

Abstract
BACKGROUND AND PURPOSE
The chemokine receptor CXCR3 directs migration of T-cells in response to the ligands CXCL9/Mig, CXCL10/IP-10 and CXCL11/I-TAC. Both ligands and receptors are implicated in the pathogenesis of inflammatory disorders, including atherosclerosis and rheumatoid arthritis. Here, we describe the molecular mechanism by which two synthetic small molecule agonists activate CXCR3.
EXPERIMENTAL APPROACH
As both small molecules are basic, we hypothesized that they formed electrostatic interactions with acidic residues within CXCR3. Nine point mutants of CXCR3 were generated in which an acidic residue was mutated to its amide counterpart. Following transient expression, the ability of the constructs to bind and signal in response to natural and synthetic ligands was examined.
KEY RESULTS
The CXCR3 mutants D112N, D195N and E196Q were efficiently expressed and responsive in chemotaxis assays to CXCL11 but not to CXCL10 or to either of the synthetic agonists, confirmed with radioligand binding assays. Molecular modelling of both CXCL10 and CXCR3 suggests that the small molecule agonists mimic a region of the ‘30s loop’ (residues 30–40 of CXCL10) which interacts with the intrahelical CXCR3 residue D112, leading to receptor activation. D195 and E196 are located in the second extracellular loop and form putative intramolecular salt bridges required for a CXCR3 conformation that recognizes CXCL10. In contrast, CXCL11 recognition by CXCR3 is largely independent of these residues.
CONCLUSION AND IMPLICATIONS
We provide here a molecular basis for the observation that CXCL10 and CXCL11 are allosteric ligands of CXCR3. Such findings may have implications for the design of CXCR3 antagonists.
LINKED ARTICLE
This article is commented on by O'Boyle, pp. 895–897 of this issue. To view this commentary visit http://dx.doi.org/10.1111/j.1476-5381.2011.01759.x
Keywords: Chemokine, chemokine receptors, molecular modelling, small molecule agonist, CXCL10, CXCL11, CXCR3
Introduction
Chemokines are low-molecular weight proteins that mediate the migration of leucocytes. In humans, they constitute a family of over 40 proteins, with the majority of chemokines falling into one of two groups, namely the CC or CXC classes, where the first two cysteine residues within the N-terminal region are either adjacent or have a single amino acid separating them (Zlotnik and Yoshie, 2000). Chemokines exert their effects by binding to specific GPCRs expressed on the leucocyte surface (Murphy et al., 2000; Murphy, 2002). The signals transduced by these receptors help to coordinate leucocyte trafficking and the establishment of lymphoid microenvironments. Their discovery has greatly increased our understanding of selective leucocyte recruitment to sites of inflammation. Indeed, the excessive or inappropriate release of chemokines has been linked with the pathogenesis of several inflammatory diseases and a variety of autoimmune disorders (Charo and Ransohoff, 2006).
The chemokine receptor CXCR3 is expressed on the surface of a substantial proportion of freshly purified T-cells (Loetscher et al., 1996), is up-regulated upon polarisation to the Th1 subset (Loetscher et al., 1998; Meiser et al., 2008) and binds the chemokines CXCL9/Mig, CXCL10/IP-10 and CXCL11/I-TAC with nanomolar affinities (Cole et al., 1998; Weng et al., 1998; receptor and ligand nomenclature follows Alexander et al.; 2011). All three CXCR3 ligands are induced by IFN-γ and therefore thought to promote Th1 immune responses (Luster and Ravetch, 1987; Farber, 1990; Cole et al., 1998;), notably in the pathogenesis of several clinically important inflammatory disorders, including rheumatoid arthritis and atherosclerosis (Tsubaki et al., 2005; Kwak et al., 2008). As GPCRs are inherently ‘druggable’, accounting for 40% of all prescribed drugs (Fredriksson et al., 2003), blockade of CXCR3 by small molecules may suggest alternative therapeutic approaches to treat inflammation and several prototypic antagonists of CXCR3 have been described in the literature (Pease and Horuk, 2009). Consequently, much effort has been undertaken to understand the events underlying CXCR3 activation. The chemokines CXCL10 and CXCL11 have distinct potencies and efficacies in a variety of assays, including internalization and cell migration (Xanthou et al., 2003; Colvin et al., 2004; 2006; Dagan-Berger et al., 2006), suggesting that they interact with CXCR3 in different ways and are likely to stabilize different conformations of the receptor. Previous work by ourselves and others has highlighted domains of CXCR3 implicated in ligand binding and receptor activation, notably the second extracellular loop (ECL) (Xanthou et al., 2003; Colvin et al., 2006). In this study, we used recently described small molecule agonists of CXCR3 to probe the structure–function relationships of ligands and receptor, and to identify key residues required for activation of CXCR3 by CXCL10 and small-molecule mimetics of CXCL10.
Methods
Generation of receptor mutants and their transient expression in the murine pre-B cell line L1.2
Previously described pcDNA3 plasmids containing human wild-type (WT) CXCR3 cDNA with an HA epitope tag encoded at the N terminus (Meiser et al., 2008) were used as a template for the generation of point mutants by PCR using the QuikChange II site-directed mutagenesis kit (Stratagene, Amsterdam, the Netherlands). All constructs were verified by DNA sequencing (Eurofins MWG Operon, Ebersberg, Germany) before use. L1.2 cells were transiently transfected by electroporation with 1 µg of vector DNA per 106 cells at 330 V, 975 µF and incubated overnight in medium supplemented with 10 mM of sodium butyrate to enhance gene expression.
Flow cytometry
Cell surface expression of CXCR3 was assessed by flow cytometry after staining with either mouse anti-human CXCR3 mAb or an anti-HA antibody and FITC-conjugated secondary antibody as described previously (Vaidehi et al., 2009). Expression was analysed using a FACSCalibur flow cytometer (Becton Dickinson, Mountain View, CA, USA). Data are presented as a percentage of the amount of WT CXCR3 expressed in control transfectants.
Chemotaxis assay
Assays of chemotactic responsiveness were carried out as previously described (Vaidehi et al., 2009) using 96-well ChemoTx® plates with 5 µm pores (Neuroprobe, Gaithersburg, MD, USA). Migrating cells were detected by the use of CellTiter Glo® Dye (Promega, Southampton, UK) and resulting luminescence measured using a TopCount scintillation counter (Perkin Elmer, Waltham, MA, USA). Basal migration of cells to buffer alone was subtracted from the resulting data, with individual results expressed as a percentage of the total cells applied to the filter. In all experiments, each data point was assayed in duplicate. In every experiment, cells transiently expressing WT CXCR3 were employed as a positive control.
Radiolabelled chemokine binding studies
Whole-cell binding assays on transiently transfected L1.2 cells were performed as described previously (Vaidehi et al., 2009) using 0.1 nM 125I-CXCL10 or 125I-CXCL11 (Perkin Elmer) and increasing concentrations of unlabelled chemokine or antagonist. Cell-associated radioactivity was counted in a Canberra Packard Cobra 5010 gamma counter (Canberra Packard, Pangebourne, UK). Curve fitting and subsequent data analysis was carried out using the program PRISM (GraphPad Software, Inc., La Jolla, CA, USA) and IC50 values were obtained by nonlinear regression analysis. In all experiments, each data point was assayed in duplicate. Background binding levels obtained in the presence of a 1000–3000 molar excess of unlabelled chemokine were subtracted from each data point and data are presented as the percentage of counts obtained in the absence of competing ligand. Kd values were calculated from homologous binding curves prepared in GraphPad Prism using the equation:
where Bmax refers to the total ligand binding, [Hot] and [Cold] refer to the concentrations of labelled and unlabelled ligands, respectively, and NSB refers to non-specific binding.
Three-dimensional alignments of CXCL10 with small molecule CXCR3 agonists
This was carried out using a novel multiple ligand alignment method as previously described (Anghelescu et al., 2008). Visualization of the CXCL10 crystal structure (Booth et al., 2002) was carried out using PyMol (DeLano, 2002), using structure 1LV9 from the Protein Data Bank (http://www.rcsb.org/pdb/home/home.do).
Modelling CXCR3 interactions with small molecule agonists
The three-dimensional model of the seven helical transmembrane bundle of human CXCR3 was predicted using the ab initio method MembStruk (Vaidehi et al., 2002; 2009). The extra- and intracellular loops were added using the method, Modeller (Fiser et al., 2000). In ECLs 2 and 3, we observed that the residues D195, R216, E196 and K125 were in proximity and therefore performed constrained minimization to bring the pairs of residues together to form salt bridges. The small molecules compounds 1 and 3 were built using the LigPrep module from the Schrodinger Glide suite (Schrodinger Inc., San Diego, CA, USA). Multiple ligand conformations were generated for the two compounds and docked using Glide XP (Schrodinger Inc). Next, a short energy minimization was performed on each docked pose and the binding energy of this optimized pose was calculated. The binding energy was calculated as BE (binding energy) = PE (ligand in fixed protein) – PE (ligand in solvation), where BE is the binding energy and PE is the potential energy. The compound poses were then sorted by binding energy and the top 20 conformations inspected visually to maximize the interactions with residues that are known to interact with ligands in chemokine receptors (Vaidehi et al., 2009).
Data analysis
Data are expressed as the mean ± SEM of at least three separate experiments, and were analysed with a relevant statistical test, as stated, using PRISM v4.03 software.
Materials
Reagents were purchased from Invitrogen (Paisley, UK), unless stated otherwise. Recombinant human CXCL10 and CXCL11 were purchased from PeproTech EC Ltd (London, UK). The mouse anti-human CXCR3 mAb (clone 49801.111) and the mouse isotype-matched control IgG1 (MOPC 21 clone) were obtained from Sigma-Aldrich (Poole, UK). The monoclonal mouse anti-haemagglutinin (HA) anti-HA.11 antibody was from Covance (Berkeley, CA, USA) and its corresponding IgG1 isotype control antibody from Sigma-Aldrich. The murine pre-B cell line L1.2 was maintained as described previously (Vaidehi et al., 2009) in suspension at 37°C with 5% CO2 at a density of no more than 1 × 106 cells·mL−1. The human lymphoma cell line H9 [Repository # 0001] was obtained from the Programme EVA Centre for AIDS Reagents, National Institute for Biological Standards and Control, Potters Bar, UK, supported by the EC FP6/7 Europrise Network of Excellence, AVIP and NGIN consortia and the Bill and Melinda Gates GHRC-CAVD Project and was donated by Dr R. Gallo, University of Maryland School of Medicine. H9 cells were maintained in suspension in RPMI medium supplemented with 10% FBS at 37°C with 5% CO2 at a density of no more than 1 × 106 cells·mL−1.
Results
Compounds #1 and #3 are partial agonists of CXCR3
Stroke et al. (2006) previously reported the identification of two small molecule agonists of CXCR3 named compound #1 (Cp#1) and compound #3 (Cp#3). Both small molecules share a similar chemical structure consisting of an N-containing bicyclic unit, a hydrophobic group and a basic amino acid (Figure 1A). In the first instance, we assessed the agonistic activities of Cp#1 and Cp#3, in chemotactic assays using the human lymphoblast line H9 (which expresses endogenous CXCR3) and previously described murine L1.2 WT CXCR3-transfectants (Xanthou et al., 2003). Both compounds induced typically bell-shaped chemotactic responses in either cell line, with 100 nM–1 µM of either small molecule inducing an optimal response. Both Cp#1 and Cp#3 were partial agonists compared with the natural ligands CXCL11 and CXCL10, which exhibited more potent and efficacious responses as previously reported (Stroke et al., 2006) (Figure 1B and C). Notably, Cp#1 and Cp#3 were an order of magnitude more potent in H9 cells, compared with CXCR3 transfectants, which may reflect their ability to induce more efficient coupling of CXCR3 to human G proteins or that CXCR3 expression levels were higher in this cell line.
Figure 1.
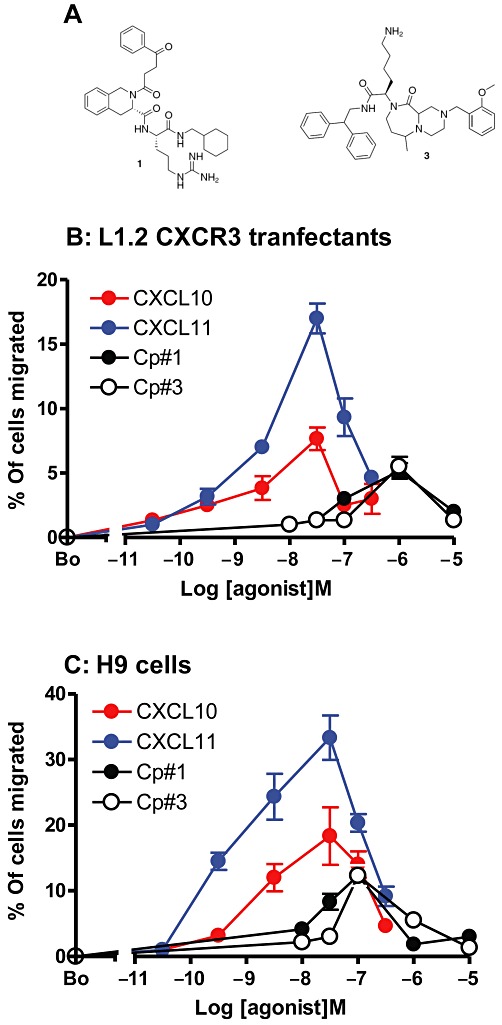
Basic small molecules are partial agonists of CXCR3. (A) The chemical structures of Cp#1 and Cp#3. The chemotactic responses of (B) the L1.2 CXCR3 transfectants and (C) the human lymphoblast cell line H9 to increasing concentrations of CXCL10, CXCL11, Cp#1 and Cp#3.
Disparate binding sites on CXCR3 for CXCL10 and CXCL11
Previous studies have described CXCL10 and CXCL11 as allosteric ligands of CXCR3, based on their respective abilities in heterologous competition binding assays. While CXCL11 is reported to completely displace CXCL10 from the receptor, a considerable proportion of bound CXCL11 is not displaced by CXCL10 (Cox et al., 2001; Xanthou et al., 2003). To examine the activity of the compounds further, we subjected L1.2 transfectants expressing WT CXCR3 to heterologous competition assays using 125I-CXCL10 and 125I-CXCL11 (Figure 2A and B). Radiolabelled CXCL10 was readily displaced from cells by CXCL11, CXCL10 and Cp#3 (respective IC50 values of 0.4 nM, 2.0 nM and 3.0 nM) while Cp#1 was unable to compete for more than 25% of the radiolabel. In the reciprocal experiment, 125I-CXCL11 was readily displaced by unlabelled CXCL11 (IC50 values of 1.2 nM) but was resistant to increasing concentrations of either CXCL10 or the two small-molecule agonists, which were unable to displace more than 50% of the 125I-CXCL11. This suggests that Cp#1 and Cp#3 mimic CXCL10 with respect to ligand binding and that CXCL10 and CXCL11 are allosteric ligands of CXCR3.
Figure 2.
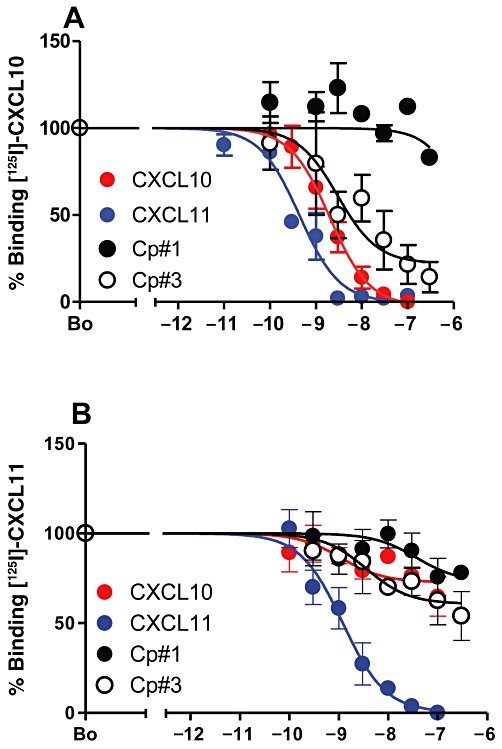
Small molecule agonists of CXCR3 mimic CXCL10 in their ability to displace CXCR3 ligands from their receptor. The relative abilities of unlabelled CXCL10, CXCL11, Cp#1 or Cp#3 to displace 125I-CXCL10 (A) or 125I-CXCL11 (B) from WT CXCR3 transfectants in heterologous competition assays.
The second ECL of CXCR3 is critical for agonist function
Previous work by ourselves using chimaeric CXCR1/CXCR3 receptors suggested a multisite model for the interaction of CXCR3 with its ligands in which multiple extracellular domains are required for chemokine binding and receptor activation, notably the second and third ECLs of CXCR3 (Xanthou et al., 2003). To assess whether the small molecule agonists activated CXCR3 in a similar fashion, we used two chimeric constructs from our previous study where the second and third ECLs of CXCR3 were exchanged with the corresponding regions of CXCR1 to generate the constructs Chi-7 and Chi-8 respectively (Figure 3A). Both ECL2 and ECL3 share limited sequence homology (16 and 23% identity respectively). Both Chi-7 and Chi-8 were transiently expressed as detected by flow cytometry (data not shown). The chemotactic responses of the L1.2 transfectants to increasing concentrations of Cp#1 and Cp#3 were compared with that of WT-CXCR3. Replacement of ECL2 of CXCR3 with that of CXCR1 (Chi-7) resulted in a loss of chemotactic responses to both Cp#1 and Cp#3 (Figure 3B and C), while the replacement of ECL3 (Chi-8) markedly reduced the efficacy of the chemotactic responses compared with WT-CXCR3 transfectants but did not ablate them. Thus, as is the case for the natural agonists, ECL2 and ECL3 of CXCR3 appear important for functional responses to synthetic agonists.
Figure 3.
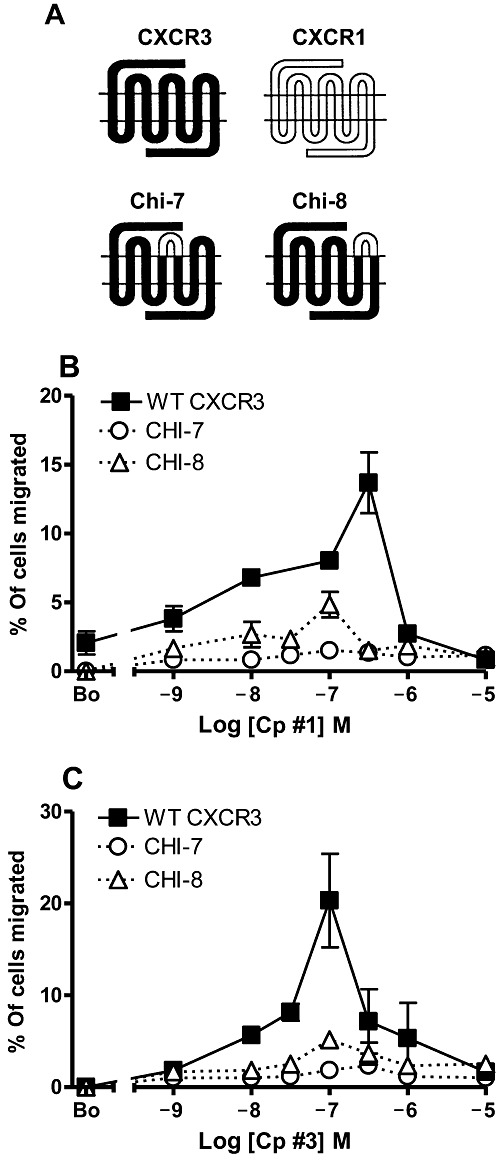
The ECL2 of CXCR3 appears critical for small molecule agonist function. (A) The structure of previously described CXCR3 chimeras containing ELC2 or ECL3 from CXCR1. The chemotactic responses of L1.2 transfectants expressing WT CXCR3 or chimeric constructs to increasing concentrations of Cp#1 (B) and Cp#3 (C).
Small molecule agonists of CXCR3 appear to mimic a portion of the ‘30s loop’ (residues 30-40) of the natural ligand CXCL10
We subsequently compared the structure of Cp#1 and Cp#3 with the natural CXCR3 agonists CXCL10 and CXCL11. As the tetrahydroisoquinoline ring in Cp#1 imposes conformational restraints similar to proline residues in peptide chains, the amino acid sequences of CXCR3 ligands were searched for candidate sequences containing a Pro-Arg-X or Pro-Lys-X motif (X corresponding to a hydrophobic residue). One such five-residue sequence of CXCL10, spanning amino acid residues 35–39 of CXCL10 (Phe-Cys-Pro-Arg-Val), matched these requirements and the coordinates for these atoms were taken from the previously solved NMR structure of CXCL10 (Booth et al., 2002). Keeping the atomic coordinates of the CXCL10 atoms fixed, there was an attempt to align both compounds to the peptide sequence of CXCL10 but only Cp#1 was successfully aligned (Figure 4A), revealing obvious structural similarities with the ‘30s loop’ region of CXCL10, which connects the first two strands of the anti-parallel β-pleated sheet. (Figure 4B).
Figure 4.
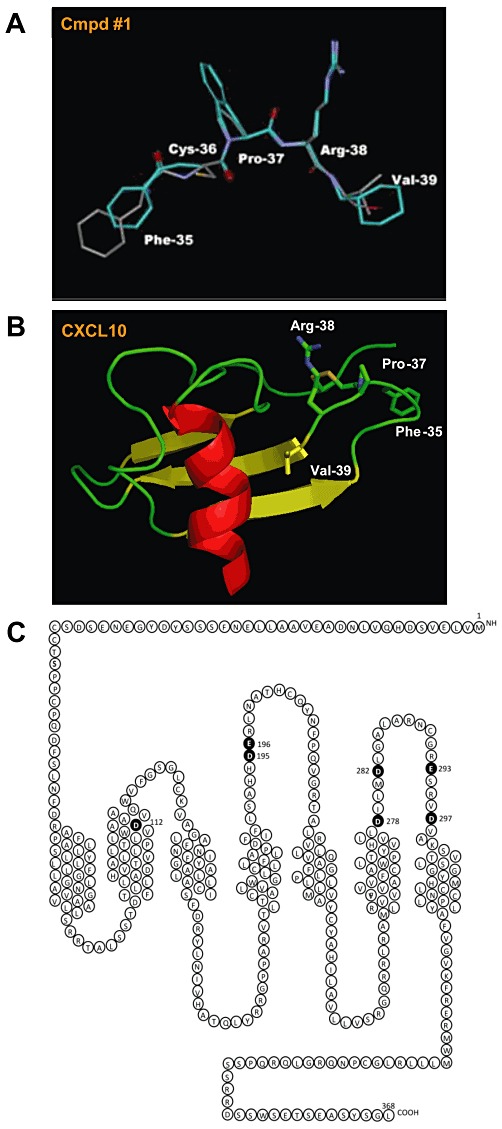
Small molecule agonists of CXCR3 appear to mimic the ‘30s loop’ of the natural ligand CXCL10. (A) The alignment of a five-residue sequence of CXCL10 spanning amino acid residues 35–39 (Phe-Cys-Pro-Arg-Val) with Cp#1. CXCL10 is depicted in grey and is overlaid with Cp#1 (cyan). (B) Modelling of the CXCL10 crystal structure, with the pertinent side chains in the ‘30s loop’ highlighted. (C) Acidic residues in CXCR3 (filled circles) mutated to their amide counterpart.
Biological responses to CXCL10 but not CXCL11 are susceptible to mutation of acidic residues in the second transmembrane helix and the second ECL
Examination of the CXCR3 primary sequence highlighted seven acidic residues within CXCR3, which we postulated might act as counter-ions to the basic moieties of Cp#1 and Cp#3 (Figure 4C). To test this hypothesis, point mutagenesis of these acidic residues was undertaken, generating a panel of seven mutant cDNA constructs in which each acidic residue (aspartate or glutamate) was mutated to its amide counterpart (asparagine or glutamine). Transient transfection of all seven constructs suggested that they were expressed at levels not significantly different from that of WT CXCR3 (Figure 5A, Table 1). The same transfectants were initially subjected to homologous competition radiolabelled binding, using either 0.1 nM 125I-CXCL11 (Figure 5B) or 0.1 nM 125I-CXCL10 (Figure 5C) and a single 1000-fold excess of unlabelled ligand. 125I-CXCL11 binding was robust among all mutants, except for the D112N mutant, which bound 125I-CXCL11 at detectable but significantly reduced levels (Figure 4B) compared with WT CXCR3. In contrast, 125I-CXCL10 binding was extremely sensitive to mutation, with several mutants displaying significantly reduced ligand binding, notably the D112N, D195N and E196Q mutants (Figure 5C). Homologous competition assays employing a series of unlabelled ligand concentrations were subsequently used to determine the relative affinity of each ligand for the mutants (Figure 6A and B and Table 1).
Figure 5.
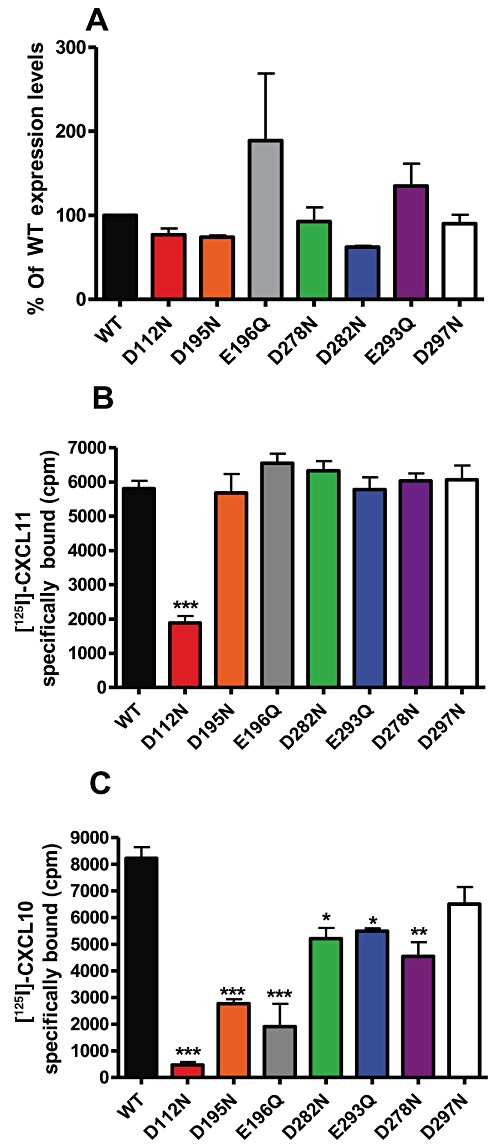
CXCL10 but not CXCL11 binding is susceptible to mutation of D112, D195 and E196 residues in CXCR3. (A)The relative expression profiles of mutant CXCR3 constructs compared with WT CXCR3. Specific binding of 125I-CXCL11 (B) or 125I-CXCL10 (C) to the mutant CXCR3 constructs.
Table 1.
Expression, chemotaxis and binding properties of CXCR3 mutants
| Construct | % of WT CXCR3 surface expression | % of WT chemotaxis to 30 nM CXCL11 | % of WT chemotaxis to 30 nM CXCL10 | KD CXCL11 binding (nM) | KD CXCL10 binding (nM) |
|---|---|---|---|---|---|
| WT CXCR3 | 100 | 100 | 100 | 10.9 ± 5.6 | 4.7 ± 0.5 |
| D112N | 76.8 ± 7.6 | 80.2 ± 24.5 | 17.8 ± 1.5*** | 3.8 ± 0.7 | ND |
| D195N | 74.2 ± 2.1 | 90.3 ± 19.7 | 14.7 ± 3.9** | 16.0 ± 3.4 | 4.7 ± 1.1 |
| E196Q | 188.0 ± 79.9 | 76.0 ± 18.9 | 9.5 ± 4.1** | 0.4 ± 0.1 | 7.6 ± 3.4 |
| D278N | 92.6 ± 1.4 | 80.7 ± 27.6 | 49.3 ± 5.9* | 2.6 ± 1.0 | 30.9 ± 3.2* |
| D282N | 62.1 ± 26.9 | 87.5 ± 17.5 | 84.2 ± 18.9 | 1.4 ± 0.2 | 2.9 ± 1.3 |
| E293Q | 134.8 ± 16.8 | 77.6 ± 23.1 | 49.8 ± 4.4** | 3.7 ± 0.9 | 1.2 ± 0.0* |
| D297N | 90.2 ± 10.4 | 44.1 ± 6.1* | 72.9 ± 25.7 | 26.3 ± 0.5* | 7.1 ± 0.5* |
Surface expression and chemotaxis are expressed relative to values obtained for transfectants expressing WT CXCR3. ND indicates not determined. The mean values ± SEM from at least three experiments are shown. ***P < 0.001, **P < 0.01 and *P < 0.05, significantly different from corresponding values for the WT CXCR3; Student's t-test.
Figure 6.
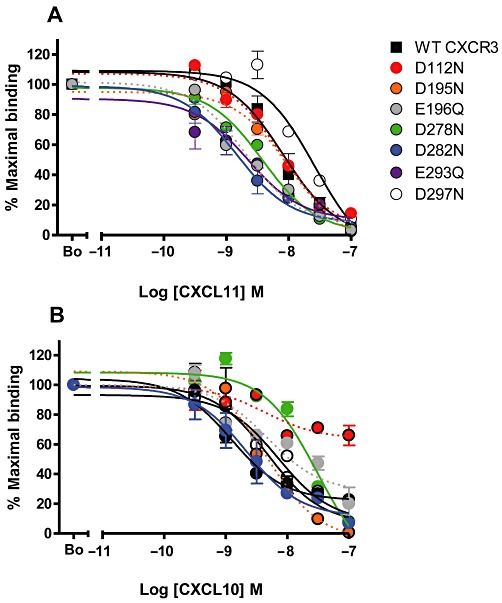
Mutation of several acidic residues in the first and second ECLs of CXCR3 significantly increases affinity for CXCL11 but not for CXCL10. The relative abilities of increasing concentrations of unlabelled ligand to displace 0.1 nM 125I-CXCL11 (A) or 0.1 nM 125I-CXCL10 (B) in homologous competition assays using CXCR3 transfectants.
D112, E195 and E196 of CXCR3 are specifically required for receptor activation by CXCL10 but not CXCL11
We subsequently examined the activities of each CXCR3 mutant in chemotaxis assays, examining migratory responses to increasing amounts of CXCL11, CXCL10, Cp#1 or Cp#3 (Figure 7A–D). In keeping with the 125I-CXCL11 binding data, all of the CXCR3 mutants allowed cells to migrate in a dose-dependent fashion in response to CXCL11 (Figure 7A), with optimal chemotaxis for the majority of constructs observed at 30 nM, identical to that of WT CXCR3 transfectants. The CXCL11-mediated responses of D282N, E293Q and D297N were shifted to the right, suggesting that these residues play a role in receptor activation by CXCL11. In the case of the D297N construct, the reduced potency of CXCL11 in chemotaxis assays correlated with a significant decrease in affinity for CXCR3, compared with WT CXCR3 (Kd of 10.9 nM cf. Kd of 26.3 nM). The majority of CXCR3 mutants also responded to CXCL10 in the same manner as WT CXCR3, exhibiting the bell-shaped dose-response curve typical of these assays, with a maximum response to 30 nM CXCL10 (Figure 7B). Unlike the robust CXCL11 responses, chemotaxis to CXCL10 was significantly impaired for several mutants. Notably, cells expressing the D112N, D195N and E196Q mutants were unresponsive to CXCL10. In the main, this correlated with a decreased ability to bind 125I-CXCL10, although for the D195N mutant, the affinity was almost identical to that of WT CXCR3, suggesting that D195 is critical for receptor activation but not binding of CXCL10. As was the case for CXCL10, transfectants expressing the D112N, D195N and E196Q mutants were also unresponsive to Cp#1 (Figure 7C) and Cp#3 (Figure 7D), suggesting that with respect to their biological activity, the compounds mimic CXCL10.
Figure 7.
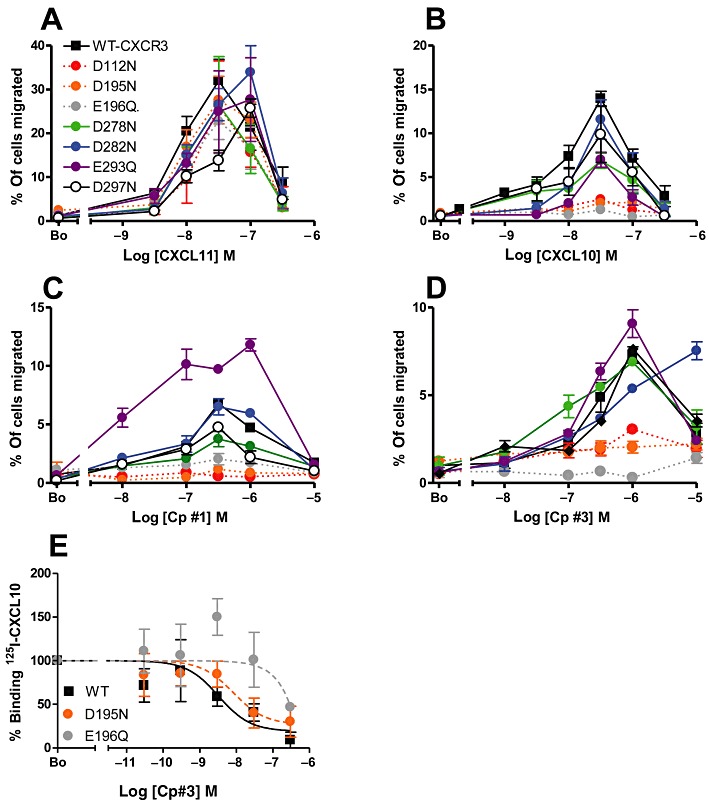
Mutation of D112, D195 and E196 in CXCR3 abolishes chemotaxis to CXCL10 and small molecule CXCL10 mimetics but has little effect on CXCL11 responses. The relative abilities of WT and mutant CXCR3 transfectants to migrate in response to increasing concentrations of CXCL11 (A), CXCL10 (B), Cp#1 (C) and Cp#3 (D) in chemotaxis assays. Panel E shows the relative abilities of increasing concentrations of Cp#3 to displace 125I-CXCL10 from transfectants expressing WT CXCR3 or the mutant constructs D125N and E196Q.
To assess the effects of the D112N, D195N and E196Q mutations on the ligand binding sites of Cp#1 and Cp#3, we examined the ability of the compounds to displace 125I-CXCL10 from transfectants expressing the mutant constructs, albeit with some experimental limitations. Firstly, 125I-CXCL11 was omitted from these experiments as we had previously shown that neither compound was able to effectively displace the radioligand (Figure 2B). Secondly, the D112N mutation was omitted from these experiments as this construct was unable to effectively bind CXCL10 (Figure 6B). Thirdly, the significant drop in Bmax for 125I-CXCL10 binding at the D195N and E196Q mutants, coupled with the relatively poor ability of Cp#1 to displace 125I-CXCL10, leads us to omit Cp#1 from these experiments. This left us with experimental determination of the relative Kd values for Cp#3 at cells expressing WT CXCR3 and the D195N and E196Q variants (Figure 7E). While the D195N mutant behaved essentially as WT CXCR3 in these assays (EC50 values of 3.2 nM and 8.2 nM respectively), the ability of Cp#3 to displace 125I-CXCL10 from cells expressing the E196Q mutant was significantly impaired, with less than 50% of the radiolabel displaced with a 3000-fold molar excess of Cp#3.
Discussion and conclusions
Current models of chemokine receptor activation are based predominantly on the two-step model of receptor activation, in which the chemokine is initially tethered by the receptor N-terminus and orientated such that key regions of the chemokine can then make productive interactions with other regions of the receptor (Monteclaro and Charo, 1996; Pease et al., 1998). Several studies have implicated the N-terminus and ‘30's loop’ regions of chemokines as playing critical roles in receptor activation, with truncation and mutation of either chemokine domain typically resulting in a loss of activity (Clark-Lewis et al., 1994; Crump et al., 1997; Gong and Clark-Lewis, 1995; Jarnagin et al., 1999). The discovery of small molecule agonists of chemokine receptors has allowed us to bypass the N-terminal tethering event and ask questions about which residues of the receptor are involved in activation. We describe here the characterization of two small molecule agonists of CXCR3, Cp#1 and Cp#3, which activate CXCR3 by binding to an intrahelical pocket. Such compounds are currently under investigation as potential therapies for transplant rejection, where the desensitization of CXCR3 may be envisaged to dampen the recruitment of T-cells to the allograft (O’Boyle et al., 2010) or as agents for promoting bone marrow regeneration following chemotherapy (Han et al., 2010).
In the case of Cp#1, it appears that the activity of the molecule results from its ability to successfully mimic the ‘30s loop’ of the natural chemokine agonist, CXCL10. This is supported by alignment of the small molecule agonists with the solution structure of CXCL10, with Cp#1 mimicking the Pro-Arg-Val sequence, formed by residues 37–39 within the ‘30s loop’ region of the chemokine. Lending credence to our findings, Arg-38 is one of several CXCL10 residues reported to undergo a conformational change upon binding of the chemokine to a peptide corresponding to the CXCR3 N-terminus (Booth et al., 2002). Collectively, our data is reminiscent of studies of the urotensin receptor in which a peptidomimetic was generated from the Trp-7, Lys-8 and Tyr-9 triplet of urotensin (Flohr et al., 2002) and supports the notion that virtual screening of pharmocophores comprising the ‘30s loop’ region of chemokines could be a general strategy for the identification of compounds with agonist activity.
We have identified three residues of CXCR3 that were critical for receptor activation by Cp#1 and Cp#3, namely D112, D195 and E196. Of significance was our finding that mutation of these three acidic residues ablated responses to CXCL10 but not responses to CXCL11. This suggests that D112, D195 and E196 of CXCR3 are required to bind and be activated by CXCL10, resulting in productive signalling, as measured by chemotaxis. In contrast, mutation of D112, D195 and E196 does not preclude binding and stabilization of an active CXCR3 conformation by CXCL11. Interestingly, the equivalent of the Pro-Arg-Val sequence of the ‘30s loop’ region of CXCL11 is Asp-Lys-Ile, suggesting that in order to activate CXCR3, the basic charge at this position has to be in the context of the conformational restraints imposed by Pro-37 in CXCL10 or the tetrahydroisoquinoline ring found in Cp#1.
Combining our experimental data with ab initio modelling of CXCR3, we find that D112 is located at the extracellular end of the transmembrane helix 2, (TM2) and acts as a counter-ion for the arginine moiety of the small molecules (Figure 8A and B), which mimics the interaction of Arg-38 of the natural ligand CXCL10 with the receptor. Previous mutagenesis of D112 to lysine or alanine was reported to result in a loss of both CXCL10 and CXCL11 binding and chemotactic responses to both ligands (Colvin et al., 2006). Here, we show that mutation of D112 to asparagine, which preserves some features of the aspartate side chain but loses the negative charge, results in a selective loss of CXCL10 binding and chemotactic response. Comparison of the binding site of the compound called ‘1t’ in the CXCR4 crystal structure (Wu et al., 2010) to the predicted binding site of Cp#3 in CXCR3 shows that the two compounds bind in similar locations in their respective receptors (Figure 8C). However, as Cp#3 is larger than 1t, its binding site extends more towards TM1 and TM7, as shown in Figure 8C. The CXCR4 crystal structure reveals a severely tilted TM3 that is not observed in our model of CXCR3. The tilt accounts for the differences in the respective position of the ligands in our model.
Figure 8.
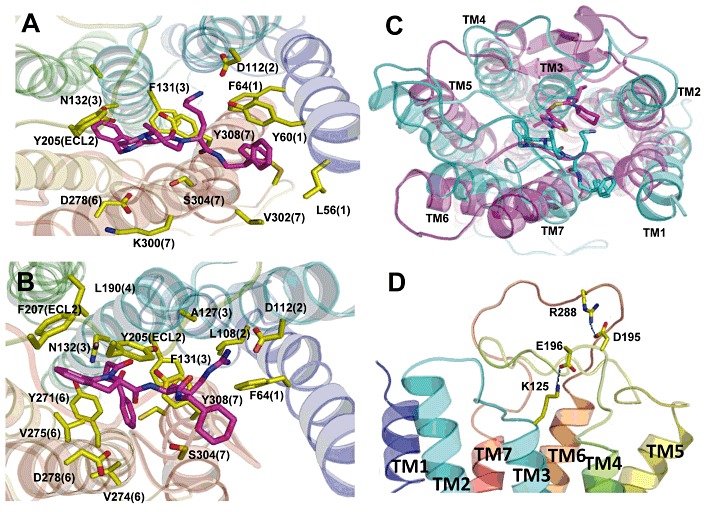
Ab initio modelling of CXCR3. (A) and (B) Top views of a model of human CXCR3 predicted using MembStruk showing the small molecule agonists Cp#1 (A) and Cp#3 (B) residing in a binding site predicted using Glide XP. In both models, D112 acts as a counter-ion for the basic moiety of either compound, with a predicted cluster of predominantly hydrophobic residues interacting with the ligand in the binding site. Numbers in parenthesis refer to the TM helix (1–7) in which the residue resides. (C) Comparison of the binding site of the compound called ‘1t’ in the CXCR4 crystal structure (pink) to the predicted binding site of Cp#3 in CXCR3 (cyan) shows that the two compounds bind in similar locations in their respective receptors. (D) The two predicted salt bridges within the extracellular domains of CXCR3 between K125 (ECL1) and D196 (ECL2) and between R288 (ECL3) and D195 (ECL2).
D195 and E196 are located within the second ECL (ECL2), a region of CXCR3 that ourselves and others have previously shown to be critical for activation of CXCR3 (Xanthou et al., 2003; Colvin et al., 2006). Modelling of the ECLs (Figure 8D) suggests that these residues form putative intermolecular salt bridges that stabilize CXCR3 and presumably gate entry of the ligand into the binding pocket as has been previously shown for the β2-adrenoceptor (Wang and Duan, 2009). In our model, D195 and E196 of ECL2 form salt bridges with R288 of ECL3 and K125 of ECL1 respectively. It is noteworthy that when we previously characterized the Chi-8 construct in which ECL3 of CXCR3 was replaced by the corresponding region of CXCR1 (Figure 3A), although CXCL11 binding was well preserved, CXCL10 binding was abolished (Xanthou et al., 2003). With reference to our model described here, we can now attribute the lack of CXCL10 binding to the loss of the salt bridge between D195 and R288, as a glutamate residue (E275) is found in the analogous position within the ECL3 of CXCR1.
The contrasting binding profiles of CXCL10 and CXCL11 in heterologous competition assays (Figure 2A and B) coupled with the mutagenesis of D112, D195 and E196, which abolished CXCL10, but not CXCL11, function, provide more detailed information regarding the previous observation that the two chemokines are allosteric ligands of CXCR3 (Cox et al., 2001). How the two ligands interact with CXCR3 is undoubtedly more complex than a simple allosteric mode of binding in which both chemokines would displace each other to a similar degree. One possible explanation is that CXCL11 binds to a population of CXCR3 molecules, which is inaccessible to CXCL10, perhaps higher order oligomers of CXCR3 or receptors pre-coupled to intracellular molecules. Alternatively, CXCL10 and CXCL11 may stabilize distinct receptor conformations. This raises the possibility that the two chemokines may induce ligand-selective bias at CXCR3 (Kenakin 2011) with the different CXCR3 conformations stabilized by either ligand coupling to distinct intracellular signalling pathways. In this study our readout of biological activity was chemotaxis, which is the final downstream function of CXCR3 activation and may be attained by several signalling pathways. To further examine such possibilities, pathway-specific assays would need to utilized.
Ligand binding data similar to that shown here has been published regarding the binding of another pair of chemokine agonists, CCL5 and CCL3, at the same CC chemokine receptor, CCR1 (Jensen et al., 2008). This raises an interesting question as to whether there is an evolutionary benefit to be able to activate signalling pathways via distinct chemokine receptor conformations. The promiscuous nature of many chemokines (activating several different receptors) has led to the proposition that the apparent redundancy in the system affords a host the possibility of mounting robust immune responses in the face of pressure from microorganisms (Mantovani, 1999). As pathogens are known to produce chemokine mimetics, which can directly antagonize chemokine receptors (Damon et al., 1998; Luttichau et al., 2000), it may be beneficial for a host to be able to activate receptors by distinct mechanisms, enhancing the prospects that at least one cognate ligand may signal under conditions of biological duress. From a pharmacological perspective, it raises the intriguing possibility that specific compounds could be developed, which block the activation of an undesirable signalling pathway at a single chemokine receptor but leave intact the ability to activate a desirable signalling pathway. In therapeutic terms, such molecules would serve to dampen down an inflammatory response without compromising host immunity.
Acknowledgments
This work was supported by Arthritis Research UK Project Grant to JEP (#18303) and the British Heart Foundation (Project Grant PG/08/031/24823 to JEP). We thank Jon Viney for help with ligand binding assays.
Glossary
- ECL
extracellular loop
- HA
haemagglutinin
- TM
transmembrane region of GPCR
- WT
wild type
Conflicts of interest
Several of the authors listed (ILS, MLW, JRM, IH, AEK and AGC) were credited with the original discovery of the small-molecule agonists while working at Pharmacopoeia Inc. (Stroke et al., 2006). Of these, IH is an employee of Ligand Pharmaceuticals Inc., which acquired Pharmacopoeia Inc. in 2008.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th Edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anghelescu AV, DeLisle RK, Lowrie JF, Klon AE, Xie X, Diller DJ. Technique for generating three-dimensional alignments of multiple ligands from one-dimensional alignments. J Chem Inf Model. 2008;48:1041–1054. doi: 10.1021/ci700395f. [DOI] [PubMed] [Google Scholar]
- Booth V, Keizer DW, Kamphuis MB, Clark-Lewis I, Sykes BD. The CXCR3 binding chemokine IP-10/CXCL10: structure and receptor interactions. Biochemistry. 2002;41:10418–10425. doi: 10.1021/bi026020q. [DOI] [PubMed] [Google Scholar]
- Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. 2006;354:610–621. doi: 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- Clark-Lewis I, Dewald B, Loetscher M, Moser B, Baggiolini M. Structural requirements for interleukin-8 function identified by design of analogs and CXC chemokine hybrids. J Biol Chem. 1994;269:16075–16081. [PubMed] [Google Scholar]
- Cole KE, Strick CA, Paradis TJ, Ogborne KT, Loetscher M, Gladue RP, et al. Interferon-inducible T cell alpha chemoattractant (I-TAC): a novel non-ELR CXC chemokine with potent activity on activated T cells through selective high affinity binding to CXCR3. J Exp Med. 1998;187:2009–2021. doi: 10.1084/jem.187.12.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colvin RA, Campanella GS, Sun J, Luster AD. Intracellular domains of CXCR3 that mediate CXCL9, CXCL10, and CXCL11 function. J Biol Chem. 2004;279:30219–30227. doi: 10.1074/jbc.M403595200. [DOI] [PubMed] [Google Scholar]
- Colvin RA, Campanella GS, Manice LA, Luster AD. CXCR3 requires tyrosine sulfation for ligand binding and a second extracellular loop arginine residue for ligand-induced chemotaxis. Mol Cell Biol. 2006;26:5838–5849. doi: 10.1128/MCB.00556-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox MA, Jenh CH, Gonsiorek W, Fine J, Narula SK, Zavodny PJ, et al. Human interferon-inducible 10-kDa protein and human interferon-inducible T cell alpha chemoattractant are allotopic ligands for human CXCR3: differential binding to receptor states. Mol Pharmacol. 2001;59:707–715. doi: 10.1124/mol.59.4.707. [DOI] [PubMed] [Google Scholar]
- Crump MP, Gong JH, Loetscher P, Rajarathnam K, Amara A, Arenzana-Seisdedos F, et al. Solution structure and basis for functional activity of stromal cell-derived factor-1; dissociation of CXCR4 activation from binding and inhibition of HIV-1. EMBO J. 1997;16:6996–7077. doi: 10.1093/emboj/16.23.6996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagan-Berger M, Feniger-Barish R, Avniel S, Wald H, Galun E, Grabovsky V, et al. Role of CXCR3 carboxyl terminus and third intracellular loop in receptor-mediated migration, adhesion and internalization in response to CXCL11. Blood. 2006;107:3821–3831. doi: 10.1182/blood-2004-01-0214. [DOI] [PubMed] [Google Scholar]
- Damon I, Murphy PM, Moss B. Broad spectrum chemokine antagonistic activity of a human poxvirus chemokine homolog. Proc Natl Acad Sci U S A. 1998;95:6403–6407. doi: 10.1073/pnas.95.11.6403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano WL. 2002. The PyMOL Molecular Graphics System.
- Farber JM. A macrophage mRNA selectively induced by gamma-interferon encodes a member of the platelet factor 4 family of cytokines. Proc Natl Acad Sci USA. 1990;87:5238–5242. doi: 10.1073/pnas.87.14.5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiser A, Do RK, Sali A. Modeling of loops in protein structures. Protein Sci. 2000;9:1753–1773. doi: 10.1110/ps.9.9.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flohr S, Kurz M, Kostenis E, Brkovich A, Fournier A, Klabunde T. Identification of nonpeptidic urotensin II receptor antagonists by virtual screening based on a pharmacophore model derived from structure-activity relationships and nuclear magnetic resonance studies on urotensin II. J Med Chem. 2002;45:1799–1805. doi: 10.1021/jm0111043. [DOI] [PubMed] [Google Scholar]
- Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol. 2003;63:1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- Gong J-H, Clark-Lewis I. Antagonists of monocyte chemoattractant protein 1 identified by modification of functionally critical NH2-terminal Residues. J Exp Med. 1995;181:631–640. doi: 10.1084/jem.181.2.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W, Liu H, Xiang D. 2010. US Patent – Methods for protecting and regenerating bone marrow using CXCR3 agonists and antagonists. Vol. US 2010/0080756 A1. USA.
- Jarnagin K, Grunberger D, Mulkins M, Wong B, Hemmerich S, Paavola C, et al. Identification of surface residues of the monocyte chemotactic protein 1 that affect signaling through the receptor CCR2. Biochemistry. 1999;38:16167–16177. doi: 10.1021/bi9912239. [DOI] [PubMed] [Google Scholar]
- Jensen PC, Thiele S, Ulven T, Schwartz TW, Rosenkilde MM. Positive versus negative modulation of different endogenous chemokines for CC-chemokine receptor 1 by small molecule agonists through allosteric versus orthosteric binding. J Biol Chem. 2008;283:23121–23128. doi: 10.1074/jbc.M803458200. [DOI] [PubMed] [Google Scholar]
- Kwak HB, Ha H, Kim HN, Lee JH, Kim HS, Lee S, et al. Reciprocal cross-talk between RANKL and interferon-gamma-inducible protein 10 is responsible for bone-erosive experimental arthritis. Arthritis Rheum. 2008;58:1332–1342. doi: 10.1002/art.23372. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Functional selectivity and biased receptor signalling. J Pharmacol Exp Ther. 2011;336:296–302. doi: 10.1124/jpet.110.173948. [DOI] [PubMed] [Google Scholar]
- Loetscher M, Gerber B, Loetscher P, Jones SA, Piali L, Clark-Lewis I, et al. Chemokine receptor specific for IP10 and mig: structure, function, and expression in activated T-lymphocytes. J Exp Med. 1996;184:963–969. doi: 10.1084/jem.184.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loetscher M, Loetscher P, Brass N, Meese E, Moser B. Lymphocyte-specific chemokine receptor CXCR3: regulation, chemokine binding and gene localization. Eur J Immunol. 1998;28:3696–3705. doi: 10.1002/(SICI)1521-4141(199811)28:11<3696::AID-IMMU3696>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Luster AD, Ravetch JV. Biochemical characterization of a gamma-interferon-inducible cytokine (tIP-10) J Exp Med. 1987;166:1084–1097. doi: 10.1084/jem.166.4.1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luttichau HR, Stine J, Boesen TP, Johnsen AH, Chantry D, Gerstoft J, et al. A highly selective CC chemokine receptor (CCR)8 antagonist encoded by the poxvirus molluscum contagiosum. J Exp Med. 2000;191:171–180. doi: 10.1084/jem.191.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A. The chemokine system: redundancy for robust outputs. Immunol Today. 1999;20:254–257. doi: 10.1016/s0167-5699(99)01469-3. [DOI] [PubMed] [Google Scholar]
- Meiser A, Mueller A, Wise EL, McDonagh EM, Petit SJ, Saran N, et al. The chemokine receptor CXCR3 is degraded following internalization and is replenished at the cell surface by de novo synthesis of receptor. J Immunol. 2008;180:6713–6724. doi: 10.4049/jimmunol.180.10.6713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteclaro FS, Charo IF. The amino-terminal extracellular domain of the MCP-1 receptor, but not the RANTES/MIP-1alpha receptor, confers chemokine selectivity. Evidence for a two-step mechanism for MCP-1 receptor activation. J Biol Chem. 1996;271:19084–19092. doi: 10.1074/jbc.271.32.19084. [DOI] [PubMed] [Google Scholar]
- Murphy PM. International Union of Pharmacology. XXX. Update on chemokine receptor nomenclature. Pharmacol Rev. 2002;54:227–229. doi: 10.1124/pr.54.2.227. [DOI] [PubMed] [Google Scholar]
- Murphy PM, Baggiolini M, Charo IF, Hebert CA, Horuk R, Matsushima K, et al. International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol Rev. 2000;52:145–176. [PubMed] [Google Scholar]
- O'Boyle G, Newton P, Jenkins Y, Ali S, Kirby JA. A strategy to inhibit effector T cell infiltration of allograft tissues by stimulation of the chemokine receptor CXCR3. Transplantation. 2010;90:144. [Google Scholar]
- Pease JE, Horuk R. Chemokine receptor antagonists: part 2. Expert Opin Ther Pat. 2009;19:199–221. doi: 10.1517/13543770802641353. [DOI] [PubMed] [Google Scholar]
- Pease JE, Wang J, Ponath PD, Murphy PM. The N-terminal extracellular segments of the chemokine receptors CCR1 and CCR3 are determinants for MIP-1a and eotaxin binding, respectively, but a second domain is essential for receptor activation. J Biol Chem. 1998;273:19972–19976. doi: 10.1074/jbc.273.32.19972. [DOI] [PubMed] [Google Scholar]
- Stroke IL, Cole AG, Simhadri S, Brescia MR, Desai M, Zhang JJ, et al. Identification of CXCR3 receptor agonists in combinatorial small molecule libraries. Biochem Biophys Res Commun. 2006;349:221–228. doi: 10.1016/j.bbrc.2006.08.019. [DOI] [PubMed] [Google Scholar]
- Tsubaki T, Takegawa S, Hanamoto H, Arita N, Kamogawa J, Yamamoto H, et al. Accumulation of plasma cells expressing CXCR3 in the synovial sublining regions of early rheumatoid arthritis in association with production of Mig/CXCL9 by synovial fibroblasts. Clin Exp Immunol. 2005;141:363–371. doi: 10.1111/j.1365-2249.2005.02850.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidehi N, Floriano WB, Trabanino R, Hall SE, Freddolino P, Choi EJ, et al. Prediction of structure and function of G protein-coupled receptors. Proc Natl Acad Sci U S A. 2002;99:12622–12627. doi: 10.1073/pnas.122357199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidehi N, Pease JE, Horuk R. Modeling small molecule-compound binding to G-protein-coupled receptors. Methods Enzymol. 2009;460:263–288. doi: 10.1016/S0076-6879(09)05213-6. [DOI] [PubMed] [Google Scholar]
- Wang T, Duan Y. Ligand entry and exit pathways in the beta2-adrenergic receptor. J Mol Biol. 2009;392:1102–1115. doi: 10.1016/j.jmb.2009.07.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng Y, Siciliano SJ, Waldburger KE, Sirotina-Meisher A, Staruch MJ, Daugherty BL, et al. Binding and functional properties of recombinant and endogenous CXCR3 chemokine receptors. J Biol Chem. 1998;273:18288–18291. doi: 10.1074/jbc.273.29.18288. [DOI] [PubMed] [Google Scholar]
- Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, et al. Structures of the CXCR4 chemokine GPCR with small molecule and cyclic peptide antagonists. Science. 2010;330:1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xanthou G, Williams TJ, Pease JE. Molecular characterization of the chemokine receptor CXCR3: evidence for the involvement of distinct extracellular domains in a multi-step model of ligand binding and receptor activation. Eur J Immunol. 2003;33:2927–2936. doi: 10.1002/eji.200324235. [DOI] [PubMed] [Google Scholar]
- Zlotnik A, Yoshie O. Chemokines: a new classification system and their role in immunity. Immunity. 2000;12:121–127. doi: 10.1016/s1074-7613(00)80165-x. [DOI] [PubMed] [Google Scholar]