases promote Wnt/beta-catenin signalling via phosphorylation of LRP6. Mol Cell Biol. 2010;31:179–189. doi: 10.1128/MCB.00550-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan WW, Wise SC, Kaufman MD, Ahn YM, Ensinger CL, Haack T, et al. Conformational control inhibition of the BCR-ABL1 tyrosine kinase, including the gatekeeper T315I mutant, by the switch-control inhibitor DCC-2036. Cancer Cell. 2011;19:556–568. doi: 10.1016/j.ccr.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Changelian PS, Moshinsky D, Kuhn CF, Flanagan ME, Munchhof MJ, Harris TM, et al. The specificity of JAK3 kinase inhibitors. Blood. 2008;111:2155–2157. doi: 10.1182/blood-2007-09-115030. [DOI] [PubMed] [Google Scholar]
- Cheng AC, Eksterowicz J, Geuns-Meyer S, Sun Y. Analysis of kinase inhibitor selectivity using a thermodynamics-based partition index. J Med Chem. 2010;53:4502–4510. doi: 10.1021/jm100301x. [DOI] [PubMed] [Google Scholar]
- Cheng JQ, Lindsley CW, Cheng GZ, Yang H, Nicosia SV. The Akt/PKB pathway: molecular target for cancer drug discovery. Oncogene. 2005;24:7482–7492. doi: 10.1038/sj.onc.1209088. [DOI] [PubMed] [Google Scholar]
- Christopher JA, Bamborough P, Alder C, Campbell A, Cutler GJ, Down K, et al. Discovery of 6-aryl-7-alkoxyisoquinoline inhibitors of IκB kinase-β (IKK-β. J Med Chem. 2009;52:3098–3102. doi: 10.1021/jm9000117. [DOI] [PubMed] [Google Scholar]
- Cohen P. Protein kinases – the major drug targets of the twenty-first century? Nat Rev Drug Discov. 2002;1:309–315. doi: 10.1038/nrd773. [DOI] [PubMed] [Google Scholar]
- Conway JG, McDonald B, Parham J, Keith B, Rusnak DW, Shaw E. Inhibition of colony-stimulating-factor-1 signalling in vivo with the orally bioavailable cFMS kinase inhibitor GW-2580. Proc Natl Acad Sci U S A. 2005;102:16078–16083. doi: 10.1073/pnas.0502000102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MI, Hunt JP, Herrgard S, Ciceri P, Wood LM, Pallares G, et al. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1046–1051. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- Deguchi Y, Kimura S, Ashihara E, Niwa T, Hodohara K, Fujiyama Y, et al. Comparison of imatinib, dasatinib, nilotinib and INNO-406 in imatinib-resistant cell lines. Leuk Res. 2008;32:980–983. doi: 10.1016/j.leukres.2007.11.008. [DOI] [PubMed] [Google Scholar]
- Ditchfield C, Johnson V, Tighe A, Ellston R, Haworth C, Johnson T, et al. Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2 and Cenp-E to kinetochores. J Cell Biol. 2003;161:267–280. doi: 10.1083/jcb.200208091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–1037. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc Natl Acad Sci U S A. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar KA, Wallin JJ, Berry M, Lee LB, Prior WW, Sampath D, et al. Isoform-specific phosphoinositide 3-kinase inhibitors exert distinct effects in solid tumors. Cancer Res. 2010;70:1164–1172. doi: 10.1158/0008-5472.CAN-09-2525. [DOI] [PubMed] [Google Scholar]
- Emanuel S, Rugg CA, Gruninger RH, Lin R, Fuentes-Pesquera A, Connolly PJ, et al. The in vitro and in vivo effects of JNJ-7706621: a dual inhibitor of cyclin-dependent kinases and aurora kinases. Cancer Res. 2005;65:9038–9046. doi: 10.1158/0008-5472.CAN-05-0882. [DOI] [PubMed] [Google Scholar]
- Emmitte KA, Adjebang GM, Andrews CW, Badiang Alberti JG, Bambal R, Chamberlain SD, et al. Design of potent thiophene inhibitors of polo-like kinase 1 with improved solubility and reduced protein binding. Bioorg Med Chem Lett. 2009;19:1694–1697. doi: 10.1016/j.bmcl.2009.01.094. [DOI] [PubMed] [Google Scholar]
- Fabian MA, Biggs WH, III, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG, et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol. 2005;23:329–336. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- Fedorov O, Marsden B, Pogagic V, Rellos P, Müller S, Bullock AN, et al. A systematic interaction map of validated kinase inhibitors with Ser/Thr kinases. Proc Natl Acad Sci U S A. 2007;104:20525–20527. doi: 10.1073/pnas.0708800104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;36:809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade MA, Trachet E, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004;3:1427–1438. [PubMed] [Google Scholar]
- Galkin AV, Melnick JS, Kim S, Hood TL, Li N, Li L, et al. Identification of NVP-TAE684, a potent, selective and efficacious inhibitor of NPM-ALK. Proc Natl Acad Sci U S A. 2006;104:270–275. doi: 10.1073/pnas.0609412103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese MC. Inhibition of p38: has the fat lady sung? Arthritis Rheum. 2009;60:317–320. doi: 10.1002/art.24264. [DOI] [PubMed] [Google Scholar]
- Gnoni A, Marech I, Silvestris N, Vacca A, Lorusso V. Dasatinib: an anti-tumour agent via Src inhibition. Curr Drug Targets. 2011;12:563–578. doi: 10.2174/138945011794751591. [DOI] [PubMed] [Google Scholar]
- Godden JW, Stahura FL, Bajorath J. Variability of molecular descriptors in compound databases revealed by Shannon entropy calculations. J Chem Inf Comput Sci. 2000;40:796–800. doi: 10.1021/ci000321u. [DOI] [PubMed] [Google Scholar]
- Goldstein DM, Gray NS, Zarrinkar PP. High-throughput kinase profiling as a platform for drug discovery. Nat Rev Drug Discov. 2008;7:391–397. doi: 10.1038/nrd2541. [DOI] [PubMed] [Google Scholar]
- Gozgit JM, Bebernitz G, Patil P, Ye M, Parmentier J, Wu J, et al. Effects of the JAK2 inhibitor AZ960 on Pim/BAD/BCL-xL survival signalin in the human JAK2 V617F cell line SET-2. J Biol Chem. 2008;283:32334–32343. doi: 10.1074/jbc.M803813200. [DOI] [PubMed] [Google Scholar]
- Graczyk P. Gini coefficient: a new way to express selectivity of kinase inhibitors against a family of kinases. J Med Chem. 2007;50:5773–5779. doi: 10.1021/jm070562u. [DOI] [PubMed] [Google Scholar]
- Green TP, Fennel M, Whittaker R, Curwen J, Jacobs V, Allen J, et al. Preclinical anticancer activity of the potent, oral Src inhibitor AZD0530. Mol Oncol. 2009;3:248–261. doi: 10.1016/j.molonc.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimshaw KM, Hunter LJ, Yap TA, Heaton SP, Walton MI, Woodhead SJ, et al. AT7867 is a potent and oral inhibitor of AKT and p70 S6 kinase that induces pharmacodynamic changes and inhibits tumor xenograft growth. Mol Cancer Therap. 2010;9:1100–1110. doi: 10.1158/1535-7163.MCT-09-0986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton JA. Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol. 2008;8:533–544. doi: 10.1038/nri2356. [DOI] [PubMed] [Google Scholar]
- Hamon J, Whitebread S, Techer-Etienne V, Le Coq H, Azzoui K, Urban L. In vitro safety pharmacology profiling: what else beyond hERG. Future Med Chem. 2009;1:645–665. doi: 10.4155/fmc.09.51. [DOI] [PubMed] [Google Scholar]
- Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, et al. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J Biol Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- Hansen JD, Grina J, Newhouse B, Welch M, Topalov G, Littman N, et al. Potent and selective pyrazole-based inhibitors of B-Raf kinase. Bioorg Med Chem Lett. 2008;18:4692–4695. doi: 10.1016/j.bmcl.2008.07.002. [DOI] [PubMed] [Google Scholar]
- Hantschel O, Superti-Furga G. Regulation of the c-Abl and Bcr-Abl tyrosine kinases. Nat Rev Mol Cell Biol. 2004;5:33–44. doi: 10.1038/nrm1280. [DOI] [PubMed] [Google Scholar]
- Hart S, Goh KC, Novotny-Diermayr V, Hu CY, Hentze H, Tan YC, et al. SB1518, a novel macrocyclic pyrimidine-based JAK2 inhibitor for the treatment of myeloid and lymphoid malignancies. Leukemia. 2011;25:1751–1759. doi: 10.1038/leu.2011.148. [DOI] [PubMed] [Google Scholar]
- Hartnett JC, Barnett SF, Bilodeau MT, Defeo-Jones D, Hartman GD, Huber E, et al. Optimization of 2,3,5-trisubstituted pyridine derivatives as potent allosteric Akt1 and Akt2 inhibitors. BioOrg Med Chem Lett. 2008;18:2194–2197. doi: 10.1016/j.bmcl.2007.12.040. [DOI] [PubMed] [Google Scholar]
- Hedvat M, Huszar D, Herrmann A, Gozgit JM, Schroeder A, Sheehy A, et al. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell. 2009;16:487–497. doi: 10.1016/j.ccr.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilker R, Wolff M, Tautermann CS, Bieler M. G-protein-coupled receptor-focused drug discovery using a target class platform approach. Drug Discov Today. 2009;14:231–240. doi: 10.1016/j.drudis.2008.11.011. [DOI] [PubMed] [Google Scholar]
- Hideshima T, Neri P, Tassone P, Yasui H, Ishitsuka K, Raje N, et al. MLN120B, a novel IκB kinase β inhibitor, blocks multiple myelome cell growth in vitro and in vivo. Clin Cancer Res. 2006;12:5887–5894. doi: 10.1158/1078-0432.CCR-05-2501. [DOI] [PubMed] [Google Scholar]
- Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther. 2010;9:1956–1967. doi: 10.1158/1535-7163.MCT-09-1012. [DOI] [PubMed] [Google Scholar]
- Hodgson DR, Schröder M. Chemical approaches towards unravelling kinase-mediated signalling pathways. Chem Soc Rev. 2011;40:1211–1223. doi: 10.1039/c0cs00020e. [DOI] [PubMed] [Google Scholar]
- Hoeflich KP, Herter S, Tien J, Wong L, Berry L, Chan J, et al. Antitumor efficacy of the novel RAF inhibitor GDC-0879 is predicted by BRAFV600E mutational status and sustained extracellular signal-regulated kinase/mitogen activated protein kinase pathway suppression. Cancer Res. 2009;69:3042–3051. doi: 10.1158/0008-5472.CAN-08-3563. [DOI] [PubMed] [Google Scholar]
- Hopkins AL. Network pharmacology: the next paradigm in drug discovery. Nature Chem Biol. 2008;4:682–690. doi: 10.1038/nchembio.118. [DOI] [PubMed] [Google Scholar]
- Huber MA, Maier HJ, Alacakaptan M, Wiedemann E, Braunger J, Boehmelt G, et al. BI5700 a selective chemical inhibitor of IκB kinase 2, specifically suppresses epithelial-mesenchymal transition and metastasis in mouse models of tumor progression. Genes Cancer. 2010;1:101–114. doi: 10.1177/1947601910361749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SP, Schoenwaelder SM, Goncalves I, Nesbitt WS, Yap CL, Wright CE, et al. PI3-kinase p110β: a new target for antithrombotic therapy. Nat Med. 2005;11:507–511. doi: 10.1038/nm1232. [DOI] [PubMed] [Google Scholar]
- Jamieson S, Flanagan JU, Kolekar S, Buchanan C, Kendall JD, Lee WJ, et al. A drug targeting only p110α can block phosphoinositide 3-kinase signalling and tumour growth in certain cell types. Biochem J. 2011;438:53–62. doi: 10.1042/BJ20110502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jani JP, Finn RS, Campbell M, Coleman KG, Connell RD, Currier N, et al. Discovery and pharmacologic characterization of CP-724,714, a selective ErbB2 tyrosine kinase inhibitor. Cancer Res. 2007;67:9887–9893. doi: 10.1158/0008-5472.CAN-06-3559. [DOI] [PubMed] [Google Scholar]
- Jiang J, Greulich H, Jänne PA, Sellers WR, Meyerson M, Griffin JD. Epidermal growth factor-independent transformation of Ba/F3 cells with cancer-derived epidermal growth factor receptor mutants induces gefitinib-sensitive cell cycle progression. Cancer Res. 2005;65:8968–8974. doi: 10.1158/0008-5472.CAN-05-1829. [DOI] [PubMed] [Google Scholar]
- Johnson LN. Protein kinase inhibitors: contributions from structure to clinical compounds. Q Rev Biophys. 2009;42:1–40. doi: 10.1017/S0033583508004745. [DOI] [PubMed] [Google Scholar]
- Joshi KS, Rathos MJ, Joshi RD, Sivakumar M, Mascarenhas M, Kamble S, et al. Antitumor properties of a novel cyclin-dependent kinase inhibitor, P276-00. Mol Cancer Ther. 2007;6:918–925. doi: 10.1158/1535-7163.MCT-06-0613. [DOI] [PubMed] [Google Scholar]
- Kamenecka T, Jiang R, Song X, Duckett D, Chen W, Ling YY, et al. Synthesis, biological evaluation, X-ray structure, and pharmacokinetics of aminopyrimidine c-jun-N-terminal kinase (JNK) inhibitors. J Med Chem. 2010;53:419–431. doi: 10.1021/jm901351f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26:127–131. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- Katayama H, Sen S. Aurora kinase inhibitors as anticancer molecules. Biochim Biophys Acta. 2010;1799:829–839. doi: 10.1016/j.bbagrm.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama R, Khan TM, Benes C, Lifshits E, Ebi H, Rivera VM. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc Natl Acad Sci U S A. 2011;18:7535–7540. doi: 10.1073/pnas.1019559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katso R, Okkenhaug K, Ahmadi K, White S, Timms J, Waterfield MD. Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis, and cancer. Annu Rev Cell Dev Biol. 2001;17:615–675. doi: 10.1146/annurev.cellbio.17.1.615. [DOI] [PubMed] [Google Scholar]
- Knight ZA, Shokat KM. Features of selective kinase inhibitors. Chem Biol. 2005;12:621–637. doi: 10.1016/j.chembiol.2005.04.011. [DOI] [PubMed] [Google Scholar]
- Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, et al. A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling. Cell. 2006;125:733–747. doi: 10.1016/j.cell.2006.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R, Crouthamel MC, Rominger DH, Gontarek RR, Tummino PJ, Levin RA, et al. Myelosuppression and kinase selectivity of multikinase angiogenesis inhibitors. Br J Cancer. 2009;101:1717–1723. doi: 10.1038/sj.bjc.6605366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lannutti BJ, Meadows SA, Herman SE, Kashishian A, Steiner B, Johnson AJ, et al. CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. 2011;117:591–594. doi: 10.1182/blood-2010-03-275305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsley CW, Zhao Z, Leister WH, Robinson RG, Barnett SF, Defeo-Jones D, et al. Allosteric Akt (PKB) inhibitors: discovery and SAR of isozyme selective inhibitors. Bioorg Med Chem Lett. 2005;15:761–764. doi: 10.1016/j.bmcl.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Lindstrom TM, Robinson WH. A multitude of kinases – which are the best targets in treating rheumatoid arthritis? Rheum Dis Clin North Am. 2010;36:367–383. doi: 10.1016/j.rdc.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippa B, Kauffman GS, Arcari J, Kwan T, Chen J, Hungerford W, et al. The discovery of highly selective erbB2 (Her2) inhibitors for the treatment of cancer. Bioorg Med Chem Lett. 2007;17:3081–3086. doi: 10.1016/j.bmcl.2007.03.046. [DOI] [PubMed] [Google Scholar]
- Logan CY, Nusse R. The Wnt signalling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- Lovely CM, Heuckmann JM, de Stanchina E, Chen H, Thomas RK, Liang C, et al. Insights into ALK-driven cancers revealed through development of novel ALK tyrosine kinase inhbitors. Cancer Res. 2011;71:4920–4931. doi: 10.1158/0008-5472.CAN-10-3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma FY, Flanc RS, Tesch GH, Han Y, Atkins RC, Bennett BL, et al. A pathogenic role for c-Jun amino-terminal kinase signaling in renal fibrosis and tubular cell apoptosis. J Am Soc Nephrol. 2007;18:472–484. doi: 10.1681/ASN.2006060604. [DOI] [PubMed] [Google Scholar]
- Ma JG, Huang H, Chen SM, Chen Y, Xin XL, Lin LP, et al. PH006, a novel and selective Src kinase inhibitor, suppresses human breast cancer growth and metastasis in vitro and in vivo. Breast Cancer Res Treat. 2010;130:85–96. doi: 10.1007/s10549-010-1302-4. [DOI] [PubMed] [Google Scholar]
- MacDonald BT, Tamai K, He X. Wnt/β-catenin signalling: components, mechanisms, and diseases. Dev Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153–166. doi: 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- Manley PW, Drueckes P, Fendrich G, Furet P, Liebetanz J, Martiny-Baron G, et al. Extended kinase profile and properties of the protein kinase inhibitor nilotinib. Biochim Biophys Acta. 2010;1804:445–453. doi: 10.1016/j.bbapap.2009.11.008. [DOI] [PubMed] [Google Scholar]
- Manning AM, Davis RJ. Targeting JNK for therapeutic benefit: from junk to gold? Nat Rev Drug Discov. 2003;2:554–565. doi: 10.1038/nrd1132. [DOI] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- Mbalaviele G, Sommers CD, Bonar SL, Mathialagan S, Schindler JF, Guzova JA, et al. A novel, highly selective, tight binding IkB kinase-2 (IKK-2) inhibitor: a tool to correlated IKK-2 activity to the fate and functions of the components of the nucelar factor-kB pathway in arthritis-relevant cells and animal models. J Pharmacol Exp Therap. 2009;329:14–25. doi: 10.1124/jpet.108.143800. [DOI] [PubMed] [Google Scholar]
- Meijer L, Borgne A, Mulner O, Chong JPJ, Blow JJ, Inagaki N, et al. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur J Biochem. 1997;243:527–536. doi: 10.1111/j.1432-1033.1997.t01-2-00527.x. [DOI] [PubMed] [Google Scholar]
- Metz J, Johnson EF, Soni NB, Merta PJ, Kifle L, Hajduk PJ. Navigating the kinome. Nature Chem Biol. 2011;7:200–202. doi: 10.1038/nchembio.530. [DOI] [PubMed] [Google Scholar]
- Miduturu CV, Deng X, Kwiatkowski N, Yang W, Brault L, Filippakopoulos P, et al. High-throughput kinase profiling: a more efficient approach toward the discovery of new kinase inhibitors. Chem Biol. 2011;18:868–879. doi: 10.1016/j.chembiol.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihara K, Almansa C, Smeets RL, Loomand EE, Dulos JJ, Vink PM, et al. A potent and selective p38 inhibitor protects against bone damage in murine collagen-induced arthritis: a comparison with neutralization of mouse TNFα. Br J Pharmacol. 2008;154:153–164. doi: 10.1038/bjp.2008.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsiades CS, Mitsiades N, Koutsilieris M. The Akt pathway: molecular targets for anti-cancer drug development. Curr Cancer Drug Targets. 2004;4:235–256. doi: 10.2174/1568009043333032. [DOI] [PubMed] [Google Scholar]
- Moasser MM. Targeting the function of the HER2 oncogene in human cancer therapeutics. Oncogene. 2007;26:6577–6592. doi: 10.1038/sj.onc.1210478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munshi N, Jeay S, Li Y, Chen CR, France DS, Ashwell MA. ARQ197, a novel and selective inhibitor of the human cMET receptor tyrosine kinase with antitumor activity. Mol Cancer Ther. 2010;9:1544–1553. doi: 10.1158/1535-7163.MCT-09-1173. [DOI] [PubMed] [Google Scholar]
- Nagasawa J, Mizokami A, Koshida K, Yoshida S, Naito K, Namiki M. Novel HER2 selective tyrosine kinase inhibitor, TAK-165, inhibits bladder, kidney and androgen-independent prostate cancer in vitro and in vivo. Int J Urol. 2006;13:587–592. doi: 10.1111/j.1442-2042.2006.01342.x. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Taguchi E, Miura T, Yamamoto A, Takahashi K, Bichat F, et al. KRN951, a highly potent inhibitor of vascular endothelial growth factor receptor tyrosine kinases, has antitumor activities and affects functional vascular properties. Cancer Res. 2006;66:9134–9142. doi: 10.1158/0008-5472.CAN-05-4290. [DOI] [PubMed] [Google Scholar]
- Ohren JF, Chen H, Pavlovsky A, Whitehead C, Zhang E, Kuffa P. Structures of human MAP kinase kinase 1 (MEK1) and MEK2 describe novel noncompetitive kinase inhibition. Nat Struct Mol Biol. 2004;11:1192–1197. doi: 10.1038/nsmb859. [DOI] [PubMed] [Google Scholar]
- Pardanani A, Lasho T, Smith G, Burns CJ, Fantino E, Tefferi A. CYT387, a selective JAK1/JAK2 inhibitor: in vitro assessment of kinase selectivity and preclinical studies using cell lines and primary cells from polycythemia vera patients. Leukemia. 2009;23:1441–1445. doi: 10.1038/leu.2009.50. [DOI] [PubMed] [Google Scholar]
- Patricelli MP, Nomanbhoy TK, Wu J, Brown H, Zhou D, Zhang J, et al. In situ kinase profiling reveals functionally relevant properties of native kinases. Chem Biol. 2011;18:699–710. doi: 10.1016/j.chembiol.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payton M, Bush TL, Chung G, Ziegler B, Eden P, McElroy P, et al. Preclinical evaluation of AMG900, a novel potent and highly selective pan-Aurora kinase inhibitor with activity in taxane-resistant tumor cell lines. Cancer Res. 2010;70:9846–9854. doi: 10.1158/0008-5472.CAN-10-3001. [DOI] [PubMed] [Google Scholar]
- Peters JM, McKay RM, McKay JP, Graff JM. Casein kinase I transduces Wnt signals. Nature. 1999;401:345–350. doi: 10.1038/43830. [DOI] [PubMed] [Google Scholar]
- Peters J-U, Schnider P, Mattei P, Kansy M. Pharmacological promiscuity: dependence on compound properties and target specificity in a set of recent Roche compounds. ChemMedChem. 2009;4:680–686. doi: 10.1002/cmdc.200800411. [DOI] [PubMed] [Google Scholar]
- Posy SL, Hermsmeier MA, Vaccaro W, Ott K-H, Todderud G, Lippy JS, et al. Trends in kinase selectivity: insights for target class-focused library screening. J Med Chem. 2011;54:54–66. doi: 10.1021/jm101195a. [DOI] [PubMed] [Google Scholar]
- Quintás-Cardama A, Vaddi K, Liu P, Manshouri T, Li J, Scherle PA, et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of neoproliferative diseases. Blood. 2010;115:3109–3117. doi: 10.1182/blood-2009-04-214957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raynaud FI, Eccles S, Clarke PA, Hayes A, Nutley B, Alix S, et al. Pharmacologic characterization of a potent inhibitor of class I phosphatidylinositide 3-kinases. Cancer Res. 2007;67:5840–5850. doi: 10.1158/0008-5472.CAN-06-4615. [DOI] [PubMed] [Google Scholar]
- Remsing-Rix LL, Rix U, Colinge J, Hantschel O, Bennett KL, Stranzl T, et al. Global target profile of the kinase inhibitor bosutinib in primary chronic myeloid leukemia cells. Leukemia. 2009;23:477–485. doi: 10.1038/leu.2008.334. [DOI] [PubMed] [Google Scholar]
- Rhodes N, Heerding DA, Duckett DR, Eberwein DJ, Knick VB, Lansing TJ, et al. Characterization of an Akt kinase inhibitor with potent pharmacodynamic and antitumor activity. Cancer Res. 2008;68:2366–2374. doi: 10.1158/0008-5472.CAN-07-5783. [DOI] [PubMed] [Google Scholar]
- Rini BI. Vascular endothelial growth factor-targeted therapy in renal cell carcinoma: current status and future directions. Clin Cancer Res. 2007;13:1098–1106. doi: 10.1158/1078-0432.CCR-06-1989. [DOI] [PubMed] [Google Scholar]
- Ritzeler O. 2011. The discovery of SAR119345 an IKK inhibitor for intra-articular administration in patients with knee osteoarthritis. Informa kinase meeting, Berlin, Germany.
- Rix U, Superti-Furga G. Target profiling of small molecules by chemical proteomics. Nat Chem Biol. 2009;5:616–624. doi: 10.1038/nchembio.216. [DOI] [PubMed] [Google Scholar]
- Rix U, Remsing-Rix LL, Terker AS, Fernbach NV, Hantschel O, Planyavsky M, et al. A comprehensive target selectivity survey of the BCR-ABL kinase inhibitor INNO-406 by kinase profiling and chemical proteomics in chronic myeloid leukemia cells. Leukemia. 2010;24:44–50. doi: 10.1038/leu.2009.228. [DOI] [PubMed] [Google Scholar]
- Rommel C, Camps M, Ji H. PI3K delta and PI3K gamma: partners in crime in inflammation in rheumatoid arthritis and beyond? Nat Rev Immunol. 2007;7:191–201. doi: 10.1038/nri2036. [DOI] [PubMed] [Google Scholar]
- Rudolph D, Steegmaier M, Hoffmann M, Grauert M, Baum A, Quant J, et al. BI6727, a polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin Cancer Res. 2009;15:3094–3102. doi: 10.1158/1078-0432.CCR-08-2445. [DOI] [PubMed] [Google Scholar]
- Sakamoto H, Tsukaguchi T, Hiroshima S, Kodama T, Kobayashi T, Fukami TA, et al. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell. 2011;19:679–690. doi: 10.1016/j.ccr.2011.04.004. [DOI] [PubMed] [Google Scholar]
- Sakanaka C, Leong P, Xu L, Harrison SD, Williams LT. Casein kinase epsilon in the Wnt pathway: regulation of β-catenin. Proc Natl Acad Sci U S A. 1999;96:12548–12552. doi: 10.1073/pnas.96.22.12548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- Scapin G, Patel SB, Lisnock J, Becker JW, LoGrasso PV. The structure of JNK3 in complex with small molecule inhibitors: structural basis for potency and selectivity. Chem Biol. 2003;10:705–712. doi: 10.1016/s1074-5521(03)00159-5. [DOI] [PubMed] [Google Scholar]
- Schöffski P. Polo-like kinase (PLK) in preclinical and early clinical development in oncology. Oncologist. 2009;14:559–570. doi: 10.1634/theoncologist.2009-0010. [DOI] [PubMed] [Google Scholar]
- Shide K, Kameda T, Markovtsov V, Shimoda HK, Tonkin E, Fang S, et al. R723, a selective JAK2 inhibitor, effectively treats JAK2V617F-induced murine myoproliferative neoplasm. Blood. 2011;117:6866–6875. doi: 10.1182/blood-2010-01-262535. [DOI] [PubMed] [Google Scholar]
- Shimomura T, Hasako S, Nakatsuru Y, Mita T, Ichikawa K, Kodera T, et al. MK-5108, a highly selective Aurora-A kinase inhibitor, shows antitumor activity alone and in combination with docetaxel. Mol Cancer Ther. 2010;9:157–166. doi: 10.1158/1535-7163.MCT-09-0609. [DOI] [PubMed] [Google Scholar]
- Shokat K, Velleca M. Novel chemical genetic approaches to the discovery of signal transduction inhibitors. Drug Discov Today. 2002;16:872–879. doi: 10.1016/s1359-6446(02)02391-7. [DOI] [PubMed] [Google Scholar]
- Solit DB, Sawyers CL. Drug discovery: how melanomas bypass new therapy. Nature. 2010;468:902–903. doi: 10.1038/468902a. [DOI] [PubMed] [Google Scholar]
- Spencer J, Mendham AP, Kotha AK, Richardson SC, Hillard EA, Jaouen G, et al. Structural and biological investigation of ferrocene-substituted 3-methylidene-1,3-dihydro-2H-indol-2-ones. Dalton Trans. 2009;6:918–921. doi: 10.1039/b816249b. [DOI] [PubMed] [Google Scholar]
- Squires MS, Feltell RE, Wallis NG, Lewis EJ, Smit DM, Cross DM, et al. Biological characterization of AT7519, a small molecule inhibitor of cyclin-dependent kinases, in human tumor cell lines. Mol Cancer Ther. 2009;8:324–332. doi: 10.1158/1535-7163.MCT-08-0890. [DOI] [PubMed] [Google Scholar]
- Steegmaier M, Hoffmann M, Baum A, Lénart P, Pertronczki M, Krššák M, et al. BI2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr Biol. 2007;17:316–322. doi: 10.1016/j.cub.2006.12.037. [DOI] [PubMed] [Google Scholar]
- Summy JM, Trevino JG, Lesslie DP, Baker CH, Shakespeare WC, Wang Y, et al. AP23846, a novel and highly potent Src family kinase inhibitor, reduces vascular endothelial growth factor and interleukin-8 expression in human solid tumor cell lines and abrogates downstream angiogenic processes. Mol Cancer Ther. 2005;4:1900–1911. doi: 10.1158/1535-7163.MCT-05-0171. [DOI] [PubMed] [Google Scholar]
- Swahn BM, Xue Y, Arzel E, Kallin E, Magnus A, Plobeck N, et al. Design and synthesis of 2′-anilino-4,4′-bipyridines as selective inhibitors of c-Jun N-terminal kinase-3. Bioorg Med Chem Lett. 2006;16:1397–1401. doi: 10.1016/j.bmcl.2005.11.039. [DOI] [PubMed] [Google Scholar]
- Szczepankiewicz BG, Kosogof C, Nelson LT, Liu G, Liu B, Zhao H, et al. Aminopyridine-based c-Jun N-terminal kinase inhibitors with cellular activity and minimal cross-kinase activity. J Med Chem. 2006;49:3563–3580. doi: 10.1021/jm060199b. [DOI] [PubMed] [Google Scholar]
- Takle AK, Bamford MJ, Davies S, Davis RP, Dean DK, Gaiba A, et al. The identification of potent, selective and CNS penetrant furan-based inhibitors of B-Raf kinase. Bioorg Med Chem Lett. 2008;18:4373–4376. doi: 10.1016/j.bmcl.2008.06.070. [DOI] [PubMed] [Google Scholar]
- Tan S, Ng Y, James DE. Next-generation Akt inhibitors provide greater specificity: effects on glucose metabolism in adipocytes. Biochem J. 2011;435:539–544. doi: 10.1042/BJ20110040. [DOI] [PubMed] [Google Scholar]
- Tegeder I, Niederberger E, Schmidt R, Kunz S, Gühring H, Ritzeler O, et al. Specific inhibition of IκB kinase reduces hyperalgesia in inflammatory and neuropathic pain models in rats. J Neurosci. 2004;24:1637–1645. doi: 10.1523/JNEUROSCI.3118-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian G, Cory M, Smith AA, Knight WB. Structural determinants for potent, selective dual site inhibition of human pp60c-src by 4-anilinoquinazolines. Biochemistry. 2001;40:7084–7091. doi: 10.1021/bi0100586. [DOI] [PubMed] [Google Scholar]
- Trujillo JL. MEK inhibitors: a patent review 2008-2010. Expert Opin Ther Pat. 2011;21:1045–1069. doi: 10.1517/13543776.2011.577068. [DOI] [PubMed] [Google Scholar]
- Tsai J, Lee JT, Wang W, Zhang J, Cho H, Mamo S, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci U S A. 2008;105:3041–3046. doi: 10.1073/pnas.0711741105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uckun FM, Dibirdik I, Qazi S, Vassilev A, Ma H, Mao C, et al. Anti-breast cancer activity of LFM-A13, a potent inhibitor of polo-like kinase (PLK) Bioorg Med Chem. 2007;15:800–814. doi: 10.1016/j.bmc.2006.10.050. [DOI] [PubMed] [Google Scholar]
- Uitdehaag JCM. Screening for Selectivity. Barcelona: Screening Europe; 2010. [Google Scholar]
- Uitdehaag JCM, Zaman GJ. A theoretical entropy score as a single value to express inhibitor selectivity. BMC Bioinformatics. 2011;12:94. doi: 10.1186/1471-2105-12-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uitdehaag JCM, Sünnen CM, van Doornmalen AM, de Rouw N, Oubrie A, Azevedo R, et al. Multidimensional profiling of CSF1R screening hits and inhibitors: assessing cellular activity, target residence time and selectivity in a higher throughput way. J Biomol Screen. 2011;16:1007–1017. doi: 10.1177/1087057111418113. [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Leevers SJ, Ahmadi K, Timms J, Katso R, Driscoll PC, et al. Synthesis and function of 3-phosphorylated inositol lipids. Annu Rev Biochem. 2001;70:535–602. doi: 10.1146/annurev.biochem.70.1.535. [DOI] [PubMed] [Google Scholar]
- Verkaar F, van der Doelen AA, Smits JF, Blankensteijn WM, Zaman GJ. Inhibition of Wnt/beta-catening signalling by p38 MAP kinase inhibitors is explained by cross reactivity with casein kinase I δ/ε. Chem Biol. 2011;18:485–494. doi: 10.1016/j.chembiol.2011.01.015. [DOI] [PubMed] [Google Scholar]
- Vieth M, Higgs RE, Robertson DH, Shapiro M, Gragg EA, Hemmerle H. Kinomics- structural biology and chemogenomics of kinase inhibitors and targets. Biochim Biophys Acta. 2004;1697:243–267. doi: 10.1016/j.bbapap.2003.11.028. [DOI] [PubMed] [Google Scholar]
- Wang WY, Hsieh PW, Wu YC, Wu CC. Synthesis and pharmacological evaluation of novel beta-nitrostyrene derivatives as tyrosine kinase inhibitors with potent antiplatelet activity. Biochem Pharmacol. 2007;74:601–611. doi: 10.1016/j.bcp.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Wedge SR, Kendrew J, Hennequin LF, Valentine PJ, Barry ST, Brave SR, et al. AZD2171: a highly potent, orally bioavailable, vascular endothelial growth factor receptor-2 tyrosine kinase inhibitor for the treatment of cancer. Cancer Res. 2005;65:4389–4400. doi: 10.1158/0008-5472.CAN-04-4409. [DOI] [PubMed] [Google Scholar]
- Wen D, Nong Y, Morgan JG, Gangurde P, Bielecki A, DaSilva J, et al. A selective small molecule IκB kinase β inhibitor blocks nuclear factor κB-mediated inflammatory responses in human fibroblast-like synoviocytes, chondrocytes and mast cells. J Pharmacol Exp Ther. 2006;317:989–1001. doi: 10.1124/jpet.105.097584. [DOI] [PubMed] [Google Scholar]
- Whittles CE, Pocock TM, Wedge SR, Kendrew J, Hennequin LF, Harper SJ, et al. ZM323881, a novel inhibitor of vascular endothelial growth factor-receptor-2 tyrosine kinase activity. Microcirculation. 2002;9:513–522. doi: 10.1038/sj.mn.7800164. [DOI] [PubMed] [Google Scholar]
- Wilson MB, Schreiner SJ, Choi HJ, Kamens J, Smithgall TE. Selective pyrrolo-pyrimidine inhibitors reveal a necessary role for Src family kinases in Bcr-Abl signal transduction and oncogenesis. Oncogene. 2002;21:8075–8088. doi: 10.1038/sj.onc.1206008. [DOI] [PubMed] [Google Scholar]
- Wood JM, Bold G, Buchdunger E, Cozens R, Ferrari S, Frei J, et al. PTK787/ZK 222584, a novel and potent inhibitor of vascular endothelial growth factor receptor tyrosine kinases, impairs vascular endothelial growth factor-induced responses and tumor growth after oral administration. Cancer Res. 2000;60:2178–2189. [PubMed] [Google Scholar]
- Wu Z, Hartnett JC, Neilson LA, Robinson RG, Fu S, Barnett SF, et al. Development of pyridopyrimidines as potent Akt1/2 inhibitors. Bioorg Med Chem Lett. 2008;18:1274–1279. doi: 10.1016/j.bmcl.2008.01.054. [DOI] [PubMed] [Google Scholar]
- Yan A, Wang L, Xu S, Xu J. Aurora-A kinase inhibitor scaffolds and binding modes. Drug Discov Today. 2011;16:260–269. doi: 10.1016/j.drudis.2010.12.003. [DOI] [PubMed] [Google Scholar]
- Yap TA, Walton MI, Hunter LJ, Valenti M, de Haven Brandon A, Eve PD, et al. Preclinical pharmacology, antitumor activity, and development of pharmacodynamic markers for the novel, potent AKT inhibitor CCT128930. Mol Cancer Ther. 2011;10:360–371. doi: 10.1158/1535-7163.MCT-10-0760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh TC, Marsh V, Bernet BA, Ballard J, Colwell H, Evans RJ. Biological characterization of ARRY-142886 (AZD6244) a potent highly selective mitogen activated protein kinase 1/2 inhibitor. Clin Cancer Res. 2007;13:1576–1583. doi: 10.1158/1078-0432.CCR-06-1150. [DOI] [PubMed] [Google Scholar]
- Yezhelyev MV, Koehl G, Guba M, Brabletz T, Jauch KW, Ryan A. Inhibition of SRC tyrosine kinase as treatment for human pancreatic cancer growing orthotopically in nude mice. Clin Cancer Res. 2004;10:8026–8036. doi: 10.1158/1078-0432.CCR-04-0621. [DOI] [PubMed] [Google Scholar]
- Zarrinkar PP, Ruwanthi NG, Cramer MD, Gardner MF, Brigham D, Belli B. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML) Blood. 2009;114:2984–2992. doi: 10.1182/blood-2009-05-222034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Berezov A, Wang Q, Zhang G, Drebin J, Murali R, et al. ErbB receptors: from oncogenes to targeted cancer therapies. J Clin Invest. 2007;117:2051–2058. doi: 10.1172/JCI32278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Robinson RG, Barnett SF, Defeo-Jones D, Jones RE, Hartman GD, et al. Development of potent, allosteric dual Akt1 and Akt2 inhibitors with improved physical properties and cell activity. Bioorg Med Chem Lett. 2008;18:49–53. doi: 10.1016/j.bmcl.2007.11.015. [DOI] [PubMed] [Google Scholar]
- Zou HY, Li Q, Lee JH, Arango ME, McDonnell SR, Yamazaki S, et al. An orally available small-molecule inhibitor of c-Met, PF-2341066 exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer Res. 2007;67:4408–4417. doi: 10.1158/0008-5472.CAN-06-4443. [DOI] [PubMed] [Google Scholar]