

Abstract
BACKGROUND AND PURPOSE
The amphibian peptide Bv8 induces potent nociceptive sensitization in rodents. Its mammalian homologue, prokineticin 2 (PROK2), is strongly up-regulated in inflamed tissues and is a major determinant in triggering inflammatory pain. Bv8 and PROK2 activate two closely related GPCRs, PK1 and PK2, in a relatively non-selective fashion. To characterize better the roles of the two receptors in hyperalgesia and to obtain ligands whose binding affinity and efficacy differed for the two receptors, we modified the Bv8 molecule in regions essential for receptor recognition and activation.
EXPERIMENTAL APPROACH
We modified the Bv8 molecule by substituting Trp in position 24 with Ala (A-24) and compared it with Bv8 for binding and activating PK1 and PK2 receptors in cell preparations and in affecting nociceptive thresholds in rodents.
KEY RESULTS
A-24 preferentially bound to PK2 receptors and activated them with a lower potency (5-fold) than Bv8. When systemically injected, A-24 induced Bv8-like hyperalgesia in rats and in mice, at doses 100 times higher than Bv8. Locally and systemically injected at inactive doses, A-24 antagonized Bv8-induced hyperalgesia. In rat and mouse models of inflammatory and post-surgical pain, A-24 showed potent and long-lasting anti-hyperalgesic activity. Unlike Bv8, A-24 increased β-endorphin levels in mouse brain.
CONCLUSIONS AND IMPLICATIONS
A-24 induced its anti-hyperalgesic effect in rodents by directly blocking nociceptor PK1 receptors and by activating the central opioid system and the descending pain control pathway through brain PK2 receptors.
Keywords: prokineticins, Bv8, Bv8 analogues, prokineticin receptors, hyperalgesia, anti-hyperalgesic effect, chemotaxis, cytokine release
Introduction
Bv8 is a large peptide (77 amino acid residues) secreted by the skin of the frog Bombina variegata (Mollay et al., 1999; Kaser et al., 2003). Bv8 homologues are present in the secretion from other Bombina species, in black mamba snake venom (MIT-1, mamba intestinal toxin-1), in lizards and in fishes (Negri and Lattanzi, 2011). Research over the past decade has identified mammalian Bv8 orthologues, the prokineticins PROK1 (or endocrine gland-VEGF) and PROK2 (or mammalian Bv8) (LeCouter et al., 2001; Li et al., 2001), and their corresponding GPCRs, PK1 and PK2 (Lin et al., 2002; Masuda et al., 2002; Soga et al., 2002; receptor nomenclature follows Alexander et al., 2011) and linked them to several biological effects such as gut motility, circadian rhythms, neurogenesis, angiogenesis and cancer progression, haematopoiesis and nociception. Non-mammalian Bv8 is a valuable research tool for characterizing prokineticin pharmacology in rats and mice. Spinal, systemic and intraplantar injections of Bv8 lower nociceptor thresholds to widely differing physical and chemical stimuli and do so by acting on PK1 and PK2 receptors present in primary senory neurones and in the dorsal horn of the spinal cord (Negri et al., 2002; 2005; 2006). Bv8 injected into the periaqueductal grey (PAG) exerts a pro-nociceptive action, increasing GABAergic tone within the PAG, leading to inhibition of output excitatory neurones in the PAG and consequent PAG-mediated hyperalgesia. This action is mediated by activation of PK2 receptors present in the dorso-medial and lateral PAG (Cheng et al., 2006; de Novellis et al., 2007; Zhang et al., 2009). Studies conducted in our group suggest that mammalian Bv8 (i.e., prokineticin 2; PROK2), which is strongly up-regulated in neutrophils and other inflammatory cells, may be a major determinant in triggering inflammatory pain. Bv8 produced by inflammatory cells is released at the site of inflammation where it sensitizes peripheral nociceptors, stimulates chemotaxis and modulates the release of inflammatory and pro-nociceptive cytokines (Martucci et al., 2006; Franchi et al., 2008; Giannini et al., 2009).
The prokineticin receptors (PK1 and PK2) are Gq-coupled receptors that promote intracellular Ca2+ mobilization (Lin et al., 2002; Masuda et al., 2002; Negri et al., 2002; Soga et al., 2002; Vellani et al., 2006). Both receptors (especially PK2) also couple to Gi and Gs proteins, indicating that PK receptors activate multiple intracellular signal transduction pathways (Negri et al., 2007). PK1 and PK2 receptor sequences differ most at the N-terminal, whereas the sequences in the transmembrane domains are almost identical (Masuda et al., 2002; Soga et al., 2002). The mammalian PROKs and amphibian Bv8 activate both receptors in a relatively non-selective fashion, whereas MIT-1 is a PK2 receptor preferring ligand (Negri et al., 2007). Identifying the structural determinants required for Bv8/PROK receptor binding is a prerequisite for designing selective PK ligands that may be useful in treating and preventing various disease states.
Peptides belonging to the Bv8/PROK family share the same N-terminal sequence (AVIT) and have a compact structure, stabilized by five disulphide bridges. Studies from our group suggested that members of the AVIT peptide family interact with PK1 or PK2 receptors by orienting the protein region that comprises the AVIT sequence and the conserved tryptophan residue in position 24 (Trp24) (Miele et al., 2010). Analysing the evolutionary conservation grade of each residue in Bv8 homologous peptides on the modelled Bv8 structure (PDB ID: 2kra; Morales et al., 2010) suggests that modifying the primary Bv8 structure in any position from 6 to 40 will produce molecules with altered affinity or efficacy for PK receptors, or both. To verify this hypothesis, in earlier studies, we produced amphibian Bv8 analogues with the N-terminus altered (Negri et al., 2005) or Trp24 substituted (Miele et al., 2010).
In this in vitro and in vivo study, we investigated the PK receptor affinity and pharmacological activity of the Bv8 variant obtained by substituting Trp with Ala ([Ala24]Bv8), hereafter called A-24. To do so, we compared the effects of A-24 with those of Bv8 in in vitro studies conducted on cell lines transfected with PK1 or PK2 receptors (affinity and potency) and on cultured mouse macrophages (chemotaxis and cytokine release). In in vivo studies in rats and in mice, we then evaluated whether A-24, injected by different routes, altered nociceptive thresholds and whether it acted through peripheral or central mechanisms or both.
Methods
In vitro experiments
Receptor binding assay
The affinity of A-24 for prokineticin receptors was assayed on membrane preparations from CHO cells transfected with PK1 or PK2 receptors, as previously described (Negri et al., 2005). The prokineticin binding sites were labelled with 125I-MIT (4 pM; PerkinElmer, Membrane Target Systems, Waltham, MA, USA). Non-specific binding was determined in the presence of 0.1 mM Bv8. Displacement curves and IC50 values were calculated with the PRISM software (GraphPad Software, San Diego, CA, USA).
Intracellular Ca2+ imaging
CHO cells transfected with PK1 or PK2 receptors were loaded for 50 min at room temperature with 2 µM Fura-2-AM (Invitrogen, Molecular Probes, Eugene, OR, USA) in a balanced saline solution (140 mM NaCl, 2.5 mM KCl, 1 mM CaCl2, 10 mM MgCl2, 10 mM d-glucose and 10 mM HEPES/NaOH, pH 7.4). Cells were then washed, mounted onto an inverted microscope and illuminated with a xenon lamp to excite fluorescence. The fluorescence ratio at the two excitation wavelengths (340 and 380 nm) was recovered with a monochromator (Polychrome II, Till Photonics, Munich, Germany); the emission light was collected by a digital camera (Sensicam, PCO, Kelheim Germany) and recorded on the hard disk of a PC, as previously described (Balboni et al., 2008). The effect of Bv8 or A-24 was evaluated as percent responding cells (responding cells divided by total × 100).
Animals
All animal care and experimental procedures were approved by the Animal Care and Use Committee of the Italian Ministry of Health, according to EC directives (O.J. of E.C. L358/1 18/12/86), and performed during the light cycle.
Chemotaxis
Peritoneal macrophages from Balb/C male mice, 18–20 g body weight (Charles River, Calco, Italy), were collected as previously described (Martucci et al., 2006; 2007) and pooled. Chemotaxis was measured using a Boyden modified 48-well microchemotaxis chamber. Bv8 or A-24 at concentrations from 0.0001 to 10 nM was added to the lower chamber. The chambers were incubated for 100 min at 37°C in an atmosphere of 5% CO2, then the migrated cells adhering to the distal part of the filters were fixed, stained and quantitated by microscopically counting random fields (Martucci et al., 2006). When indicated, Bv8 at the fixed concentration of 0.1 nM was added to A-24 at the concentrations of 0.1, 0.01 and 0.001 nM. Results are reported as the chemotactic index, that is, cells that migrated in the presence of the chemoattractant: cells that migrated with medium alone. All experiments were repeated two to four times.
Cytokine production and measurement
Macrophages were collected as described in the foregoing section and pooled. Macrophages, isolated and purified by adherence to culture plates, were primed with 1 µg·mL−1 of LPS (Sigma, St Louis, MO, USA) for IL-1 and IL-10 production or with 1 µg·mL−1 LPS and 50 U·mL−1 interferon (IFN-γ) (Pharmingen, San Diego, CA, USA) for IL-12 stimulation (Martucci et al., 2006; 2007). Bv8 or A-24 was added to macrophage cultures at the same time as LPS at concentrations ranging from 0.0001 to 10 nM. In the antagonism experiments, Bv8 and A-24 were added together at the concentrations of 0.01 nM. The plates were incubated at 37°C and 5% CO2. Supernatants for cytokine evaluation were collected 24 h later (Sacerdote et al., 2000).
IL-12 p70 and IL-10 protein levels were determined by an elisa protocol standardized by Pharmingen (BD, San Diego, CA, USA). IL-1β levels were measured with the CytoSet Elisa kit for mouse IL-1 β (Biosource, Prodotti Gianni, Milan, Italy).
In vivo experiments
Animals
All behavioural experiments were carried out in male Sprague-Dawley rats (250–300 g, n = 120 rats) as well as in C57Bl/6 mice (n = 130 mice) and in pk1 and pk2 null mice (n = 10 mice) (Negri et al., 2006). Each animal was used only once and immediately killed by CO2 inhalation after the experiment ended. Electrophysiological evaluations were conducted in adult male Sprague-Dawley rats (250–300 g, n = 70 rats) as previously described (de Novellis et al., 2007).
Measurement of nociception
Thermal hypersensitivity was evaluated with the hot-plate test. The nociceptive withdrawal threshold was assessed by placing mice on a 48°C heated surface and recording the time to licking the hind paws or to an escape jump. The paw immersion test was performed by dipping one mouse hind paw into hot water (48°C) and measuring the latency to paw withdrawal. For the plantar test (Hargraves), hind paws of rats acclimatized to an enclosure on a pre-warmed clear glass plate were exposed to infrared thermal stimuli, and withdrawal latency was determined with a motion detector (Ugo Basile, Comerio, Italy). Mechanical sensitivity (Randall-Selitto) was tested in rats by measuring paw pressure (g) using an analgesy meter (Ugo Basile). Tactile sensitivity was tested on the plantar aspect of rat and mouse hind paws by calibrated von Frey filaments (0.41 to 15.1 g) and evaluated with the up–down method (Dixon, 1980).
Drug injections and drugs
A-24 (m.w. = 7915, produced in Pichia pastoris as described by Miele et al., 2010) and amphibian Bv8 (m.w = 8030, purified from the skin secretion of Bombina variegata as described by Mollay et al., 1999) were injected in rats by s.c., i.v., intrathecal (i.t.), intracerebral (i.c.) or intra-plantar (i.pl.) routes and in mice by s.c and i.pl. routes. For i.pl. injections, the drugs dissolved in saline were injected into the plantar region of the paw using a microsyringe fitted with a 30-gauge needle (20 µL for mice, 100 µL for rats). For systemic administration, compounds dissolved in saline solution were injected in a volume of 2 mL·kg−1 by the s.c. route. For i.t. applications, chronic lumbar i.t. catheters were implanted in rats under ketamine–xylazine anaesthesia (60 mg·kg−1 + 10 mg·kg−1, i.p.; Sigma-Aldrich, Milan, Italy) as previously described (Negri et al., 2002). The vehicle for i.t. or i.c. injections was artificial CSF, and each rat received 5 µL of the vehicle or solution of tested compounds in vehicle, followed by a 5 µL vehicle flush. Control animals were injected s.c, i.pl., i.c. or i.t. with an equal volume of vehicle. Naloxone HCl and naltrexone HCL were bought from SALARS (Como, Italy), naloxone methiodide from RBI (Natick, MA, USA).
Animal pain models
CFA-induced paw inflammation
The right hind paw of mice or rats was inflamed by injecting complete Freund's adjuvant (CFA, 20 or 100 µL),and the left hind paw acted as control. Paw oedema elicited by CFA was evaluated by measuring paw volume with a plethysmometer (7140, Ugo Basile). At 12 or 24 h after CFA injection, separate groups of rats or mice underwent testing for thermal, tactile and mechanical hypersensitivities, followed by systemic injection of the appropriate dose of A-24 or saline (control rats), and were tested at 30 min intervals to determine the time course of A-24 activity. Separate groups of animals were tested for each dose. Two groups of five rats were injected with 100 µL CFA in the right paw 12 h before killing and paw tissue collection. Starting 1 h before CFA injection, rats received A-24 (20 µg·kg−1 s.c., treated rats) or saline (2 mL·kg−1 s.c., control rats) every 5 h. PROK2 mRNA levels in the paw skin were quantified by RT-PCR as previously described (Giannini et al., 2009).
Incisional model of post-operative pain
Rats were anaesthetized with isoflurane (1.3% ; Abbott Labs, Chicago, IL) delivered through a nose cone. The plantar aspect of the left hind paw was prepared, and a 1 cm longitudinal incision was made through the skin fascia and muscle of the plantar surface. The skin was closed with two 5-0 nylon sutures and the wound was covered with antibiotic ointment just before recovery. Sutures were removed on post-operative day 2. Control rats underwent anaesthesia and surgical preparations but did not receive the incision. Baseline sensory thresholds of the left hind paw were determined for tactile (von Frey), thermal (plantar test) and noxious mechanical (Randall Selitto) stimuli as detailed in the Methods section. At 2 h, 1 day and 4 days after incision, separate groups of rats were tested for thermal, tactile and mechanical hypersensitivities, followed by the appropriate A-24 dose given by s.c. injection and were then tested at 30 min intervals to determine the time course of drug activity.
β-Endorphin measurements
Bv8 and A-24 were injected s.c (2 µg·kg−1) and two hours later, mice were killed by decapitation, brains were removed, and hypothalamus and midbrain were dissected out. β-Endorphin levels were measured by radioimmunoassay according to a method previously described (Bianchi et al., 1999). The β-endorphin antiserum against synthetic β-endorphin1–27, directed towards the C-terminal peptide, was obtained in the rabbit. [125I-tyrosyl]β-endorphin was purchased from Bachem (Merseyside, UK). The sensitivity of the method was 10 pg·per tube, the intra-assay coefficient was 8% and the inter-assay variation coefficient was 11%.
Electrophysiological studies
Surgical preparation for intra-PAG microinjections and RVM extracellular recordings and tail-flick test
All the procedures for in vivo electrophysiological recordings-tail flick combined experiments in the anaesthetized rats have been described in our previous experiments (de Novellis et al. 2007). In brief, the guide cannula was implanted into the ventrolateral PAG on the day of the experiments, and anaesthesia was maintained with a continuous infusion of propofol (5–10 mg·kg·h−1, i.v.; Sigma-Aldrich, Milan, Italy) and adjusted so that tail flicks (Ugo Basile) were elicited at a constant latency of 4–5 s. A glass-insulated tungsten filament electrode (3–5 MΩ) (FHC Frederick Haer & Co., Bowdoinham, ME, USA) was lowered into the RVM [2.8–3.3 mm caudal to lambda, 0.4–0.9 mm lateral and 9.9–11 mm in depth from the surface of the brain (Paxinos and Watson, 1986)]. RVM noxious stimuli-responding neurons were identified by the characteristic OFF cell pause and ON cell burst activity immediately before tail-flick responses (Fields et al., 1991; Fields and Basbaum, 1994). To ensure that the unit under study was unambiguously discriminated throughout the experiment, the recorded signals were amplified and displayed on an analogue and a digital storage oscilloscope. Signals were also fed into a window discriminator, whose output was processed by an interface (CED 1401) (Cambridge Electronic Design Ltd., Cambridge, UK) connected to a Pentium III PC. Spike2 software (version 4, Cambridge Electronic Design Ltd.) was then used to create peristimulus rate histograms online and to store and analyse digital records of single-unit activity off-line. This study included only ON or OFF cells whose spike configuration remained constant and could clearly be discriminated from the background activity throughout the entire experiment. For each neurone, the ongoing activity was obtained by averaging the firing rate (spikes·per second) for 50 s before the tail-flick trials (carried out every 5 min).
Statistical analysis
For in vitro experiments, results are expressed as the mean ± SEM. Data were analysed by one-way anova followed by Bonferroni's t-test for multiple comparisons. For behavioural and electrophysiological studies two-way anova was used for repeated measures followed by the Student–Newman–Keul test for multiple comparisons. P values < 0.05 were considered to indicate statistical significance.
Results
In vitro
Receptor binding assay and intracellular calcium imaging
In displacement binding assays, substituting Trp24 with Ala reduced Bv8 affinity for PK1 receptors 30-fold but only eightfold for PK2 receptors, thus obtaining a peptide that preferentially binds PK2 receptors (Table 1). In PK2-transfected CHO cells, A-24 at 0.1–10 nM induced transient, concentration-dependent increases of [Ca2+]i (Figure 1A), yielding an EC50 six times higher than that of Bv8 (0.97 ± 0.13 nM vs. 0.16 ± 0.02 nM). Non-linear regression analysis (log agonist vs. response, three parameters, Prism5) indicated a lower maximum value for the A-24 curve than for the Bv8 curve [mean (95% confidence limits), 82(73–91) vs. 113(94–132) P < 0.0001], suggesting lower efficacy. Conversely in CHO cells transfected with PK1 receptors, the A-24 EC50 was 50 times higher than that of Bv8 (5.2 ± 0.71 vs. 0.1 ± 0.02 nM). These data showed a higher loss of affinity and potency for PK1 than for PK2 receptors (30 to 50-fold vs. six- to eightfold) (Table 1). A-24, therefore, preferentially bound to PK2 receptors and was only slightly (five times) less potent than Bv8 in activating these receptors, whereas it was 50 times less potent than Bv8 in activating PK1 receptors.
Table 1.
Affinity and potency of A-24 and Bv8 for the prokineticin receptors, PK1 and PK2, stably expressed in CHO cells
| Inhibition of 125I-MIT binding | [Ca++]i mobilization | |||
|---|---|---|---|---|
| (IC50, nM) | (EC50, nM) | |||
| Compounds | PK1 receptors | PK2 receptors | PK1 receptors | PK2 receptors |
| Bv8 | 1.07 ± 0.13 | 1.00 ± 0.15 | 0.10 ± 0.02 | 0.16 ± 0.02 |
| A-24 | 33.00 ± 4.30 | 8.00 ± 1.10 | 5.20 ± 0.70 | 0.97 ± 0.10 |
In binding experiments, PK receptors were labelled with 4 pM [125I]MIT.
Values are mean ± SEM of five determinations.
Figure 1.
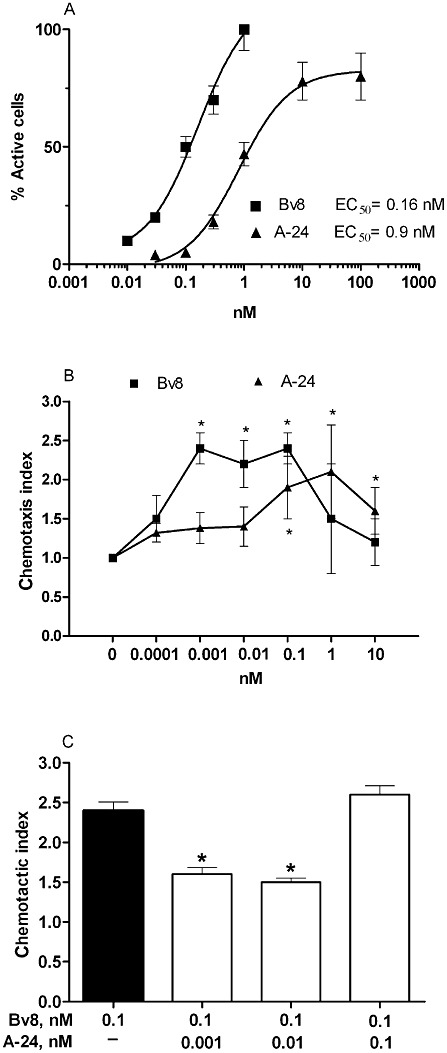
(A) Transient increase in intracellular Ca2+ concentrations induced by different concentrations of Bv8 or A-24 in CHO cells transfected with PK2 receptors. The responding cells are shown as active cells (as % of total cells in the field). Values are mean ± SEM of five experiments. (B) Macrophage chemotactic activity of Bv8 or A-24, added in the lower Boyden chamber at the concentrations indicated. Results are reported as chemotactic index, that is, the number of cells migrated with Bv8 or A-24/number of cells migrated in the presence of medium alone. Mean ± SEM of four experiments. *P < 0.05 versus background migration (chemotactic index 1). (C) Macrophage chemotactic activity of Bv8 at the concentration of 0.1 nM, combined with A-24 at 0.001 nM, 0.01 nM and 0.1 nM. Mean ± SEM of four experiments. *P < 0.05 versus Bv8 alone.
Chemotaxis
In mouse macrophage preparations, Bv8-induced chemotaxis reached maximum at concentrations from 0.001 to 0.1 nM whereas A-24 induced significant chemotaxis at concentrations of 0.1 to 10 nM, indicating that A-24 is approximately 100-fold less potent than Bv8 (Figure 1B). When Bv8 was combined with A-24 at effective doses (0.1 nM Bv8 + 0.1 nM A-24), its chemotactic effect matched that elicited by the two compounds alone. Conversely, A-24 at low, inactive doses (0.01 nM and 0.001 nM) combined with an effective Bv8 dose significantly reduced Bv8-induced chemotaxis (Figure 1C), suggesting that despite differing significantly in potency, the two ligands occupied the same macrophage receptor.
Cytokines
When Bv8 was added to mouse macrophage cultures in concentrations ranging from 0.001 to 10 nM, cell production of the pro-inflammatory cytokine IL-1 and the typical Th1 cytokine IL-12 increased, whereas production of the anti-inflammatory cytokine IL-10 decreased. A-24 induced similar cytokine changes starting from concentrations higher than 0.1 nM (Figure 2A–C). It was nevertheless less effective than Bv8 in increasing IL-1 and IL-12 (Figure 2A,C). When Bv8 at the concentration of 0.01 nM was added to macrophage cultures together with an inactive concentration of 0.01 nM A-24, the Bv8-induced effect on IL-1 and IL-10 was diminished significantly (Figure 2D,E).
Figure 2.
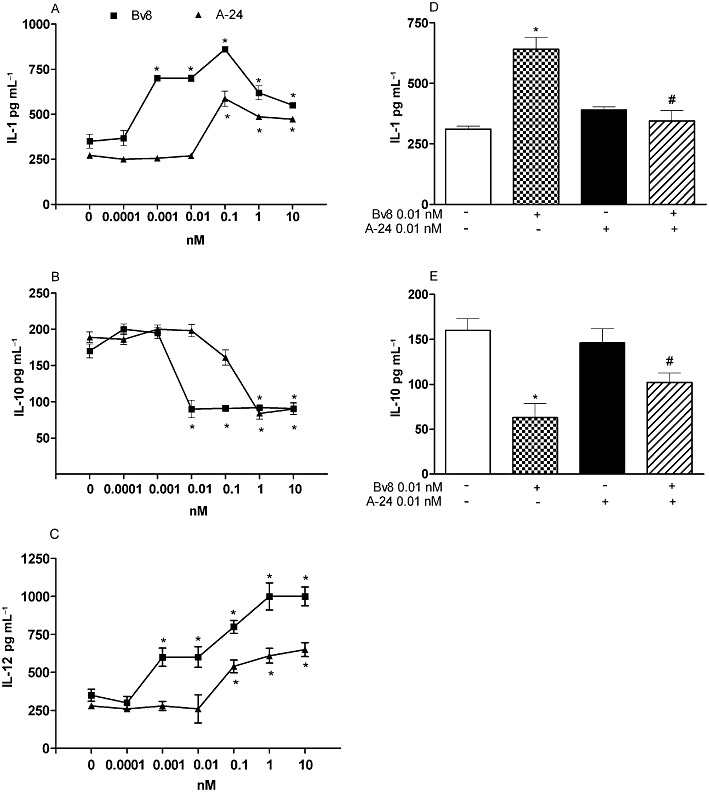
Effect of adding Bv8 and A-24 alone on IL-1β (A), IL-10 (B) and IL-12 (C) release by murine macrophages. Panels D and E: effect of combining Bv8 (0.01 nM) and A-24 (0.01 nM) on IL-1 and IL-10 release by murine macrophages. Bv8 and A-24 were added together with stimuli to induce cytokine production. IL-1β and IL-10 were stimulated with 1 µg·mL−1 of LPS. IL-12 production was stimulated with 1 µg·mL−1 of LPS + 50 U·mL−1 INF-γ. After 24 h incubation, culture media were collected and cytokines were measured by specific elisa. Mean ± SEM of four experiments. *P < 0.01 versus control (i.e., macrophage cultures without Bv8 or A-24 addition). #P < 0.01 versus Bv8 (0.01 nM) alone.
In vivo
Antinociceptive activity of A-24 in Bv8-induced hyperalgesia models
In rats, A-24 (50 ng·per rat) injected i.t. induced biphasic hyperalgesia as did Bv8 (0.5 ng). Although A-24 at lower doses left the nociceptive threshold unchanged, when pre-injected i.t. (−5 min, at the ineffective dose 10 ng), it abolished the first and second mechanical hypersensitivity phase induced by i.t. Bv8 (0.5 ng) (Figure 3A). By contrast, A-24 (10 ng) given i.t. did not antagonize hyperalgesia induced by i.pl. injection of Bv8 (0.5 ng)(Figure 3B). Systemically injected Bv8 produced characteristic biphasic hyperalgesia that within 1 h reached its first peak, depending on PK receptor activation on peripheral nerves, and in 4–5 h reached its second peak depending on central sensitization. At very low doses (0.5 ng), intraplantar injection of Bv8 induced only one hyperalgesic peak, only in the injected paw, and did so by activating PK receptors on peripheral nociceptors. A-24 10 ng i.t. pre-injected was unable to antagonize the first hyperalgesic phase (peripheral) induced by systemically injected 200 ng·kg−1 Bv8 but it abolished the second (central) hyperalgesic phase (Figure 3C). At this small dose, A-24 therefore acted only topically at spinal level but induced a long-lasting (central) effect antagonising the second hyperalgesic phase 4–5 h later. When injected s.c. at doses up to 20 µg·kg−1, A-24 left the mechanical nociceptive threshold unchanged. A-24 preinjected (−15 min) at 2–20 µg·kg−1, however, abolished the first and second mechanical hypersensitivity phases induced by 0.5 ng i.t. and 200 ng·kg−1 s.c. injected Bv8 (Figure 3D,E). A significantly lower dose (0.1 µg·kg−1, s.c., −15 min) sufficed to abolish the local hyperalgesia induced by intraplantar Bv8 injected at 0.5 ng (Figure 3F), indicating that this low s.c. dose was enough to antagonize Bv8-induced effects at peripheral sites, whereas a dose at least 20 times higher (2 µg·kg−1, s.c.) was needed to antagonize the effects of Bv8 at spinal level. When injected s.c. at doses of 2 µg·kg−1 or higher, A-24 therefore reached sites within the CNS.
Figure 3.
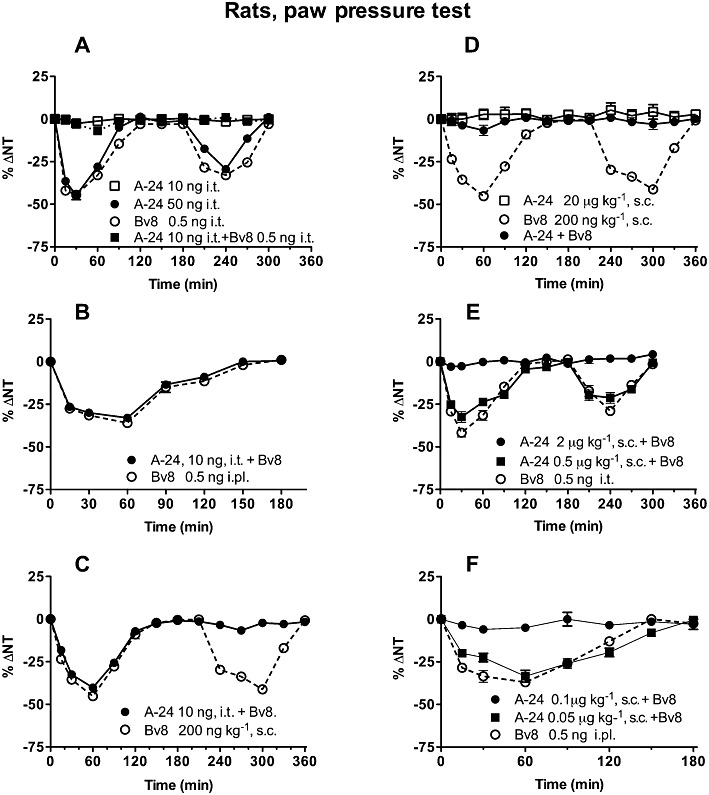
Effects of A-24 and Bv8, given centrally (i.t.0 systemically (s.c.) or locally (i.pl), on nociceptive thresholds (ΔNT) to mechanical stimuli (paw pressure) in rats. In (A), A-24 injected i.t. at the dose of 50 ng induces biphasic hyperalgesia as does Bv8 injected i.t. at 0.5 ng; but the ineffective A-24 dose (10 ng) antagonizes the hyperalgesic effect induced by i.t. Bv8; P < 0.005, two-way anova. In (B) the same dose is unable to antagonize the hyperalgesia induced by i.pl. injection of 0.5 ng Bv8, P > 0.05 and (C) the first hyperalgesic phase induced by s.c. injection of 200 ng·kg−1 Bv8. Conversely, it abolished the second hyperalgesic phase induced by systemically injected Bv8, P < 0.05 (C). In (D), s.c. injected A-24 (20 µg·kg−1; 2 µg·kg−1; 0.1 µg·kg−1) antagonizes the hyperalgesic effect produced by Bv8 given s.c. (200 ng·kg−1, P < 0.005), given i.t. (0.5 ng·rat−1, P < 0.005) (E) or given i.pl. (0.5 ng·paw−1, P < 0.005) (F). There were five to six rats in each group; total number of rats: 70.
In mice, as in rats, A-24 acted as an agonist or antagonist depending on the dose. A-24 s.c. injected at 50 µg·kg−1, like Bv8 at 1 µg·kg−1, induced biphasic thermal and tactile hyperalgesia (Figure 4A,B). Although lower doses (2–10 µg·kg−1) were unable to induce hyperalgesia, they antagonized thermal and tactile hypersensitivity induced by s.c. injected Bv8, 1 µg·kg−1 (Figure 4B). A-24 injected s.c. at 20 µg·kg−1 significantly increased the thermal nociceptive threshold evaluated with hot-plate test. This analgesic effect lasted about 90 min and was abolished by pretreatment with naloxone (10 mg·kg−1, s.c., −15 min), which disclosed a weak hyperalgesic effect. The peripherally restricted opioid antagonist, naloxone methiodide, (10 mg·kg−1, s.c., −15 min) was ineffective (Figure 4C). Conversely, A-24 injected s.c. at 20 µg·kg−1 left the thermal threshold measured by the paw-immersion test unchanged but completely antagonized thermal (Figure 4E) and tactile (not shown) hyperalgesia induced by locally injected Bv8. This anti-hyperalgesic effect was partially antagonized by naloxone pretreatment (10 mg·kg−1, i.p., −15 min) (Figure 4E), confirming that A-24 at systemic anti-hyperalgesic doses in some way activated the endogenous opioid system, thus strengthening its anti-hyperalgesic or analgesic effect, even though the paw-immersion test, unlike the hot-plate test, provided no evidence of a nociceptive threshold increase. Given by intraplantar injection, A-24 induced local thermal hyperalgesia at doses 100 times higher than those of Bv8 (50 ng vs. 0.5 ng of Bv8). But an ineffective dose of 5 ng antagonized Bv8-induced local hyperalgesia. Because this A-24-induced local anti-hyperalgesic effect was not antagonised by naloxone pretreatment (Figure 4D), it presumably depended directly on a block of peripheral PK1 receptors and was not mediated by the endogenous opioid system.
Figure 4.
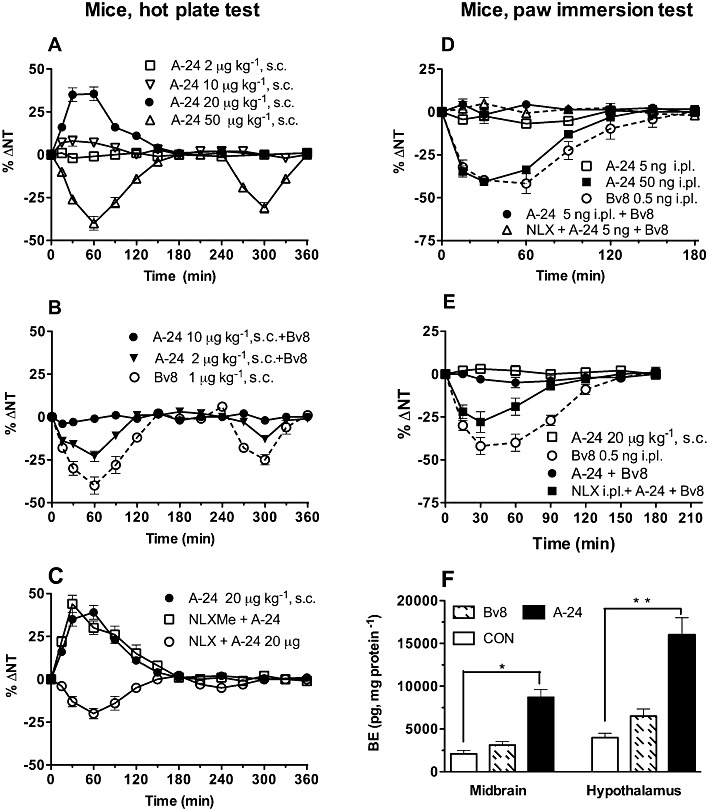
Nociceptive effects of A-24 and Bv8 in mice. In (A) dose-response data for A-24 (2 to 50 µg·kg−1 s.c.). In (B) A-24 injected at ineffective doses dose-dependently antagonizes the biphasic hyperalgesia induced by Bv8 (1 µg·kg−1). In (C) the analgesia induced by A-24 at 20 µg·kg−1 is antagonized by pretreatment with naloxone (NLX; P < 0.005, two-way anova) but not with naloxone methiodide (NLXMe; P > 0.05). (D) A-24 induces local hyperalgesia at doses 100 times higher than Bv8 but antagonizes the Bv8-induced local hyperalgesia (P < 0.005) at lower ineffective doses and the anti-hyperalgesic effect of A-24 is naloxone insensitive (P > 0.05). (E) Systemically injected A-24 antagonizes Bv8-induced local hyperalgesia (P < 0.005) in a naloxone-dependent manner (P < 0.05). There were four to five mice in each group; total number of mice: 74. In (F) effect of injecting s.c. the same dose (2 µg·kg−1) of Bv8 and A-24 on β-endorphin (BE) levels in mouse midbrain and hypothalamus. There were six mice in each group. *P < 0.05 **P < 0.01.
β-Endorphin levels measured in midbrain and hypothalamus removed from mice 2 h after receiving Bv8 or A-24 given at the same dose (2 µg·kg−1) (Figure 4F) increased in A-24- treated mice but increased slightly though not significantly also in Bv8-treated mice.
A-24 (20 µg·kg−1 s.c., hot-plate test) induced biphasic hyperalgesia when injected in mice lacking the PK2 receptor but slightly increased the thermal nociceptive threshold in mice lacking PK1 receptors (PK1-KO; Figure 5A). Naloxone pretreatment abolished the analgesic effect in PK1-KO mice, but left the hyperalgesic effect in PK2-KO mice unchanged (data not shown).
Figure 5.
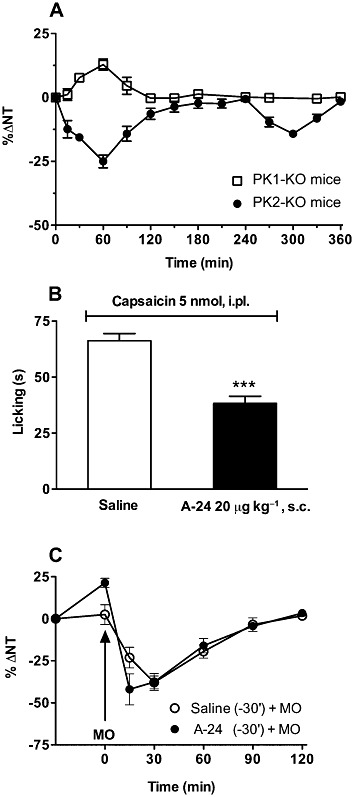
In (A) systemically injected A-24 (20 µg·kg−1) induces biphasic hyperalgesia in PK2-KO mice but the thermal nociceptive threshold in PK1-KO mice slightly increases (hot-plate test, four mice in each group). (B) A-24 significantly reduces capsaicin-induced licking (***P < 0.001; six mice in each group) but (C) has no effect on mustard oil (MO at arrow)-induced thermal hypersensitivity (hot-plate test, five mice in each group in mice.
Systemically injected A-24 significantly reduced capsaicin-induced licking (Figure 5B), a response depending on cooperativity between PK1 and TRPV1 receptors (Negri et al., 2006) but did not change mustard oil-induced thermal hypersensitivity, a response depending on cooperativity between PK2 and TRPA1 receptors (L Negri et al., unpublished data;) (Figure 5C), again underlining that at the peripheral nociceptor level, A-24 induced anti-hyperalgesia by blocking PK1 receptors.
Antinociceptive activity in inflammatory pain model (CFA-induced paw inflammation)
Thermal, tactile and mechanical hypersensitivities developed within 6 h post-CFA injection, peaked at 12–24 h and returned to pre-injection baseline values by 4 days post-injection.
In rats, systemic A-24 injected s.c and i.v. dose-dependently abolished the established CFA-induced mechanical hyperalgesia (paw-pressure test) within 30 min after injection: after 20 µg·kg−1 injected s.c., the anti-hyperalgesic effect lasted for 8 h, after 5 µg·kg−1 it lasted about 5 h and after 2 µg·kg−1 it lasted more than 3 h. The 0.5 µg·kg−1 dose was ineffective when injected s.c., but abolished hyperalgesia for more than 3 h when injected i.v. (Figure 6A). The i.v. dose of 0.1 µg·kg−1 was ineffective.
Figure 6.
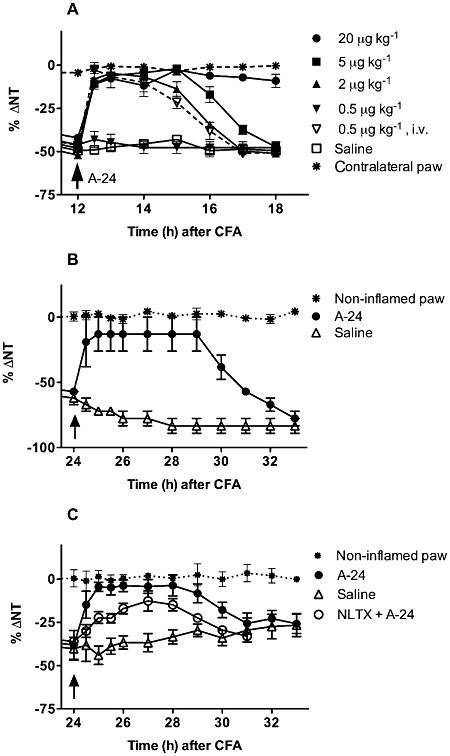
In (A), systemically (s.c and i.v.) injected A-24 dose-dependently abolished CFA-induced mechanical hyperalgesia (paw-pressure test) in rats. The dose of 0.5 µg·kg−1, ineffective by the s.c. route, still abolishes hyperalgesia for more than 3 h by the i.v. route. There were four to six rats in each group, total number of rats: 30 (A). In mice (B), A-24 abolishes the tactile (von Frey filaments) and (C) thermal hyperalgesia (paw-immersion test, at 48°C) for more than 6 h (P < 0.005 by two-way anova). Naltrexone (NLTX) partially antagonizes A-24-induced hyperalgesia (P < 0.05) (C). There were five rats in each group.
Also in mice, A-24 given at 20 µg·kg−1 abolished CFA-induced tactile and thermal hyperalgesia (von Frey filaments, paw-immersion test at 48°C) for more than 6 h (Figure 6B,C). In rats (data not shown) and in mice (Figure 6C), the A-24-induced anti-hyperalgesic effect was partially antagonized by naltrexone pretreatment (2 mg·kg−1, i.p., −30 min).
Antinociceptive activity in the post-surgery pain model (plantar incision)
Thermal and mechanical hypersensitivities developed within 2 h post-incision diminished at 4 days post-incision and returned to pre-incision baseline values by 7 days post-incision. A-24 (20 µg·kg−1 s.c.) given 1 day post-incision reversed the incision-induced thermal and mechanical hypersensitivities within 30 min of injection (Figure 7A,B). This reversal was long lasting and diminished at 7 h post-injection.
Figure 7.
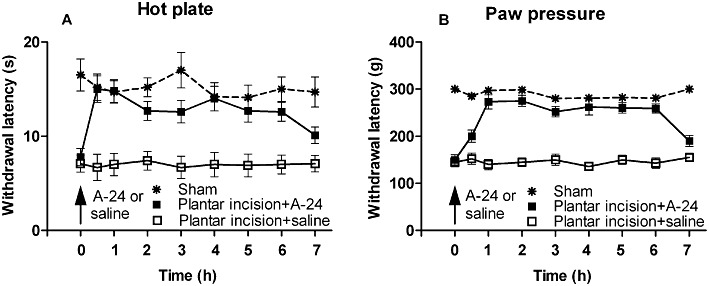
In (A), A-24 (20 µg·kg−1 s.c.) reverses intraplantar incision-induced thermal and (B) mechanical hypersensitivities in rats, for about 7 h (P < 0.005, two-way anova, six rats in each group).
Preventive treatment of inflammation with A-24
Repeated treatment of CFA-injected rats with 20 µg·kg−1 A-24 abolished the inflammation-induced hyperalgesia and significantly decreased the inflammation-induced PROK2 mRNA up-regulation in the paw skin (Figure 8A,B).
Figure 8.
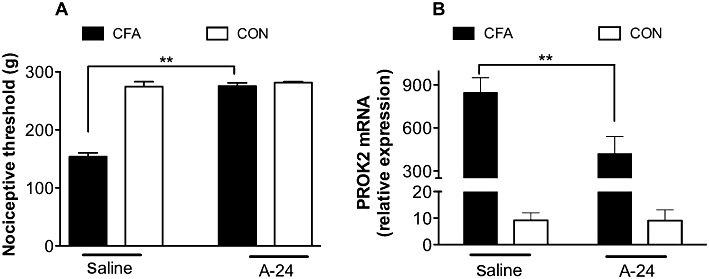
In (A) the mechanical nociceptive threshold (g) in the CFA-inflamed rat paw is about half that in the contralateral (CON) non-inflamed paw (saline); repeated treatment of rats with 20 µg·kg−1 A-24 s.c. brings the nociceptive threshold in the inflamed paw to the same values in the non-inflamed paw (A-24). In (B) the PROK2 mRNA level in the CFA-inflamed paw is dramatically higher than that in CON non-inflamed paw (saline); repeated treatment of rats with 20 µg·kg−1 A-24 s.c. significantly decreases CFA-induced PROK2 mRNA up-regulation in the paw skin (A-24). There were five rats in each group. **P < 0.01.
Effect of A-24 and Bv8 on the ongoing activities of RVM ON and OFF cells
All RVM noxious stimuli-responding neurons here recorded (Figure 9) displayed spontaneous electrical activity and discharged at a mean frequency of 7.2 ± 0.4 (ON cells) and 8.3 ± 0.5 (OFF cells) spikes·per second. Bv8 (1.6 µg per rat) microinjected into the ventrolateral PAG caused a pronociceptive effect characterized by an increase in ON cell activity and a rapid decrease in OFF cell activity. When rats were treated with A-24 (1.6, 3.2 and 6.4 µg), ON cell activity immediately increased (n = 9 rats; P < 0.05) and OFF cell activity rapidly decreased (n = 8 rats; P < 0.05) in a dose-dependent manner (Figure 10A,C). When Bv8 (1.6 µg) and A-24 (1.6 µg) were combined, both changes reversed (Figure 10B,D). Bv8 and A-24 microinjected caused no change in spontaneous activities in RVM neutral neurons (n = 8) as identified by their non-responsiveness to the tail-flick test (data not shown). Neither Bv8 nor A-24 accidentally injected outside the PAG altered firing in ON, OFF and neutral cells (data not shown).
Figure 9.
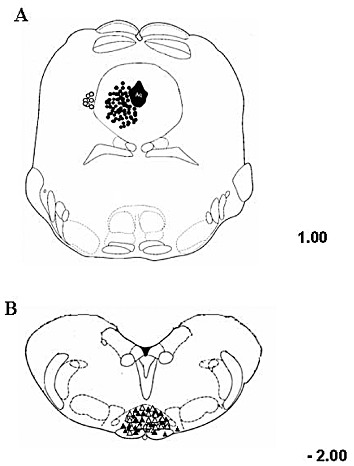
Schematic illustration showing the ventrolateral PAG microinjection sites (A) and RVM ON- or OFF-cell recording sites (B). In (A) vehicle or drug was microinjected into the ventrolateral PAG (filled circles). The open circles indicate microinjections given outside the investigated area, whose effects were not considered in the study (A). In (B) cell responses were recorded by lowering a tungsten electrode into the RVM. ON cells (filled triangles) or OFF cells (open triangles) recording sites, as shown. Many sites are not shown because of symbol overlapping. Distances are indicated in mm from the inter-aural line. The results were based on RVM neurons (one cell recorded from each rat per treatment) at a depth of 9.900–10.955 mm from the brain surface.
Figure 10.
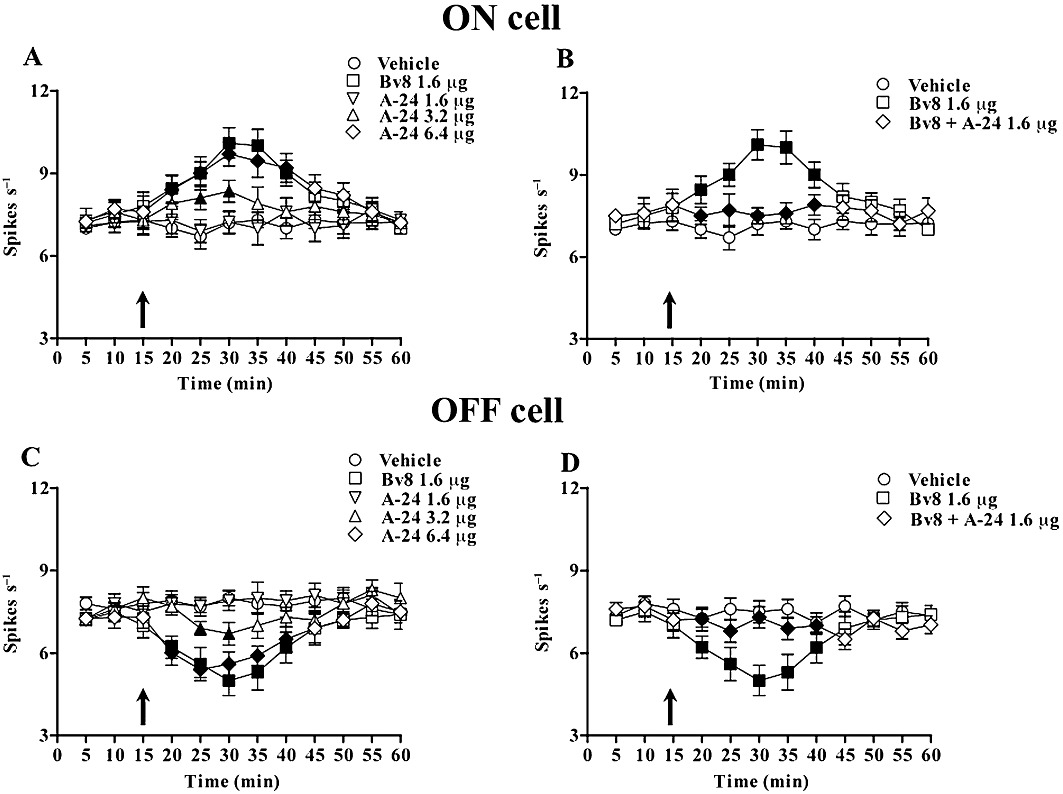
Effects of ventrolateral PAG microinjections of vehicle, Bv8 (1.6 µg), A-24 (1.6, 3.2 and 6.4 µg) and Bv8 (1.6 µg) + A-24 (1.6 µg) on the spontaneous activity of RVM ON (A,C) or OFF (B,D) cells. Filled symbols indicate significant differences versus respective controls (P < 0.05). Microinjections of vehicle, Bv8, A-24 or Bv8 + A-24 are indicated by black arrow. Each point represents the mean ± SEM of 8–10 neurones.
Effect of A-24 and Bv8 on the tail- flick latencies
Vehicle (n = 12 rats) or A-24 (1.6 µg) (n = 10) microinjected into the ventrolateral PAG left the tail-flick latency unchanged from baseline values (5.03 ± 0.4 s). But A-24 at higher doses (3.2 µg and 6.4 µg) significantly decreased the tail-flick latencies (P < 0.05; n = 16) (Figure 11A). As expected, Bv8 microinjections (1.6 µg) decreased the tail-flick response latency (P < 0.05, n = 15 rats). Again, when Bv8 (1.6 µg) was co-injected with the A-24 ineffective dose (1.6 µg) (n = 15), tail-flick latencies remained unchanged as compared with vehicle (Figure 11B).
Figure 11.
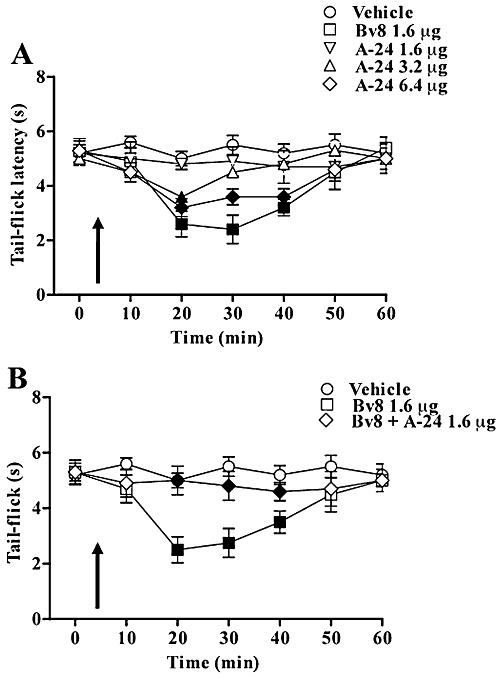
Tail-flick latencies after vehicle, Bv8 (1.6 µg), A-24 (1.6, 3.2 and 6.4 µg) and Bv8 (1.6 µg) + A-24 (1.6 µg) microinjected into the ventrolateral PAG. Each point represents the mean ± SEM for 10–16 rats per group. Filled symbols indicate significant differences versus respective controls (P < 0.05).
Discussion
In this in vivo and in vitro study, we show that A-24, the Bv8 analogue we obtained by substituting Trp24 with Ala, exhibited lower affinity for PK receptors and weaker agonist activity, than Bv8. It abolished for many hours the thermal, mechanical and tactile hypersensitivities produced by Bv8 itself, by paw inflammation or by paw incision, and did so through peripheral and central mechanisms.
Our experiments assessing its PK1 receptor-mediated effects show that A-24 is 50- to 100-fold less potent than Bv8 in inducing calcium mobilization in CHO cells transfected with PK1 receptors and in inducing macrophage chemotaxis and cytokine release (Martucci et al., 2006). Conversely, in in vitro and in vivo tests involving the PK2 receptor, A-24 shows slightly (four- to fivefold) lower potency than Bv8 in mobilizing calcium in CHO cells transfected with PK2 receptors and in stimulating ongoing RVM ON and OFF cell activities mediated by PK2, the only receptor present in rodent PAG (Cheng et al., 2006; de Novellis et al., 2007). In our in vitro experiments, the dose-response curves for calcium imaging and for IL-12 release suggested that A-24 exhibited lower efficacy than Bv8. Ineffective concentrations of A-24 prevented the Bv8-induced effect on chemotaxis and cytokine release, showing that A-24 exerted strong antagonist activity in the presence of high Bv8/prokineticin levels. Accordingly, in vivo experiments in rats and mice showed that A-24 injected systemically and topically into the spinal cord or into the paw, at a dose 100- fold higher than Bv8, produced hyperalgesia as Bv8 did, whereas pretreatment with ineffective A-24 doses antagonized Bv8-induced hyperalgesia. Similarly, A-24 injected into the PAG induced the same effects as Bv8 at a dose four times higher, but when co-injected at an ineffective dose antagonized the effects of Bv8.
A distinctive finding was that A-24 differentially altered the nociceptive threshold in rats and in mice depending on the test used. Whereas A-24 (20 µg·kg−1, s.c.) did not change the paw withdrawal latency to pressure in rats or to hot water in mice, the same dose enhanced the thermal nociceptive threshold evaluated with the hot-plate test in mice. This unexpected result fits in with our previous observation that in PK1-KO mice, the hot-plate test, but not the hot water immersion, test discloses a large nociceptive deficit (Negri et al., 2006), suggesting that A-24 at the 20 µg·kg−1 dose blocks PK1 receptors, thus mimicking the result obtained by disrupting the PK1 receptor gene. Pretreatment with naloxone, but not with naloxone methiodide, prevented the increase in nociceptive thresholds, indicating that A-24 activated the central opioid system and probably did so through PK2, the receptor that A-24 preferentially binds to and activates. PK2 is the main prokineticin receptor type in rodent brain and, besides being widely distributed in the olfactory bulb, PK2 receptors are highly expressed in amygdala, thalamus, hypothalamus, mesencephalon and brain stem (Cheng et al., 2006; Negri et al., 2007). PK1, the main receptor in the peripheral nervous system (Negri et al., 2002; 2006), is hardly expressed at all in some brain nuclei such as the olfactory bulb, hippocampus, nucleus arcuatus, zona incerta and dorsal motor nucleus of vagus. Evidence linking the A-24-induced increase in nociceptive threshold and PK2 receptors, comes from experiments in mice lacking the PK2 receptor gene. In this genotype, although A-24 injection did not change the nociceptive threshold, it induced marked hyperalgesia, mediated by the remaining PK1 receptors. Activation of the central opioid system activation appears to be a characteristic action of A-24. When we injected mice with A-24 at a dose of 2 µg·kg−1, β-endorphin content increased strongly in the hypothalamus and midbrain, whereas Bv8, injected at the same dose, left brain β-endorphincontent almost unchanged. Whether A-24 exerts this effect directly on β-endorphin producing neurones or indirectly by stimulating corticotropin-releasing hormone release upstream remains unclear. Unpublished data from our group suggest that Bv8 and A-24 could also modulate the hypothalamus-pituitary axis and increase corticosterone release. This A-24-induced analgesic effect accords with our studies using other Bv8 analogues modified at the N-terminus (R Lattanzi et al., unpubl. data) and merits further investigation.
Whatever the underlying pharmacological mechanism, the anti-hyperalgesic effect of systemically injected A-24 (20 µg·kg−1, a dose that induced no detectable increase in the nociceptive threshold) partially diminished after naloxone or naltrexone pretreatment. When injected i.pl. in low doses (less than 10 ng), A-24 produced naloxone-insensitive anti-hyperalgesic effects, indicating that local A-24 in low doses antagonized Bv8/PROK2-induced hyperalgesia by directly blocking PK receptors on peripheral nociceptors. Further evidence that the anti-hyperalgesic effect results from blocking PK1 receptors comes from our experiments showing that the same A-24 systemically injected dose (20 µg·kg−1) reduced capsaicin-induced licking, a response that depends on TRPV1-PK1 receptor cooperativity (Negri et al., 2006), but not mustard oil-induced thermal hypersensitivity, a response depending on TRPA1-PK2 receptor cooperation (R Lattanzi et al., unpublished data).
Evidence suggesting that the roles of PK1 and PK2 receptors differ in regulating pain perception receive support from the results we obtained with A-24 in PK-KO mice. The opposing effects, analgesia or hyperalgesia, induced by the same A-24 dose in PK1- or PK2-KO mice, indicate that the final nociceptive threshold depends on the fine-tuning of activation and inhibition of PK1 and PK2 receptors.
Our study also provides some evidence suggesting that A-24 injected s.c. at doses higher than 2 µg·kg−1 may act within the CNS. For example, this dose antagonized hyperalgesia induced by Bv8 injected i.t and the same dose increased hypothalamic β-endorphin.. To investigate possible A-24 supraspinal actions, we therefore compared A-24-induced and Bv8-induced effects on the endogenous descending pain pathway. Specifically, we evaluated A-24-induced changes in RVM ON and OFF cell spontaneous activity by microinjecting the drug directly into the ventrolateral PAG, an important centre for processing descending pain mechanisms (Fields and Basbaum, 1994). In accordance with behavioural data, we found that A-24, at a very high dose, increased spontaneous activities in ON cells and decreased them in OFF cells in a way similar to Bv8, whereas at a lower, ineffective, dose, it prevented Bv8-induced hyperalgesia (i.e. tail-flick latency).
When we compared potency ratios for Bv8 and A-24, the electrophysiological experiments yielded a ratio of 2:8 whereas the behavioural and immunological experiments yielded a ratio of 1:100. Binding and Ca2+-imaging data indicate a Bv8/A-24 affinity ratio of about 1:8 on PK2 and 1:30–1:100 on PK1 receptors. These pharmacological data confirm that A-24 acts on PK2 receptors and strongly support a role for these receptors in inhibiting the descending pathway of pain control (de Novellis et al., 2007). PK2 receptors are the only prokineticin receptors present in rodent PAG (Cheng et al., 2006; de Novellis et al., 2007) and this area receives PROK2-ergic fibres from hypothalamic nuclei that highly express PROK2, such as the preoptic nucleus and suprachiasmatic nucleus (Zhang et al., 2009). The A-24 dose inducing hyperalgesia by this route is about 6 µg, a dose level impossible to achieve in the PAG by systemic injections of 20 µg·kg−1. Because A-24 at an ineffective dose antagonizes the effect produced by exogenously administered Bv8 on the ongoing activities in RVM ON and OFF cells, we assume that the portion of A-24 reaching the central sites (surely far lower than the dose here used to produce agonistic effects) prevents endogenous PROK2-induced PK2 receptor activation. Hence, A-24 antagonizes endogenous inhibition in descending pain control pathway.
The ability of A-24 to block Bv8-induced hyperalgesia indicates that compounds with this anti-hyperalgesic profile could be important therapeutic tools in chronic diseases in which the prokineticin system is involved and perhaps up-regulated (Giannini et al., 2009; Kurosaka et al., 2009). In this study, we evaluated its effect in a rat paw incision pain model and in the CFA inflammatory model, in order to characterize the pharmacology of A-24 further. In these models, endogenous PROK2 released by infiltrating granulocytes at the site of injury decreased the nociceptor excitability threshold acting on PK receptors on peripheral nerves and in spinal cord dorsal horns. Systemically injected A-24 significantly reduced or abolished for 6–9 h thermal and mechanical hypersensitivities induced by skin inflammation or incision. The rapid onset of the anti-hyperalgesic effect suggests that A-24 directly antagonised PK receptors in primary nociceptors. The long-lasting anti-hyperalgesic effect may depend on the physico-chemical characteristics of A-24, because it is highly resistant to enzymic degradation and binds strongly to the extracellular matrix (LeCouter et al., 2003; Monnier and Samson, 2008). But it might also depend partly on central actions: the increase in β-endorphin brain concentrations persists up to and beyond 4 h (data not shown). Whether A-24 acts directly in modulating the descending pain control pathway and for how long it acts is open to question.
Besides abolishing inflammation-induced pain in rats, repeatedly injected A-24 also significantly reduced the inflammation-induced PROK2 up-regulation. Because we showed here that A-24 reduced macrophage chemotaxis and pro-inflammatory cytokine release in vitro and, given that inflammation-derived PROK2 stimulates macrophage chemotaxis and cytokine release (Martucci et al., 2006), reducing PROK2 faster at the site of inflammation may promote faster healing.
In conclusion, our results have confirmed the key role of the surface-exposed tryptophan hydrophobic residue (Trp24) in Bv8. Substituting Trp with Ala in position 24 of the Bv8 molecule changed its potency and PK receptor affinity. Depending on the dose used, this analogue behaved as a PK receptor agonist or prevented the Bv8/PROK2-mediated hyperalgesic effects. These findings suggest that A-24 mainly antagonized peripheral Bv8/PROK2-induced activation of PK1 receptors, and could therefore have a therapeutic application in disease states in which this system is up-regulated, such as chronic inflammatory diseases. The anti-hyperalgesic effects of A-24 depend also on central mechanisms including increased β-endorphin levels in midbrain and hypothalamus and possibly on inhibiting the putative endogenous inhibition of the descending pain pathway induced by PROK2 on PK2 receptors in the PAG.
Further investigation into the prokineticin system is needed to clarify the possible role of these peptides in pathological pain states, such as neuropathic pain, and to study their role in regulating neuronal and non-neuronal phenotypical changes.
Acknowledgments
This work was supported by grants from the Italian Ministry of University and Scientific Research and Sapienza University of Rome.
Glossary
- PROK2
prokineticin 2
- PAG
periaqueductal grey
- PK1
prokineticin receptor 1
- PK2
prokineticin receptor 2
- RVM
rostro ventral medulla
Conflict of interest
None.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th Edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balboni G, Lazzari I, Trapella C, Negri L, Lattanzi R, Giannini E, et al. Triazine compounds as antagonists at Bv8-prokineticin receptors. J Med Chem. 2008;51:7635–7639. doi: 10.1021/jm800854e. [DOI] [PubMed] [Google Scholar]
- Bianchi M, Maggi R, Pimpinelli F, Rubino T, Parolaro D, Poli V, et al. Presence of a reduced opioid response in interleukin-6 knock out mice. Eur J Neurosci. 1999;11:1501–1507. doi: 10.1046/j.1460-9568.1999.00563.x. [DOI] [PubMed] [Google Scholar]
- Cheng MY, Leslie FM, Zhou QY. Expression of prokineticins and their receptors in the adult mouse brain. J Comp Neurol. 2006;498:796–809. doi: 10.1002/cne.21087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol. 1980;20:441–462. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- Fields HL, Basbaum AI. Central nervous system mechanisms of pain modulation. In: Wall PD, Melzack R, editors. Textbook of Pain. Edinburgh, UK: Harcourt Publisher Limited; 1994. pp. 243–257. [Google Scholar]
- Fields HL, Heinricher MM, Mason P. Neurotransmitters in nociceptive modulatory circuits. Annu Rev Neurosci. 1991;4:219–245. doi: 10.1146/annurev.ne.14.030191.001251. [DOI] [PubMed] [Google Scholar]
- Franchi S, Giannini E, Lattuada D, Lattanzi R, Melchiorri P, Negri L, et al. The prokineticin receptor agonist Bv8 decreases IL-10 and IL-4 production in mice splenocytes by activating prokineticin receptor-1. BMC Immunol. 2008;9:60. doi: 10.1186/1471-2172-9-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannini E, Lattanzi R, Nicotra A, Campese AF, Grazioli P, Screpanti I, et al. The chemokine Bv8/prokineticin 2 is up-regulated in inflammatory granulocytes and modulates inflammatory pain. Proc Natl Acad Sci USA. 2009;106:14646–14651. doi: 10.1073/pnas.0903720106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaser A, Winklmayr M, Lepperdinger G, Kreil G. The AVIT protein family. Secreted cysteine-rich vertebrate proteins with diverse functions. EMBO Rep. 2003;4:469–473. doi: 10.1038/sj.embor.embor830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosaka D, Noda K, Yoshida K, Furuya K, Ukichi T, Takahashi E, et al. Elevation of Bombina variegate peptide 8 in mice with collagen-induced arthritis. BMC Musculoskelet Disord. 2009;10:45. doi: 10.1186/1471-2474-10-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeCouter J, Kowalski J, Foster J, Hass P, Zhang Z, Dillard-Telm L, et al. Identification of an angiogenic mitogen selective for endocrine gland endothelium. Nature. 2001;412:876–884. doi: 10.1038/35091000. [DOI] [PubMed] [Google Scholar]
- LeCouter J, Lin R, Tejada M, Frantz G, Peale F, Hillan KJ, et al. The endocrine-glandderived VEGF homologue Bv8 promotes angiogenesis in the testis: localization of Bv8 receptors to endothelial cells. Proc Natl Acad Sci USA. 2003;100:2685–2690. doi: 10.1073/pnas.0337667100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Bullock C, Dj K, Fj E, Qy Z. Identification of two prokineticin cDNAs: recombinant proteins potently contract gastrointestinal smooth muscle. Mol Pharmacol. 2001;59:692–698. doi: 10.1124/mol.59.4.692. [DOI] [PubMed] [Google Scholar]
- Lin D, Bullock C, Ehlert F, Chen J, Thian H, Zhou Q. Identification and molecular characterization of two closely related G-protein coupled receptors activated by prokineticins/EG-VEGF. J Biol Chem. 2002;277:19276–19280. doi: 10.1074/jbc.M202139200. [DOI] [PubMed] [Google Scholar]
- Martucci C, Franchi S, Giannini E, Tian H, Melchiorri P, Negri L, et al. Bv8, the amphibian homologue of the mammalian prokineticins, induces a proinflammatory phenotype of mouse macrophages. Br J Pharmacol. 2006;147:225–234. doi: 10.1038/sj.bjp.0706467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martucci C, Franchi S, Lattuada D, Panerai AE, Sacerdote P. Differential involvement of relb in morphine-induced modulation of chemotaxis, NO and cytokine production in murine macrophages and lymphocytes. J Leukoc Biol. 2007;81:344–354. doi: 10.1189/jlb.0406237. [DOI] [PubMed] [Google Scholar]
- Masuda Y, Takatsu Y, Terao Y, Kumano S, Ishibashi Y, Suenaga M, et al. Isolation and identification of EG-VEGF/prokineticins as cognate ligands for two orphan G-protein-coupled receptors. Biochem Biophys Res Commun. 2002;293:396–402. doi: 10.1016/S0006-291X(02)00239-5. [DOI] [PubMed] [Google Scholar]
- Miele R, Lattanzi R, Bonaccorsi di Patti MC, Paiardini A, Negri L, Barra D. Expression of Bv8 in Pichia pastoris to identify structural features for receptor binding. Protein Expr Purif. 2010;73:10–14. doi: 10.1016/j.pep.2010.04.012. [DOI] [PubMed] [Google Scholar]
- Mollay C, Wechselberger C, Mignogna G, Negri L, Melchiorri P, Barra D, et al. Bv8, a small protein from frog skin and its homolog from snake venom induce hyperalgesia in rats. Eur J Pharmacol. 1999;374:189–196. doi: 10.1016/s0014-2999(99)00229-0. [DOI] [PubMed] [Google Scholar]
- Monnier J, Samson M. Cytokine properties of prokineticins. FEBS J. 2008;275:4014–4021. doi: 10.1111/j.1742-4658.2008.06559.x. [DOI] [PubMed] [Google Scholar]
- Morales RAV, Daly NL, Vetter I, Mobli M, Napier IA, Craik DJ, et al. Chemical synthesis and structure of the prokineticin Bv8. Chembiochem. 2010;11:1882–1888. doi: 10.1002/cbic.201000330. [DOI] [PubMed] [Google Scholar]
- Negri L, Lattanzi R, Giannini E, Metere A, Colucci M, Barra D, et al. Nociceptive sensitization by the secretory protein Bv8. Br J Pharmacol. 2002;137:1147–1154. doi: 10.1038/sj.bjp.0704995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negri L, Lattanzi R, Giannini E, Colucci A, Mignogna G, Barra D, et al. Biological activities of Bv8 analogues. Br J Pharmacol. 2005;146:625–632. doi: 10.1038/sj.bjp.0706376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negri L, Lattanzi R, Giannini E, Colucci M, Margheriti F, Melchiorri P, et al. Impaired nociception and inflammatory pain sensation in mice lacking the prokineticin receptor PKR1: focus on interaction between PKR1 and the capsaicin receptor TRPV1 in pain behavior. J Neurosci. 2006;26:6716–6727. doi: 10.1523/JNEUROSCI.5403-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negri L, Lattanzi R, Giannini E, Melchiorri P. Bv8/Prokineticin proteins and their receptors. Life Sci. 2007;81:1103–1116. doi: 10.1016/j.lfs.2007.08.011. [DOI] [PubMed] [Google Scholar]
- Negri L, Lattanzi R. Bv8-Prokineticins and their receptors: modulators of pain. Curr Pharm Biotechnol. 2011;12:1720–1727. doi: 10.2174/138920111798357410. [DOI] [PubMed] [Google Scholar]
- de Novellis V, Negri L, Lattanzi R, Rossi F, Palazzo E, Marabese I, et al. The prokineticin receptor agonist Bv8 increases GABA release in the periaqueductal grey and modifies RVM cell activities and thermoceptive reflexes in the rat. Eur J Neurosci. 2007;26:3068–3078. doi: 10.1111/j.1460-9568.2007.05910.x. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Worcester, MA: Academic press; 1986. [Google Scholar]
- Sacerdote P, Manfredi B, Gaspani L, Panerai AE. The opioid antagonist naloxone induces a shift from type 1 cytokine pattern to type 2 cytokine pattern in BALB/CJ mice. Blood. 2000;95:2031–2036. [PubMed] [Google Scholar]
- Soga T, Matsumoto S, Oda T, Saito T, Hiyama H, Takasaki J, et al. Molecular cloning and characterization of prokineticin receptors. Biochem Biophys Acta. 2002;1579:173–179. doi: 10.1016/s0167-4781(02)00546-8. [DOI] [PubMed] [Google Scholar]
- Vellani V, Colucci M, Lattanzi R, Giannini E, Negri L, Melchiorri P, et al. Sensitization of transient receptor potential vanilloid 1 by the prokineticin receptor agonist Bv8. J Neurosci. 2006;26:5109–5116. doi: 10.1523/JNEUROSCI.3870-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Truong KK, Zhou QY. Efferent projections of prokineticin 2 expressing neurons in the mouse suprachiasmatic nucleus. PLoS ONE. 2009;4:1–12. doi: 10.1371/journal.pone.0007151. [DOI] [PMC free article] [PubMed] [Google Scholar]