

Abstract
BACKGROUND AND PURPOSE
Neurocognitive disorders afflict approximately 20% of HIV-infected patients. HIV-1-infected cells in the brain shed viral proteins such as transactivator of transcription (Tat). Tat elicits cell death and synapse loss via processes initiated by NMDA receptor activation but mediated by separate downstream signalling pathways. Subunit selective NMDA receptor antagonists may differentially modulate survival relative to synaptic changes.
EXPERIMENTAL APPROACH
Tat-evoked cell death was quantified by measuring propidium iodide uptake into rat hippocampal neurons in culture. The effects of Tat on synaptic changes were measured using an imaging-based assay that quantified clusters of the scaffolding protein postsynaptic density 95 fused to green fluorescent protein.
KEY RESULTS
Dizocilpine, a non-competitive NMDA receptor antagonist, inhibited Tat-induced synapse loss, subsequent synapse recovery and Tat-induced cell death with comparable potencies. Memantine (10 µM) and ifenprodil (10 µM), which preferentially inhibit GluN2B-containing NMDA receptors, protected from Tat-induced cell death with no effect on synapse loss. Surprisingly, memantine and ifenprodil induced synapse recovery in the presence of Tat. In contrast, the GluN2A-prefering antagonist TCN201 prevented synapse loss and recovery with no effect on cell death.
CONCLUSIONS AND IMPLICATIONS
Synapse loss is a protective mechanism that enables the cell to cope with excess excitatory input. Thus, memantine and ifenprodil are promising neuroprotective drugs because they spare synaptic changes and promote survival. These GluN2B-preferring drugs induced recovery from Tat-evoked synapse loss, suggesting that synaptic pharmacology changed during the neurotoxic process. NMDA receptor subtypes differentially participate in the adaptation and death induced by excitotoxic insult.
Keywords: HIV Tat, neurotoxicity, dizocilpine, memantine, ifenprodil, TCN201, synapse loss, NMDA receptor, synapse recovery, HIV-associated neurocognitive disorders (HAND)
Introduction
AIDS affects over 30 million people worldwide. Approximately 20% of HIV-infected patients suffer from neurological symptoms ranging from mild cognitive and motor impairment to dementia, known collectively as HIV-associated neurocognitive disorders (HAND) (Ellis et al., 2007). HAND is an important complication of HIV infection of the brain because cognitive decline in some individuals progresses to where they are unable to perform basic functions of daily living (Kaul and Lipton, 2006; Hult et al., 2008; Minagar et al., 2008), and HIV patients showing neurocognitive impairment have shorter lifespans (Tozzi et al., 2005). Despite the reduced incidence of severe dementia with widespread use of combined anti-retroviral therapies, increased diagnosis and prolonged lifespan of HIV patients increases the need for improved therapies to cope with the rising prevalence of HAND.
Cognitive decline in HAND patients correlates with dendritic pruning and loss of synaptic spines (Wiley et al., 1999; Sa et al., 2004). The mechanism by which HIV infection leads to neurocognitive dysfunction is largely indirect because the virus infects macrophages and microglia in the CNS, but not neurons. Infected cells within the brain secrete inflammatory cytokines and shed viral proteins, which in turn exert toxic effects on neurons (Genis et al., 1992; Speth et al., 2001). The HIV protein transactivator of transcription (Tat) is shed by infected cells and induces neuronal injuries that include dendritic pruning, loss of spine density, synapse loss, and overt neuronal death (Liu et al., 2000; Eugenin et al., 2007; Kim et al., 2008; Fitting et al., 2010). Moreover, introducing Tat into the brain produces neuropathological symptoms in vivo (Kim et al., 2003; Maragos et al., 2003), and Tat mRNA is found in the brains of HAND patients (Wiley et al., 1996; Hudson et al., 2000).
Tat induces synapse loss via a pathway distinct from that leading to neuronal death (Kim et al., 2008). Both processes are initiated by NMDA receptor activity, but synapse loss is mediated by the ubiquitin-proteasome pathway and cell death is mediated by activation of NOS. Synapses lost following treatment with Tat can be recovered by treating with an antagonist to the lipoprotein receptor (Kim et al., 2008). We hypothesize that synapse loss is a reversible mechanism that protects the neuron from excitotoxic stimuli such as Tat. Indeed, preventing synapse loss actually sensitizes neurons to Tat-induced cell death (Kim et al., 2008). AIDS patients with HAND symptoms display cognitive improvement after starting treatment with antiretroviral therapy, suggesting that impairment is initially reversible (McArthur, 2004). Furthermore, lithium treatment increases the number of synapses in vitro (Kim and Thayer, 2009), and can improve neurological symptoms of HIV infection in vivo (Dou et al., 2005; Letendre et al., 2006). Perhaps inducing recovery of synapses can improve cognition in HAND patients.
The location and subunit composition of NMDA receptors determines the survival versus death-promoting effects of glutamate. Activation of GluN2A-containing NMDA receptors, which are preferentially localized to synapses, exerts pro-survival effects. In contrast, NMDA receptors that contain GluN2B subunits are predominantly extrasynaptic and their activation initiates cell death (Hardingham et al., 2002; Liu et al., 2004; 2007). The activity of NMDA receptors at synaptic relative to extrasynaptic sites may be vital for the initiation of synapse loss, or alternatively, inducing recovery of synapses. However, the role of NMDA receptor subunit composition in the mechanism of Tat-induced synapse loss, and subsequent recovery, has not been studied.
Dizocilpine (MK801) is a potent and efficacious NMDA receptor antagonist that affords short-term protection from acute excitotoxicity, but is poorly tolerated in vivo because of its psychotomimetic effects (Muir and Lees, 1995; Manahan-Vaughan et al., 2008). Memantine blocks NMDA receptors with rapid binding kinetics that enable it to preferentially block extrasynaptic NMDA receptors (Xia et al., 2010). It is well tolerated in humans and is clinically approved to improve cognition in Alzheimer's patients (Reisberg et al., 2003). GluN2B-selective NMDA receptor antagonists such as ifenprodil (Tovar and Westbrook, 1999; Williams, 1993) can selectively target receptors that participate in the toxic effects of NMDA receptor stimulation while sparing survival-promoting GluN2A-containing receptors (Hardingham et al., 2002; Liu et al., 2004; 2007). In contrast, GluN2A-selective NMDA receptor antagonists such as TCN201 (Bettini et al., 2010) might reduce survival.
Here we examined the effects of dizocilpine, memantine, ifenprodil and TCN201 on death and synapse loss and recovery following HIV-1 Tat in rat hippocampal neurons in vitro. With the exception of TCN201, all the NMDA receptor antagonists protected from Tat-induced cell death. Memantine and ifenprodil failed to affect synapse loss, while dizocilpine and TCN201 were protective. Surprisingly, memantine and ifenprodil, but not dizocilpine or TCN201, induced recovery of synapses following exposure to Tat.
Methods
Materials
Materials were obtained from the following sources: the PSD95-enhanced green fluorescent protein expression vector (pGW1-CMV-PSD95-EGFP) was kindly provided by Donald B. Arnold (Arnold and Clapham, 1999); the expression vector for DsRed2 (pDsRed2-N1) from Clontech (Mountain View, CA, USA); HIV-1 Tat (Clade B, recombinant) from Prospec Tany TechnoGene Ltd. (Rehovot, Israel); recombinant rat receptor-associated protein (RAP) from Fitzgerald Industries International (Concord, MA, USA); Dulbecco's modified Eagle medium (DMEM), fetal bovine serum, horse serum and propidium iodide (PI) from Invitrogen (Carlsbad, CA, USA); memantine HCl, ifenprodil hemitartrate, and TCN201 from Tocris (Ellsville, MO, USA); penicillin/streptomycin, dizocilpine (MK801) and all other reagents from Sigma (St. Louis, MO, USA).
Cell culture
Rat hippocampal neurons were grown in primary culture as described previously (Shen and Thayer, 1998) with minor modifications. Fetuses were removed on embryonic day 17 from maternal rats, anaesthetized with CO2 and killed by decapitation. Hippocampi were dissected and placed in Ca2+ and Mg2+-free HEPES-buffered Hanks salt solution (HHSS), pH 7.45. HHSS was composed of the following (in mM): HEPES 20, NaCl 137, CaCl2 1.3, MgSO4 0.4, MgCl2 0.5, KCl 5.0, KH2PO4 0.4, Na2HPO4 0.6, NaHCO3 3.0 and glucose 5.6. Cells were dissociated by trituration through flame-narrowed Pasteur pipettes of decreasing aperture, pelleted and resuspended in DMEM without glutamine, supplemented with 10% fetal bovine serum and penicillin/streptomycin (100 U·mL−1 and 100 µg·mL−1, respectively). Dissociated cells were then plated at a density of 80 000–120 000 cells per dish onto a 25 mm round cover glass (#1) glued to cover a 19 mm diameter opening drilled through the bottom of a 35 mm Petri dish. The cover glass was precoated with Matrigel (200 µL, 0.2 mg·mL−1) (BD Biosciences, Billerica, MA, USA). Neurons were grown in a humidified atmosphere of 10% CO2 and 90% air (pH 7.4) at 37°C, and fed at days 1 and 6 by exchange of 75% of the media with DMEM, supplemented with 10% horse serum and penicillin/streptomycin. Cells used in these experiments were cultured without mitotic inhibitors for at least 12 days, resulting in a mixed glial-neuronal culture. Immunocytochemistry experiments demonstrated that these cultures were composed of 18 ± 2% neurons, 70 ± 3% astrocytes and 9 ± 3% microglia (Kim et al., 2011).
Transfection
Rat hippocampal neurons were transfected between 10 and 12 days in vitro using a modification of a protocol described previously (Kim et al., 2008). Briefly, hippocampal cultures were incubated for at least 20 min in DMEM supplemented with 1 mM kynurenic acid, 10 mM MgCl2, and 5 mM HEPES, to reduce neurotoxicity. A DNA/calcium phosphate precipitate containing 1 µg total plasmid DNA per well was prepared, allowed to form for 30 min at room temperature, and added to the culture. After a 90 min incubation period, cells were washed once with DMEM supplemented with MgCl2 and HEPES and then returned to conditioned media, saved at the beginning of the procedure. Transfected neurons were imaged 48–96 h post-transfection. Transfection efficiency ranged from 1% to 5%.
Confocal imaging
Petri dishes containing transfected neurons were sealed with Parafilm, transferred to the stage of an inverted confocal microscope (Olympus Fluoview 300, Melville, NY, USA) and viewed through a 60X oil-immersion objective (NA = 1.4). To enable repeated imaging of the same cell over a 24 h period, the location of the cell was recorded using micrometers attached to the stage of the microscope. Multiple optical sections spanning 8 µm in the z-dimension were collected (1 µm steps), and these optical sections were combined through the z-axis into a compressed z stack. Green fluorescent protein (GFP) was excited at 488 nm with an argon ion laser and emission collected at 530 nm (10 nm band pass). DsRed2 was excited at 543 nm with a green HeNe laser and emission collected at >605 nm. The cell culture dish was returned to the CO2 incubator between image collections. Experiments studying synapse recovery were performed for 24 h in the continuous presence of Tat, with or without the specified drugs added at 16 h.
Image processing
To count and label PSD95-GFP puncta, an automated algorithm was created using MetaMorph 6.2 image processing software described previously (Waataja et al., 2008). Briefly, maximum z-projection images were created from the DsRed2 and GFP image stacks. Next, a threshold set 1 SD above the image mean was applied to the DsRed2 image. This created a 1 bit image, which was used as a mask via a logical AND function with the GFP maximum z-projection. A top-hat filter (80 pixels) was applied to the masked PSD95-GFP image. A threshold set 1.5 SD above the mean intensity inside the mask was then applied to the contrast enhanced image. Structures between 8 and 80 pixels (approximately 0.37–3.12 µm in diameter) were counted as PSDs. The structures were then dilated and superimposed on the DsRed2 maximum z-projection for visualization.
Toxicity
Cell death was quantified using PI fluorescence as previously described (Kim et al., 2008). Cell culture was performed as described earlier except that 50 000–60 000 cells per well were plated in 96-well plates and grown for 12–14 days in vitro. The experiment was started after 12 days in vitro by replacing 100 µL (approximately two-thirds volume) of the cell culture medium with fresh DMEM containing 10% horse serum, penicillin/streptomycin, 70 µM PI and either Tat (50 ng·mL−1) or vehicle. The plate was placed in a FluoStar Galaxy multiwell fluorescent plate scanner (BMG Technologies GmbH, Offenburg, Germany) and maintained at 37°C. PI fluorescence intensity measurements (excitation 544 nm ± 15 nm, emission 620 nm ± 15 nm) were taken at time 0 and 48 h. Between measurements, cells were returned to the incubator and kept at 37°C in 10% CO2. Drugs, when present, were applied 15 min before application of Tat and included in the media exchange. Each treatment was performed in triplicate; a set of 3 wells from a single plating of cells was defined as an individual experiment (n= 1).
Immunocytochemistry
Hippocampal cultures prepared as described earlier were transfected with pGW1-CMV-PSD95-EGFP on day 12 in culture. Forty-eight hours after transfection, cover slips with cells were washed with tris-buffered saline (TBS) and fixed with Lana's fixative (8% paraformaldehyde, 14% picrate, 0.16 M phosphate) for 10 min. The cells were washed with TBS, then permeabilized in TBS + 0.2% Triton X100 (Sigma) for 10 min at room temperature. After permeabilization, the cells were incubated with mouse anti-GluN2A (1:200, Chemicon/Millipore, Billerica, MA, USA) or mouse anti-GluN2B (1:200, BD Transduction Laboratories, San Jose, CA, USA) in TBS + 0.2% Tween-20 (Sigma) for 1 h at room temperature. Cells were washed with TBS and labelled proteins visualized with tetramethyl rhodamine isothiocyanate (TRITC)-conjugated goat anti-mouse antibody (Millipore, Billerica, MA, USA, 1:400) in TBS + 0.2% Tween-20. Cover slips were mounted with Fluoromount (Southern Biotech, Birmingham, AL, USA) and imaged on an inverted confocal microscope (Olympus Fluoview 300, Melville, NY, USA) using a 60X oil-immersion objective (NA = 1.4). TRITC was excited at 543 nm, and emission collected at >605 nm.
Statistics
For synapse loss and recovery studies, an individual experiment (n= 1) was defined as the change in the number of PSDs from a single cell from a single coverslip. PSD counts were presented as mean ± SEM. Each experiment was replicated over at least three separate cultures. For cell survival studies, each treatment was performed in triplicate; thus, a set of three wells from a single plating of cells was defined as an individual experiment (n= 1). In all statistical analyses we used Student's t-test for single or anova with Tukey's post hoc test for multiple statistical comparisons (OriginPro v8.5; Northampton, MA, USA).
Results
Changes in the number of synapses between rat hippocampal neurons in culture were monitored by imaging neurons expressing PSD95-GFP and DsRed2, as previously described (Waataja et al., 2008). The PSD95-GFP fusion construct expressed in a punctate pattern. Processing of PSD95-GFP images identified PSDs as fluorescent puncta meeting intensity and size criteria (average puncta size = 0.52 µm2) and in contact with a mask derived from the DsRed2 image (Figure 1A). We have previously shown that fluorescent puncta co-localized with synaptically evoked local Ca2+ increases, functional neurotransmitter release sites and NMDA receptor immunoreactivity, indicating that they represent functional post-synaptic sites (Waataja et al., 2008). The number of PSD95-GFP puncta was calculated for a single neuron over multiple time points to assess changes in synapse number (Figure 1B). In untreated neurons, the number of synaptic sites increased by 24 ± 5% (n= 66) over 24 h. Treatment with 50 ng·mL−1 Tat for 24 h evoked a 24 ± 4% (n= 37) decrease in the number of postsynaptic sites (Figure 1B).
Figure 1.
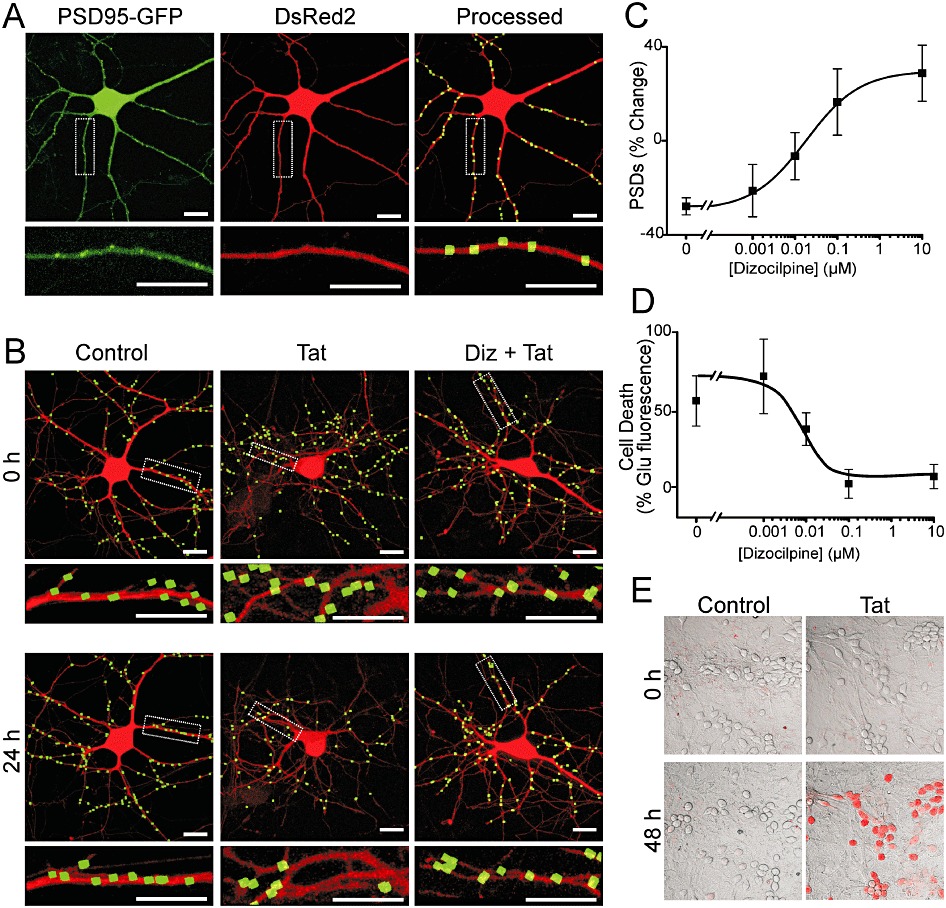
NMDA receptor activity is required for Tat-mediated neuronal death and synapse loss. (A) Representative confocal images of neurons were collected and processed as described in Methods. Maximum projection images show a neuron expressing PSD95-GFP and DsRed2. PSD95-GFP puncta were identified by filtering compressed z-stacks (8 µm) of confocal images for fluorescent intensity and size and counted when in contact with a mask derived from the DsRed2 image. Labelled PSDs were dilated and overlaid on the DsRed2 image for visualization purposes (processed). The insets are enlarged images of the boxed region. Scale bars represent 10 µm. (B) Processed images show neurons before (0 h) and after (24 h) no treatment (control), treatment with 50 ng·mL−1 Tat (Tat), or treatment with 10 µM dizocilpine 15 min before and during treatment with Tat (Diz + Tat). The insets are enlarged images of the boxed regions. Scale bars represent 10 µm. (C) Graph shows the % change in the number of PSD95-GFP puncta (mean ± SEM) for cells treated with 50 ng·mL−1 Tat for 24 h in the presence of the indicated concentrations of dizocilpine (n≥ 7 for each data point). The curve was fit with a logistic equation of the form %PSD change = A2+ (A1− A2)/(1 + (X/EC50)p) where X = dizocilpine concentration, A1=−28 ± 2% PSD change without dizocilpine, A2= 17 ± 4% PSD change at a maximally effective dizocilpine concentration and p = slope factor. EC50 was calculated using a nonlinear, least squares curve-fitting programme. EC50 and p were 9.6 nM and 0.5, respectively. (D) Graph shows cell death in cultures treated with 50 ng·mL−1 Tat for 48 h in the presence of the indicated concentrations of dizocilpine (n≥ 5 for each data point). Cell death was quantified by PI fluorescence as described in Methods. PI fluorescence was normalized to that induced by treatment with 1 mM glutamate for 48 h, which kills virtually all neurons in this culture. The mean ± SEM % change in PI fluorescence is plotted against increasing concentrations of dizocilpine. The curve was fit with a logistic equation of the form % change in PI fluorescence = A1+ (A2− A1)/(1 + (X/EC50)p) where X = dizocilpine concentration, A1=−1 ± 5% PI fluorescence change at a maximally effective dizocilpine concentration, A2= 54 ± 9% PI fluorescence change without dizocilpine, and p = slope factor. EC50 was calculated using a nonlinear, least squares curve-fitting programme. EC50 and p were 10.4 nM and – 0.7, respectively. (E) Representative images show differential-interference-contrast micrographs of hippocampal neurons in culture with PI fluorescence (red) superimposed. Images from control and Tat-treated (50 ng·mL−1) cultures are shown before (0 h) and after (48 h) treatment.
Dizocilpine blocks Tat-induced changes in synapses and survival
Synapse loss induced by Tat is mediated by the NMDA receptor (Kim et al., 2008). In Figure 1B, we show that treatment with dizocilpine prevented synapse loss evoked by 24 h treatment with 50 ng·mL−1 Tat. This protection was concentration-dependent with an EC50 of 9.6 nM (Figure 1C).
NMDA receptor activity is also required for Tat-induced cell death (Eugenin et al., 2007). Treatment with Tat induced neuronal death by 48 h, which we quantified by uptake of PI. Tat (50 ng·mL−1) increased PI fluorescence to 52 ± 15% (n= 13) of that evoked by 1 mM glutamate. Treatment with dizocilpine 15 min prior to and during 48 h exposure to Tat afforded a concentration-dependent protection with an EC50 of 10.4 nM (Figure 1D). Figure 1E shows representative images of PI fluorescence superimposed on differential-interference-contrast images of the hippocampal culture under control conditions and following 48 h treatment with Tat. Treatment with Tat for 24 h, the time at which synapse loss was determined (Figure 1B–C), did not significantly affect viability as determined by retention of the DsRed2 protein (Figure 1B) and failure to take up PI (Kim et al., 2008). Thus, synapse loss precedes neuronal death and both synapse loss and death are reduced by similar concentrations of dizocilpine.
Tat-induced loss of PSD95-GFP puncta is mediated by the low-density lipoprotein receptor-related protein (LRP) (Kim et al., 2008). The loss of postsynaptic sites was reversed by treatment with RAP, a competitive antagonist of LRP. Treating neurons for 16 h with 50 ng·mL−1 Tat induced significant synapse loss (Figure 2A) that was sustained for 24 h (Figure 2B). Addition of dizocilpine (10 µM) at 16 h to cells treated with Tat did not affect the sustained loss (Kim et al., 2008). In contrast, adding RAP to Tat-treated cells at 16 h in the continuous presence of Tat produced a marked recovery of synaptic sites (n= 52) (Figure 2A and B). Interestingly, the recovery of PSD95-GFP puncta induced by RAP was inhibited by adding 10 µM dizocilpine 15 min before the addition of RAP at 16 h (n= 29) (Figure 2A and B), indicating that NMDA receptor activity was necessary for the recovery process. We next determined the concentration-dependence for dizocilpine's inhibition of synapse recovery. As shown in Figure 2C, dizocilpine impaired synapse recovery with an EC50 of 24 nM. Taken together, these results show that NMDA receptor activity is not only required for Tat-induced synapse loss and neuronal death, it is also required for recovery of synapses following Tat-induced loss. Moreover, dizocilpine attenuated Tat-induced synapse loss, Tat-induced cell death and RAP-induced synapse recovery with comparable potencies.
Figure 2.
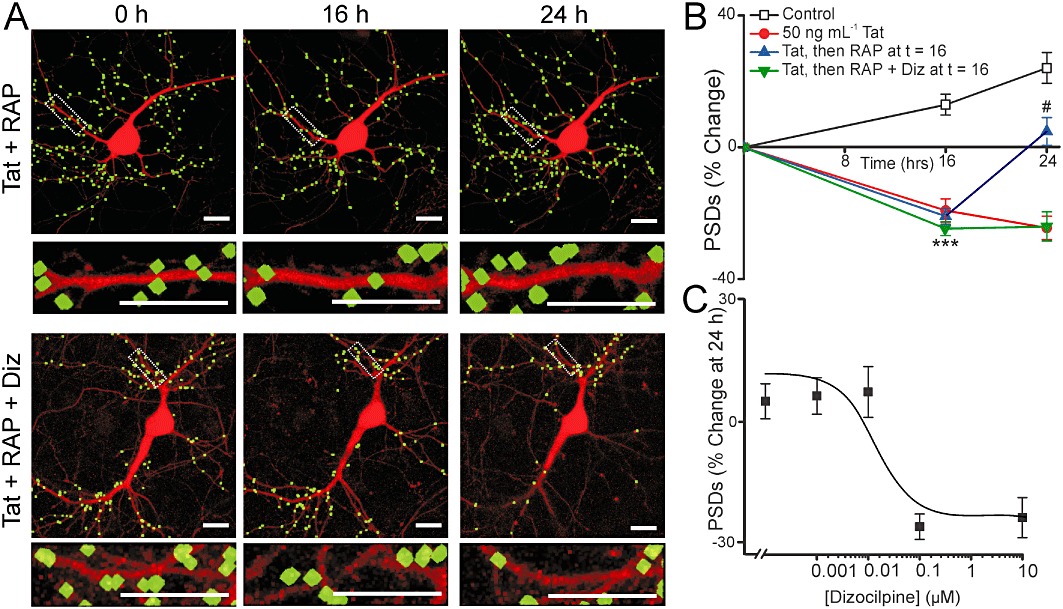
NMDA receptor activity is required for RAP-induced synapse recovery. (A) Representative processed images of neurons before (0 h) and 16 and 24 h after treatment with 50 ng·mL−1 Tat. After 16 h exposure to Tat the cells were treated with 50 nM RAP alone (Tat + RAP) or RAP + 10 µM dizocilpine (Tat + RAP + Diz) (dizocilpine was applied 15 min before adding RAP). The insets are enlarged images of the boxed region. Scale bars represent 10 µm. (B) Graph summarizes % change in number of PSDs in the absence (control) or presence of 50 ng·mL−1 Tat for 24 h. After 16 h exposure to Tat cells were then left untreated or treated with 50 nM RAP alone or RAP + 10 µM dizocilpine. Data are expressed as mean ± SEM. ***P < 0.001 relative to control at 16 h; #P < 0.05 relative to 50 ng·mL−1 Tat at 24 h (anova with Tukey's post-test). (C) Graph shows the % change in the number of PSD95-GFP puncta (mean ± SEM) for cells treated with 50 ng·mL−1 Tat for 24 h. 50 nM RAP was applied at 16 h in the presence of the indicated concentrations of dizocilpine (n≥ 5 for each data point). The curve was fit with a logistic equation of the form % PSD change = A1+ (A2− A1)/[1 + (X/EC50)p] where X = dizocilpine concentration, A1=−26 ± 3% PSD change at a maximally effective dizocilpine concentration, A2= 5 ± 1% PSD change without dizocilpine and p = slope factor. EC50 was calculated using a nonlinear, least squares curve-fitting programme. EC50 and p were 24.3 nM and – 0.9, respectively.
Memantine differentially affects survival and synapse loss
Dizocilpine is a high affinity, non-competitive NMDA receptor antagonist developed as a neuroprotective agent, but not used clinically because of psychotomimetic side effects (Muir and Lees, 1995). Memantine is a non-competitive NMDA receptor antagonist that, in contrast to dizocilpine, is well-tolerated and used clinically to improve cognition in patients with Alzheimer's disease (Reisberg et al., 2003). Therefore, we decided to test the effects of memantine on Tat-induced cell death and synapse loss. Memantine (10 µM) prevented Tat-induced neuronal death (Figure 3A), similar to dizocilpine (n= 9). However, in contrast to dizocilpine, 10 µM memantine did not inhibit synapse loss induced by Tat (n= 13) (Figure 3B–C). Even at 100 µM, a concentration 10-fold higher than that needed to prevent Tat-induced cell death, memantine had no effect on Tat-induced synapse loss (n= 5) (Figure 3C).
Figure 3.
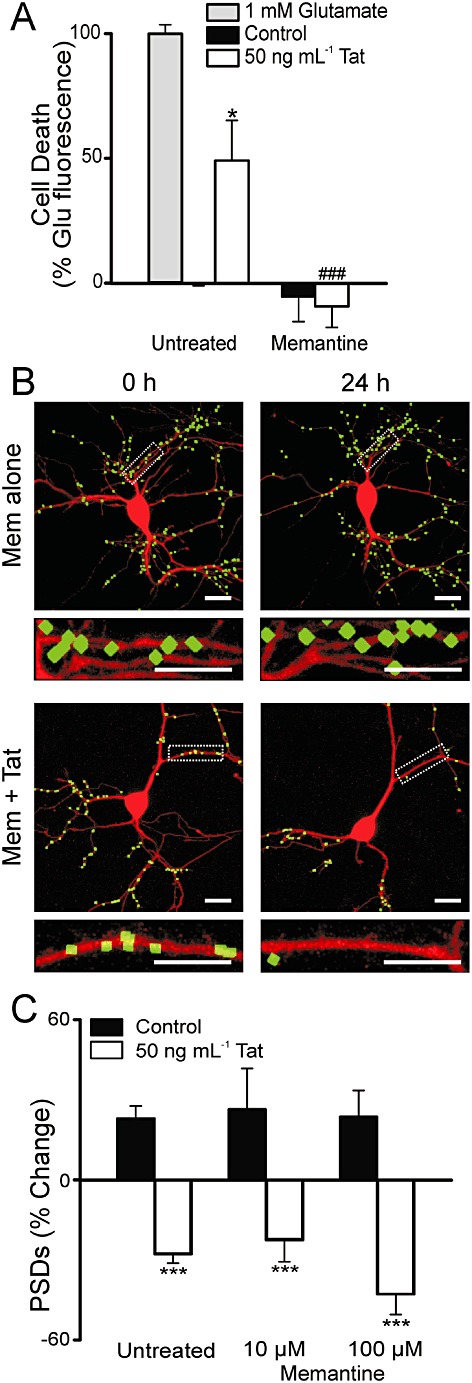
Memantine prevents Tat-induced cell death without affecting synapse loss. (A) Cell death was measured with the PI fluorescence assay described in Methods. Bar graph summarizes PI uptake in neurons 48 h after no treatment (control), treatment with 1 mM glutamate, or treatment with 50 ng·mL−1 Tat in the absence (untreated) or presence of 10 µM memantine as indicated. PI fluorescence was normalized to 1 mM glutamate treatment. Data are expressed as mean ± SEM. *P < 0.05 relative to control, ###P < 0.001 relative to Tat treatment alone (anova with Tukey's post-test). (B) Representative processed images of neurons incubated with 10 µM memantine before (0 h) and after (24 h) no treatment (Mem alone) or treatment with 50 ng·mL−1 Tat (Mem + Tat). The insets are enlarged images of the boxed region. Scale bars represent 10 µm. (C) Bar graph summarizes changes in PSD-GFP puncta (PSDs) after 24 h treatment under control conditions (solid bars) or following treatment with 50 ng·mL−1 Tat in the absence or presence of the indicated concentration of memantine. Data are expressed as mean ± SEM. ***P < 0.001 relative to control (Student's t-test).
Memantine induces recovery of synapses
We next examined the effect of memantine on the recovery of synaptic sites following Tat-induced loss. In contrast to dizocilpine, memantine significantly increased the number of synapses in Tat-treated cells. As shown in Figure 4A and B, addition of 10 µM memantine to cells treated with 50 ng·mL−1 Tat for 16 h evoked a recovery in the number of PSD95-GFP puncta (n= 13). Furthermore, memantine did not inhibit recovery of synapses induced by RAP. When memantine was applied 15 min before the addition of RAP at 16 h, the number of synapses still increased similar to cells treated with RAP alone at 16 h (n= 14) (Figure 4C). Memantine induced synapse recovery following Tat-induced synapse loss and did not interfere with synapse recovery in the presence of RAP.
Figure 4.
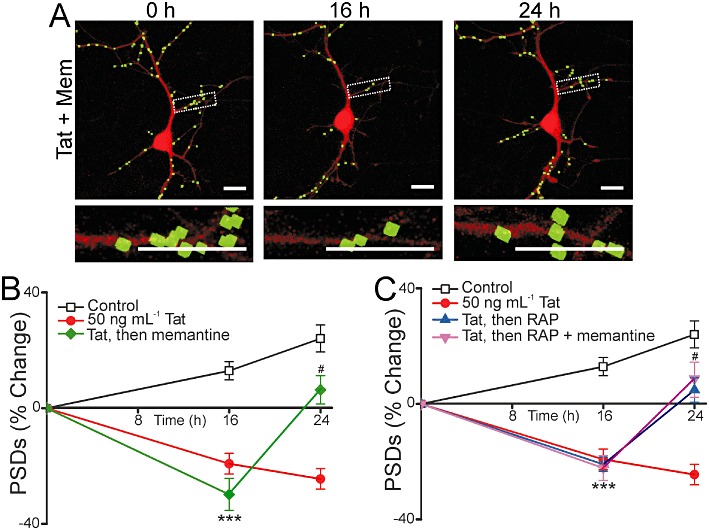
Memantine evokes synapse recovery after Tat-induced loss. (A) Representative processed images of a neuron before (0 h) and 16 or 24 h after treatment with 50 ng·mL−1 Tat. 10 µM memantine was applied after 16 h in the presence of Tat. The insets are enlarged images of the boxed region. Scale bars represent 10 µm. (B–C) Graphs summarize % change in number of PSDs in the absence (control) or presence of 50 ng·mL−1 Tat for 24 h. (B) After 16 h exposure to Tat, cells were then left untreated or treated with 10 µM memantine. (C) After 16 h exposure to Tat, cells were then left untreated, treated with 50 nM RAP alone or treated with RAP + 10 µM memantine. Memantine was added to neurons 15 min before 50 nM RAP. Data are expressed as mean ± SEM. ***P < 0.001 relative to control at 16 h, #P < 0.05 relative to 50 ng·mL−1 Tat at 24 h (anova with Tukey's post-test).
One mechanism by which memantine might selectively inhibit NMDA receptor-mediated cell death while sparing NMDA receptor-induced synapse loss would be if these two processes were mediated by different subsets of NMDA receptors. As shown in Figure 5, immunocytochemistry experiments demonstrate that these hippocampal neurons in culture express both GluN2A and GluN2B immunoreactivity. Neurons expressing PSD95-GFP (Figure 5 green) were fixed and labelled with GluN2A and GluN2B-selective antibodies (Figure 5 red). Diffuse labelling is apparent on the soma and dendrites with intense punctate labelling also present along the dendrites. PSD95-GFP puncta co-localized with GluN2A and GluN2B immunoreactive puncta (Figure 5, merged yellow). Note that the antibodies labelled non-transfected cells in the field and thus, not all NMDA receptor immunoreactive puncta corresponded to GFP puncta.
Figure 5.
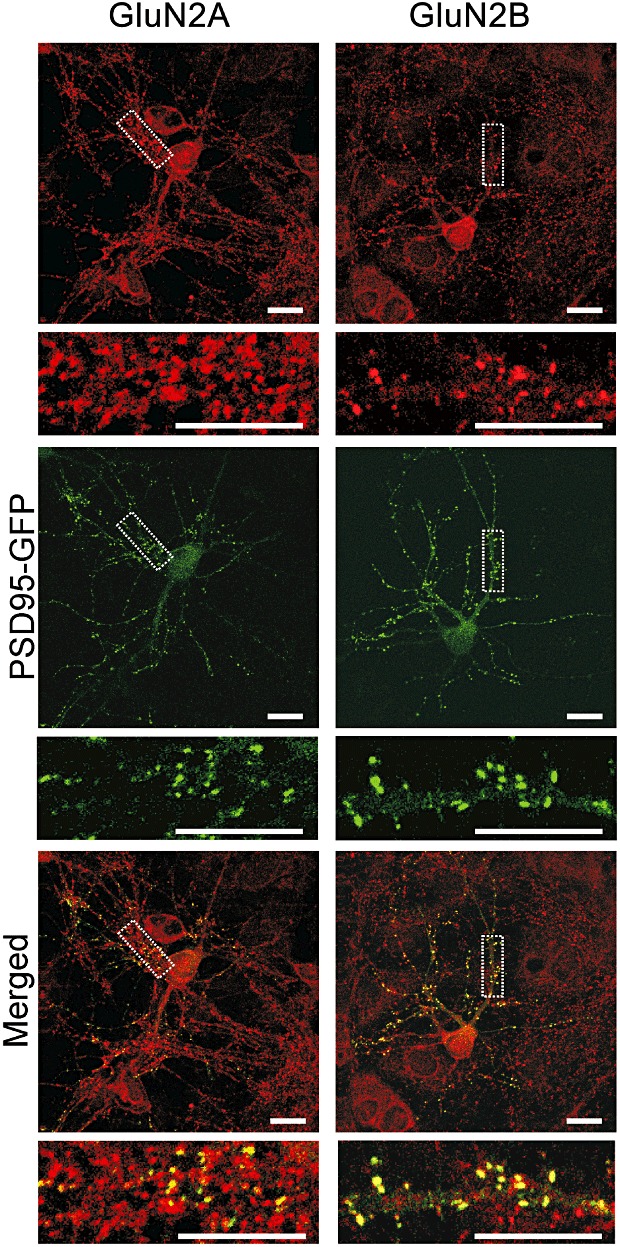
Hippocampal neurons in culture exhibit GluN2A and GluN2B immunoreactivity. Forty-eight hours after transfection with the PSD95-GFP expression vector, hippocampal cultures were fixed and labelled with GluN2A and GluN2B-selective antibodies (red) as described in Methods. PSD95-GFP puncta (green) co-localized with GluN2A and GluN2B immunoreactive puncta (merged, yellow). Note that non-transfected cells were also present in the field and thus not all immunoreactive puncta (red) co-localize with a PSD95-GFP puncta. Scale bars represent 10 µm.
Ifenprodil differentially affects survival and synapse loss
Memantine differs from dizocilpine in that it preferentially inhibits NMDA receptor activity at extrasynaptic sites (Xia et al., 2010). Because GluN2B subunits are present at extrasynaptic sites and their activation triggers cell death (Liu et al., 2007), we hypothesized that memantine protected from Tat-induced neuronal death by preferentially inhibiting GluN2B-containing NMDA receptors while sparing synaptic plasticity induced by GluN2A-containing NMDA receptor activity. We tested this hypothesis with the use of the NMDA receptor antagonist ifenprodil.
Ifenprodil is a non-competitive antagonist that is selective for GluN2B-containing NMDA receptors at a concentration of 10 µM (Williams, 1993; Avenet et al., 1996). Ifenprodil (10 µM) prevented Tat-induced neuronal death (n= 13) (Figure 6A). However, this concentration of ifenprodil did not inhibit synapse loss induced by Tat (n= 9) (Figure 6B–C). This result is similar to that seen with memantine, indicating that GluN2B-containing, or extrasynaptic NMDA receptors were not required for Tat-induced synapse loss.
Figure 6.
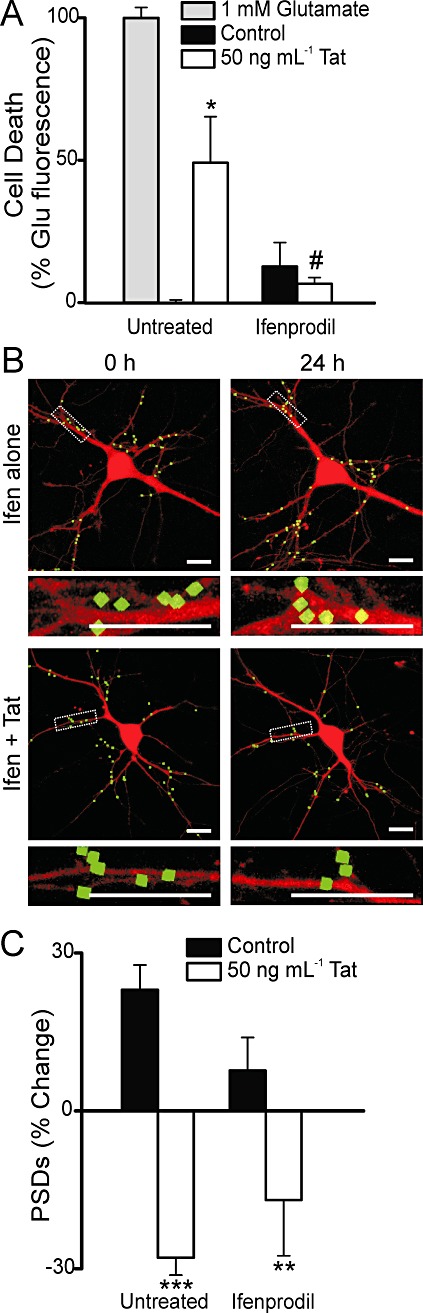
Ifenprodil improves survival without inhibiting Tat-induced synapse loss. (A) Cell death was measured with the PI fluorescence assay described in Methods. Bar graph summarizes PI uptake in neurons 48 h after no treatment (control), treatment with 1 mM glutamate, or treatment with 50 ng·mL−1 Tat in the absence (untreated) or presence of 10 µM ifenprodil as indicated. PI fluorescence was normalized to 1 mM glutamate treatment. Data are expressed as mean ± SEM. *P < 0.05 relative to control, #P < 0.05 relative to Tat treatment alone (anova with Tukey's post-test). (B) Representative processed images of neurons incubated with 10 µM ifenprodil before (0 h) and after (24 h) no treatment (Ifen alone) or treatment with 50 ng·mL−1 Tat (Ifen + Tat). The insets are enlarged images of the boxed region. Scale bars represent 10 µm. (C) Bar graph summarizes changes in PSD-GFP puncta (PSDs) after 24 h treatment under control conditions or following treatment with 50 ng·mL−1 Tat in the absence (untreated) or presence of 10 µM ifenprodil. Data are expressed as mean ± SEM. **P < 0.01 relative to control; ***P < 0.001 relative to control (Student's t test).
Ifenprodil induces synaptic recovery
We next examined the effect of ifenprodil on the recovery of synaptic sites following Tat-induced loss. Similar to memantine, 10 µM ifenprodil significantly increased the number of synapses in Tat-treated cells (Figure 7A and B). As shown in Figure 7A, addition of 10 µM ifenprodil after a 16 h treatment with 50 ng·mL−1 Tat evoked recovery in the number of PSD95-GFP puncta (n= 11). Furthermore, 10 µM ifenprodil did not inhibit recovery of synapses when applied 15 min before the addition of RAP at 16 h (n= 9) (Figure 7C). This result is similar to that seen with memantine. Ifenprodil has non-selective effects at high concentrations (Williams, 1993). However, we were unable to study the effects of ifenprodil at concentrations greater than 10 µM because 24 h exposure to a high concentration of ifenprodil (100 µM) was directly toxic to the neuronal culture.
Figure 7.
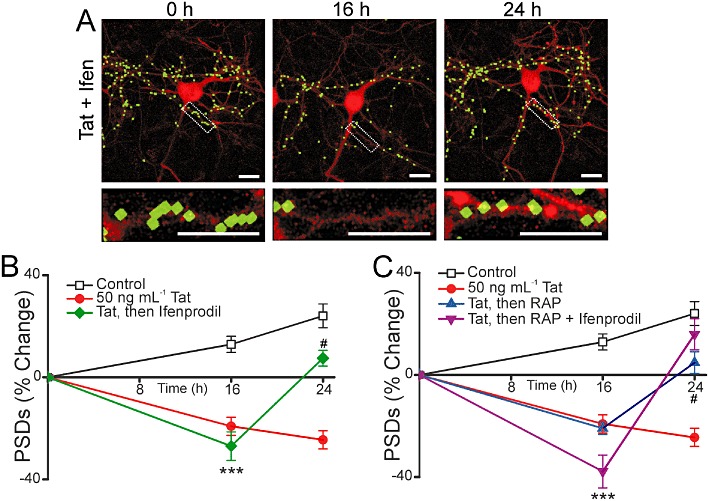
Ifenprodil evokes synapse recovery after Tat-induced loss. (A) Representative processed images of a neuron before (0 h) and 16 or 24 h after treatment with 50 ng·mL−1 Tat; 10 µM ifenprodil was applied after 16 h in the presence of Tat. The insets are enlarged images of the boxed region. Scale bars represent 10 µm. (B–C) Graphs summarize % change in number of PSDs in the absence (control) or presence of 50 ng·mL−1 Tat for 24 h. (B) After 16 h exposure to Tat cells were then left untreated or treated with 10 µM ifenprodil. (C) After 16 h exposure to Tat cells were then left untreated, treated with 50 nM RAP alone or treated with RAP + 10 µM ifenprodil. Ifenprodil was added to neurons 15 min before 50 nM RAP. Data are expressed as mean ± SEM. ***P < 0.001 relative to control at 16 h; #P < 0.05 relative to 50 ng·mL−1 Tat at 24 h (anova with Tukey's post-test).
TCN201 differentially affects survival and synapse loss
If the effects of memantine and ifenprodil on synapse loss and recovery differed from dizocilpine because of their GluN2B selective actions, then a GluN2A-selective antagonist should produce opposite effects. TCN201 is selective for GluN2A-containing NMDA receptors at concentrations less than 50 µM (Bettini et al., 2010). TCN201 at a concentration of 10 µM did not protect against Tat-induced cell death (n= 11) (Figure 8A), in contrast to ifenprodil and memantine. However, this same concentration of TCN201 prevented Tat-induced synapse loss (n= 10) (Figure 8B–C). These results suggest that activation of GluN2A-containing NMDA receptors are required for Tat-induced synapse loss, but these receptors do not participate in Tat-induced cell death.
Figure 8.
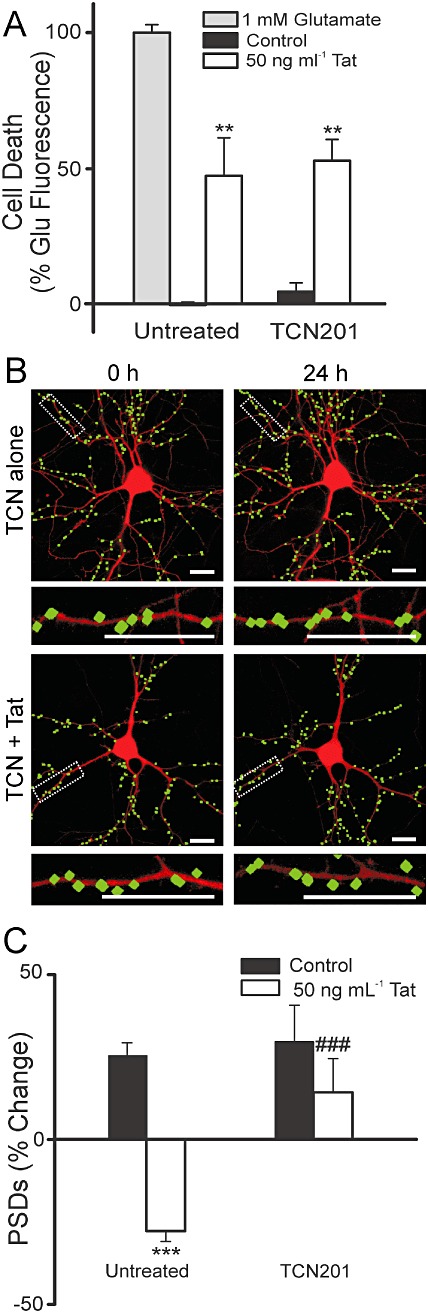
TCN201 inhibits Tat-induced synapse loss without affecting survival. (A) Cell death was measured with the PI fluorescence assay described in Methods. Bar graph summarizes PI uptake in neurons 48 h after no treatment (control), treatment with 1 mM glutamate, or treatment with 50 ng·mL−1 Tat in the absence (untreated) or presence of 10 µM TCN201 as indicated. PI fluorescence was normalized to 1 mM glutamate treatment. Data are expressed as mean ± SEM. **P < 0.01 relative to control (anova with Tukey's post-test). (B) Representative processed images of neurons incubated with 10 µM TCN201 before (0 h) and after (24 h) no treatment (TCN alone) or treatment with 50 ng·mL−1 Tat (TCN + Tat). The insets are enlarged images of the boxed region. Scale bars represent 10 µm. (C) Bar graph summarizes changes in PSD-GFP puncta (PSDs) after 24 h treatment under control conditions or following treatment with 50 ng·mL−1 Tat in the absence or presence of 10 µM TCN201. Data are expressed as mean ± SEM. ***P < 0.001 relative to control; ###P < 0.001 relative to 50 ng·mL−1 Tat alone (anova with Tukey's post-test).
TCN201 does not induce synapse recovery
We next examined the effects of TCN201 on synapse recovery. When applied 16 h after the application of 50 ng·mL−1 Tat, 10 µM TCN201 showed no effect (n= 13) (Figure 9A–B). The significant reduction in the number of synapses observed at the time of TCN201 application (t= 16 h) was sustained in the presence of the drug, in contrast to the effects of ifenprodil and memantine, which induced synapse recovery. Furthermore, when 10 µM TCN201 was applied 15 min before RAP, it prevented RAP-induced recovery of synapses (n= 10) (Figure 9C), similar to the effects of dizocilpine.
Figure 9.
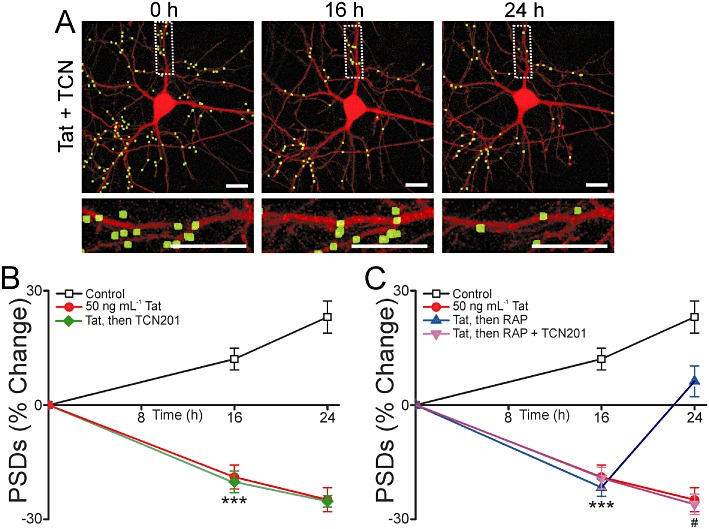
TCN201 inhibits synapse recovery after Tat-induced loss. (A) Representative processed images of a neuron before (0 h) and 16 or 24 h after treatment with 50 ng·mL−1 Tat. 10 µM TCN201 was applied after 16 h in the presence of Tat. The insets are enlarged images of the boxed region. Scale bars represent 10 µm. (B–C) Graphs summarize % change in number of PSDs in the absence (control) or presence of 50 ng·mL−1 Tat for 24 h. (B) After 16 h exposure to Tat cells were then left untreated or treated with 10 µM TCN201. (C) After 16 h exposure to Tat cells were then left untreated, treated with 50 nM RAP alone or treated with RAP + 10 µM TCN201. TCN201 was added to neurons 15 min before 50 nM RAP. Data are expressed as mean ± SEM. ***P < 0.001 relative to control at 16 h; #P < 0.05 relative to 50 ng·mL−1 Tat, then RAP at 24 h (anova with Tukey's post-test).
Taken together, these results indicate that memantine, as well as GluN2B-selective concentrations of ifenprodil protect from Tat-induced cell death, fail to affect Tat-induced synapse loss and will precipitate a recovery of synapses lost following Tat treatment. In contrast, GluN2A-selective concentrations of TCN201 do not protect from Tat-induced cell death, yet inhibit Tat-induced synapse loss. TCN201 inhibited RAP-induced synapse recovery. The NMDA receptor antagonist dizocilpine, which lacks subunit selectivity, inhibited all aspects of Tat-induced toxicity, synapse loss and subsequent recovery.
Discussion and conclusions
Dendritic pruning and loss of synaptic spines correlates with cognitive decline in patients with HIV-associated dementia (Masliah et al., 1997; Everall et al., 1999). HIV neurotoxicity is mediated in part by viral proteins, such as Tat, shed by infected non-neuronal cells (King et al., 2006). In this study, we used a live cell, confocal imaging assay to track dynamic changes in synapse number in neurons exposed to the HIV protein Tat. Tat induced a marked loss of excitatory synapses by 24 h. Synapse loss following 16 h exposure to Tat was reversible, although prolonged exposure led to cell death by 48 h. All three events – death, synapse loss and synapse recovery – required NMDA receptor activity (Figure 10). We examined the effects of four NMDA receptor antagonists with distinct mechanisms of action on these processes. Dizocilpine blocked all three processes. Memantine and ifenprodil prevented Tat-induced cell death. These drugs failed to affect Tat-induced synapse loss but, if applied late after significant synapse loss was evident, memantine and ifenprodil induced a recovery of synapse number. In contrast, TCN201 inhibited Tat-induced synapse loss as well as RAP-induced synapse recovery, but could not induce recovery, nor protect against Tat-induced cell death. These results suggest specific roles for NMDA receptor subtypes in these processes. Furthermore, the pharmacology of synapse loss and recovery changed during the course of the neurodegenerative process.
Figure 10.
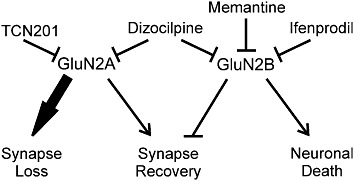
Modulation of HIV Tat-induced changes in synapses and survival by subtype specific NMDA receptor antagonists. Summary scheme links effects of pharmacological agents to hypothesized functions mediated by GluN2A- and GluN2B-containing NMDA receptors.
Considerable evidence implicates HIV-1 Tat as a significant contributing factor in HAND. It is present in the serum of HAND patients, and Tat protein and mRNA levels in the parenchyma correlate with the severity of neurological symptoms (Wiley et al., 1996; Hudson et al., 2000). Intracerebroventricular injection of Tat induced behavioural alterations, impaired long-term potentiation and produced neuronal and glial toxicity (Bruce-Keller et al., 2003; Li et al., 2004). In vitro and in vivo studies have demonstrated that Tat induces dendritic pruning (Fitting et al., 2010), synapse loss (Kim et al., 2008) and cell death (Eugenin et al., 2007). The imaging-based assay employed for this study is well suited for evaluating synaptic changes induced by neurotoxins such as Tat. In addition, because the assay tracks the number of synapses on the same cell before and during the course of toxin exposure, it is especially useful for studying the recovery of synapses. Thus, this cell culture model is suitable for studying the effects of neuroprotective agents on cell survival and synapse loss and recovery.
The non-competitive NMDA receptor antagonist dizocilpine inhibited Tat-induced neuronal death, Tat-induced synapse loss and RAP-induced synapse recovery in a concentration-dependent manner. The EC50 values for all three effects were in the 10–25 nM range, consistent with those reported previously for dizocilpine inhibition of synaptic transmission (Wong et al., 1986), and NMDA-induced cell death (Gill et al., 1987). Dizocilpine does not distinguish among NMDA receptor subtypes and while it is use-dependent (Foster and Wong, 1987; Huettner and Bean, 1988), the prolonged exposures employed in this study would effectively block all receptors in the spontaneously active network that forms in culture. Thus, we conclude from the dizocilpine experiments that NMDA receptor activity is necessary for Tat-induced cell death, and for synapse loss and recovery. Tat-induced cell death was previously shown to require NMDA receptor-mediated Ca2+ entry (Bonavia et al., 2001; Eugenin et al., 2007). Tat-induced synapse loss was also previously shown to be triggered by NMDA receptor activity (Kim et al., 2008). NMDA receptor activity is required to remodel synapses (Shi and Ethell, 2006), and can promote synaptogenesis and stabilize synapses during development in a subunit-specific manner (Gambrill and Barria, 2011). Here we showed that NMDA receptor activity was required for the recovery of synapses lost during a neurodegenerative process. That synapse recovery might follow some of the same processes responsible for synapse formation during development is plausible. Dizocilpine is highly efficacious in acute neurotoxicity models (Gill et al., 1987). However, it is poorly tolerated in humans because of psychotomimetic side effects (Muir and Lees, 1995; Manahan-Vaughan et al., 2008) consistent with inhibition of synaptic plasticity. Dizocilpine blocks long-term potentiation (Coan et al., 1987), long-term depression (Massey et al., 2004) and, as shown here, synapse loss and recovery. Thus, dizocilpine inhibits the homeostatic regulation of synapses after toxic environmental changes.
Memantine, like dizocilpine, is a non-competitive NMDA receptor antagonist. However, it is of lower potency and displays higher binding off-rate kinetics (Parsons et al., 1993; Chen and Lipton, 1997). The binding kinetics in particular are thought to confer selectivity to extrasynaptic NMDA receptors (Xia et al., 2010). The rapid kinetics spare transiently activated receptors involved in synaptic transmission while inhibiting extrasynaptic receptors preferentially activated following sustained elevation of glutamate. Extrasynaptic NMDA receptors that are thought to preferentially activate cell death processes can be activated by excess synaptic activity that produces glutamate spillover (Rusakov and Kullmann, 1998), or the tonic increase in extracellular glutamate produced by enhanced release or impaired uptake by astrocytes (Pasti et al., 2001). Sparing of synaptic NMDA receptors by memantine is consistent with our observation that it does not affect Tat-induced synapse loss but does prevent Tat-induced cell death. Reported EC50 values for memantine inhibition of NMDA-induced responses are in the low micromolar range (Chen and Lipton, 1997). However, the lower potency of memantine relative to dizocilpine cannot explain the failure of memantine to prevent Tat-induced synapse loss, because even at 100 µM, a concentration 10 times higher than that needed to prevent Tat-induced cell death, memantine failed to affect synapse loss. Memantine is approved for use in patients with Alzheimer's disease to improve cognition. The drug is well-tolerated, presumably because it spares synaptic plasticity, but is of modest efficacy. Perhaps analogues with altered binding kinetics can be developed that spare synaptic activation of NMDA receptors, but display greater neuroprotective efficacy. Our observation that memantine reversed synapse loss induced by Tat is intriguing. Clearly, the pharmacology of synapse loss and recovery has changed during the course of exposure to Tat. It is possible that the pattern of NMDA receptor activation changes following exposure to Tat such that it favours memantine binding. Alternatively, excessive activation of extrasynaptic NMDA receptors might lead to receptor internalization (Roche et al., 2001; Nong et al., 2003) changing the relative balance of extrasynaptic to synaptic NMDA receptors.
Ifenprodil exerts its selective effects via a mechanism different from that of memantine, although the functional outcome in our assays was comparable. Ifenprodil is a GluN2B-selective NMDA receptor antagonist; its affinity for GluN2B-containing NMDA receptors is 10 times higher than for GluN2A-containing NMDA receptors (Williams, 1993). Ifenprodil and dizocilpine have similar binding kinetics (Black et al., 1996). Thus, subunit selectivity is responsible for its profile of antagonism, one that is distinct from dizocilpine and similar to memantine. The preferential localization of GluN2B subunits to NMDA receptors at extrasynaptic sites (Liu et al., 2007) is consistent with the similar effects observed with memantine. Activation of GluN2B-containing NMDA receptors triggers cell death processes possibly because binding sites for death-inducing signalling molecules such as NOS are present on the carboxyl tail of GluN2B subunits (Christopherson et al., 1999). GluN2A-containing NMDA receptors are highly localized to synapses, and their activation has been shown to promote survival (Liu et al., 2007). Sparing GluN2A-containing NMDA receptors might account for the failure of ifenprodil to prevent Tat-induced synapse loss if the pro-survival GluN2A subtype of receptor mediates loss of synapses as a protective mechanism. However, the ifenprodil-evoked increase in synapses following Tat-induced loss suggests that the abundance of GluN2B-relative to GluN2A-containing NMDA receptors changes over the course of exposure to Tat. A changing pharmacological profile over the course of disease has important implications for when to administer and how to design neuroprotective drugs.
TCN201 exerted effects opposite to those elicited by the GluN2B-preferring antagonists, memantine and ifenprodil. This observation is consistent with the selectivity of TCN201 for GluN2A-containing NMDA receptors (Bettini et al., 2010). Previous reports demonstrated differential effects of GluN2A- versus GluN2B-prefering antagonists on synaptic plasticity (Ge et al., 2010) and a prominent role for NMDA receptors containing GluN2B, but not GluN2A, subunits in initiating cell death (Hardingham et al., 2002; Liu et al., 2004; 2007). GluN2A-containing NMDA receptors participate in anxiety- and depression-like behaviours (Boyce-Rustay and Holmes, 2006) suggesting that they may be useful pharmacological targets. Our results caution that drugs targeting GluN2A-containing NMDA receptors may interfere with the synaptic changes that enable neurons to adapt to synaptically driven excitotoxicity.
We hypothesize that synapse loss is a protective mechanism that enables the cell to cope with excitotoxic stimuli such as Tat by down-regulating excitatory input. Synapse loss is triggered by Ca2+ influx via NMDA receptors with subsequent activation of an ubiquitin ligase (Colledge et al., 2003; Kim et al., 2008). This pathway is separate from the one driving cell death, which also requires NMDA receptor activation but is mediated by Ca2+-dependent nNOS activation (Kim et al., 2008). Tat-induced, NO-mediated neuronal death is an apoptotic process (Kruman et al., 1998). Using the same hippocampal cultures and synapse and survival assays described here, we previously reported that inhibiting the ubiquitin-proteasome pathway with nutlin-3 prevented Tat-induced synapse loss, but not Tat-induced neuronal death. Conversely, L-NAME, an nNOS inhibitor, did not inhibit synapse loss, but prevented neuronal death induced by Tat. In the presence of nutlin-3, the concentration response curve for Tat-induced neuronal death was shifted to the left. Thus, the inhibition of PSD95 degradation by the ubiquitin proteasome pathway increases the sensitivity to Tat-induced death. The coupling of nNOS to GluN2B subunits is consistent with the ability of dizocilpine, memantine and ifenprodil, but not TCN201, to prevent Tat-induced cell death (Christopherson et al., 1999). The pro-survival role previously attributed to GluN2A-containing NMDA receptors is consistent with the idea that synaptic activity drives synapse loss by activation of GluN2A-containing NMDA receptors and that this loss improves survival. We speculate that Tat and NMDA receptor antagonists shift the delicate balance between survival and full integration into synaptic circuits.
Linking GluN2B subunits to NOS-mediated death and GluN2A to the ubiquitin-proteasome pathways and synapse loss does not adequately explain the recovery of synapses induced by application of memantine or ifenprodil after Tat-induced synapse loss. The recovery of synapses induced by a block of GluN2B-containing NMDA receptors suggests that activation of these receptors suppresses new synapse formation. The failure of these drugs to affect Tat-induced synapse loss when given before Tat and the induced recovery produced by their late application, suggest that the composition of functional NMDA receptors changes during exposure to Tat. NMDA receptors internalize following sustained activation (Roche et al., 2001; Nong et al., 2003) and synapse loss clearly reduces NMDA receptor levels. The mechanism by which these drugs can induce recovery has yet to be elucidated. Indeed, the mechanism by which RAP can induce synapse recovery is not yet known; RAP is classified as a competitive antagonist for LRP, but this mechanism alone cannot explain how synapse recovery occurs at a time point when Tat is presumably already internalized.
Conclusions
We propose that memantine and ifenprodil, by inhibiting a select subset of NMDA receptors as compared with the broad actions of dizocilpine, display several properties desirable for neuroprotective drugs. They improve survival by preventing Tat-induced activation of cell death pathways. Memantine and ifenprodil did not interfere with Tat-induced synapse loss, which allows a potentially beneficial reduction in excitatory input. Surprisingly, these drugs induced the recovery of synapses lost during exposure to Tat. These findings may be broadly applicable to other neurodegenerative processes with an excitotoxic component.
Acknowledgments
The National Institute on Drug Abuse (grants DA07304 and DA11806) and the National Science Foundation (grant IOS0814549) supported this work. National Institute on Drug Abuse Training Grant (grant DA07234) supported AS.
Glossary
- DMEM
Dulbecco's modified Eagles media
- HAND
HIV-associated neurocognitive disorders
- HIV-1
human immunodeficiency virus type 1
- LRP
low-density lipoprotein receptor-related protein
- PI
propidium iodide
- PSD95
post-synaptic density protein 95
- RAP
receptor-associated protein
- Tat
transactivator of transcription
- TBS
tris-buffered saline
Conflicts of interest
None.
References
- Arnold DB, Clapham DE. Molecular determinants for subcellular localization of PSD-95 with an interacting K+ channel. Neuron. 1999;23:149–157. doi: 10.1016/s0896-6273(00)80761-8. [DOI] [PubMed] [Google Scholar]
- Avenet P, Leonardon J, Besnard F, Graham D, Frost J, Depoortere H, et al. Antagonist properties of the sterioisomers of ifenprodil at NR1A/NR2A and NR1A/NR2B subtypes of the NMDA receptor expressed in Xenopos oocytes. Eur J Pharmacol. 1996;296:209–213. doi: 10.1016/0014-2999(95)00700-8. [DOI] [PubMed] [Google Scholar]
- Bettini E, Sava A, Griffante C, Carignani C, Buson A, Capelli AM, et al. Identification and characterization of novel NMDA receptor antagonists selective for NR2A- over NR2B-containing receptors. J Pharmacol Exp Ther. 2010;335:636–644. doi: 10.1124/jpet.110.172544. [DOI] [PubMed] [Google Scholar]
- Black M, Lanthorn T, Small D, Mealing G, Lam V, Morley P. Study of potency, kinetics of block and toxicity of NMDA receptor antagonists using fura-2. Eur J Pharmacol. 1996;317:377–381. doi: 10.1016/s0014-2999(96)00730-3. [DOI] [PubMed] [Google Scholar]
- Bonavia R, Bajetto A, Barbero S, Albini A, Noonan DM, Schettini G. HIV-1 Tat causes apoptotic death and calcium homeostasis alterations in rat neurons. Biochem Biophys Res Commun. 2001;288:301–308. doi: 10.1006/bbrc.2001.5743. [DOI] [PubMed] [Google Scholar]
- Boyce-Rustay JM, Holmes A. Genetic inactivation of the NMDA receptor NR2A subunit has anxiolytic- and antidepressant-like effects in mice. Neuropsychopharmacology. 2006;31:2405–2414. doi: 10.1038/sj.npp.1301039. [DOI] [PubMed] [Google Scholar]
- Bruce-Keller AJ, Chauhan A, Dimayuga FO, Gee J, Keller JN, Nath A. Synaptic transport of human immunodeficiency virus-Tat protein causes neurotoxicity and gliosis in rat brain. J Neurosci. 2003;23:8417–8422. doi: 10.1523/JNEUROSCI.23-23-08417.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HSV, Lipton SA. Mechanism of memantine block of NMDA-activated channels in rat retinal ganglion cells: uncompetitive antagonism. J Physiol. 1997;499:27–46. doi: 10.1113/jphysiol.1997.sp021909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopherson KS, Hillier BJ, Lim WA, Bredt DS. PSD-95 assembles a ternary complex with the N-methyl-D-aspartic acid receptor and a bivalent neuronal NO synthase PDZ domain. J Biol Chem. 1999;274:27467–27473. doi: 10.1074/jbc.274.39.27467. [DOI] [PubMed] [Google Scholar]
- Coan EJ, Saywood W, Collingridge GL. MK-801 blocks NMDA receptor-mediated synaptic transmission and long term potentiation in rat hippocampals slices. Neurosci Lett. 1987;80:111–114. doi: 10.1016/0304-3940(87)90505-2. [DOI] [PubMed] [Google Scholar]
- Colledge M, Snyder EM, Crozier RA, Soderling JA, Jin Y, Langeberg LK, et al. Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron. 2003;40:595–607. doi: 10.1016/s0896-6273(03)00687-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou H, Ellison B, Bradley J, Kasiyanov A, Poluektova LY, Xiong H, et al. Neuroprotective mechanisms of lithium in murine human immunodeficiency virus-1 encephalitis. J Neurosci. 2005;25:8375–8385. doi: 10.1523/JNEUROSCI.2164-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis R, Langford D, Masliah E. HIV and antiretroviral therapy in the brain: neuronal injury and repair. Nat Rev Neurosci. 2007;8:33–44. doi: 10.1038/nrn2040. [DOI] [PubMed] [Google Scholar]
- Eugenin EA, King JE, Nath A, Calderon TM, Zukin RS, Bennett MV, et al. HIV-tat induces formation of an LRP-PSD-95- NMDAR-nNOS complex that promotes apoptosis in neurons and astrocytes. Proc Natl Acad Sci U S A. 2007;104:3438–3443. doi: 10.1073/pnas.0611699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everall IP, Heaton RK, Marcotte TD, Ellis RJ, McCutchan JA, Atkinson JH, et al. Cortical synaptic density is reduced in mild to moderate human immunodeficiency virus neurocognitive disorder. HNRC Group. HIV Neurobehavioral Research Center. Brain Pathol. 1999;9:209–217. doi: 10.1111/j.1750-3639.1999.tb00219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitting S, Xu R, Bull C, Buch S, El-Hage N, Nath A, et al. Interactive comorbidity between opioid drug abuse and HIV-1 Tat: chronic exposure augments spine loss and sublethal dendritic pathology in striatal neurons. Am J Pathol. 2010;177:1397–1410. doi: 10.2353/ajpath.2010.090945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster AC, Wong EHF. The novel anticonvulsant MK-801 binds to the activated state of the N-methyl-D-aspartate receptor in rat brain. Br J Pharmacol. 1987;91:403–409. doi: 10.1111/j.1476-5381.1987.tb10295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambrill AC, Barria A. NMDA receptor subunit composition controls synaptogenesis and synapse stabilization. Proc Natl Acad Sci U S A. 2011;108:5855–5860. doi: 10.1073/pnas.1012676108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y, Dong Z, Bagot RC, Howland JG, Phillips AG, Wong TP, et al. Hippocampal long-term depression is required for the consolidation of spatial memory. Proc Natl Acad Sci U S A. 2010;107:16697–16702. doi: 10.1073/pnas.1008200107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genis P, Jett M, Bernton EW, Boyle T, Gelbard HA, Dzenko K, et al. Cytokines and arachidonic metabolites produced during human immunodeficiency virus (HIV)-infected macrophage interactions: implications for the neuropathogenesis of HIV disease. J Exp Med. 1992;176:1703–1718. doi: 10.1084/jem.176.6.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill R, Foster AC, Woodruff G. Systemic administration of MK-801 protects against ischemia-induced hippocampal neurodegeneration in the gerbil. J Neurosci. 1987;7:3343–3349. doi: 10.1523/JNEUROSCI.07-10-03343.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Hudson L, Liu J, Nath A, Jones M, Raghavan R, Narayan O, et al. Detection of the human immunodeficiency virus regulatory protein tat in CNS tissues. J Neurovirol. 2000;6:145–155. doi: 10.3109/13550280009013158. [DOI] [PubMed] [Google Scholar]
- Huettner JE, Bean BP. Block of N-methyl-D-aspartate-activated current by the anticonvulsant MK-801: selective binding to open channels. Proc Natl Acad Sci U S A. 1988;85:1307–1311. doi: 10.1073/pnas.85.4.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hult B, Chana G, Masliah E, Everall I. Neurobiology of HIV. Int Rev Psychiatry. 2008;20:3–13. doi: 10.1080/09540260701862086. [DOI] [PubMed] [Google Scholar]
- Kaul M, Lipton SA. Mechanisms of neuronal injury and death in HIV-1 associated dementia. Curr HIV Res. 2006;4:307–318. doi: 10.2174/157016206777709384. [DOI] [PubMed] [Google Scholar]
- Kim BO, Liu Y, Ruan Y, Xu ZC, Schantz L, He JJ. Neuropathologies in transgenic mice expressing human immunodeficiency virus type 1 Tat protein under the regulation of the astrocyte-specific glial fibrillary acidic protein promoter and doxycycline. Am J Pathol. 2003;162:1693–1707. doi: 10.1016/S0002-9440(10)64304-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Thayer SA. Lithium increases synapse formation between hippocampal neurons by depleting phosphoinositides. Mol Pharmacol. 2009;75:1021–1030. doi: 10.1124/mol.108.052357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Martemyanov KA, Thayer SA. Human immunodeficiency virus protein Tat induces synapse loss via a reversible process that is distinct from cell death. J Neurosci. 2008;28:12604–12613. doi: 10.1523/JNEUROSCI.2958-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Shin AH, Thayer SA. Activation of cannabinoid type 2 receptors inhibits HIV-1 envelope glycoprotein gp120-induced synapse loss. Mol Pharmacol. 2011;80:357–366. doi: 10.1124/mol.111.071647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King JE, Eugenin EA, Buckner CM, Berman JW. HIV tat and neurotoxicity. Microbes Infect. 2006;8:1347–1357. doi: 10.1016/j.micinf.2005.11.014. [DOI] [PubMed] [Google Scholar]
- Kruman II, Nath A, Mattson MP. HIV-1 protein Tat induces apoptosis of hippocampal neurons by a mechanism involving caspase activation, calcium overload, and oxidative stress. Exp Neurol. 1998;154:276–288. doi: 10.1006/exnr.1998.6958. [DOI] [PubMed] [Google Scholar]
- Letendre SL, Woods SP, Ellis RJ, Atkinson JH, Masliah E, van den Brande G, et al. Lithium improves HIV-associated neurocognitive impairment. AIDS. 2006;20:1885–1888. doi: 10.1097/01.aids.0000244208.49123.1b. [DOI] [PubMed] [Google Scholar]
- Li ST, Matsushita M, Moriwaki A, Saheki Y, Lu YF, Tomizawa K, et al. HIV-1 Tat inhibits long-term potentiation and attenuates spatial learning. Ann Neurol. 2004;55:362–371. doi: 10.1002/ana.10844. [DOI] [PubMed] [Google Scholar]
- Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, et al. Role of NMDA Receptor Subtypes in Governing the Direction of Hippocampal Synaptic Plasticity. Science. 2004;304:1021–1024. doi: 10.1126/science.1096615. [DOI] [PubMed] [Google Scholar]
- Liu Y, Jones M, Hingtgen CM, Bu G, Laribee N, Tanzi RE, et al. Uptake of HIV-1 tat protein mediated by low-density lipoprotein receptor-related protein disrupts the neuronal metabolic balance of the receptor ligands. Nat Med. 2000;6:1380–1387. doi: 10.1038/82199. [DOI] [PubMed] [Google Scholar]
- Liu Y, Wong TP, Aarts M, Rooyakkers A, Liu L, Lai TW, et al. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J Neurosci. 2007;27:2846–2857. doi: 10.1523/JNEUROSCI.0116-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manahan-Vaughan D, von Haebler D, Winter C, Juckel G, Heinemann U. A single application of MK801 causes symptoms of acute psychosis, deficits in spatial memory, and impairment of synaptic plasticity in rats. Hippocampus. 2008;18:125–134. doi: 10.1002/hipo.20367. [DOI] [PubMed] [Google Scholar]
- Maragos WF, Tillman P, Jones M, Bruce-Keller AJ, Roth S, Bell JE, et al. Neuronal injury in hippocampus with human immunodeficiency virus transactivating protein, Tat. Neuroscience. 2003;117:43–53. doi: 10.1016/s0306-4522(02)00713-3. [DOI] [PubMed] [Google Scholar]
- Masliah E, Heaton RK, Marcotte TD, Ellis RJ, Wiley CA, Mallory M, et al. Dendritic injury is a pathological substrate for human immunodeficiency virus-related cognitive disorders. Ann Neurol. 1997;42:963–972. doi: 10.1002/ana.410420618. [DOI] [PubMed] [Google Scholar]
- Massey PV, Johnson BE, Moult PR, Auberson YP, Brown MW, Molnar E, et al. Differential roles of NR2A and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression. J Neurosci. 2004;24:7821–7828. doi: 10.1523/JNEUROSCI.1697-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArthur JC. HIV dementia: an evolving disease. J Neuroimmunol. 2004;157:3–10. doi: 10.1016/j.jneuroim.2004.08.042. [DOI] [PubMed] [Google Scholar]
- Minagar A, Commins D, Alexander JS, Hoque R, Chiappelli F, Singer EJ, et al. NeuroAIDS: characteristics and diagnosis of the neurological complications of AIDS. Mol Diagn Ther. 2008;12:25–43. doi: 10.1007/BF03256266. [DOI] [PubMed] [Google Scholar]
- Muir KW, Lees KR. Clinical experience with excitatory amino acid antagonist drugs. Stroke. 1995;26:503–513. doi: 10.1161/01.str.26.3.503. [DOI] [PubMed] [Google Scholar]
- Nong Y, Huang YQ, Ju W, Kalia LV, Ahmadian G, Wang YT, et al. Glycine binding primes NMDA receptor internalization. Nature. 2003;422:302–307. doi: 10.1038/nature01497. [DOI] [PubMed] [Google Scholar]
- Parsons CG, Gruner R, Rozental J, Millar J, Lodge D. Patch clamp studies on the kinetics and selectivity of N-methyl-D-aspartate receptor antagonism by memantine (1-amino-3,5-dimethyladamantan) Neuropharmacology. 1993;32:1337–1350. doi: 10.1016/0028-3908(93)90029-3. [DOI] [PubMed] [Google Scholar]
- Pasti L, Zonta M, Pozzan T, Vicini S, Carmignoto G. Cytosolic calcium oscillations in astrocytes may regulate exocytotic release of glutamate. J Neurosci. 2001;21:477–484. doi: 10.1523/JNEUROSCI.21-02-00477.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisberg B, Doody R, Stoffler A, Schmitt F, Ferris S, Mobius HJ. Memantine in moderate-to-severe Alzheimer's disease. N Engl J Med. 2003;348:1333–1341. doi: 10.1056/NEJMoa013128. [DOI] [PubMed] [Google Scholar]
- Roche KW, Standley S, McCallum J, Ly CD, Ehlers MD, Wenthold RJ. Molecular determinants of NMDA receptor internalization. Nat Neurosci. 2001;4:794–802. doi: 10.1038/90498. [DOI] [PubMed] [Google Scholar]
- Rusakov DA, Kullmann DM. Extrasynaptic glutamate diffusion in the hippocampus – ultrastructural constraints, uptake, and receptor activation. J Neurosci. 1998;18:3158–3170. doi: 10.1523/JNEUROSCI.18-09-03158.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sa MJ, Madeira MD, Ruela C, Volk B, Mota-Miranda A, Paula-Barbosa MM. Dendritic changes in the hippocampal formation of AIDS patients: a quantitative Golgi study. Acta Neuropathol. 2004;107:97–110. doi: 10.1007/s00401-003-0781-3. [DOI] [PubMed] [Google Scholar]
- Shen M, Thayer SA. The cannabinoid agonist Win55,212-2 inhibits calcium channels by receptor-mediated and direct pathways in cultured rat hippocampal neurons. Brain Res. 1998;783:77–84. doi: 10.1016/s0006-8993(97)01195-5. [DOI] [PubMed] [Google Scholar]
- Shi Y, Ethell IM. Integrins control dendritic spine plasticity in hippocampal neurons through NMDA receptor and Ca2+/calmodulin-dependent protein kinase II-mediated actin reorganization. J Neurosci. 2006;26:1813–1822. doi: 10.1523/JNEUROSCI.4091-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speth C, Stockl G, Mohsenipour I, Wurzner R, Stoiber H, Lass-Florl C, et al. Human immunodeficiency virus type 1 induces expression of complement factors in human astrocytes. J Virol. 2001;75:2604–2615. doi: 10.1128/JVI.75.6.2604-2615.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci. 1999;19:4180–4188. doi: 10.1523/JNEUROSCI.19-10-04180.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tozzi V, Balestra P, Serraino D, Bellagamba R, Corpolongo A, Piselli P, et al. Neurocognitive impairment and survival in a cohort of HIV-infected patients treated with HAART. AIDS Res Hum Retroviruses. 2005;21:706–713. doi: 10.1089/aid.2005.21.706. [DOI] [PubMed] [Google Scholar]
- Waataja JJ, Kim HJ, Roloff AM, Thayer SA. Excitotoxic loss of post-synaptic sites is distinct temporally and mechanistically from neuronal death. J Neurochem. 2008;104:364–375. doi: 10.1111/j.1471-4159.2007.04973.x. [DOI] [PubMed] [Google Scholar]
- Wiley CA, Baldwin M, Achim CL. Expression of regulatory and structural mRNA in the central nervous system. AIDS. 1996;10:943–947. doi: 10.1097/00002030-199607000-00007. [DOI] [PubMed] [Google Scholar]
- Wiley CA, Masliah E, Achim CL. Measurement of CNS HIV burden and its association with neurologic damage. Adv Neuroimmunol. 1999;4:319–325. doi: 10.1016/s0960-5428(06)80272-x. [DOI] [PubMed] [Google Scholar]
- Williams K. Ifenprodil discriminates subtypes of the N-methyl-D-aspartate receptor – selectivity and mechanisms at recombinant heteromeric receptors. Mol Pharmacol. 1993;44:851–859. [PubMed] [Google Scholar]
- Wong EHF, Kemp JA, Priestley T, Knight AR, Woodruff GN, Iverson LL. The anticonvulsant MK-801 is a potent N-methyl-D-aspartate antagonist. Proc Natl Acad Sci U S A. 1986;83:7104–7108. doi: 10.1073/pnas.83.18.7104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia P, Chen HSV, Zhang D, Lipton SA. Memantine preferentially blocks extrasynaptic over synaptic NMDA receptor currents in hippocampal autapses. J Neurosci. 2010;30:11246–11250. doi: 10.1523/JNEUROSCI.2488-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]