

Abstract
BACKGROUND AND PURPOSE
Prostaglandin E2 (PGE2) stimulation of the G protein-coupled prostanoid EP1 receptor was found to up-regulate the expression of Nur-related factor 1 (Nurr1) (NR4A2), a transcription factor in the NR4A subfamily of nuclear receptors. The present studies characterize the molecular mechanism of this up-regulation.
EXPERIMENTAL APPROACH
The expression of Nurr1 was examined by immunoblot analysis, the polymerase chain reaction and reporter gene assays in human embryonic kidney (HEK) cells stably expressing the recombinant EP1 receptor and in SH-SY5Y neuroblastoma cells expressing endogenous EP1 receptors. Signalling pathway inhibitors were used to examine the roles of Rho, PKA, the cAMP response element binding protein (CREB) and NF-κB on the PGE2 stimulated up-regulation of Nurr1. CREB and NF-κB signalling were also examined by immunoblot analysis and reporter gene assays.
KEY RESULTS
The EP1 receptor mediated up-regulation of Nurr1 was blocked with inhibitors of Rho, PKA, NF-κB and CREB; but PGE2 failed to significantly stimulate intracellular cAMP formation. PGE2 stimulation of the EP1 receptor induced the phosphorylation and activation of CREB and NF-κB, which could be blocked by inhibition of PKA.
CONCLUSIONS AND IMPLICATIONS
PGE2 stimulation of the human EP1 receptor up-regulates the expression of Nurr1 by a mechanism involving the sequential activation of the Rho, PKA, CREB and NF-κB signalling pathways. EP1 receptors are implicated in tumorigenesis and the up-regulation of Nurr1 may underlie the anti-apoptotic effects of PGE2.
Keywords: EP1 prostanoid receptor, G-protein coupled receptors, Nurr1, NR4A2, orphan nuclear receptors, prostaglandin E2, PGE2, PKA, CREB, NF-κB
Introduction
Prostaglandin E2 (PGE2) is one of the major products of the COX pathway that exerts its actions through the activation of four different E-type prostaglandin receptors (EP). These receptors are members of the G protein-coupled family of receptors and are designated EP1, EP2, EP3 and EP4, (receptor nomenclature follows Alexander et al., 2011) and are the products of four separate genes that have evolved to subserve the activation of unique signal transduction pathways leading to the regulation of different physiological responses (Coleman et al., 1994; Regan, 2003; Sugimoto and Narumiya, 2007). Classically, these receptors were distinguished by their differential coupling to G proteins and their effects on smooth muscle. Thus, EP1 receptors were generally acknowledged to couple to Gq resulting in the activation of Ca2+ signalling and the contraction of smooth muscle. The EP2 and EP4 receptors, on the other hand, were associated with coupling to Gs, resulting in the stimulation of intracellular cAMP formation and smooth muscle relaxation. EP3 receptors were initially recognized for coupling to Gi, resulting in the inhibition of intracellular cAMP formation and smooth muscle contraction.
It is now appreciated that the EP receptor subtypes can couple to multiple G proteins leading to the activation of diverse, but nevertheless unique, signalling networks. For example, the EP3 receptors were found to undergo extensive alternative mRNA splicing resulting in isoforms that could differentially couple to Gi and Gs and Gq (Kotani et al., 1997). Although EP2 receptors to date have only been shown to couple to Gs, they can also activate β-catenin dependent Tcf/Lef signalling in addition to activating a traditional cAMP/protein kinase A (PKA) pathway (Fujino et al., 2002). EP4 receptors can activate β-catenin-dependent Tcf/Lef signalling as well (Fujino et al., 2002), but in contrast to the EP2 receptors, the EP4 receptors do this through coupling to Gi and the activation of a PI3K signalling pathway (Fujino and Regan, 2006).
We have recently found that EP1 receptors can couple to Gi in addition to coupling to Gq (Ji et al., 2010). Coupling of the EP1 receptor to Gi results in the activation of a PI3K, Akt and mTOR signalling cascade leading to the up-regulation of hypoxia-inducible factor-1α (HIF-1α). This up-regulation of HIF-1α occurred under normoxic conditions and involved increased translation rather than increased transcription. We further showed that the EP1 receptor-mediated up-regulation of HIF-1α in HepG2 hepatocellular carcinoma cells was associated with the up-regulation of mRNA-encoding lymphatic vascular endothelial growth factor-C (VEGF-C). These and other findings (Huang et al., 2005; Han et al., 2006) strongly suggest that the EP1 receptor-mediated up-regulation of HIF-1α and VEGF-C could be driving tumour angiogenesis and metastasis in hepatocellular carcinoma.
EP1 receptors have also been implicated in the development and/or progression of colon, lung, breast and skin cancer (Chell et al., 2006; Fulton et al., 2006). Interestingly, while the role of EP1 receptors in various epithelial cell cancers suggest anti-apoptotic effects and the promotion of cell survival, in the central nervous system (CNS), the activation of EP1 receptors by PGE2 can induce apoptosis and mediate neurotoxicity. For example, genetic ablation or pharmacological blockade of the EP1 receptor in mice dramatically decreased excitotoxic and ischemia-induced brain injury (Ahmad et al., 2006; Kawano et al., 2006). Similarly, in rabbits, pharmacological blockade of the EP1 receptor significantly decreased the extent of brain damage caused by intraventricular haemorrhage and was associated with decreased apoptosis and neuronal degeneration (Vinukonda et al., 2010). In primary cultures of rat mesencephalic neurons, stimulation of the EP1 receptor selectively killed dopaminergic neurons and pharmacological blockade of the EP1 receptor protected dopaminergic neurons against 6-hydroxydopamine-induced neurotoxicity (Carrasco et al., 2007). Although inhibition of the Na+–Ca2+ exchanger has been implicated in the excitotoxic brain injury caused by EP1 receptor activation (Kawano et al., 2006), the molecular mechanisms that could potentially explain EP1 receptor-mediated neuronal degeneration and apoptosis are unknown.
Recently, using DNA microarray analysis, we discovered that PGE2 treatment of HEK cells stably expressing the recombinant human EP1 receptor resulted in a strong up-regulation of Nurr1 (XB Chen and JW Regan, unpublished). Nurr1, also known as NR4A2, is a transcription factor in the NR4A subfamily of nuclear receptors, which includes two additional members, Nur77 and Nor-1 (Pearen and Muscat, 2010). These so-called orphan receptors are interesting because they activate gene expression in a constitutive ligand-independent manner, and thus, are essentially proxies for those factors that can up-regulate their expression. In the present case, the up-regulation of Nurr1 and activation of Nurr1-dependent transcriptional activity is controlled by PGE2 signalling through the EP1 receptor. The NR4A receptor subfamily function as immediate early response genes and have a wide variety of physiological and pathophysiological actions (Zhao and Bruemmer, 2010). Nurr1 itself is essential for the development and maintenance of midbrain dopaminergic neurons and has anti-apoptotic actions in colon (Holla et al., 2006), cervical (Ke et al., 2004) and squamous cell carcinomas (Shigeishi et al., 2011).
The purpose of this study was to understand the signalling mechanisms responsible for the up-regulation of Nurr1 by the human EP1 prostanoid receptor. We have found that PGE2 stimulation of the EP1 receptor up-regulates the expression of Nurr1 by a cAMP-independent activation of PKA, cAMP response element binding protein (CREB) and nuclear factor-kappa B (NF-κB). Additionally, this up-regulation of Nurr1 involves the activation of Rho signalling, which is likely to occur through the coupling of the EP1 receptor to G12/13. These findings provide new knowledge concerning the functional regulation of Nurr1 by PGE2 and have implications for understanding the role of prostanoid EP1 receptors in cancer and neuronal cell death.
Methods
Cell culture
HEK cells stably expressing the recombinant human EP1 prostanoid receptor (HEK-EP1) or expressing the empty vector (HEK-pCEP4) were prepared as previously described (Ji et al., 2010). The cells were maintained in DMEM supplemented with 10% fetal bovine serum (FBS), 250 µg·mL−1 geneticin, 100 µg·mL−1 gentamicin and 200 µg·mL−1 hygromycin B. A human neuroblastoma cell line (SH-SY5Y) was obtained from the American Type Culture Collection (Manassas, VA, USA) and cultured in Eagle's minimal essential medium/F12 containing 10% FBS. The cells were cultured in a humidified incubator at 37°C in 5% CO2/95% air.
Immunoblotting
The cells were plated at a density of 106 cells per well in six-well plates and incubated overnight at 37°C. The cells were then incubated with PGE2 as described in the figure legends, and immunoblotting was performed as previously described (Ji et al., 2010). Briefly, whole cell lysates were prepared and ∼50 µg of protein was analysed by electrophoresis on 10% SDS-PAGE gels and transferred to PVDF membranes. Following an overnight incubation at 4°C with the primary antibodies, the membranes were incubated for 1 h at room temperature with the secondary antibodies. Antibodies against phospho-CREB/phospho-ATF-1, phospho-I-κB, phospho-NF-κB and Nurr1 were used at a final dilution of 1:1000 in 3% non-fat milk. Horseradish peroxidase-conjugated secondary antibodies were used at a final dilution of 1:10 000 in 3% non-fat milk. Following incubation with the secondary antibodies, the membranes were washed and immunoreactivity was detected by enhanced chemiluminescence. To ensure equal loading of proteins, the membranes were stripped and re-probed with antibodies to either vinculin or ERK 1/2.
Luciferase reporter gene assay
Cells were split into six-well plates and grown to ∼75% confluence. Approximately 24 h later, the cells were co-transfected with 2 µg/well of a firefly luciferase reporter plasmid (pGL3/HRE-Luc27) under the control of a Nurr1 binding response element (NBRE) or a cAMP response element (CRE) or a NF-κB response element and 10 ng per well of the Renilla luciferase reporter, pRL-CMV, using 5 µL FuGENE-HD. Approximately 18 h later, the cells were treated with either vehicle (0.1% dimethyl sulfoxide in phosphate-buffered saline solution) or 1 µM PGE2. The next day, cell lysates were prepared and 2 µL were used to measure luciferase activity using the Dual Luciferase Reporter Assay System according to the manufacturer's instructions. The data were normalized by calculating ratios of firefly luciferase scores to the corresponding Renilla luciferase values.
Quantitative real-time PCR (qPCR)
qPCR was performed as previously described (Ji et al., 2010) using TaqMan Gene Expression Assay primers for Nurr1 (assay ID no. Hs00428691_m1) from Applied Biosystems (Foster City, CA, USA). PCR reactions were composed of 40 cycles of 95°C for 15 s and 60°C for 45 s in an ABI 7500 thermal cycler (Applied Biosystems, Foster City, CA, USA). Threshold values (Ct) were determined automatically by the system software and relative mRNA expression was analysed by the comparative ΔΔCt method. Data were normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH).
cAMP assay
The cells were cultured in six-well plates as above, and 16 h before the assay, the regular culture medium was replaced with Opti-MEM containing 250 µg·mL−1 geneticin and 100 µg·mL−1 gentamicin. Following a 15 min pretreatment with 3-isobutyl-1-methylxanthine (IBMX; 0.1 mg·mL−1), the cells were incubated with either vehicle (0.1% dimethyl sulfoxide) or 1 µM PGE2 for 10 min at 37°C. The media were removed and the cells were placed on ice in preparation for the cAMP assay, which was performed as previously described (Fujino and Regan, 2006).
Data analysis
Unless otherwise stated, data are expressed as the means ± S.E.M. Differences between means were analysed by one-way ANOVA, followed by Bonferroni post test using GraphPad Prism version 4.00 for Windows (San Diego, CA). P values less than 0.05 were taken as showing significant differences between means.
Materials
Trizol® Reagent, Dulbecco's modified Eagle's medium (DMEM), Eagle's minimum essential medium/F12 (MEM/F12), Opti-MEM, hygromycin B, geneticin and gentamicin were from Invitrogen (Carlsbad, CA, USA). Absolutely RNA Miniprep kit was from Strategene (La Jolla, CA, USA). Antibodies against Nurr1, phospho-IκBα protein (Ser32/36; product no. sc-101713) were from Santa Cruz (San Jose, CA, USA). Anti-mouse IgG conjugated with horseradish peroxidase, vinculin antibodies, IBMX and PKA were from Sigma-Aldrich (St Louis, MO, USA). iScript cDNA kit, PVDF membranes and Transfectin reagent were from Bio-Rad Laboratories (Hercules, CA, USA). Cell lysis buffer and antibodies against phospho-CREB/phospho-activating transcription factor-1 (ATF-1) (Ser133; product no. 9191), phospho-PKAc (Thr197; product no. 4781), phospho-NF-κB p65 (Ser276; product no. 3037) were from Cell Signaling Technology (Waltham, MA, USA). PGE2 was from Cayman Chemical Company (Ann Arbor, MI, USA). 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid-acetoxymethyl ester (BAPTA/AM), bisindolylmaleimide I (BIM), H89, protein kinase A inhibitor (PKI) and C3 toxin were from Calbiochem (San Diego, CA, USA). FuGENE 6 was from Roche (San Francisco, CA, USA). The Dual Luciferase Reporter Assay System and the Renilla luciferase reporter (pRL-CMV) were from Promega (Madison, WI, USA). [3H]cAMP was from PerkinElmer Life & Analytical Sciences (Boston, MA, USA).
Results
Up-regulation of Nurr1 mRNA and protein expression by PGE2 in HEK cells stably expressing the human EP1 receptor
Using DNA microarray analysis, we had previously found that mRNA encoding the orphan nuclear receptor Nurr1 (NR4A2) was strongly up-regulated by PGE2 stimulation of HEK cells stably expressing the recombinant human EP1 receptor (XB Chen and JW Regan, unpublished observations). qPCR analysis and immunoblotting were therefore used to examine the time course and concentration response of Nurr1 expression following the treatment of HEK-EP1 cells with PGE2. As shown in Figure 1A, there was a strong induction of Nurr1 mRNA expression within 1 h of treatment with 1 µM PGE2, which decreased but was still elevated over pretreatment levels after 6 h. Figure 1B shows that Nurr1 protein expression was strongly induced after 3 and 6 h of treatment with 1 µM PGE2 and that it was less but still clearly elevated over pretreatment levels after 12 h. Figure 1C shows the concentration-dependent response of the up-regulation of Nurr1 protein expression following treatment of HEK-EP1 cells with either vehicle or 10−9−10−5 M PGE2 for 3 h. As compared with treatment with vehicle, there was already a significant up-regulation of Nurr1 expression at 10−9 M PGE2. Indeed, treatment with 10−9 M PGE2 induced roughly half the maximal expression of Nurr1 observed at 10−5 M PGE2, which compares favourably to the binding of PGE2 to HEK-EP1 cells (IC50= 3.6 nM) or to the stimulation of inositol phosphates formation by PGE2 in these cells (EC50= 4.8 nM; Ji et al., 2010). These findings indicate that PGE2 stimulation of the human EP1 receptor involves an initial increase in Nurr1 gene transcription followed by increased translation and up-regulation of Nurr1 protein expression.
Figure 1.
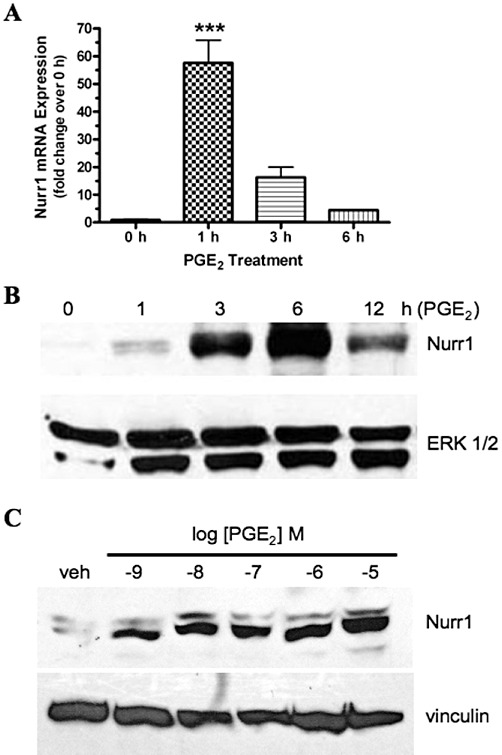
Time course for the PGE2-stimulated up-regulation of Nurr1 mRNA (A) and concentration-response (B) and time course for the protein expression (C) of Nurr1 in HEK cells stably expressing the human EP1 receptor. (A) HEK-EP1 cells were incubated with 1 µM PGE2 at 37°C for the indicated times and then RNA was isolated and used for quantitative real-time PCR with primers specific for either Nurr1 or GAPDH. Data were analysed by the comparative ΔΔCt method, relative to the expression of GAPDH. Data are the means ± SEM (n= 4) of the pooled data from two independent experiments each done in duplicate. ***P < 0.001; compared with time 0; one-way anova, followed by Bonferroni post-test. (B) HEK-EP1 cells were incubated with 1 µM PGE2 at 37°C for the indicated times and were subjected to immunoblot analysis using antibodies against human Nurr1 or ERK 1/2 as described in the methods section. (C) HEK-EP1 cells were incubated with either vehicle (veh) or the indicated concentrations of PGE2 for 3 h at 37°C and were subjected to immunoblot analysis using antibodies against human Nurr1 or vinculin. Immunoblots are representative from one of three independent experiments.
The up-regulation of Nurr1 mediated by the EP1 receptor involves the activation of NF-κB and CREB
It has been previously reported that in synovial tissue from patients with rheumatoid arthritis, pro-inflammatory mediators can up-regulate the expression of Nurr1 by increased transcription involving interactions of NF-κB and CREB with the proximal promoter of the Nurr1 gene (McEvoy et al., 2002). The family of NF-κB proteins are transcription factors that bind to NF-κB response elements in target genes (Hayden and Ghosh, 2008; Vallabhapurapu and Karin, 2009). The binding of inhibitory subunits, referred to as I-κB, regulates NF-κB activity. In the classical pathway of activation, the phosphorylation of I-κB leads to the dissociation of the NF-κB/I-κB complex and translocation of NF-κB to the nucleus. We therefore examined the activation of NF-κB-mediated transcriptional activity and the phosphorylation of I-κB following the treatment of HEK-EP1 cells with PGE2. As shown in Figure 2A, treatment of HEK-EP1 cells with 1 µM PGE2 strongly stimulated NF-κB responsive luciferase reporter gene activity but had no effect in control HEK cells stably transfected with empty vector (HEK-pCEP4). In Figure 2B, immunoblot analysis was used to examine the phosphorylation of I-κB following treatment of HEK-EP1 cells with 1 µM PGE2 from 0–12 h. The results show increased phosphorylation of I-κB as early as 1 h following treatment of the cells with PGE2. The PGE2-induced phosphorylation of I-κB was maximal at 3 h and was still evident following 12 h of treatment. PGE2 stimulation of the human EP1 receptor can therefore activate NF-κB transcriptional activity by a mechanism involving the phosphorylation of I-κB.
Figure 2.
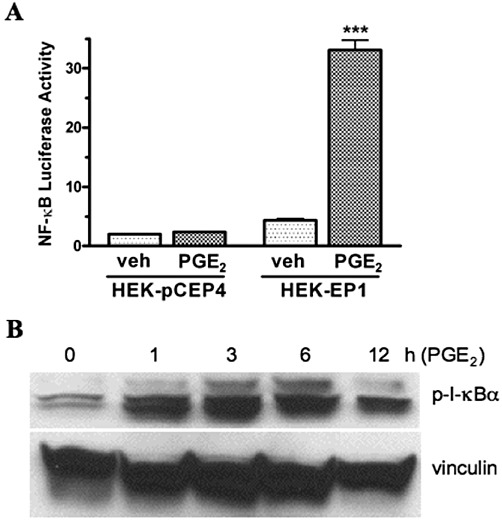
PGE2-stimulated NF-κB responsive luciferase reporter gene activity (A) and time course of phosphorylation of I-κBα (B) in HEK cells stably transfected with the human EP1 receptor (HEK-EP1). (A) HEK-EP1 cells or HEK cells stably transfected with empty vector (HEK-pCEP4) were transiently transfected with an NF-κB luciferase reporter plasmid as described in the methods section and ∼18 h later, were treated either vehicle (veh) or 1 µM PGE2 at 37°C. Luciferase activity was determined the next day. Data are the means ± SEM of triplicate measurements from a representative experiment that was repeated three times. ***P < 0.001 compared with the corresponding vehicle treatment; one-way anova, followed by Bonferroni post-test. (B) HEK-EP1 cells were treated with 1 µM PGE2 for the indicated times at 37°C and lysates were prepared and subjected to immunoblot analysis with antibodies against either phospho-I-κBα (p-I-κBα) or vinculin. A representative immunoblot is shown from one of three independent experiments.
Next, we examined the potential involvement of NF-κB and CREB signalling in the PGE2-stimulated up-regulation of Nurr1 by the EP1 receptor. Figure 3A shows an immunoblot for the PGE2-stimulated expression of Nurr1 in HEK-EP1 cells under control conditions or following pretreatment with the NF-κB inhibitor, BAY 11–7082 or dominant negative CREB. It is evident that under control conditions, treatment of cells with 1 µM PGE2 for 3 h caused a robust up-regulation of Nurr1 expression, whereas following pretreatment with either BAY 11–7082 or dominant negative CREB, the PGE2-stimulated up-regulation of Nurr1 was completely blocked.
Figure 3.
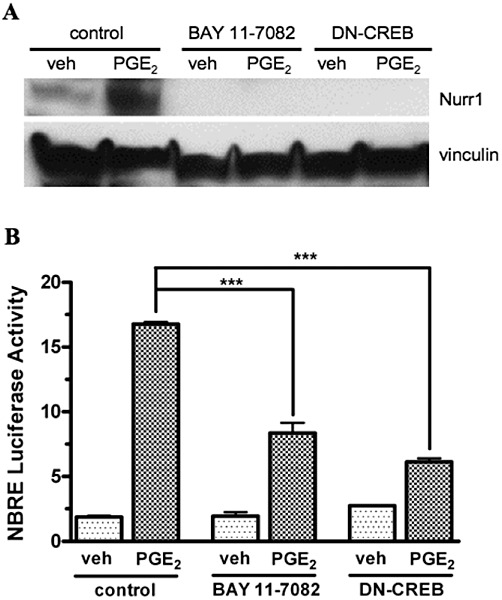
The effects of the NF-κB inhibitor, BAY 11–7082, and dominant negative CREB (DN-CREB) on PGE2-stimulated Nurr1 protein expression (A) and on PGE2-stimulated Nurr1 responsive (NBRE) luciferase reporter gene activity (B) in HEK cells stably expressing the human EP1 receptor. (A) HEK-EP1 cells were transiently transfected with DN-CREB ∼18 h prior to the experiment or were pretreated the day of the experiment with either vehicle (control) or 10 µM BAY 11–7082 for 30 min at 37°C, followed by treatment with either vehicle (veh) or 1 µM PGE2 for 3 h at 37°C. Lysates were prepared and subjected to immunoblot analysis using antibodies against either Nurr1 or vinculin. A representative immunoblot is shown from one of three independent experiments. (B) HEK-EP1 cells were transiently transfected either alone with a NBRE luciferase reporter plasmid or together with a plasmid encoding DN-CREB as described in the methods section and ∼18 h later were pretreated with either vehicle (control) or 10 µM BAY 11–7082 for 30 min at 37°C, followed by treatment with either vehicle (veh) or 1 µM PGE2. Luciferase activity was determined the next day. Data are the means ± SEM of quadruplicate measurements from a representative experiment that was repeated three times. ***P < 0.001; one-way anova, followed by Bonferroni post-test.
A luciferase reporter gene under the control of a NBRE was further used to examine the role of NF-κB and CREB on the PGE2-stimulated up-regulation of Nurr1 by the EP1 receptor. Figure 3B shows PGE2-stimulated NBRE-luciferase reporter activity in HEK-EP1 cells under control conditions or following pretreatment with BAY 11–7082 or dominant negative CREB. It can be seen that under control conditions, treatment of the cells with 1 µM PGE2 produced a nine-fold induction of luciferase transcriptional activity, reflecting the activation of the NBRE following the PGE2 mediated up-regulation of Nurr1. However, following pretreatment of the cells with the NF-κB inhibitor, BAY 11–7082, the PGE2 stimulation of luciferase reporter activity was inhibited by ∼50%; and following pretreatment with dominant negative CREB, the PGE2 stimulation of luciferase activity was decreased by ∼60%.
The CREB family of proteins are also transcription factors that produce their actions through binding to CREs in target genes (Mayr and Montminy, 2001). Classically, the transcriptional activity of CREB is induced after phosphorylation of Ser133 by cAMP-dependent protein kinase (PKA). However, a number of other kinases may also phosphorylate Ser133 leading to the activation of CREB. To further explore the role of CREB in EP1 receptor signalling, we investigated the activation of CREB transcriptional activity and the PKA-dependent phosphorylation of CREB in HEK-EP1 cells treated with PGE2. Figure 4A shows that the transcriptional activity of a CREB responsive luciferase reporter gene was stimulated nearly 20-fold in HEK-EP1 cells by PGE2, whereas in control HEK cells, treatment with PGE2 produced a two-fold increase that was not statistically significant.
Figure 4.
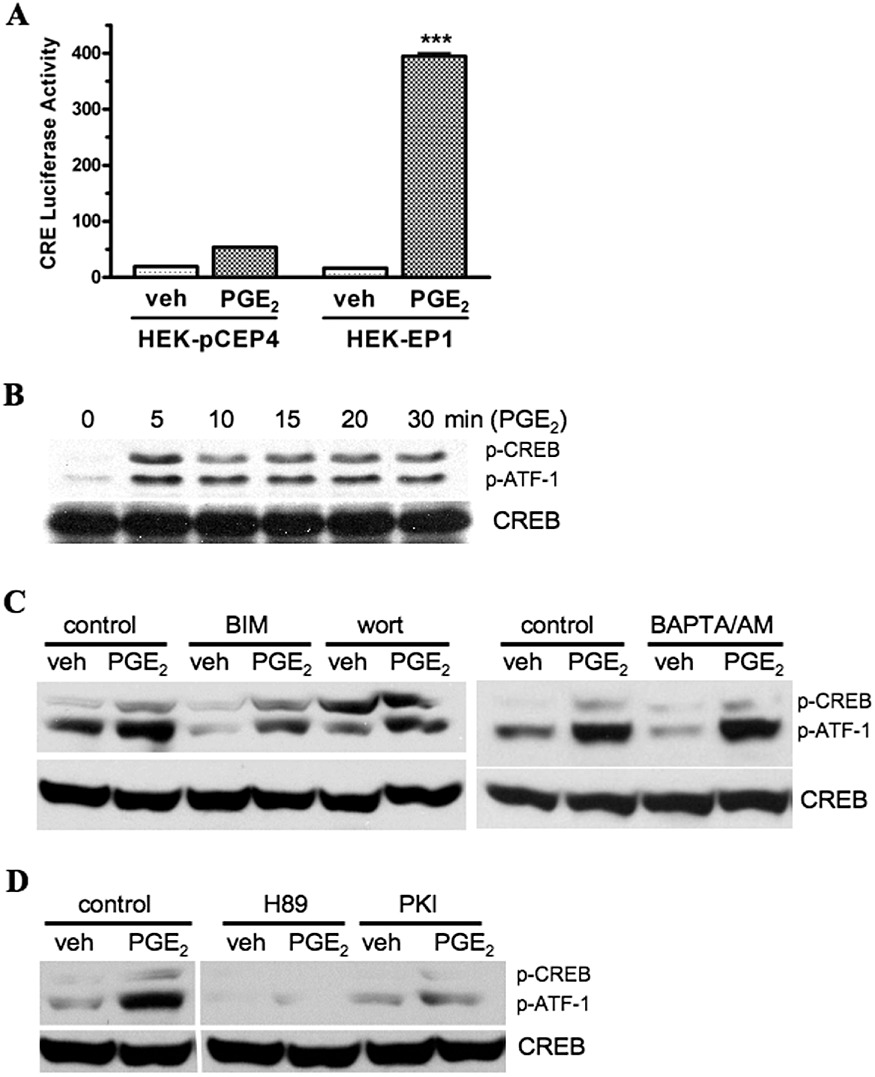
PGE2-stimulated CREB responsive (CRE) luciferase reporter gene activity (A) and PGE2-stimulated phosphorylation of CREB and ATF-1 either alone or following pretreatment with various signalling pathway inhibitors (B, C, D) in HEK cells stably transfected with the human EP1 receptor (HEK-EP1). (A) HEK-EP1 cells or HEK cells stably transfected with empty vector (HEK-pCEP4) were transiently transfected with a CREB responsive (CRE) luciferase reporter plasmid and ∼18 h later were treated either vehicle (veh) or 1 µM PGE2 at 37°C. Luciferase activity was determined the next day. Data are the means ± SEM of quadruplicate measurements from a representative experiment that was repeated three times. ***P < 0.001 compared with the corresponding vehicle treatment; one-way anova, followed by Bonferroni post-test. (B) HEK-EP1 cells were treated with 1 µM PGE2 for indicated times at 37°C and lysates were prepared and subjected to immunoblot analysis with antibodies against either phospho-CREB/ATF-1 (p-CREB/p-ATF-1) or CREB. (C, D) HEK-EP1 cells were pretreated for 30 min 37°C with either vehicle (control), or 10 µM of the PKC inhibitor, bisindolylmaleimide I (BIM), or 10 µM of the PI3K inhibitor wortmannin (wort), or 10 µM of the Ca2+ signalling inhibitor, BAPTA/AM, or 10 and 5 µM of the PKA inhibitors, H89 and PKI, respectively. They were then treated with either vehicle (veh) or 1 µM PGE2 for 5 min at 37°C. Lysates were prepared and subjected to immunoblot analysis as in panel B. Shown are representative immunoblots that were repeated at least three times for each antibody and condition.
In Figure 4B, C and D, immunoblot analysis was used to examine the PGE2-induced phosphorylation of CREB in HEK-EP1 cells under control conditions or following pretreatment with various signalling pathway inhibitors. As shown in Figure 4B, both CREB and its closely related family member, activating transcription factor-1 (ATF-1), were rapidly phosphorylated after treatment with 1 µM PGE2. Figure 4C shows neither BIM, wortmannin, nor BAPTA/AM, which respectively inhibit PKC, PI3K and Ca2+ signalling, had little effect on the PGE2 stimulated phosphorylation of either CREB or ATF-1 as compared with the controls. In contrast, Figure 4D shows that pretreatment of HEK-EP1 cells with PKA inhibitors, H89 or PKI markedly reduced the PGE2-stimulated phosphorylation of CREB and ATF-1. These results suggest that PKA is the primary kinase responsible for the PGE2-stimulated phosphorylation and activation of CREB by the human EP1 receptor.
Nurr1 up-regulation by the EP1 receptor involves the activation of PKA but is independent of cAMP formation
As noted in the introduction, agonist stimulation of the EP1 receptor leads to the activation of Ca2+ signalling, although it is not associated with a robust activation of inositol phosphates turnover, nor with any changes in intracellular cAMP formation. Nonetheless, our findings suggested a possible induction of cAMP formation given the PKA-dependent phosphorylation of CREB demonstrated previously in Figure 4. We therefore examined the formation of intracellular cAMP in HEK-EP1 cells in response to treatment with PGE2, as well as the potential involvement of PKA in the up-regulation of Nurr1 expression. Using HEK cells stably expressing the cAMP-stimulatory EP2 receptor (HEK-EP2) as a positive control, Figure 5A shows that 1 µM PGE2 strongly stimulated intracellular cAMP formation in HEK-EP2 cells but had no effect on the formation of intracellular cAMP in either control HEK cells or HEK-EP1 cells.
Figure 5.
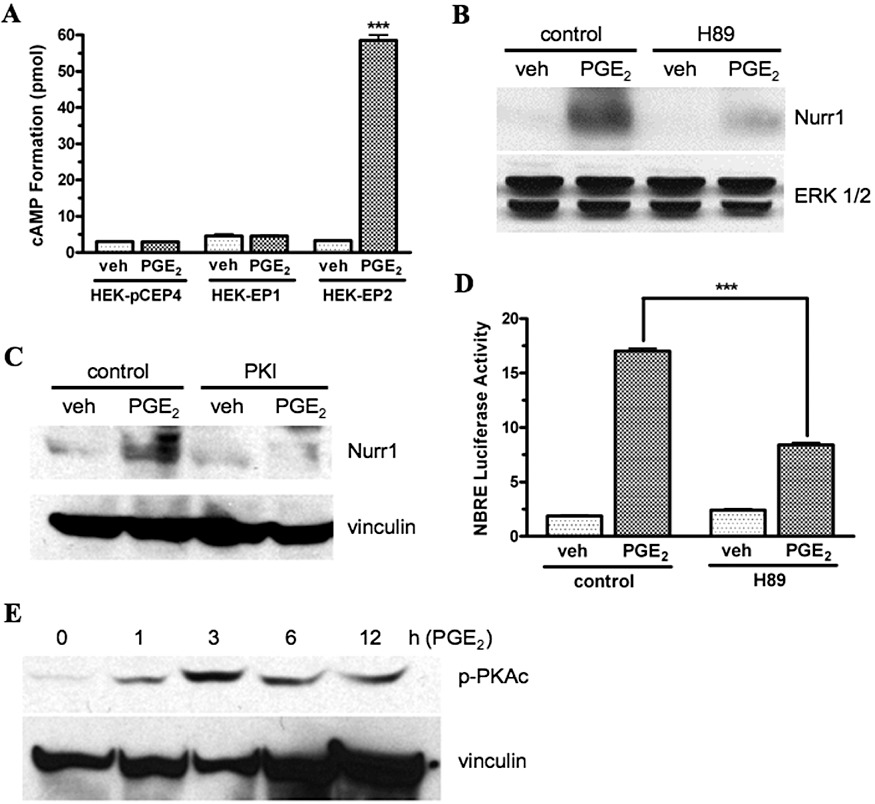
PGE2-stimulated cAMP formation (A); effects of the PKA inhibitors, H89 and PKI on PGE2-stimulated Nurr1 protein expression (B, C) and on Nurr1-stimulated (NBRE) luciferase reporter gene activity (D); and PGE2-stimulated phosphorylation of the catalytic subunit of PKA (E) in HEK cells stably transfected with the human EP1 receptor (HEK-EP1). (A) HEK-EP1 cells, HEK cells stably transfected with empty vector (HEK-pCEP4), and HEK cells stably transfected with the cAMP stimulatory EP2 receptor (HEK-EP2), were treated with either vehicle (veh) or 1 µM PGE2 at 37°C for 60 min and intracellular cAMP formation was measured. Data are the means ± SEM of duplicate measurements from a representative experiment that was repeated three times. ***P < 0.001 compared with the corresponding vehicle treatment; one-way anova, followed by Bonferroni post-test. (B, C) HEK-EP1 cells were pretreated with vehicle (control) or 10 µM H89 or 5 µM PKI, for 30 min 37°C and were then treated with either vehicle (veh) or 1 µM PGE2 for 3 h. Lysates were prepared and subjected to immunoblot analysis using antibodies against either Nurr1, ERK 1/2 or vinculin. Shown are representative immunoblots that were repeated at least three times for each antibody and condition. (D) HEK-EP1 cells were transfected with a NBRE luciferase reporter plasmid and ∼18 h later were pretreated for 30 min 37°C with either vehicle (control) or 10 µM H89 followed by treatment with either vehicle (veh) or 1 µM PGE2. Luciferase activity was determined the next day. Data are the means ± SEM of quadruplicate measurements from a representative experiment that was repeated three times. ***P < 0.001; one-way anova, followed by Bonferroni post-test. (E) HEK-EP1 cells were treated with 1 µM PGE2 for indicated times at 37°C and lysates were prepared and subjected to immunoblot analysis with antibodies against either the phosphorylated catalytic subunit of PKA (p-PKAc) or vinculin. A representative immunoblot is shown from one of three independent experiments.
The PKA inhibitors, H89 and PKI, were then used to examine the potential involvement of PKA in the PGE2-mediated up-regulation of Nurr1 by the EP1 receptor. The immunoblots shown in Figure 5B and C, respectively, show that pretreatment of HEK-EP1 cells with either H89 or PKI blocked the PGE2-stimulated up-regulation of Nurr1 as compared with the controls. Using a Nurr1 responsive luciferase reporter gene, Figure 5D shows that pretreatment of HEK-EP1 cells with H89 also inhibited the PGE2 stimulation of Nurr1 transcriptional activity by 50%.
The phosphorylation of Thr197 in the catalytic subunit of PKA (PKAc) is associated with the activation of PKA (Steinberg et al., 1993). Thus, using immunoblot analysis, we examined the potential phosphorylation of PKAc in HEK-EP1 cells after treatment with 1 µM PGE2. Figure 5E shows that treatment of HEK-EP1 cells with 1 µM PGE2 resulted in a time-dependent phosphorylation of PKAc that was maximal at 3 h. Together, these data indicate that the up-regulation of Nurr1 following PGE2 stimulation of the human EP1 receptor involves an activation of PKA that does not require the induction of intracellular cAMP formation.
Agonist stimulation of the EP1 receptor results in the phosphorylation of the p65 subunit of NF-κB, which involves the activation of PKA
In addition to its association with the regulatory subunit of PKA, the catalytic subunit of PKA (PKAc) is known to exist in an inactive complex with I-κB and NF-κB (Zhong et al., 1997; Gambaryan et al., 2010). Following the phosphorylation of I-κB, this complex dissociates yielding active PKAc and NF-κB. Active PKAc can then phosphorylate the p65 subunit of NF-κB at Ser276, which increases the transcriptional activity of NF-κB (Zhong et al., 1997). Consequently, we analysed the phosphorylation of the p65 subunit of NF-κB in whole cell lysates from HEK-EP1 cells after treatment with PGE2 as well as the potential role of PKAc in this process. In Figure 6A and B, immunoblot analysis was used to determine the phosphorylation of the p65 subunit of NF-κB under control conditions or following pretreatment with the PKA inhibitors H89 and PKI. Figure 6A shows that the p65 subunit of NF-κB was phosphorylated within 1 h following treatment of HEK-EP1 cells with 1 µM PGE2 and was maintained for at least 6 h. Figure 6B shows that the PGE2-stimulated phosphorylation of p65 in HEK-EP1 cells was almost completely blocked by pretreatment with either H89 or PKI. Using a NF-κB responsive luciferase reporter gene, Figure 6C shows that pretreatment of HEK-EP1 cells with H89 decreased PGE2-stimulated NF-κB transcriptional activity by approximately 70%, compared with the control cells. Together, these data show that agonist stimulation of the human EP1 receptor results in a PKA-dependent increase in the phosphorylation and transcriptional activity of NF-κB.
Figure 6.
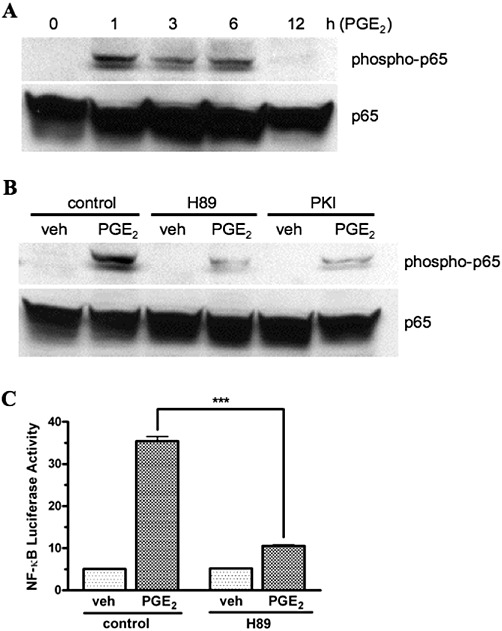
Time course of the PGE2-stimulated phosphorylation of the p65 subunit of NF-κB (A) and effects of the PKA inhibitors, H89 and PKI on the PGE2-stimulated phosphorylation of p65 (B) and on PGE2-stimulated NF-κB responsive luciferase reporter gene activity (C) in HEK cells stably transfected with the human EP1 receptor (HEK-EP1). (A) HEK-EP1 cells were treated with 1 µM PGE2 for indicated times at 37°C and whole cell lysates were prepared and subjected to immunoblot analysis with antibodies against either phospho-p65 or total p65. (B) HEK-EP1 cells were pretreated with vehicle (control) or 10 µM H89 or 5 µM PKI for 30 min 37°C and were then treated with either vehicle (veh) or 1 µM PGE2 for 3 h. Lysates were prepared and subjected to immunoblot analysis as in panel A. Shown are representative immunoblots that were repeated at least three times for each antibody and condition. (C) HEK-EP1 cells were transiently transfected with a NF-κB luciferase reporter plasmid and ∼18 h later were pretreated for 30 min 37°C with either vehicle (control) or 10 µM H89 followed by treatment with either vehicle (veh) or 1 µM PGE2. Luciferase activity was determined the next day. Data are the means ± SEM of triplicate measurements from a representative experiment that was repeated three times. ***P < 0.001; one-way anova, followed by Bonferroni post-test.
Up-regulation of Nurr1 by the EP1 receptor involves PGE2-mediated activation of RhoA
Stimulation of the PI3K and Rho signalling pathways are among the mechanisms known to induce the phosphorylation of I-κB and activation of NF-κB transcriptional activity (Perona et al., 1997; Vallabhapurapu and Karin, 2009). We have previously shown that agonist stimulation of the human EP1 receptor can up-regulate the expression of HIF-1α by a mechanism involving coupling to Gi and activation of a PI3K, Akt and mTOR signalling cascade (Ji et al., 2010). It is therefore possible that the activation of NF-κB signalling that is necessary for the PGE2-stimulated up-regulation of Nurr1 is through the EP1 receptor-mediated activation of PI3K signalling. However, pretreatment of HEK-EP1 cells with the Gi inhibitor, Pertussis toxin, had no effect on the PGE2-stimulated up-regulation of Nurr1, suggesting that the activation of NF-κB, which precedes the up-regulation of Nurr1, does not involve the activation of PI3K (data not shown). Furthermore, Figure 4C shows that the PI3K inhibitor, wortmannin, did not block the PGE2-mediated phosphorylation of CREB, which is also required for the PGE2-stimulated up-regulation of Nurr1 by the EP1 receptor.
C3 exoenzyme, which is a potent inhibitor of RhoA, was then used to examine the potential role of Rho signalling in the up-regulation of Nurr1 by the EP1 receptor. Figure 7A and B, respectively, show immunoblots for the PGE2-stimulated expression of Nurr1 and phospho-I-κB following pretreatment of HEK-EP1 cells with C3 toxin. In Figure 7A, pretreatment of HEK-EP1 cells with C3 toxin completely blocked the PGE2-stimulated up-regulation of Nurr1 as compared with control. Figure 7B shows that pretreatment of HEK-EP1 cells with C3 toxin also decreased the PGE2-induced phosphorylation of I-κB. Together, these results suggest a mechanism for the PGE2-stimulated up-regulation of Nurr1 that involves sequential activation of the EP1 receptor, Rho, phosphorylation of I-κB, dissociation and activation of PKAc from the I-κB/NF-κB/PKAc complex, phosphorylation of NF-κB and CREB by PKAc and transcriptional up-regulation of Nurr1 through the combined actions of phospho-NF-κB and phospho-CREB.
Figure 7.
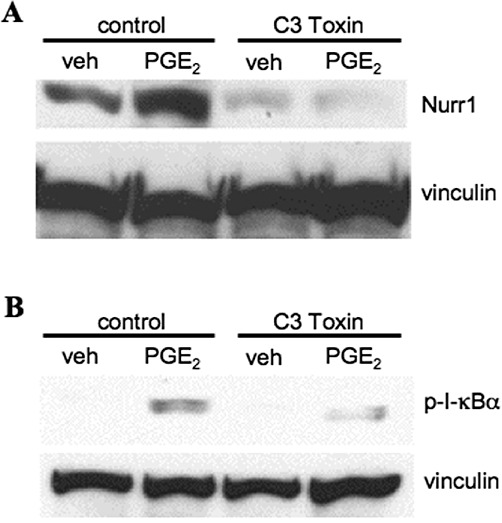
The effects of the Rho inhibitor, C3 toxin, on PGE2-stimulated Nurr1 protein expression (A) and the phosphorylation of I-κBα (B) in HEK cells stably transfected with the human EP1 receptor (HEK-EP1). (A, B) HEK-EP1 cells were pretreated with either vehicle (control) or 5 µM C3 toxin overnight and were then incubated with either vehicle (veh) or 1 µM PGE2 for 3 h at 37°C. Lysates were prepared and subjected to immunoblot analysis with antibodies against either Nurr1, phospho-I-κBα (p-I-κBα) or vinculin. Representative immunoblots are shown that were repeated at least three times for each antibody and condition.
PGE2 up-regulates Nurr1 protein and mRNA expression in SH-SY5Y neuroblastoma cells through activation of the EP1 receptor
We have previously documented the expression of EP1 receptor mRNA in SH-SY5Y human neuroblastoma cells (Ji et al., 2010). Nurr1 is also expressed in these cells and has been shown to be up-regulated by stimulation of the dopamine D3 receptors (Pan et al., 2005). We, therefore, decided to examine the possible up-regulation of Nurr1 by PGE2 through the activation of endogenous EP1 receptors in SH-SY5Y cells. Figure 8A shows an immunoblot for the PGE2-stimulated expression of Nurr1 in SH-SY5Y cells either under control conditions or following pretreatment with the EP1 receptor antagonist SC-51322. The data show that the expression of Nurr1 increased at 3 h and 6 h after treatment with 1 µM PGE2 and that this up-regulation of Nurr1 expression could be blocked by pretreatment with SC-51322. The PGE2-stimulated expression of Nurr1 mRNA in SH-SY5Y cells was also examined by qPCR and the results are shown in Figure 8B. Nurr1 mRNA expression was strongly induced as early as 1 h after treatment with 1 µM PGE2 and was maintained up to 6 h.
Figure 8.
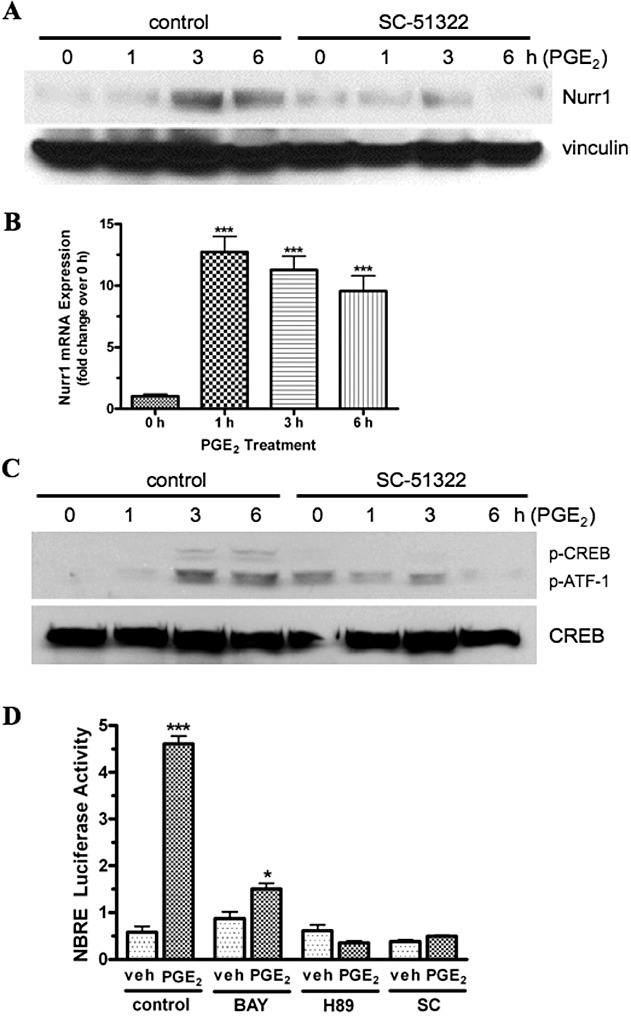
PGE2-stimulated Nurr1 protein expression (A) and mRNA expression (B); PGE2-stimulated phosphorylation of CREB and ATF-1 (C); and PGE2-stimulated Nurr1 responsive (NBRE) luciferase reporter gene activity (D) in SH-SY5Y neuroblastoma cells in the presence and absence of the EP1 receptor antagonist, SC-51322 and various signalling pathway inhibitors. (A) SH-SY5Y cells were pretreated with either vehicle (control) or 1 µM SC-51322 for 30 min at 37°C and were then incubated with 1 µM PGE2 for the indicated times. Lysates were prepared and subjected to immunoblot analysis using antibodies against Nurr1 or vinculin. (B) SH-SY5Y cells were incubated with 1 µM PGE2 at 37°C for the indicated times and then RNA was isolated and used for quantitative real-time PCR with primers specific for either Nurr1 or GAPDH as described in the methods section. Data were analysed by the comparative ΔΔCt method, relative to the expression of GAPDH. Data are the means ± SEM (n= 4) of the pooled data from two independent experiments, each in duplicate. (C) SH-SY5Y cells were pretreated with either vehicle (control) or 1 µM SC-51322 for 30 min at 37°C and were then incubated with 1 µM PGE2 for indicated times. Lysates were prepared and subjected to immunoblot analysis with antibodies against either phospho-CREB/ATF-1 (p-CREB/p-ATF-1) or CREB. (D) SH-SY5Y cells were transiently transfected with an NBRE luciferase reporter plasmid and ∼18 h later were pretreated with vehicle (control) or 10 µM of the NF-κB inhibitor BAY 11–7082 (BAY) or 10 µM of the PKA inhibitor H89 or 1 µM SC-51322 (SC) for 30 min at 37°C, followed by treatment with either vehicle (veh) or 1 µM PGE2. Luciferase activity was determined the next day. Data are the means ± SEM of quadruplicate measurements from a representative experiment that was repeated three times. ***P < 0.001; *P < 0.05 compared with time 0 or with the corresponding vehicle control; one-way anova, followed by Bonferroni post-test. Immunoblots are representative from one of at least three independent experiments.
PGE2 stimulation of the EP1 receptor in SH-SY5Y neuroblastoma cells induces CREB/ATF-1 phosphorylation and increases Nurr1 transcriptional activity that is dependent upon the activation of NF-κB and PKA. Figure 8C shows an immunoblot for the PGE2-stimulated expression of phospho-CREB and phospho-ATF-1 in SH-SY5Y cells either under control conditions of following pretreatment with the EP1 receptor antagonist, SC-51322. After 3 and 6 h of treatment with PGE2, there was an up-regulation of the expression of phospho-CREB and phospho-ATF-1 that was decreased by pretreatment with SC-51322. A Nurr1 responsive (NBRE) luciferase reporter gene was used to examine PGE2-stimulated Nurr1 transcriptional activity in SH-SY5Y cells either under control conditions or following pretreatment with various signalling pathway inhibitors, and the results are shown in Figure 8D. Under control conditions, incubation of SH-SY5Y cells with 1 µM PGE2 resulted in a nearly eight-fold increase in Nurr1 transcriptional activity. This PGE2-stimulated increase in Nurr1 transcriptional activity was decreased ∼80% following pretreatment with the NF-κB inhibitor, BAY 11–7082, and it was completed blocked by pretreatment with the PKA inhibitor, H89. Pretreatment with the EP1 receptor antagonist, SC-51322, also completely blocked the PGE2-stimulated up-regulation of Nurr1 transcriptional activity in SH-SY5Y cells. Together, these data show that activation of endogenous EP1 receptors in SH-SY5Y cells by PGE2 can up-regulate the expression of Nurr1 by a transcriptional mechanism that involves the activation of NF-κB and PKA.
Discussion
As depicted in Figure 9, the prostanoid EP1 receptor has been classically thought to couple to Gq/11 to activate Ca2+ signalling and smooth muscle contraction. While there is no doubt regarding the activation of Ca2+ signalling, it is unclear if this is mediated by conventional activation of phospholipase Cβ, as the stimulation of inositol phosphates formation by the EP1 receptor is modest as compared with other receptors that are known to couple to Gq/11 (see Sugimoto and Narumiya, 2007). We have recently shown that the human EP1 receptor also couples to Gi to activate a PI3K, Akt and mTOR signalling cascade that results in the up-regulation of HIF-1α (Ji et al., 2010). We now report that PGE2 stimulation of the human EP1 receptor can up-regulate the expression of the orphan nuclear receptor, Nurr1, both in HEK cells stably expressing the EP1 receptor and in SH-SY5Y human neuroblastoma cells expressing endogenous EP1 receptors. Figure 9 shows that the mechanism of this up-regulation involves the activation of Rho followed by a novel cAMP-independent activation of PKAc. Thus, PGE2 stimulation of the EP1 receptor activates Rho, leading to the phosphorylation of I-κB and subsequent dissociation and activation of PKAc from an I-κB/NF-κB/PKAc complex. Activated PKAc then phosphorylates NF-κB and CREB, resulting in the transcriptional up-regulation of Nurr1 mRNA and protein expression, as well as increased Nurr1-mediated transcriptional activity. It is generally recognized that receptors that couple to Gq/11 frequently couple to G12/13, which in turn are well known to activate Rho signalling pathways (Kurose, 2003; Juneja and Casey, 2009). It seems likely, therefore, that the activation of Rho by the EP1 receptor involves coupling to G12/13. In addition to the up-regulation of Nurr1, the activation of Rho, PKA, NF-κB and CREB are signalling properties that have not been previously ascribed to agonist stimulation of the EP1 receptor.
Figure 9.
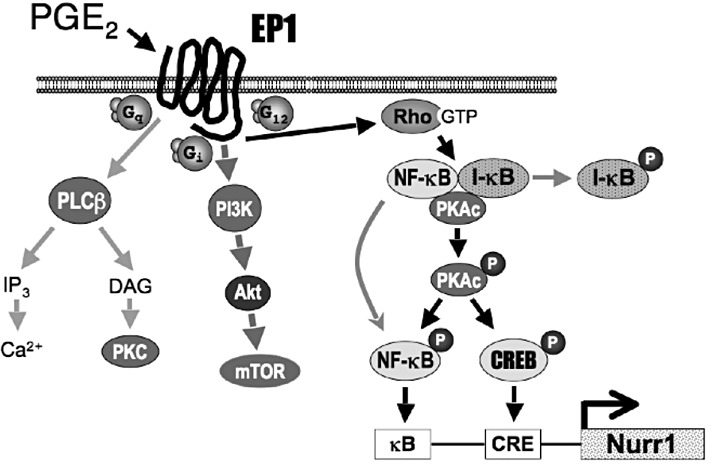
Model for the signalling pathways activated by the human prostanoid EP1 receptor. PGE2 stimulation of the EP1 receptor has typically been associated with the activation of Ca2+ signalling through coupling to Gq and the activation of phopholipase-Cβ (PLCβ). Recently, PGE2 stimulation of the EP1 receptor has been shown to result in coupling to Gi and activation of a PI3K, PKB (Akt) and mammalian target of rapamycin (mTOR) signalling cascade (Ji et al., 2010). The present study shows that PGE2 stimulation of the EP1 receptor can up-regulate the expression of Nurr1 through the sequential activation of Rho, phosphorylation of I-κB, dissociation and activation of the catalytic subunit of PKA (PKAc) from an IκB/NF-κB/PKAc complex, followed by PKAc-dependent phosphorylation and activation of NF-κB and CREB.
The up-regulation of Nurr1 by PGE2 was first described in colon cancer cells (Holla et al., 2006) and later in rheumatoid arthritis synovial tissue (McEvoy et al., 2002); however, neither study identified the nature of the specific EP receptor subtype mediating this response. Furthermore, although both studies suggested that this up-regulation involved the activation of cAMP/PKA, neither study measured PGE2-stimulated cAMP formation so the actual role of cAMP was unclear. Nevertheless, Holla et al. (2006) clearly established a role for PKA by showing that the PKA inhibitor, H-89, completely blocked the PGE2-stimulated up-regulation of Nurr1. Importantly, these authors also demonstrated that the anti-apoptotic effects of PGE2 on colon cancer cells could be blocked with a dominant negative construct of Nurr1, suggesting that ability of PGE2 to protect cells from undergoing apoptosis is achieved through the up-regulation of Nurr1.
The importance of NF-κB and CREB in the transcriptional up-regulation of Nurr1 in synovial tissue was clearly delineated by McEvoy et al., (2002). They found that the up-regulation of Nurr1 by IL-1β and TNF-α involved the binding of NF-κB to a consensus site in the proximal promoter of the Nurr1 gene. Likewise, the up-regulation of Nurr1 by PGE2 required the binding of CREB to a CRE slightly downstream of the NF-κB binding site. In contrast to the present findings, however, the two sites appeared to function independently of one another; that is, activation of either site increased Nurr1 transcriptional activity, and the activities were additive. We have observed that the up-regulation of Nurr1 by PGE2 in HEK cells requires the activation of both NF-κB and CREB, which can be provided entirely by agonist stimulation of the EP1 receptor. McEvoy et al., (2002) found that stimulation of intracellular cAMP formation by forskolin increased the expression of Nurr1 via CREB activation; but their data also show that the effects of forskolin were markedly less than those produced by PGE2, suggesting the involvement of additional signalling pathways in the up-regulation of Nurr1 by PGE2.
Activation of the thromboxane A2 receptor (TP) has recently been shown to up-regulate the expression of Nurr1 in human lung cancer cells (Li and Tai, 2009). This up-regulation of Nurr1 was brought about through the activation of PKA and CREB, combined with activation of the PKC and MAPK/ERK pathways. Li and Tai (2009) also found that stimulation of intracellular cAMP formation with forskolin up-regulated the expression of Nurr1, but since they did not measure intracellular cAMP formation, it is not known if the TP receptor-mediated activation of PKA is cAMP-dependent or not. Li and Tai (2009), however, did provide evidence that NF-κB was not involved in the up-regulation of Nurr1 by the TP receptor. These authors also found that PGE2 stimulated the up-regulation of Nurr1 in some lung cancer cell lines but not others. In responsive cell lines, the PGE2-stimulated up-regulation of Nurr1 was blocked by the PKA inhibitor H-89, but in contrast to TP receptor activation, the up-regulation of Nurr1 by PGE2 was not affected by inhibition of the MAPK/ERK pathway. Because of the involvement of PKA, Li and Tai (2009) attributed the up-regulation of Nurr1 by PGE2 to possible activation of the cAMP stimulatory EP2 and/or EP4 receptors. Interestingly, however, the two lung cancer cell lines that did not respond to PGE2 did not appear to express mRNA encoding the EP1 receptor, whereas they did express mRNA encoding either the EP2 or EP4 receptors, suggesting that the up-regulation of Nurr1 by PGE2 in the responsive cell lines was mediated by activation of EP1 receptors.
Pathophysiological involvement of the EP1 receptor has been implicated in the development and/or progression of colon, lung, breast and skin cancer (Chell et al., 2006; Fulton et al., 2006). Our finding of the up-regulation of Nurr1 by the EP1 receptor provides a potential mechanism that is consistent with this role. Thus, Nurr1 has been shown to have anti-apoptotic effects in colon cancer cells (Holla et al., 2006), as well as in cervical carcinoma cells where it also increases anchorage-independent growth (Ke et al., 2004). The anti-apoptotic effects of Nurr1 appear to involve an interaction with p53 that inhibits its transcriptional activity and is associated with decreased expression of the pro-apoptotic protein, Bax (Zhang et al., 2009). We have previously shown that PGE2 stimulation of the human EP1 receptor up-regulates the expression of HIF-1α, a transcription factor that has also been associated with tumour angiogenesis and metastasis (Ji et al., 2010). Thus, pathophysiological activation of the EP1 receptor could promote tumorigenesis by potentially up-regulating the expression of both Nurr1 and HIF-1α.
The ability of Nurr1 to promote cell survival is perhaps best recognized in the CNS where the expression of Nurr1 is essential for the development and maintenance of midbrain dopaminergic neurons (Zetterstrom et al., 1997) and has been associated with neuroprotection (Volakakis et al., 2010). For example, genetic ablation of Nurr1 in the adult mouse brain resulted in a progressive loss of tyrosine hydroxylase activity and dopamine that is reminiscent of the changes seen in Parkinson's disease (Kadkhodaei et al., 2009). Similarly, Nurr1 expression in astrocytes and microglia has been shown to protect dopaminergic neurons from cell death by suppressing the production of pro-inflammatory neurotoxic mediators (Saijo et al., 2009). It is puzzling, therefore, why activation of EP1 receptors in the CNS is strongly associated with neurotoxicity (reviewed in the Introduction) when our present findings would suggest that the up-regulation of Nurr1 by the EP1 receptor would have neuroprotective effects in the brain.
In short, we do not have a good answer to this potential paradox except that EP1 receptor activation in vivo may simply not up-regulate the expression of Nurr1 in the brain. Another possibility is that activation of the EP1 receptor in the brain may up-regulate the expression of the other NR4A family members (Nur77 and Nor-1) besides, or in addition to, the up-regulation of Nurr1. The up-regulation of Nur77 in particular has been shown to be strongly pro-apoptotic in a number of cells and tissues where it acts by a transcriptionally independent mechanism to convert Bcl-2 from an anti-apoptotic molecule into a pro-apoptotic one (Li et al., 2000; Thompson and Winoto, 2008; Cheng et al., 2011). Additionally, the up-regulation of Nurr1 by PGE2 is often accompanied by the simultaneous up-regulation of Nur77. For example, PGE2 has been shown to up-regulate the expression of both Nurr1 and Nur77 in synoviocytes (McEvoy et al., 2002) and in chondrocytes (Mix et al., 2007). In cementoblastic cells, PGE2 was found to up-regulate the expression of Nur77 through specific activation of the EP1 receptor (Moldovan et al., 2009). It is also possible that the effect of the induced expression of Nurr1 in brain cells is different from the constitutive expression of Nurr1 that is required for the maintenance of dopaminergic neurons. Thus, it has been reported that ectoptic over-expression of Nurr1 increased apoptosis in SK-N-SH neuroblastoma cells exposed to the neurotoxin, 6-hydroxydopamine (Liu et al., 2005).
In summary, we have found that activation of the human EP1 receptor by PGE2 leads to a cAMP-independent activation of PKA resulting in transcriptional up-regulation of Nurr1 through the actions of CREB and NF-κB. This up-regulation of Nurr1 may underlie the known tumorigenic potential of EP1 receptors in cancer and inflammation. In the brain, activation of EP1 receptors is known to mediate neurotoxicity; however, whether the potential up-regulation of Nurr1 facilitates or offsets this neurotoxicity is presently unknown and would be interesting to address in future studies.
Acknowledgments
This work was supported in part by grants from the National Institutes of Health (NIH) (EY11291) and by Allergan Inc. Portions of this work were also made possible by grants to the Arizona Cancer Center Genomics Facility Core (5P30 ES06694-14900209).
Glossary
- CREB
cAMP response element binding protein
- EP
E-type prostanoid receptor
- Nurr1
Nur-related factor 1 or NR4A2
- PKAc
the catalytic subunit of PKA
Conflict of interest
The authors state no conflict of interest.
References
- Ahmad AS, Saleem S, Ahmad M, Doré S. Prostaglandin EP1 receptor contributes to excitotoxicity and focal ischemic brain damage. Toxicol Sci. 2006;89:265–270. doi: 10.1093/toxsci/kfj022. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. (5th Edition) 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco E, Casper D, Werner P. PGE2 receptor EP1 renders dopaminergic neurons selectively vulnerable to low-level oxidative stress and direct PGE2 neurotoxicity. J Neurosci Res. 2007;85:3109–3117. doi: 10.1002/jnr.21425. [DOI] [PubMed] [Google Scholar]
- Chell S, Kadi A, Williams AC, Paraskeva C. Mediators of PGE2 synthesis and signalling downstream of COX-2 represent potential targets for the prevention/treatment of colorectal cancer. Biochim Biophys Acta. 2006;1766:104–119. doi: 10.1016/j.bbcan.2006.05.002. [DOI] [PubMed] [Google Scholar]
- Cheng Z, Völkers M, Din S, Avitabile D, Khan M, Gude N, et al. Mitochondrial translocation of Nur77 mediates cardiomyocyte apoptosis. Eur Heart J. 2011;32:2179–2188. doi: 10.1093/eurheartj/ehq496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman RA, Smith WL, Narumiya S. VIII. International Union of Pharmacology classification of prostanoid receptors: properties, distribution, and structure of the receptors and their subtypes. Pharmacol Rev. 1994;46:205–229. [PubMed] [Google Scholar]
- Fujino H, Regan JW. EP4 prostanoid receptor coupling to a pertussis toxin-sensitive inhibitory G-protein. Mol Pharmacol. 2006;69:13–18. doi: 10.1124/mol.105.017749. [DOI] [PubMed] [Google Scholar]
- Fujino H, West KA, Regan JW. Phosphorylation of glycogen synthase kinase-3 and stimulation of T-cell factor signaling following activation of EP2 and EP4 prostanoid receptors by prostaglandin E2. J Biol Chem. 2002;277:2614–2619. doi: 10.1074/jbc.M109440200. [DOI] [PubMed] [Google Scholar]
- Fulton AM, Ma X, Kundu N. Targeting prostaglandin E EP receptors to inhibit metastasis. Cancer Res. 2006;66:9794–9797. doi: 10.1158/0008-5472.CAN-06-2067. [DOI] [PubMed] [Google Scholar]
- Gambaryan S, Kobsar A, Rukoyatkina N, Herterich S, Geiger J, Smolenski A, et al. Thrombin and collagen induce a feedback inhibitory signaling pathway in platelets involving dissociation of the catalytic subunit of protein kinase A from an NFκB-IκB complex. J Biol Chem. 2010;285:18352–18363. doi: 10.1074/jbc.M109.077602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han C, Michalopoulos GK, Wu T. Prostaglandin E2 receptor EP1 transactivates EGFR/MET receptor tyrosine kinases and enhances invasiveness in human hepatocellular carcinoma cells. J Cell Physiol. 2006;207:261–270. doi: 10.1002/jcp.20560. [DOI] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Shared principles in NF-κB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- Holla VR, Mann JR, Shi Q, DuBois RN. Prostaglandin E2 regulates the nuclear receptor NR4A2 in colorectal cancer. J Biol Chem. 2006;281:2676–2682. doi: 10.1074/jbc.M507752200. [DOI] [PubMed] [Google Scholar]
- Huang GW, Yang LY, Lu WQ. Expression of hypoxia-inducible factor 1α and vascular endothelial growth factor in hepatocellular carcinoma: impact on neovacularization and survival. World J Gastroenterol. 2005;11:1705–1708. doi: 10.3748/wjg.v11.i11.1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji R, Chou CL, Xu W, Chen XB, Woodward DF, Regan JW. EP1 prostanoid receptor coupling to Gi/o upregulates the expression of hypoxia-inducible factor-1α through activation of a phosphoinositide-3 kinase signaling pathway. Mol Pharmacol. 2010;77:1025–1036. doi: 10.1124/mol.110.063933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juneja J, Casey PJ. Role of G12 proteins in oncogenesis and metastasis. Br J Pharmacol. 2009;158:32–40. doi: 10.1111/j.1476-5381.2009.00180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadkhodaei B, Ito T, Joodmardi E, Mattsson B, Rouillard C, Carta M, et al. Nurr1 is required for maintenance of maturing and adult midbrain dopamine neurons. J Neurosci. 2009;29:15923–15932. doi: 10.1523/JNEUROSCI.3910-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano T, Anrather J, Zhou P, Park L, Wang G, Frys KA, et al. Prostaglandin E2 EP1 receptors: downstream effectors of COX-2 neurotoxicity. Nat Med. 2006;12:225–229. doi: 10.1038/nm1362. [DOI] [PubMed] [Google Scholar]
- Ke N, Claassen G, Yu DH, Albers A, Fan W, Tan P, et al. Nuclear hormone receptor NR4A2 is involved in cell transformation and apoptosis. Cancer Res. 2004;64:8208–8212. doi: 10.1158/0008-5472.CAN-04-2134. [DOI] [PubMed] [Google Scholar]
- Kotani M, Tanaka I, Ogawa Y, Usui T, Tamura N, Mori K, et al. Structural organization of the human prostaglandin EP3 receptor subtype gene (PTGER3) Genomics. 1997;40:425–434. doi: 10.1006/geno.1996.4585. [DOI] [PubMed] [Google Scholar]
- Kurose H. Galpha12 and Galpha13 as key regulatory mediator in signal transduction. Life Sci. 2003;74:155–161. doi: 10.1016/j.lfs.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Li X, Tai HH. Activation of thromboxane A2 receptors induces orphan nuclear receptor Nurr1 expression and stimulates cell proliferation in human lung cancer cells. Carcinogenesis. 2009;30:1606–1613. doi: 10.1093/carcin/bgp161. [DOI] [PubMed] [Google Scholar]
- Li H, Kolluri SK, Gu J, Dawson MI, Cao X, Hobbs PD, et al. Cytochrome c release and apoptosis induced by mitochondrial targeting of nuclear orphan receptor TR3. Science. 2000;289:1159–1164. doi: 10.1126/science.289.5482.1159. [DOI] [PubMed] [Google Scholar]
- Liu Y, Zhao Y, Zhang H, Liw E, Xu Q. The effects of overexpression of Nurr1 on vulnerability of SK-N-SH cells to neurotoxin 6-OHDA. Chin Pharmacological Bulletin. 2005;21(10):1171–1176. [Google Scholar]
- Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. 2001;2:599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- McEvoy AN, Murphy EA, Ponnio T, Conneely OM, Bresnihan B, FitzGerald O, et al. Activation of nuclear orphan receptor NURR1 transcription by NF-κB and cyclic adenosine 5'-monophosphate response element-binding protein in rheumatoid arthritis synovial tissue. J Immunol. 2002;168:2979–2987. doi: 10.4049/jimmunol.168.6.2979. [DOI] [PubMed] [Google Scholar]
- Mix KS, Attur MG, Al-Mussawir H, Abramson SB, Brinckerhoff CE, Murphy EP. Transcriptional repression of matrix metalloproteinase gene expression by the orphan nuclear receptor NURR1 in cartilage. J Biol Chem. 2007;282:9492–9504. doi: 10.1074/jbc.M608327200. [DOI] [PubMed] [Google Scholar]
- Moldovan SM, Nervina JM, Tetradis S, Camargo PM. Regulation of Nur77 gene expression by prostanoids in cementoblastic cells. Arch Oral Biol. 2009;54:412–419. doi: 10.1016/j.archoralbio.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan T, Xie W, Jankovic J, Le W. Biological effects of pramipexole on dopaminergic neuron-associated genes: relevance to neuroprotection. Neurosci Lett. 2005;377:106–109. doi: 10.1016/j.neulet.2004.11.080. [DOI] [PubMed] [Google Scholar]
- Pearen M, Muscat GEO. Minireview: nuclear hormone receptor 4A signaling: implications for metabolic disease. Mol Endocrinol. 2010;24:1891–1903. doi: 10.1210/me.2010-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perona R, Montaner S, Saniger L, Sanchez-Perez I, Bravo R, Lacal JC. Activation of the nuclear factor-κB by Rho, CDC42, and Rac-1 proteins. Genes Dev. 1997;11:463–475. doi: 10.1101/gad.11.4.463. [DOI] [PubMed] [Google Scholar]
- Regan JW. EP2 and EP4 prostanoid receptor signaling. Life Sci. 2003;74:143–153. doi: 10.1016/j.lfs.2003.09.031. [DOI] [PubMed] [Google Scholar]
- Saijo K, Winner B, Carson CT, Collier JG, Boyer L, Rosenfeld MG, et al. A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell. 2009;137:47–59. doi: 10.1016/j.cell.2009.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigeishi H, Higashikawa K, Hatano H, Okui G, Tanaka F, Tran TT, et al. PGE2 targets squamous cell carcinoma cell with the activated epidermal growth factor receptor family for survival against 5-fluorouracil through NR4A2 induction. Cancer Lett. 2011;307:227–236. doi: 10.1016/j.canlet.2011.04.008. [DOI] [PubMed] [Google Scholar]
- Steinberg RA, Cauthron RD, Symcox MM, Shuntoh H. Autoactivation of catalytic (C alpha) subunit of cyclic AMP-dependent protein kinase by phosphorylation of threonine 197. Mol Cell Biol. 1993;13:2332–2341. doi: 10.1128/mcb.13.4.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto Y, Narumiya S. Prostaglandin E receptors. J Biol Chem. 2007;282:11613–11617. doi: 10.1074/jbc.R600038200. [DOI] [PubMed] [Google Scholar]
- Thompson J, Winoto A. During negative selection, Nur77 family proteins translocate to mitochondria where they associate with Bcl-2 and expose its proapoptotic BH3 domain. J Exp Med. 2008;205:1029–1036. doi: 10.1084/jem.20080101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallabhapurapu S, Karin M. Regulation and function of NF-κB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- Vinukonda G, Csiszar A, Hu F, Dummula K, Pandey NK, Zia MT, et al. Neuroprotection in a rabbit model of intraventricular haemorrhage by cyclooxygenase-2, prostanoid receptor-1 or tumour necrosis factor-alpha inhibition. Brain. 2010;133:2264–2280. doi: 10.1093/brain/awq107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volakakis N, Kadkhodaei B, Joodmardi E, Wallis K, Panman L, Silvaggi J, et al. NR4A orphan nuclear receptors as mediators of CREB-dependent neuroprotection. Proc Natl Acad Sci U S A. 2010;107:12317–12322. doi: 10.1073/pnas.1007088107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetterstrom RH, Solomin L, Jansson L, Hoffer BJ, Olson L, Perlmann T. Dopamine neuron agenesis in Nurr1-deficient mice. Science. 1997;276:248–250. doi: 10.1126/science.276.5310.248. [DOI] [PubMed] [Google Scholar]
- Zhang T, Wang P, Ren H, Fan J, Wang G. NGFI-B nuclear receptor Nurr1 interacts with p53 and suppresses its transcriptional activity. Mol Cancer Res. 2009;7:1408–1415. doi: 10.1158/1541-7786.MCR-08-0533. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Bruemmer D. NR4A orphan nuclear receptors transcriptional regulators of gene expression in metabolism and vascular biology. Arterioscler Thromb Vasc Biol. 2010;30:1535–1541. doi: 10.1161/ATVBAHA.109.191163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H, SuYang H, Erdjument-Bromage H, Tempst P, Ghosh S. The transcriptional activity of NF-κB is regulated by the IκB-associated PKAc subunit through a cyclic AMP-independent mechanism. Cell. 1997;89:413–424. doi: 10.1016/s0092-8674(00)80222-6. [DOI] [PubMed] [Google Scholar]