

Abstract
BACKGROUND AND PURPOSE
The chemokine receptor CXCR3 is a GPCR found predominantly on activated T cells. CXCR3 is activated by three endogenous peptides; CXCL9, CXCL10 and CXCL11. Recently, a small-molecule agonist, VUF10661, has been reported in the literature and synthesized in our laboratory. The aim of the present study was to provide a detailed pharmacological characterization of VUF10661 by comparing its effects with those of CXCL11.
EXPERIMENTAL APPROACH
Agonistic properties of VUF10661 were assessed in a chemotaxis assay with murine L1.2 cells transiently transfected with cDNA encoding the human CXCR3 receptor and in binding studies, with [125I]-CXCL10 and [125I]-CXCL11, on membrane preparations from HEK293 cells stably expressing CXCR3. [35S]-GTPγS binding was used to determine its potency to induce CXCR3-mediated G protein activation and BRET-based assays to investigate its effects on intracellular cAMP levels and β-arrestin recruitment.
KEY RESULTS
VUF10661 acted as a partial agonist in CXCR3-mediated chemotaxis, bound to CXCR3 in an allosteric fashion in ligand binding assays and activated Gi proteins with the same efficacy as CXCL11 in the [35S]-GTPγS binding and cAMP assay, while it recruited more β-arrestin1 and β-arrestin2 to CXCR3 receptors than the chemokine.
CONCLUSIONS AND IMPLICATIONS
VUF10661, like CXCL11, activates both G protein-dependent and -independent signalling via the CXCR3 receptor, but probably exerts its effects from an allosteric binding site that is different from that for CXCL11. It could stabilize different receptor and/or β-arrestin conformations leading to differences in functional output. Such ligand-biased signalling might offer interesting options for the therapeutic use of CXCR3 agonists.
LINKED ARTICLE
This article is commented on by O'Boyle, pp. 895–897 of this issue. To view this commentary visit http://dx.doi.org/10.1111/j.1476-5381.2011.01759.x
Keywords: CXCR3, VUF10661, small-molecule agonist, ligand-biased signalling, β-arrestin, allosteric, BRET
Introduction
The chemokine receptor CXCR3 is an inducible chemokine receptor expressed on, for example, activated T cells of the Th1-subtype, B cells and natural killer cells. The three major CXC chemokine ligands for CXCR3 are CXCL9, CXCL10 and CXCL11 (nomenclature follows Alexander et al., 2011), of which the latter has been shown to have the highest affinity for CXCR3 (Loetscher et al., 1996; Cole et al., 1998). Stimulation of CXCR3 leads to the activation of pertussis toxin (PTX)-sensitive Gi proteins, which subsequently results in, for example, mobilization of intracellular calcium, the activation of PKB and p44/42 MAPK and chemotaxis (Bonacchi et al., 2001; Smit et al., 2003; Thompson et al., 2007).
CXCR3 chemokines are mainly secreted by activated monocytes and macrophages (Loetscher et al., 1998). As such, they direct the migration of Th1 cells to tissues that harbour inflammation or infection. Upregulation of CXCR3 ligands is found in various inflammatory disorders like allograft rejection (Hancock et al., 2000), atherosclerosis (Mach et al., 1999) and autoimmune diseases such as systemic lupus erythematosus (SLE) (Enghard et al., 2009), rheumatoid arthritis (Qin et al., 1998) and multiple sclerosis (Sorensen et al., 1999). In addition, the levels of chemokine mRNA and number of infiltrating CXCR3+ cells in tissues from transplant and SLE patients correlate with the severity of disease (Melter et al., 2001; Kao et al., 2003; Bauer et al., 2006; Lit et al., 2006). Inhibition of CXCR3 by either antibodies or small-molecule antagonists significantly delays disease progression in various mouse models (Hancock et al., 2000; Gao et al., 2003; van Wanrooij et al., 2008). As a consequence, considerable attention has been paid to the discovery and development of small-molecule CXCR3 antagonists for the treatment of chronic inflammation. To date, numerous compounds from different chemical classes have been described (Wijtmans et al., 2008).
Interestingly, during the process of screening for antagonists, two classes of small-molecule CXCR3 agonists have been identified (Stroke et al., 2006). Although the majority of CXCR3-associated disease would argue for the development of antagonists, agonists might also show therapeutic benefit in some cases, including wound healing and cancer (Walser et al., 2007; Yates et al., 2010). Moreover, small-molecule agonists are valuable tools in exploring the molecular pharmacology of the CXCR3 receptor. In the present study, we report on the pharmacological characterization of VUF10661, one of the recently discovered small-molecule CXCR3 agonists (Stroke et al., 2006). Using both G protein-dependent and -independent functional assays, we showed that VUF10661 behaves as a full CXCR3 agonist. We report for the first time that CXCL11 as well as VUF10661 recruit both β-arrestin1 and β-arrestin2 to CXCR3 receptors and regulate CXCR3 cell surface expression. In addition, radioligand binding studies indicate that VUF10661 has a different binding mode than the major endogenous chemokine ligand CXCL11. The varying functional activities of CXCL11 and VUF10661 in the different functional assays indicate ligand-biased recruitment of β-arrestins to the CXCR3 receptor.
Methods
Synthesis of small-molecule CXCR3 agonist and antagonists
The identification of several tetrahydroisoquinoline-based small-molecule non-peptide CXCR3 agonists has been reported previously by Stroke et al. (2006). The synthesis of these compounds was not disclosed. Consequently, we developed a strategy for the synthesis of one of these compounds (designated VUF10661; Figure 1), which is outlined in Figure S1 and Appendix S1 of the Supporting Information. NBI-74330, belonging to the (aza)quinazolinone class of CXCR3 antagonists, was synthesized as described previously (Storelli et al., 2007).
Figure 1.
Chemical structure of small-molecule CXCR3 agonist VUF10661.
Materials
Dulbecco's modified Eagle's medium (DMEM) and trypsin were purchased from PAA Laboratories GmbH (Pasching, Austria), poly-L-lysine, 2-mercaptoethanol, o-phenylenediamine, nonessential amino acids, formaldehyde, calcein-acetoxymethyl ester (calcein-AM), sodium pyruvate and sodium butyrate were from Sigma-Aldrich (St. Louis, MO, USA), penicillin and streptomycin were obtained from Lonza (Verviers, Belgium), fetal bovine serum (FBS) was purchased from Integro B.V. (Dieren, the Netherlands), and RPMI-1640 medium with GlutaMAX-I and 25 mM HEPES was from Invitrogen (Paisley, UK). G-418 was purchased from Duchefa Biochemie (Haarlem, the Netherlands). Coelenterazine-h was obtained from Promega (Madison, WI, USA). [125I]-CXCL10 (2200 Ci·mmol−1), [125I]-CXCL12 (2200 Ci·mmol−1) and [35S]-GTPγS were obtained from PerkinElmer Life and Analytical Sciences (Boston, MA, USA). Unlabelled chemokines were purchased from PeproTech (Rocky Hill, NJ, USA).
DNA Constructs
The cDNA of human CXCR3 inserted in pcDNA3 (Loetscher et al., 1996) was a gift from Prof Dr B. Moser (Cardiff University School of Medicine, Cardiff, UK). It was amplified by PCR and inserted into pcDEF3 (a gift from Dr Langer, Robert Wood Johnson Medical School, Piscataway, NJ, USA). cDNA encoding for the BRET-based cyclic AMP (cAMP) biosensor was purchased from American Type Culture Collection (Manassas, VA, USA) (pcDNA3.1-(L)-His-CAMYEL #ATCC-MBA-277). The pcDNA3.1(+)-β-arrestin1-eYFP and β-arrestin2-Cerulean constructs were kind gifts from Dr C. Hoffmann (University of Würzburg, Würzburg, Germany). CXCR3-Renilla luciferase (Rluc) and β-arrestin2-eYFP BRET1-fusion constructs were generated by substituting the stopcodon of CXCR3 and β-arrestin2 with a SpeI/NotI linker and fusing them in frame to Rluc and yellow fluorescent protein (YFP) respectively.
Cell culture and transfection
HEK293T cells and HEK293 cells stably expressing human CXCR3 (a gift from Dr K. Biber, University Medical Center Groningen, the Netherlands), were grown at 37°C and 5% CO2 in DMEM supplemented with 10% FBS, penicillin, streptomycin and 400 µg·mL−1 G418 for maintaining stable expression of CXCR3. For β-arrestin recruitment experiments, HEK293T cells were transfected with a 1:4 ratio of cDNA coding for CXCR3-Rluc and β-arrestin1- or 2-YFP (total DNA 5 µg for every two million cells). Cells were transfected using linear polyethyleneimine (PEI) with a molecular weight of 25 kDa (Polysciences, Warrington, PA, USA) as described previously (Verzijl et al., 2008). The day after transfection, cells were trypsinized, resuspended into culture medium and plated in poly-L-lysine-coated white-bottom 96-well assay plates. For the cAMP biosensor assay, HEK293 cells stably expressing CXCR3 were transfected with 1 ug of DNA encoding for the cAMP biosensor, added up to a total of 5 ug cDNA with empty vector.
For chemotaxis experiments, murine L1.2 cells were grown in RPMI 1640 medium with 25 mM HEPES and GlutaMAX-I, supplemented with 10% heat-inactivated FBS, penicillin, streptomycin, non-essential amino acids, sodium pyruvate and 2-mercaptoethanol. Transfection of L1.2 cells was performed with 10 µg for every 20 million cells using a Bio-Rad Gene Pulser II (330 V and 975 µF, Bio-Rad, Hemel Hempstead, UK). Cells were grown over night in culture medium supplemented with 10 mM sodium butyrate.
Chemotaxis
Twenty-four hours after transfection, the migration of L1.2 cells towards different concentrations of CXCL11 and VUF10661 was determined using ChemoTx 96-well plates with 5 µM pore size (Neuro Probe, Inc., Gaithersburg, MD, USA). Briefly, the plates were blocked with culture medium supplemented with 1% (w/v) BSA. The compound dilutions were made in culture medium with 0.1% (w/v) of BSA, and added to the wells. L1.2 cells were added on top of the filter for each well. The plate was then incubated for 5 h at 37°C in a humidified chamber. Quantification of migrated cells was done by using calcein-AM, and measuring fluorescence at 525 nm with the Victor3 plate reader.
Membrane preparation and chemokine binding
Membrane preparation and competition radioligand bindings were performed as described previously (Verzijl et al., 2008). In brief, cell membrane fractions from HEK293 cells stably expressing CXCR3 were prepared by washing the cells twice with ice-cold PBS and centrifuging them at 1500×g for 10 min. The pellet was resuspended in ice-cold membrane buffer (15 mM Tris, pH 7.5, 1 mM EGTA, 0.3 mM EDTA, and 2 mM MgCl2), and homogenized using a Teflon-glass homogenizer and rotor. The membranes were subjected to two freeze-thaw cycles using liquid nitrogen, and centrifuged at 40 000×g for 25 min. The pellet was resuspended in Tris-sucrose buffer (20 mM Tris, 250 mM sucrose, pH 7.4) and aliquots were frozen in liquid nitrogen. For 125I-CXCL10 and for 125I-CXCL11 binding, 10 and 2 µg·well−1 of membranes were used, respectively in 96-well plates. For competition-binding experiments, membranes were incubated in binding buffer [50 mM HEPES, pH 7.4, 1 mM CaCl2, 5 mM MgCl2, 100 mM NaCl, and 0.5% (w/v) BSA] with approximately 70 pM 125I-chemokine and various concentrations of displacer for 2 h at room temperature. When saturation binding analysis was performed, the membranes were incubated for 2 h with increasing concentrations of 125I-chemokine in the presence or absence of VUF10661. Subsequently, membranes were harvested by filtration through Unifilter GF/C plates (Perkin–Elmer) presoaked with 0.5% PEI, using ice-cold wash buffer (50 mM HEPES, pH 7.4, 1 mM CaCl2, 5 mM MgCl2, and 500 mM NaCl). Radioactivity was measured using a MicroBeta scintillation counter (Perkin–Elmer).
Whole cell binding
HEK293 cells stably expressing the CXCR3 receptor were plated at 100 000 cells·well−1 into a 48-well assay plate (Greiner Bio-One, Alphen a/d Rijn, the Netherlands). The next day, the medium was aspirated and the cells were incubated in binding buffer (50 mM Hepes pH 7.4, 1 mM CaCl2, 5 mM MgCl2 and 100 mM NaCl) containing ∼70 pM of 125I-CXCL10 or 125I-CXCL11 in the presence and absence of unlabeled ligands. After 4 h at 4°C, the cells were washed with ice-cold wash buffer (50 mM Hepes pH 7.4, 1 mM CaCl2, 5 mM MgCl2 and 500 mM NaCl), lysed and bound radioactivity was counted in a Wallac Compugamma counter (PerkinElmer).
[35S]-GTPγS binding assay
For this assay, performed in 96-well plates, 5 µg·well−1 of membranes from HEK293 cells stably expressing CXCR3 were incubated with CXCL11 and VUF10661 in assay buffer (50 mM HEPES, 10 mM MgCl2, 100 mM NaCl, pH 7.2) supplemented with 3 µM GDP and 500 pM of [35S]-GTPγS. When antagonism was investigated, the compound was added 30 min prior to the addition of [35S]-GTPγS. The plate was incubated at room temperature for 1 h before the membranes were harvested by filtration through Unifilter GF/B plates and [35S]-GTPγS incorporation was determined using a Microbeta scintillation counter.
cAMP biosensor assay
The experimental procedure for this assay has been adapted from Masri et al. (2008). Twenty-four hours post-transfection, cells were trypsinized and seeded in poly-L-lysine-coated white 96-well plates. The next day, cells were rinsed once with HBSS, and incubated with fresh HBSS for 30 min before being stimulated. Next, the Rluc substrate coelenterazine-h was added to reach a final concentration of 5 µM. The non-specific PDE inhibitor IBMX was added simultaneously to a final concentration of 40 µM. For measuring the effects of chemokines and VUF10661 on cAMP levels, these ligands were added 5 min after coelenterazine-h. Forskolin was added 5 min after agonists, yielding a final concentration of 10 µM. When antagonistic behaviour of compounds on agonist responses was investigated, the compounds were added 10 min before coelenterazine-h. After 5 min of incubation with forskolin the YFP emission (505–555 nm), as well as the Rluc emission (465–505 nm), were sequentially recorded using a Victor3 multilabel counter (Perkin–Elmer). The BRET signal (BRET ratio) was determined by calculating the ratio between the YFP and the Rluc emission.
β-Arrestin recruitment BRET
Cell plating and transfection was performed as mentioned earlier. To assess β-arrestin recruitment BRET, after 10 min of incubation with coelenterazine-h, agonists were added, and incubated for an additional 10 min. In the case of experiments with antagonists, these were added simultaneously with coelenterazine-h. After 10 min, the plate was measured on the Victor3 and BRET ratios were calculated. Net BRET signals were determined by subtracting the BRET ratio obtained with cells only expressing Rluc-tagged CXCR3 from BRET signals obtained with cells co-expressing both Rluc-tagged CXCR3 and YFP-tagged β-arrestin.
CXCR3 and β-arrestin translocation
HEK293 cells stably expressing CXCR3 were transfected with 1 µg of cDNA encoding for β-arrestin1- or 2-YFP added to a total of 5 µg using empty vector. After 24 h, the cells were transferred to microscope cover slips. The next day, the cells were incubated in the presence or absence of CXCL11 and VUF10661 for 1 h and subsequently acid-washed three times (DMEM pH∼2). Cells were fixed with 4% formaldehyde in PBS and blocked with 3% skim milk in PBS or simultaneously blocked and permeabilized using 3% skim milk in 0.15% Triton X-100/PBS. Then, the cells were incubated consecutively with primary (anti-CXCR3 mAb160; R&D Systems, Minneapolis, MN, USA) and secondary (anti-mouse Alexa488; Molecular Probes, Invitrogen) antibodies, each for 1 h in 3% skim milk in PBS or 3% skim milk in 0.15% Triton X100/PBS with 3 washes of ice-cold PBS after incubation with each antibody. An Olympus FSX100 BioImaging Navigator was used for detection of fluorescence and the capturing of images.
Whole cell-based elisa : cell surface receptor expression
HEK293 cells stably expressing CXCR3 were trypsinized and seeded in 48-well plates. After 24 h, the medium was replaced with fresh medium and the medium containing CXCR3 compounds was added at different time points. After treatment with CXCL11 or VUF10661, the cells were subjected to three sequential acid washes (DMEM pH∼2) and fixed with 4% formaldehyde in tris-buffered saline (TBS; 50 mM Tris, 150 mM NaCl, pH 7.5). After being blocked with 1% skim milk in 0.1 M NaHCO3 pH 8.6, cells were incubated overnight at 4°C with anti-CXCR3 antibody in TBS containing 0.1% BSA. The next day, the cells were washed three times with TBS, and incubated with goat anti-mouse horseradish peroxidase-conjugated secondary antibody (Bio-Rad Laboratories, Hercules, CA, USA). Subsequently, cells were incubated with substrate buffer containing 2 mM o-phenylenediamine, 35 mM citric acid, 66 mM Na2HPO4, and 0.015% H2O2 at pH 5.6. The colouring reaction was stopped by addition of 1 M H2SO4, and the absorption at 490 nm was determined using a Powerwave X340 absorbance plate reader (BioTek, Bad Friedrichshall, Germany).
Data analysis
Nonlinear regression analysis of the data and calculation of affinity values was performed using Prism version 4.03 (GraphPad Software Inc., San Diego, CA). The Ki values in the radioligand binding studies were calculated using the Cheng–Prusoff equation Ki= IC50/(1 +[radioligand]/Kd of radioligand) (Cheng and Prusoff, 1973). For statistical analysis, one-way anova with a Bonferroni post-test, included in the GraphPad Prism software, was used with a confidence interval of 99%. The intrinsic activity (α) of an agonist in the functional assays was calculated by dividing its maximum response by that of the maximum of the particular system (Stephenson, 1956).
Results
VUF10661 is a partial agonist in CXCR3-mediated chemotaxis
The hallmark function of chemokines is their ability to induce directional migration of cells expressing chemokine receptors. Therefore, a chemotaxis assay with L1.2 cells transiently transfected with cDNA encoding the human CXCR3 receptor was used to investigate the agonistic properties of VUF10661. Both the endogenous ligand CXCL11 and the synthetic compound VUF10661 induced a dose-dependent migration of the CXCR3-expressing cells (Figure 2A), while this did not occur in mock-transfected cells (data not shown). The concentration at which the maximum effect was attained differed between CXCL11 and VUF10661 (10 nM and 1 µM, respectively); however, this correlated with the relative difference in affinities for these ligands in competition-binding experiments (Table 1, Figure 3). In agreement with previously reported results for VUF10661 analogues by Stroke et al. (2006), the efficacies of the ligands were different. Quantitatively, this was reflected by the intrinsic activity of the two molecules, which represents the relative ability of an agonist to produce a maximum response in a functional assay, where α= 1 indicates a full agonist, and an α between 0 and 1 indicates a partial agonist. VUF10661 showed a significantly lower maximal effect than the full agonist CXCL11 (α= 1), rendering VUF10661 a partial agonist (α= 0.5) in this assay. To investigate the specificity of the observed migration, cells were simultaneously incubated with either CXCL11 (10 nM) or VUF10661 (1 µM) and the CXCR3-selective antagonist NBI-74330 (1 µM) (Verzijl et al., 2008). NBI-74330 produced complete antagonism of chemotaxis induced by both CXCL11 and VUF10661 (Figure 2B), showing that VUF10661, like CXCL11, promotes CXCR3-mediated chemotaxis.
Figure 2.
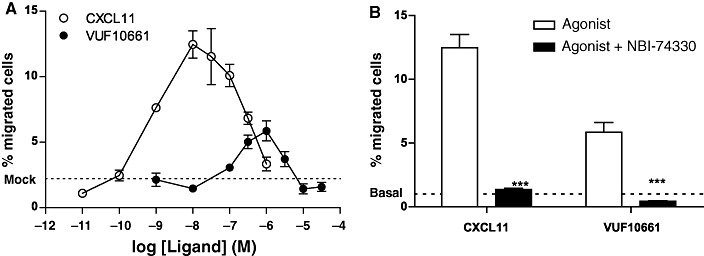
Chemotaxis assay with L1.2 cells expressing human CXCR3. (A) Migration of cells after 5 h of incubation with a concentration series of CXCL11 or VUF10661. (B) Co-incubation of CXCL11 (10 nM) and VUF10661 (1 µM) with the CXCR3 antagonist NBI-74330 (1 µM). Values are mean ± SEM, n= 3 (***P < 0.001).
Table 1.
Radioligand binding assays performed on HEK293 cells stably expressing human CXCR3
| Displacement (membranes) (pKi) | Displacement (whole cells) (pKi) | Saturation binding (membranes) | ||||
|---|---|---|---|---|---|---|
| Displacer | [125I]-CXCL10 | [125I]-CXCL11 | [125I]-CXCL10 | [125I]-CXCL11 | Kd (nM) | Bmax (pmol·mg−1 protein) |
| CXCL10 | 9.8 ± 0.1* | 8.0 ± 0.1 | 9.4 ± 0.1* | 8.0 ± 0.1 | 0.2 ± 0.02 | 0.1 ± 0.3 |
| CXCL11 | 10.4 ± 0.0 | 10.1 ± 0.1* | 9.6 ± 0.1 | 9.4 ± 0.1* | 0.08 ± 0.01 | 1.0 ± 0.3 |
| VUF10661 | 7.3 ± 0.0 | 6.2 ± 0.1 | 7.0 ± 0.0 | 5.9 ± 0.1 | – | – |
Affinities for CXCR3 ligands determined using displacement and saturation binding with [125I]-CXCL10 and [125I]-CXCL11 as radioligands on HEK293 whole cells stably expressing CXCR3 receptors or membranes prepared thereof. A ‘*’ indicates homologous displacement experiments leading to pKd values instead of pKi values. Bmax values were determined using saturation binding experiments and represent total receptor number mg−1 protein. All data are presented as mean ± SEM values from at least three independent experiments.
Figure 3.
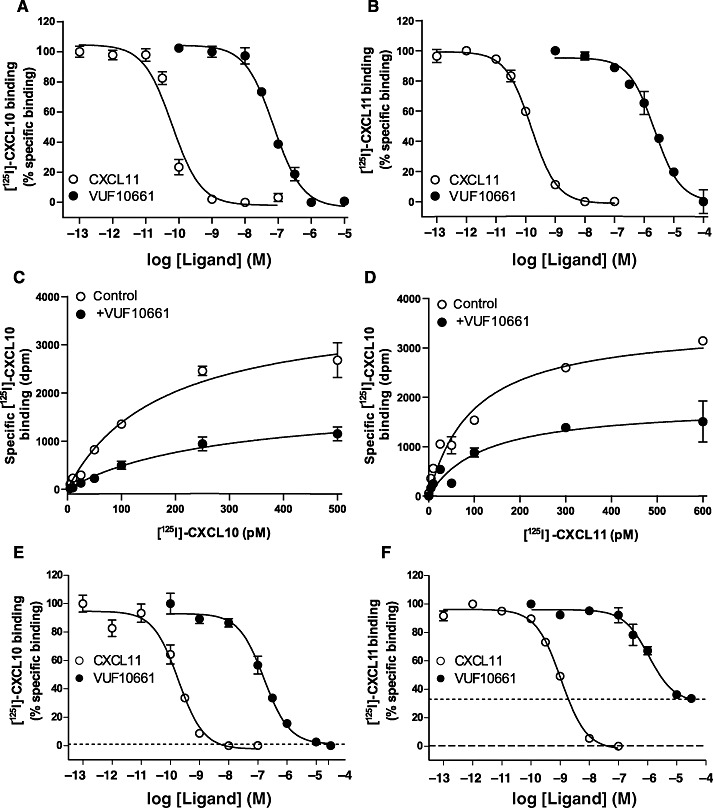
Radioligand-binding experiments. (A and B) Displacement of radioligand from HEK293 membranes prepared from HEK293 cells stably expressing CXCR3 receptors. [125I]-CXCL10 and [125I]-CXCL11 were used at a concentration of 70 pM (A) Displacement of [125I]-CXCL10 radioligand with cold CXCL11 and VUF10661. (B) Displacement of [125I]-CXCL11 with cold CXCL11 and VUF10661. (C and D) Saturation binding of [125I]-CXCL10 and [125I]-CXCL11 at HEK293 membranes expressing CXCR3 receptors. (C) Specific binding of [125I]-CXCL10 was determined in the absence or presence of 50 nM VUF10661. (D) Specific binding of [125I]-CXCL11 was determined in the absence or presence of 500 nM VUF10661. (E and F) Whole cell radioligand displacement assay using HEK293 cells stably expressing CXCR3 receptors. (E) Displacement of [125I]-CXCL10 with CXCL11 and VUF10661. Dotted line indicates level of radioligand binding after incubation with 0.3 µM of cold CXCL11 or 1 µM NBI-74330. (F) Displacement of [125I]-CXCL11 with CXCL11 and VUF10661. Dotted line indicates level of radioligand binding after incubation with 0.3 µM of cold CXCL10, and the dashed line shows specific radioligand binding after incubation with 1 µM of NBI-74330. Values shown are mean ± SEM representative of at least three independent experiments.
VUF10661 binds to CXCR3 in an allosteric fashion
To further characterize the small-molecule agonist VUF10661, the compound was subjected to radioligand binding studies with [125I]-CXCL10 and [125I]-CXCL11 on membrane preparations from HEK293 cells stably expressing the human CXCR3 receptor. Radiolabelled CXCL10 was displaced by unlabelled CXCL11, resulting in an affinity of 40 pM (pKi= 10.4 ± 0.1), while homologous [125I]-CXCL11 displacement with CXCL11 resulted in a similar affinity of 80 pM (pKd= 10.1 ± 0.1) (Figure 3A–B; Table 1), comparable with previously published data (Cox et al., 2001; Verzijl et al., 2008). Interestingly, VUF10661 inhibited the binding of both [125I]-CXCL10 and [125I]-CXCL11, although with different affinities, 50 nM (pKi= 7.3 ± 0.1) and 630 nM (pKi= 6.2 ± 0.1), respectively (Figure 3A–B and Table 1). In addition, VUF10661 was not able to displace [125I]-CXCL12 from membranes expressing CXCR4 or CXCR7 chemokine receptors (data not shown), indicating that the compound is selective for CXCR3 over CXCR4 and CXCR7.
To investigate the nature of interaction of VUF10661 with the endogenous ligands at the CXCR3 receptor, saturation binding assays with radiolabelled CXCL10 and CXCL11 were performed. Membranes were incubated with increasing concentrations of [125I]-CXCL10 or [125I]-CXCL11 in the presence or absence of a single concentration of VUF10661 (50 nM or 500 nM, respectively). The affinity for [125I]-CXCL10 of 0.20 ± 0.02 nM (n= 2) (Figure 3C and Table 1) was unaffected by the presence of VUF10661. However, in the presence of 50 nM VUF10661, the Bmax for [125I]-CXCL10 decreased approximately 50% (Figure 3C). Likewise, the Bmax of [125I]-CXCL11 decreased to a similar extent in the presence of 500 nM VUF10661, while its affinity remained constant (Kd= 0.08 ± 0.01 nM) (Figure 3D, Table 1). To investigate whether this decrease in Bmax was due to (pseudo)-irreversible binding of VUF10661, ligand washout experiments were performed (Lanzafame et al., 2004). Pre-incubation of intact HEK293/CXCR3 cells with VUF10661 for 1 h, followed by an extensive washout of 30 min, did not have an effect on maximal [125I]-CXCL11 binding (95 ± 5%, Supporting Information Figure S2: grey column) when compared with vehicle-pretreated controls (99 ± 2%, Supporting Information Figure S2: white column). This indicates that VUF10661 binds to CXCR3 in a reversible fashion. Because VUF10661 only affected the Bmax of the chemokines and not the Kd, these findings suggest an allosteric binding mode for VUF10661 (Kollias-Baker et al., 1997; de Kruijf et al., 2009). Next, we studied the interaction of VUF10661 with CXCR3 in a whole cell context. In order to do this, a series of concentrations of cold CXCL11 and VUF10661 was used to displace a fixed concentration of [125I]-CXCL10 or [125I]-CXCL11 from HEK293 cells stably expressing CXCR3 receptors. The observed affinities of CXCL11 and VUF10661 were 0.25 nM (pKi= 9.6 ± 0.1) and 100 nM (pKi= 7.0 ± 0.1), respectively, when [125I]-CXCL10 was used as radioligand, and 0.4 nM (pKi= 9.4 ± 0.1) and 1.0 µM (pKi= 6.0 ± 0.1) in the case of [125I]-CXCL11. Interestingly, whereas VUF10661 completely inhibited binding of [125I]-CXCL10 (Figure 3E), it was unable to completely displace [125I]-CXCL11 from CXCR3 in this assay (± 70%; Figure 3F). Similar behaviour was observed for CXCL10, which also partially inhibited [125I]-CXCL11 binding. In contrast, CXCL11 and NBI-74330 fully displaced both radioligands to non-specific binding levels (Figure 3E and F). Collectively, these data indicate that VUF10661 has a binding mode different from CXCL11, the major ligand for CXCR3.
VUF10661 activates Gi proteins with the same efficacy as CXCL11
Because VUF10661 showed partial agonistic behaviour in the chemotaxis assay with transfected L1.2 cells, we next determined its potency for CXCR3-mediated G protein activation using [35S]-GTPγS binding, a classical functional readout for GPCRs (Harrison and Traynor, 2003). Both CXCL11 and VUF10661 produced a dose-dependent increase in [35S]-GTPγS binding to membranes prepared from HEK293 cells stably expressing human CXCR3 receptors. The potencies for these responses were 0.6 nM (pEC50= 9.2 ± 0.1) for CXCL11 and 0.6 µM (pEC50= 6.2 ± 0.1) for VUF10661 and correlated well with their respective affinities (Figure 4A). Co-incubation of CXCL11 and VUF10661 with 1 µM NBI-74330 abolished the agonist-induced [35S]-GTPγS binding (Figure 4B), demonstrating that agonist-induced signals detected in this assay are mediated through CXCR3. Moreover, pretreatment of CXCR3-expressing cells with 25 ng·mL−1 PTX before they were harvested, resulted in a complete inhibition of agonist-induced [35S]-GTPγS binding, consistent with reports on CXCR3 coupling to Gi proteins (Figure 4C) (Smit et al., 2003; Thompson et al., 2007).
Figure 4.
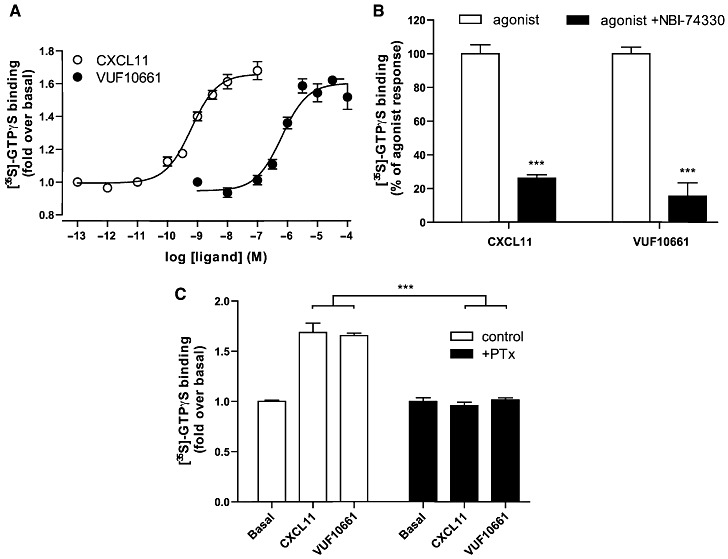
[35S]-GTPγS binding assay at membranes of HEK293 cells stably expressing CXCR3. (A) Membranes incubated with a concentration series of CXCL11 and VUF10661. Data are represented as fold over basal [35S]-GTPγS binding from three independent experiments, and values shown are mean ± SEM. (B) CXCL11 (3 nM) and VUF10661 (3 µM) were co-incubated with 1 µM of CXCR3 antagonist NBI-74330. Data are expressed as percentage of agonist response, with mean ± SEM, n= 3. (C) Cells were treated with or without PTX (25 ng·mL−1) before membrane harvesting and subsequent agonist incubations. Values are mean ± SEM, n= 3 (***P < 0.001).
VUF10661 inhibits adenylyl cyclase activity: use of a novel BRET cAMP biosensor
Inhibition of adenylyl cyclase is another well-characterized response mediated by Gi/o-coupled receptors. To investigate the effects of CXCL11 and VUF10661 on cAMP levels, a BRET-based cAMP biosensor was employed (Jiang et al., 2007). The mechanism of this biosensor is schematically shown in Figure 5A and a detailed description can be found elsewhere (Jiang et al., 2007; Barak et al., 2008; Masri et al., 2008). The biosensor was co-expressed with human CXCR3 in HEK293 cells and used to measure changes in intracellular cAMP levels. Incubation of these cells with CXCR3 agonists did not lead to an increase in cAMP production. However, stimulation with 10 µM forskolin, a direct activator of adenylyl cyclase, led to a robust response (data not shown). Addition of increasing concentrations of CXCL11 dose-dependently inhibited the forskolin-induced cAMP accumulation with a potency of 1.6 nM (pEC50= 8.8 ± 0.1) (Figure 5B). In agreement with its lower affinity for CXCR3, VUF10661 also displayed lower potency in the BRET-biosensor cAMP assay (0.5 µM; pEC50= 6.3 ± 0.1) (Figure 5B). In contrast, CXCL10 behaved as partial agonist in this assay (α= 0.7), having a potency of 13 nM (pEC50= 7.9 ± 0.2) (Figure 5B). The observed response for CXCL11 and VUF10661 reflected specific activation of CXCR3, because 1 µM of NBI-74330 inhibited the response to basal levels (Figure 5C). In accordance with the Gi-coupling properties of CXCR3, treatment of cells with 25 ng·mL−1 PTX completely inhibited the agonist-induced effects (Figure 5D).
Figure 5.
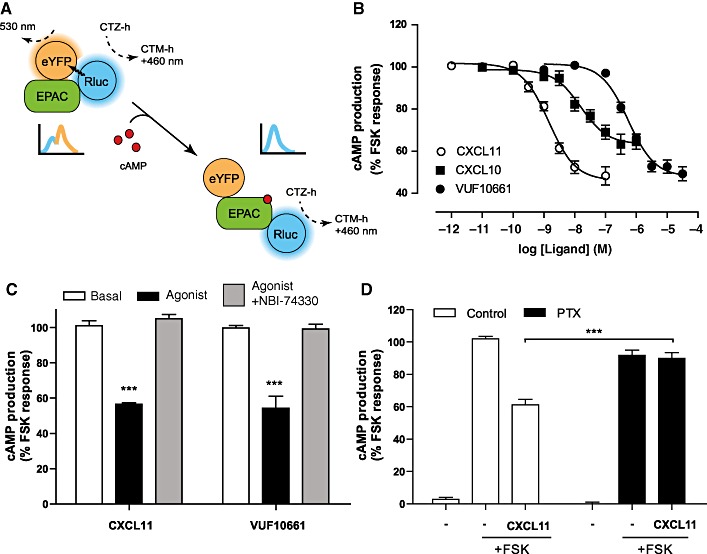
cAMP biosensor assay. (A) Schematic illustration of BRET-based cAMP biosensor assay. (B) Inhibition of forskolin-induced cAMP accumulation was determined after sequential administration of CXCL11, CXCL10 or VUF10661 and forskolin to HEK293 cells stably expressing CXCR3 and transiently transfected with the cDNA encoding the cAMP biosensor. The figure shows mean ± SEM of grouped data from three independent experiments. (C) The selective CXCR3 antagonist NBI-74330 (1 µM) was co-incubated with CXCL11 (10 nM) or VUF10661 (3 µM). (D) Influence of PTX treatment on CXCR3-induced inhibition of adenylyl cyclase. Same agonist concentrations used as for (C). Data are expressed as percentage of the maximal forskolin response. All values shown are mean ± SEM from three independent experiments (***P < 0.001).
VUF10661 induces internalization of CXCR3
Upon sustained agonist stimulation, GPCRs are generally removed from the cell surface, a process called endocytosis or internalization (Ferguson et al., 1996). CXCR3 has been shown to readily internalize, after 30 min to several hours, in response to CXCL10 or CXCL11 in various cell types, including HEK293 and peripheral blood mononuclear cells, as well as activated T lymphocytes (Dagan-Berger et al., 2006). In the present study, we used an immunocytochemistry approach to qualitatively determine the effect of CXCR3 agonists on CXCR3 surface expression and distribution. When HEK293 cells, stably expressing human CXCR3 and transiently transfected with cDNA encoding for β-arrestin1- or 2-YFP, were stained with anti-CXCR3 antibody, we observed a clear localization of CXCR3 at the plasma membrane (Figure 6A1). β-Arrestin2-YFP was homogenously distributed throughout the cells (Figure 6A2). Upon stimulation with 30 nM CXCL11 (Figure 6A4-6) or 3 µM VUF10661 (Figure 6A7-9) for 1 h, a marked redistribution of the receptor into intracellular vesicles was observed. Concomitantly, staining at the cell surface decreased, indicating CXCR3 internalization (Figure 6A4 and 7). Since β-arrestins have been shown to be partly involved in CXCR3 internalization (Colvin et al., 2004; Meiser et al., 2008), we also investigated the redistribution of these scaffolding proteins upon CXCR3 stimulation. Both CXCL11 and VUF10661 induced a pronounced punctuated intracellular redistribution of β-arrestin1 and -2-YFP (data not shown and Figure 6A5 and 8). Interestingly, CXCR3 and β-arrestin1 and -2 showed significant co-localization after agonist treatment, suggesting that both β-arrestin isoforms are involved in CXCR3 internalization (data not shown and Figure 6A6 and 9). To further quantify the internalization of CXCR3 in these cells, a whole-cell based elisa was performed. As can be seen in Figure 6B, cell surface expression of CXCR3 was significantly reduced when stimulated with either CXCL11 or VUF10661, already after 30 min, reaching a maximum after approximately one hour. CXCL11 and VUF10661-induced internalization was shown to be concentration-dependent with potencies of 2.5 nM (pEC50= 8.6 ± 0.1) and 3.2 µM (pEC50= 5.5 ± 0.1) respectively (Figure 6C). This effect was inhibited by incubation at 4°C, an approach generally used to inhibit receptor internalization. Collectively, these data show that both CXCL11 and VUF10661 induce β-arrestin1 and -2 translocation to CXCR3 and subsequent receptor internalization in HEK293 cells.
Figure 6.
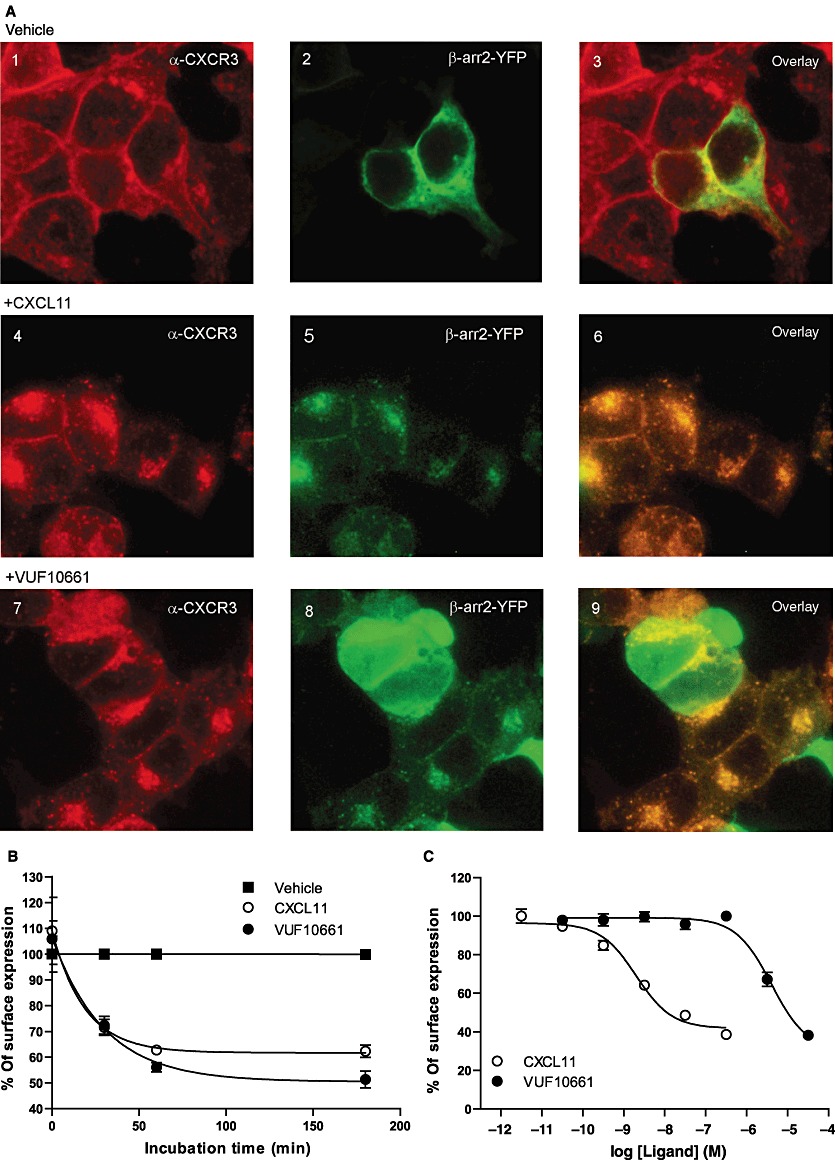
Redistribution of CXCR3 upon agonist stimulation. (A) HEK293 cells stably expressing CXCR3 and transiently transfected with cDNA encoding for β-arrestin2-YFP were incubated with or without CXCL11 (30 nM) and VUF10661 (3 µM) for 1 h. Cells were fixed and stained with anti-CXCR3 and Alexa-546 antibodies to visualize CXCR3 (images 1, 4 and 7), while β-arrestin2-YFP is shown in images 2, 5 and 8. Cells were treated with empty medium (basal; panels 1–3), CXCL11 (panels 4–6) or VUF10661 (images 7–9) for 1 h. Panels 3, 6 and 9 show an overlay of the red and green channels. Images shown are representative of three independent experiments. (B) Cell surface expression of CXCR3 was determined with elisa using the anti-CXCR3 antibody mAb160 in presence of CXCL11 (30 nM) or VUF10661 (10 µM) at different time points and compared with vehicle. (C) Effect of concentration series of CXCL11 and VUF10661 on cell surface expression of CXCR3 after 3 h of agonist incubation. Values shown are mean ± SEM, n= 3.
VUF10661 recruits β-arrestin1 and -2 to CXCR3 receptors
It is well accepted that β-arrestins are recruited to activated GPCRs after phosphorylation by GPCR kinases (GRK), leading to desensitization and ultimately to internalization of the receptor (Pierce and Lefkowitz, 2001). In addition to these properties, there is a growing body of evidence showing that β-arrestins can serve as scaffolds orchestrating the assembly of multiple proteins from several signalling pathways. Because recruitment of β-arrestins to GPCRs and its downstream effects are thought to be G protein-independent (DeWire et al., 2007), it offers a novel way to assess ligand-mediated activation of GPCRs. Following the observed co-localization of CXCR3 and β-arrestins, we decided to study CXCR3-induced β-arrestin recruitment directly. In order to investigate agonist-induced interactions of β-arrestin1 and -2 with the CXCR3 receptor we used a BRET1 approach in which β-arrestin1 and -2 were C-terminally tagged with YFP and the human CXCR3 receptor fused to Rluc. Tagging of CXCR3 with Rluc resulted in a receptor showing similar expression and functional responses as those of wild-type CXCR3 (data not shown). The cDNAs encoding for CXCR3-Rluc and either β-arrestin1-YFP or β-arrestin2-YFP were transiently co-transfected in HEK293T cells and incubated with CXCL10, CXCL11 or VUF10661. Agonist-promoted recruitment of β-arrestin concomitantly brings YFP in proximity to Rluc, facilitating BRET and leading to the emission of light at an average wavelength of 530 nm (Figure 7A). As demonstrated in Figure 7B, CXCL10, CXCL11 and VUF10661 all recruited β-arrestin2 with potencies of 32 nM (pEC50= 7.5 ± 0.3), 3 nM (pEC50= 8.5 ± 0.1) and 1 µM (pEC50= 5.9 ± 0.1), respectively. Using the same methodology, all three agonists were observed to similarly induce the interaction of human CXCR3 with β-arrestin1 (Figure 7B). Interestingly, VUF10661 showed a significantly higher efficacy (α= 1) for the recruitment of both β-arrestin1 and 2, as compared with CXCL10 (α= 0.2) and CXCL11 (α= 0.6), which both behaved as partial agonists in this assay. The CXCR3 antagonist NBI-74330 was able to completely inhibit agonist-promoted responses in this assay (Figure 7C). Moreover, the treatment of the cells with 25 ng·mL−1 PTX did not block the observed β-arrestin recruitment, suggesting that Gi proteins are not needed for the recruitment of β-arrestins to CXCR3 (Figure 7D).
Figure 7.
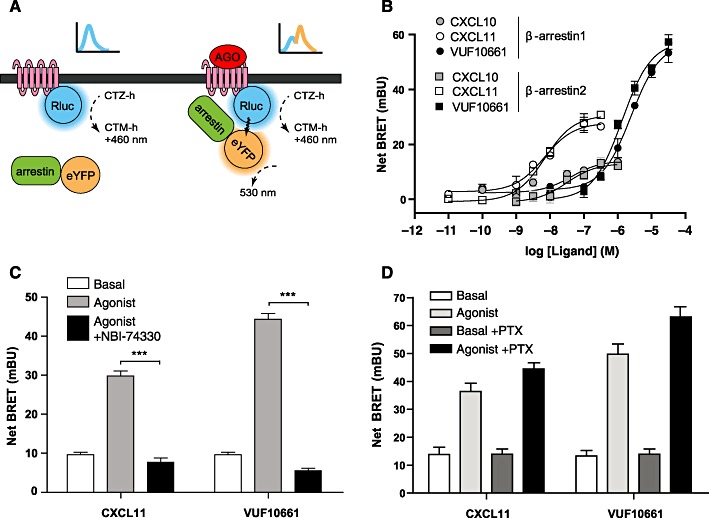
BRET-based β-arrestin recruitment assay in HEK293T cells transiently expressing CXCR3-Rluc and β-arrestin1-YFP or β-arrestin2-YFP. (A) Schematic illustration of β-arrestin recruitment to CXCR3 receptors using a BRET1 approach. (B) Effects of increasing concentrations of CXCL11, CXCL10 and VUF10661 on the recruitment of β-arrestin1-YFP or CXCL11, CXCL10 and VUF10661 on the recruitment of β-arrestin2-YFP. Data are presented as mean ± SEM, grouped from at least three independent experiments. (C) The CXCR3 antagonist NBI-74330 (1 µM) was co-incubated with 30 nM of CXCL11 or 3 µM of VUF10661. (D) PTX sensitivity of the recruitment of β-arrestin2 induced by both CXCL11 (30 nM) and VUF10661 (3 µM) was assessed with or without treatment of the cells with 25 ng·mL−1 PTX. Values are mean ± SEM, n= 3 (***P < 0.001).
Discussion
Chemokine receptor binding and activation is generally thought to occur via a two-step mechanism in which the first step is governed by binding of the large peptide ligand to the N-terminus and extracellular loops of the GPCR protein (Allen et al., 2007). Subsequently, the N-terminus of the chemokine is well-positioned to interact with the transmembrane (TM) domains, leading to activation of the receptor (Blanpain et al., 2003; Kofuku et al., 2009). In contrast, small-molecule modulators of family A GPCRs generally bind to the TM part of the GPCR, as recently shown for a small molecule CXCR4 antagonist using X-ray crystallography (Wu et al., 2010). Consequently, small-molecule chemokine receptor ligands are considered to allosterically modulate GPCR function from binding sites that are distinct or only partially overlap with the chemokine interaction points (Rosenkilde et al., 2004; Kondru et al., 2008; Verzijl et al., 2008; de Kruijf et al., 2009; Garcia-Perez et al., 2011). Recently, the first small molecule inhibitors of the chemokine receptors CXCR4 and CCR5 have reached the market for stem cell mobilization (AMD3100 or Mozobil®) or HIV infection (maraviroc or Celsentri®), indicating the huge potential of allosteric modulation of chemokine receptors.
The CXCR3 receptor is one of the members of the CXC chemokine receptor family that has attracted considerable interest as a therapeutic target in view of its role in several inflammatory conditions and (although more controversial) cancer (Wijtmans et al., 2008). Small-molecule inhibitors targeting CXCR3 act as non-competitive, potentially allosteric inhibitors (Verzijl et al., 2008) and are effective tools to study the (patho)physiological role of CXCR3 (Wijtmans et al., 2008). In a screen for CXCR3 antagonists Pharmacopeia scientists have recently identified tetrahydroisoquinolines as CXCR3 agonists (Stroke et al., 2006). They show that these compounds act as agonists, leading to calcium mobilization and chemotaxis upon binding to the CXCR3 receptor. In the present work, we confirm their initial findings and describe a further, detailed pharmacological characterization of one such small-molecule synthetic agonist for CXCR3, synthesized in-house and named VUF10661.
The tetrahydroisoquinoline VUF10661 displaces radiolabelled CXCL10 and CXCL11 with different affinities (50 and 630 nM respectively). In addition, VUF10661 shows incomplete inhibition of 125I-CXCL11 binding while completely displacing [125I]-CXCL10 binding to cells expressing CXCR3. Altogether, these data are in line with the findings of Cox and co-workers that showed that CXCL10 labels a subset of active CXCR3 conformations, whereas CXCL11 labels CXCR3 proteins in both an active and inactive conformation (Cox et al., 2001). Following this reasoning, VUF10661 has a clear preference for the active states of CXCR3, which would be in line with its reported agonistic behaviour. As CXCL11 seems to stabilize a different subset of active conformations than CXCL10 (Cox et al., 2001), it also seems likely that VUF10661 stabilizes its own subset of active conformations leading to the observed ligand-biased signalling in β-arrestin recruitment compared with G protein-dependent assays like [35S]-GTPγS and cAMP. Moreover, CXCL11 showed a 10-fold decrease in affinity in whole cells compared with membranes, while CXCL10 affinity remained constant. This again indicates that CXCL10 and CXCL11 recognize different CXCR3 conformations. In saturation binding experiments, a single concentration of VUF10661 resulted in a significant decrease in Bmax, but not in Kd, for both [125I]-CXCL10 and [125I]-CXCL11, indicating that VUF10661 interacts in a non-competitive, potential allosteric fashion with both chemokines at CXCR3. Preliminary mutagenesis studies support the hypothesis of allosteric binding of VUF10661 to CXCR3 (Scholten et al., unpubl. data). Our present findings agree with previous results with various classes of small-molecule CXCR3 inverse agonists (Verzijl et al., 2008) and have also been observed with CXCR2 antagonists (de Kruijf et al., 2009) and inverse agonists for the viral chemokine receptor US28 (Casarosa et al., 2003). It should be noted that hardly any data are available on the effect of allosteric GPCR ligands on agonist radioligand saturation curves. A simple allosteric ternary complex model cannot explain the observed decrease in Bmax and, therefore, more complex alternative hypotheses need to be considered. Washout experiments show that the decrease in Bmax is not due to irreversible binding of this compound to CXCR3. A possible explanation is that VUF10661 disrupts dimeric or higher order oligomeric CXCR3 complexes and thereby reduces the number of available radioligand-binding sites. So far, however, we have not been able to detect any biochemical evidence for ligand-induced (agonists or inverse agonist) modulation of CXCR3 oligomers (Leurs et al., unpubl. data). Alternatively, VUF10661 might enrich a population of activated, G protein-coupled CXCR3 receptors, thereby reducing the number of radioligand-receptor-G protein complexes and hence a decrease in Bmax.
To characterize the functional consequences of VUF10661 binding to the CXCR3 receptor, we investigated its ability to activate several signalling pathways, including G protein-dependent as well as G protein-independent events. Using a [35S]-GTPγS binding assay we show that VUF10661, like CXCL11, activates Gi proteins and both act as full agonists at the CXCR3 receptor. The observed difference in potencies between CXCL11 and VUF10661 reflects the relative difference in affinity as determined in the radioligand competition experiments. To assess the subsequent changes in the levels of cAMP after agonist stimulation, we used a novel BRET-based cAMP biosensor. This intracellular cAMP indicator has been successfully used for several GPCRs (Jiang et al., 2007; Barak et al., 2008; Masri et al., 2008) and allows monitoring of receptor signalling in live cells. Using this approach, we show that upon activation with CXCL10, CXCL11 or VUF10661, CXCR3 is able to inhibit the forskolin-induced adenylyl cyclase activity with different potencies that agree with the relative difference in their binding affinities. Both the small-molecule agonist and CXCL11 act as full agonists in this assay while CXCL10 is a partial agonist.
Classically, G proteins have been considered as the main interacting proteins of GPCRs. However, in recent years, the involvement of other interacting proteins, such as β-arrestins, has been emerging, both in signalling and regulation of GPCRs (Ritter and Hall, 2009). Whereas the role for β-arrestins in GPCR desensitization and internalization has been known for a while, the GPCR field is currently also appreciating their role as GPCR signalling partners (Pierce and Lefkowitz, 2001). Recruited β-arrestins act as scaffolds bringing together proteins from different signalling cascades, such as MAP kinases or PKB, in multiprotein complexes. It has previously been shown that CXCR3 internalizes after CXCL11 binding (Sauty et al., 2001; Dagan-Berger et al., 2006). In the present work, we showed that VUF10661 also induces a dose- and time-dependent internalization of CXCR3. Activation of CXCR3 by VUF10661 leads to a prolonged internalization to a similar extent as CXCL11. Moreover, as observed previously for CXCL11 (Colvin et al., 2004; Meiser et al., 2008), prolonged exposure to VUF10661 induced CXCR3 downregulation, as the total level of CXCR3 detected in the elisa, using permeabilized samples, was significantly reduced (data not shown). Fluorescence microscopy experiments revealed a significant co-localization of CXCR3 and β-arrestin1 and -2 after agonist treatment, indicating a role for β-arrestins in the redistribution and internalization of the CXCR3 receptor. Recently, GPCRs have been classified into two families with respect to their internalization profile, namely a class of receptors that have transient interaction with (mainly) β-arrestin2, leading to recycling, and a class of receptors that have more sustained interaction with β-arrestin1 and -2, generally leading to slow recycling and/or subsequent degradation (Pierce and Lefkowitz, 2001). Because CXCR3 does not seem to have a preference for either β-arrestin1 or -2, and shows prolonged co-localization with both, this suggests that CXCR3 belongs to the latter class. Moreover, this is in agreement with the presence of serine/threonine clusters in the C-terminus of CXCR3. Such motifs have been linked to stabilization of the interaction between GPCRs and β-arrestin, leading to slow recycling or degradation of the GPCR (Oakley et al., 2001). Previous reports have shown that transfection of individual β-arrestin1 or β-arrestin2 dominant negative isoforms had no effect on CXCR3 internalization in HEK293 cells (Colvin et al., 2004; Meiser et al., 2008). Our results suggest that the two isoforms exert compensatory effects and can both participate in the process of CXCR3 receptor internalization.
To directly assess the CXCR3-mediated β-arrestin recruitment, we used a BRET-based approach and showed in this study for the first time that both β-arrestins are actively recruited to CXCR3 after agonist stimulation. The agonist-induced recruitment of both arrestins is insensitive to treatment with PTX, showing that the classical signalling pathway of CXCR3 via Gi proteins (Smit et al., 2003) is not involved in this process. Both CXCL11 and VUF10661 promote recruitment of β-arrestin1 and β-arrestin2 to the CXCR3 receptor with similar potencies, suggesting that the relative expression levels of the β-arrestin isoforms in different cell types will determine the stoichiometry of coupling to CXCR3. Importantly, VUF10661 is much more efficacious in recruiting both β-arrestin isoforms than CXCL11 (α= 1 vs. α= 0.6 for CXCL11). The higher efficacy of VUF10661 in the BRET-based β-arrestin recruitment assay could reflect a difference in the relative orientation of the two BRET fluorophores, instead of a stronger β-arrestin recruitment. However, using a β-arrestin2 protein complementation assay (DiscoveRx PathHunter™), similar results were obtained, unequivocally demonstrating that VUF10661-induced CXCR3 activation results in a stronger recruitment of β-arrestin compared with the endogenous peptide CXCL11 (Scholten et al., unpubl. obs.). To exclude the possibility that the higher intrinsic activity of VUF10661 in β-arrestin recruitment compared with CXCL11 is due to differential desensitization associated with full versus partial agonism instead of functional selectivity, we also included CXCL10 in a G protein-dependent readout (cAMP) and a G protein-independent readout (β-arrestin recruitment). CXCL10 is not only a partial agonist in these cAMP experiments, but remains a partial agonist in the recruitment of β-arrestin, confirming that the higher efficacy for VUF10661 in this assay is most likely the first report of functional selectivity at the CXCR3 receptor.
One of the hallmarks of chemokine receptor signalling output is cell migration. Whereas this process is known to involve Gi proteins [see, e.g. for CXCR3 (Smit et al., 2003; Colvin et al., 2004; Thompson et al., 2007) ], it is becoming increasingly apparent that also β-arrestins play a key role in chemokine receptor-mediated cell migration. Splenocytes from β-arrestin2−/− mice are significantly impaired in their migratory responses towards CXCL12, the ligand for CXCR4 (Fong et al., 2002). In contrast, neutrophils from β-arrestin2−/− mice display an enhanced chemotaxis towards the CXCR2 ligand CXCL1 (Su et al., 2005), suggesting a complex interplay between β-arrestins and Gi proteins in migratory responses. In the present study, we showed that the small-molecule CXCR3 agonist VUF10661 induces a dose-dependent migration of CXCR3 transfected L1.2 cells, albeit to a significantly lower extent than CXCL11. This partial agonistic activity of VUF10661 contrasts with its full agonistic activity in the G protein-dependent assays (cAMP and [35S]-GTPγS) and the higher efficacy, compared with CXCL11, in the β-arrestin recruitment assay.
Based on the data from the β-arrestin recruitment assay, G-protein dependent signalling assays, the radioligand binding assays, as well as chemotaxis experiments, we propose that CXCL11 and VUF10661 stabilize different receptor- and/or β-arrestin conformations leading to differences in functional output. Such ligand-biased signalling has recently been recognized as a new opportunity for GPCR drug discovery (Urban et al., 2007; Kenakin and Miller, 2010) and might also offer interesting options for the therapeutic use of CXCR3 agonists. Based on studies with small-molecule agonists for CCR3 (Wise et al., 2007) and CCR5 (Saita et al., 2006), it has been proposed that chemokine receptor agonists able to induce downregulation of their corresponding receptor, but lacking significant chemotactic effects, can act as functional antagonists by removing the receptor from the cell surface. From a therapeutic point of view, pathway-biased agonists might therefore reveal new interesting strategies for the treatment of chemokine-associated diseases.
Acknowledgments
We would like to thank Dr G.J. Zaman from Merck Sharp & Dohme for enabling us to perform PathHunter™ experiments at their site in Oss, the Netherlands and E. Janssen for technical assistance with the synthesis of VUF10661.
This work was performed within the framework of Dutch Top Institute Pharma, project ‘The GPCR forum (D1-105).’
Glossary
- FBS
fetal bovine serum
- PEI
polyethyleneimine
- PTX
pertussis toxin
- Rluc
Renilla luciferase
- SLE
systemic lupus erythematosus
- TM
transmembrane
- VUF10661
N-(6-amino-1-(2,2-diphenylethylamino)-1-oxohexan-2-yl)-2-(4-oxo-4-phenylbutanoyl)-1,2,3,4-tetrahydroisoquinoline-3-carboxamide
Conflict of interest
The authors state no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Appendix S1 General synthetic procedures.
Figure S1 Synthesis scheme for VUF10661.
Figure S2 VUF10661 washout experiment.
Please note: Wiley–Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th Edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen S, Crown S, Handel T. Chemokine: receptor structure, interactions, and antagonism. Annu Rev Immunol. 2007;25:787–820. doi: 10.1146/annurev.immunol.24.021605.090529. [DOI] [PubMed] [Google Scholar]
- Barak L, Salahpour A, Zhang X, Masri B, Sotnikova T, Ramsey A, et al. Pharmacological characterization of membrane-expressed human trace amine-associated receptor 1 (TAAR1) by a bioluminescence resonance energy transfer cAMP biosensor. Mol Pharmacol. 2008;74:585–594. doi: 10.1124/mol.108.048884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer J, Baechler E, Petri M, Batliwalla F, Crawford D, Ortmann W, et al. Elevated serum levels of interferon-regulated chemokines are biomarkers for active human systemic lupus erythematosus. PLoS Med. 2006;3:e491. doi: 10.1371/journal.pmed.0030491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanpain C, Doranz BJ, Bondue A, Govaerts C, De Leener A, Vassart G, et al. The core domain of chemokines binds CCR5 extracellular domains while their amino terminus interacts with the transmembrane helix bundle. J Biol Chem. 2003;278:5179–5187. doi: 10.1074/jbc.M205684200. [DOI] [PubMed] [Google Scholar]
- Bonacchi A, Romagnani P, Romanelli RG, Efsen E, Annunziato F, Lasagni L, et al. Signal transduction by the chemokine receptor CXCR3: activation of Ras/ERK, Src, and phosphatidylinositol 3-kinase/Akt controls cell migration and proliferation in human vascular pericytes. J Biol Chem. 2001;276:9945–9954. doi: 10.1074/jbc.M010303200. [DOI] [PubMed] [Google Scholar]
- Casarosa P, Menge WM, Minisini R, Otto C, van Heteren J, Jongejan A, et al. Identification of the first nonpeptidergic inverse agonist for a constitutively active viral-encoded G protein-coupled receptor. J Biol Chem. 2003;278:5172–5178. doi: 10.1074/jbc.M210033200. [DOI] [PubMed] [Google Scholar]
- Cheng Y-C, Prusoff W. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Cole K, Strick C, Paradis T, Ogborne K, Loetscher M, Gladue R, et al. Interferon-inducible T cell alpha chemoattractant (I-TAC): a novel non-ELR CXC chemokine with potent activity on activated T cells through selective high affinity binding to CXCR3. J Exp Med. 1998;187:2009–2021. doi: 10.1084/jem.187.12.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colvin RA, Campanella GSV, Sun J, Luster AD. Intracellular domains of CXCR3 that mediate CXCL9, CXCL10, and CXCL11 function. J Biol Chem. 2004;279:30219–30227. doi: 10.1074/jbc.M403595200. [DOI] [PubMed] [Google Scholar]
- Cox M, Jenh C, Gonsiorek W, Fine J, Narula S, Zavodny P, et al. Human interferon-inducible 10-kDa protein and human interferon-inducible T cell alpha chemoattractant are allotopic ligands for human CXCR3: differential binding to receptor states. Mol Pharmacol. 2001;59:707–715. doi: 10.1124/mol.59.4.707. [DOI] [PubMed] [Google Scholar]
- Dagan-Berger M, Feniger-Barish R, Avniel S, Wald H, Galun E, Grabovsky V, et al. Role of CXCR3 carboxyl terminus and third intracellular loop in receptor-mediated migration, adhesion and internalization in response to CXCL11. Blood. 2006;107:3821–3831. doi: 10.1182/blood-2004-01-0214. [DOI] [PubMed] [Google Scholar]
- DeWire S, Ahn S, Lefkowitz R, Shenoy S. Beta-arrestins and cell signaling. Annu Rev Physiol. 2007;69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- Enghard P, Humrich J, Rudolph B, Rosenberger S, Biesen R, Kuhn A, et al. CXCR3+CD4+ T cells are enriched in inflamed kidneys and urine and provide a new biomarker for acute nephritis flares in systemic lupus erythematosus patients. Arthritis Rheum. 2009;60:199–206. doi: 10.1002/art.24136. [DOI] [PubMed] [Google Scholar]
- Ferguson SS, Downey WE, 3rd, Colapietro AM, Barak LS, Menard L, Caron MG. Role of beta-arrestin in mediating agonist-promoted G protein-coupled receptor internalization. Science. 1996;271:363–366. doi: 10.1126/science.271.5247.363. [DOI] [PubMed] [Google Scholar]
- Fong AM, Premont RT, Richardson RM, Yu YR, Lefkowitz RJ, Patel DD. Defective lymphocyte chemotaxis in beta-arrestin2- and GRK6-deficient mice. Proc Natl Acad Sci USA. 2002;99:7478–7483. doi: 10.1073/pnas.112198299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Zhou XY, Yashiro-Ohtani Y, Yang YF, Sugimoto N, Ono S, et al. The unique target specificity of a nonpeptide chemokine receptor antagonist: selective blockade of two Th1 chemokine receptors CCR5 and CXCR3. J Leukoc Biol. 2003;73:273–280. doi: 10.1189/jlb.0602269. [DOI] [PubMed] [Google Scholar]
- Garcia-Perez J, Rueda P, Staropoli I, Kellenberger E, Alcami J, Arenzana-Seisdedos F, et al. New insights into the mechanisms whereby low molecular weight CCR5 ligands inhibit HIV-1 infection. J Biol Chem. 2011;286:4978–4990. doi: 10.1074/jbc.M110.168955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock W, Lu B, Gao W, Csizmadia V, Faia K, King J, et al. Requirement of the chemokine receptor CXCR3 for acute allograft rejection. J Exp Med. 2000;192:1515–1520. doi: 10.1084/jem.192.10.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison C, Traynor J. The [35S]GTPgammaS binding assay: approaches and applications in pharmacology. Life Sci. 2003;74:489–508. doi: 10.1016/j.lfs.2003.07.005. [DOI] [PubMed] [Google Scholar]
- Jiang LI, Collins J, Davis R, Lin KM, DeCamp D, Roach T, et al. Use of a cAMP BRET sensor to characterize a novel regulation of cAMP by the sphingosine 1-phosphate/G13 pathway. J Biol Chem. 2007;282:10576–10584. doi: 10.1074/jbc.M609695200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao J, Kobashigawa J, Fishbein M, MacLellan W, Burdick M, Belperio J, et al. Elevated serum levels of the CXCR3 chemokine ITAC are associated with the development of transplant coronary artery disease. Circulation. 2003;107:1958–1961. doi: 10.1161/01.CIR.0000069270.16498.75. [DOI] [PubMed] [Google Scholar]
- Kenakin T, Miller LJ. Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol Rev. 2010;62:265–304. doi: 10.1124/pr.108.000992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofuku Y, Yoshiura C, Ueda T, Terasawa H, Hirai T, Tominaga S, et al. Structural basis of the interaction between chemokine stromal cell-derived factor-1/CXCL12 and its G-protein-coupled receptor CXCR4. J Biol Chem. 2009;284:35240–35250. doi: 10.1074/jbc.M109.024851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollias-Baker CA, Ruble J, Jacobson M, Harrison JK, Ozeck M, Shryock JC, et al. Agonist-independent effect of an allosteric enhancer of the A1 adenosine receptor in CHO cells stably expressing the recombinant human A1 receptor. J Pharmacol Exp Ther. 1997;281:761–768. [PubMed] [Google Scholar]
- Kondru R, Zhang J, Ji C, Mirzadegan T, Rotstein D, Sankuratri S, et al. Molecular interactions of CCR5 with major classes of small-molecule anti-HIV CCR5 antagonists. Mol Pharmacol. 2008;73:789–800. doi: 10.1124/mol.107.042101. [DOI] [PubMed] [Google Scholar]
- de Kruijf P, van Heteren J, Lim H, Conti P, van der Lee M, Bosch L, et al. Nonpeptidergic allosteric antagonists differentially bind to the CXCR2 chemokine receptor. J Pharmacol Exp Ther. 2009;329:783–790. doi: 10.1124/jpet.108.148387. [DOI] [PubMed] [Google Scholar]
- Lanzafame A, Guida E, Christopoulos A. Effects of anandamide on the binding and signaling properties of M1 muscarinic acetylcholine receptors. Biochem Pharmacol. 2004;68:2207–2219. doi: 10.1016/j.bcp.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Lit L, Wong C, Tam L, Li E, Lam C. Raised plasma concentration and ex vivo production of inflammatory chemokines in patients with systemic lupus erythematosus. Ann Rheum Dis. 2006;65:209–215. doi: 10.1136/ard.2005.038315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loetscher M, Gerber B, Loetscher P, Jones S, Piali L, Clark-Lewis I, et al. Chemokine receptor specific for IP10 and mig: structure, function, and expression in activated T-lymphocytes. J Exp Med. 1996;184:963–969. doi: 10.1084/jem.184.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loetscher M, Loetscher P, Brass N, Meese E, Moser B. Lymphocyte-specific chemokine receptor CXCR3: regulation, chemokine binding and gene localization. Eur J Immunol. 1998;28:3696–3705. doi: 10.1002/(SICI)1521-4141(199811)28:11<3696::AID-IMMU3696>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Mach F, Sauty A, Iarossi AS, Sukhova GK, Neote K, Libby P, et al. Differential expression of three T lymphocyte-activating CXC chemokines by human atheroma-associated cells. J Clin Invest. 1999;104:1041–1050. doi: 10.1172/JCI6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masri B, Salahpour A, Didriksen M, Ghisi V, Beaulieu JM, Gainetdinov RR, et al. Antagonism of dopamine D2 receptor/beta-arrestin 2 interaction is a common property of clinically effective antipsychotics. Proc Natl Acad Sci USA. 2008;105:13656–13661. doi: 10.1073/pnas.0803522105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiser A, Mueller A, Wise EL, McDonagh EM, Petit SJ, Saran N, et al. The chemokine receptor CXCR3 is degraded following internalization and is replenished at the cell surface by de novo synthesis of receptor. J Immunol. 2008;180:6713–6724. doi: 10.4049/jimmunol.180.10.6713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melter M, Exeni A, Reinders ME, Fang JC, McMahon G, Ganz P, et al. Expression of the chemokine receptor CXCR3 and its ligand IP-10 during human cardiac allograft rejection. Circulation. 2001;104:2558–2564. doi: 10.1161/hc4601.098010. [DOI] [PubMed] [Google Scholar]
- Oakley RH, Laporte SA, Holt JA, Barak LS, Caron MG. Molecular determinants underlying the formation of stable intracellular G protein-coupled receptor-beta-arrestin complexes after receptor endocytosis*. J Biol Chem. 2001;276:19452–19460. doi: 10.1074/jbc.M101450200. [DOI] [PubMed] [Google Scholar]
- Pierce KL, Lefkowitz RJ. Classical and new roles of beta-arrestins in the regulation of G-protein-coupled receptors. Nat Rev Neurosci. 2001;2:727–733. doi: 10.1038/35094577. [DOI] [PubMed] [Google Scholar]
- Qin S, Rottman JB, Myers P, Kassam N, Weinblatt M, Loetscher M, et al. The chemokine receptors CXCR3 and CCR5 mark subsets of T cells associated with certain inflammatory reactions. J Clin Invest. 1998;101:746–754. doi: 10.1172/JCI1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter SL, Hall RA. Fine-tuning of GPCR activity by receptor-interacting proteins. Nat Rev Mol Cell Biol. 2009;10:819–830. doi: 10.1038/nrm2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenkilde MM, Gerlach LO, Jakobsen JS, Skerlj RT, Bridger GJ, Schwartz TW. Molecular mechanism of AMD3100 antagonism in the CXCR4 receptor: transfer of binding site to the CXCR3 receptor. J Biol Chem. 2004;279:3033–3041. doi: 10.1074/jbc.M309546200. [DOI] [PubMed] [Google Scholar]
- Saita Y, Kodama E, Orita M, Kondo M, Miyazaki T, Sudo K, et al. Structural basis for the interaction of CCR5 with a small molecule, functionally selective CCR5 agonist. J Immunol. 2006;177:3116–3122. doi: 10.4049/jimmunol.177.5.3116. [DOI] [PubMed] [Google Scholar]
- Sauty A, Colvin RA, Wagner L, Rochat S, Spertini F, Luster AD. CXCR3 internalization following T cell-endothelial cell contact: preferential role of IFN-inducible T cell {alpha} chemoattractant (CXCL11) J Immunol. 2001;167:7084–7093. doi: 10.4049/jimmunol.167.12.7084. [DOI] [PubMed] [Google Scholar]
- Smit MJ, Verdijk P, van der Raaij-Helmer EM, Navis M, Hensbergen PJ, Leurs R, et al. CXCR3-mediated chemotaxis of human T cells is regulated by a Gi- and phospholipase C-dependent pathway and not via activation of MEK/p44/p42 MAPK nor Akt/PI-3 kinase. Blood. 2003;102:1959–1965. doi: 10.1182/blood-2002-12-3945. [DOI] [PubMed] [Google Scholar]
- Sorensen TL, Tani M, Jensen J, Pierce V, Lucchinetti C, Folcik VA, et al. Expression of specific chemokines and chemokine receptors in the central nervous system of multiple sclerosis patients. J Clin Invest. 1999;103:807–815. doi: 10.1172/JCI5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson RP. A modification of receptor theory. Br J Pharmacol. 1956;11:379–393. doi: 10.1111/j.1476-5381.1956.tb00006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storelli S, Verzijl D, Al-Badie J, Elders N, Bosch L, Timmerman H, et al. Synthesis and structure-activity relationships of 3H-quinazolin-4-ones and 3H-pyrido[2,3-d]pyrimidin-4-ones as CXCR3 receptor antagonists. Arch Pharm (Weinheim) 2007;340:281–291. doi: 10.1002/ardp.200700037. [DOI] [PubMed] [Google Scholar]
- Stroke I, Cole A, Simhadri S, Brescia M, Desai M, Zhang J, et al. Identification of CXCR3 receptor agonists in combinatorial small-molecule libraries. Biochem Biophys Res Commun. 2006;349:221–228. doi: 10.1016/j.bbrc.2006.08.019. [DOI] [PubMed] [Google Scholar]
- Su Y, Raghuwanshi SK, Yu Y, Nanney LB, Richardson RM, Richmond A. Altered CXCR2 signaling in beta-arrestin-2-deficient mouse models. J Immunol. 2005;175:5396–5402. doi: 10.4049/jimmunol.175.8.5396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson BD, Jin Y, Wu KH, Colvin RA, Luster AD, Birnbaumer L, et al. Inhibition of G alpha i2 activation by G alpha i3 in CXCR3-mediated signaling. J Biol Chem. 2007;282:9547–9555. doi: 10.1074/jbc.M610931200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban J, Clarke W, von Zastrow M, Nichols D, Kobilka B, Weinstein H, et al. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther. 2007;320:1–13. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- Verzijl D, Storelli S, Scholten DJ, Bosch L, Reinhart TA, Streblow DN, et al. Noncompetitive antagonism and inverse agonism as mechanism of action of nonpeptidergic antagonists at primate and rodent CXCR3 chemokine receptors. J Pharmacol Exp Ther. 2008;325:544–555. doi: 10.1124/jpet.107.134783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wanrooij EJ, de Jager SC, van Es T, de Vos P, Birch HL, Owen DA, et al. CXCR3 antagonist NBI-74330 attenuates atherosclerotic plaque formation in LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2008;28:251–257. doi: 10.1161/ATVBAHA.107.147827. [DOI] [PubMed] [Google Scholar]
- Walser TC, Ma X, Kundu N, Dorsey R, Goloubeva O, Fulton AM. Immune-mediated modulation of breast cancer growth and metastasis by the chemokine Mig (CXCL9) in a murine model. J Immunother. 2007;30:490–498. doi: 10.1097/CJI.0b013e318031b551. [DOI] [PubMed] [Google Scholar]
- Wijtmans M, Verzijl D, Leurs R, de Esch IJ, Smit MJ. Towards small-molecule CXCR3 ligands with clinical potential. ChemMedChem. 2008;3:861–872. doi: 10.1002/cmdc.200700365. [DOI] [PubMed] [Google Scholar]
- Wise EL, Duchesnes C, da Fonseca PC, Allen RA, Williams TJ, Pease JE. Small molecule receptor agonists and antagonists of CCR3 provide insight into mechanisms of chemokine receptor activation. J Biol Chem. 2007;282:27935–27943. doi: 10.1074/jbc.M703255200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330:1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates CC, Krishna P, Whaley D, Bodnar R, Turner T, Wells A. Lack of CXC chemokine receptor 3 signaling leads to hypertrophic and hypercellular scarring. Am J Pathol. 2010;176:1743–1755. doi: 10.2353/ajpath.2010.090564. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.