

Abstract
BACKGROUND AND PURPOSE
NMDA receptors are glutamatergic ionotropic receptors involved in excitatory neurotransmission, synaptic plasticity and excitotoxic cell death. Many allosteric modulators can influence the activity of these receptors positively or negatively, with behavioural consequences. 20-Oxo-5β-pregnan-3α-yl sulphate (pregnanolone sulphate; PA-6) is an endogenous neurosteroid that inhibits NMDA receptors and is neuroprotective. We tested the hypothesis that the interaction of PA-6 with the plasma membrane is critical for its inhibitory effect at NMDA receptors.
EXPERIMENTAL APPROACH
Electrophysiological recordings and live microscopy were performed on heterologous HEK293 cells expressing GluN1/GluN2B receptors and cultured rat hippocampal neurons.
KEY RESULTS
Our experiments showed that the kinetics of the steroid inhibition were slow and not typical of drug-receptor interaction in an aqueous solution. In addition, the recovery from steroid inhibition was accelerated by β- and γ-cyclodextrin. Values of IC50 assessed for novel synthetic C3 analogues of PA-6 differed by more than 30-fold and were positively correlated with the lipophilicity of the PA-6 analogues. Finally, the onset of inhibition induced by C3 analogues of PA-6 ranged from use-dependent to use-independent. The onset and offset of cell staining by fluorescent analogues of PA-6 were slower than those of steroid-induced inhibition of current responses mediated by NMDA receptors.
CONCLUSION AND IMPLICATIONS
We conclude that steroid accumulation in the plasma membrane is the route by which it accesses a binding site on the NMDA receptor. Thus, our results provide a possible structural framework for pharmacologically targeting the transmembrane domains of the receptor.
Keywords: neurosteroids, NMDA receptor, inhibition, patch-clamp recording, recombinant receptors, pregnane analogues, fluorescent steroid analogues
Introduction
NMDA receptors play important roles in development, synaptic plasticity, learning and memory; however, the abnormal activation of NMDA receptors is thought to mediate neuronal degeneration (Traynelis et al., 2010). NMDA receptor activity can be influenced by exogenous and endogenous ligands, including neurosteroids – endogenous steroids that are synthesized and act in the CNS (Corpechot et al., 1981; Rupprecht and Holsboer, 1999). The central effects of these compounds are mediated by interactions with ligand-gated ion channels such as glutamate, GABAA, glycine and nicotinic acetylcholine receptors (Wu et al., 1990; 1991; Bullock et al., 1997).
It has been suggested that the modulatory effect of neurosteroids on the activity of ligand-gated ion channels plays a role in a range of physiological processes such as learning, ageing and stress, as well as in certain neuropsychiatric disorders (Flood and Roberts, 1988; Nasman et al., 1991; Grobin et al., 1992; Vallee et al., 1997; Reddy, 2010). In accord with the inhibitory effect of steroids on NMDA receptors, the synthetic analogue of pregnanolone sulphate (PA-6) (PA-hemisuccinate ester) has a neuroprotective effect, in both in vitro and in vivo models of neurodegeneration, thereby indicating its potential therapeutic use (Weaver et al., 1997; Lapchak, 2004).
Our previous studies revealed that PA-6 is a use-dependent but voltage-independent inhibitor that exerts its effect by reducing the peak probability of the NMDA receptor channel opening. Furthermore, its inhibitory action is weaker for responses mediated by NMDA receptors activated by synaptically released glutamate than for those tonically activated by the agonist (Petrovic et al., 2005; Sedlacek et al., 2008). It has been proposed that PA-6 inhibits NMDA receptor activity by shifting active receptors into desensitized conformations (Kussius et al., 2009). However, it is not clear what structural determinants of this steroid are critical for its effects at NMDA receptors.
In this study, we examined the effects of PA-6 and its C3-substituted analogues on NMDA receptors. Combining electrophysiological and microscopical imaging approaches, we drew several conclusions about the steroid mode of action at NMDA receptors. First, we showed that the kinetics of the onset and offset of steroid inhibition was slow and not typical of simple drug-receptor interaction in an aqueous solution. This conclusion was also supported by the experiments in which β- and γ-cyclodextrin accelerated the kinetics of recovery from steroid-induced inhibition. Second, we showed that the onset of inhibition induced by different C3-substituted analogues ranges from use-dependent to use-independent. Third, we showed that the onset and offset of cell staining by fluorescent steroid analogues were slower than the onset and offset of inhibition induced by these steroids. Finally, we showed that there is a correlation between the inhibitory effects of steroid analogues and their lipophilicity. We conclude that steroid accumulation in the plasma membrane is the route by which the steroid accesses a site on the NMDA receptor.
Methods
All animal care and experimental procedures were carried out in accordance with the European Communities Council Directive (86/609/EEC) and with the approval of the Institutional Animal Care and Use Committee.
Transfection and maintenance of HEK293 cells
HEK 293 cells (American Type Culture Collection, ATTC No. CRL1573, Rockville, MD, USA) were cultured in Opti-MEM® I (Invitrogen, Carlsbad, CA, USA) with 5% fetal bovine serum at 37°C. The day before transfection, the cells were plated in 24-well plates at a density of 2 × 105 cells per well. The next day, cells were transfected with expression vectors containing the glutamate receptor subunits GluN1-1a (GluN1; GenBank accession no. U08261 and GluN2B (GenBank accession no. M91562; receptor subunit nomenclature follows Alexander et al., 2011)), and GFP (green fluorescent protein) (pQBI 25, Takara, Tokyo, Japan) genes, as described previously (Cais et al., 2008). Briefly, equal amounts (0.3 µg) of cDNAs encoding for GluN1, GluN2B and GFP were mixed with 0.9 µL of Matra-A Reagent (IBA, Göttingen, Germany) and added to confluent HEK293 cells on a 24 well plate. After trypsinization, the cells were resuspended in Opti-MEM® I containing 1% fetal bovine serum supplemented with 20 mmol·L−1 MgCl2, 1 mmol·L−1 D,L-2-amino-5-phosphonopentanoic acid and 3 mmol·L−1 kynurenic acid and plated on 30-mm poly-L-lysine-coated glass cover slips.
Electrophysiological recording from cultured cells
Experiments were performed 24–48 h after the end of transfection on cultured HEK293 cells transfected with vectors containing GluN1/GluN2B/GFP. Whole-cell voltage-clamp recordings were made with a patch-clamp amplifier (Axopatch 200B; Axon Instruments, Inc., Foster City, CA, USA) after a capacitance and series resistance (<10 MΩ) compensation of 80–90%. Agonist-induced responses were low-pass filtered at 1 kHz with an 8-pole Bessel filter (Frequency Devices, Haverhill, MA, USA), digitally sampled at 5 kHz, and analysed using pClamp software version 9 (Axon Instruments). Patch pipettes (3–5 MΩ) pulled from borosilicate glass were filled with Cs+-based intracellular solution (Cs-ICS) containing the following (in mmol·L−1): 125 gluconic acid, 15 CsCl, 5 EGTA, 10 HEPES, 3 MgCl2, 0.5 CaCl2, and 2 ATP-Mg salt (pH-adjusted to 7.2 with CsOH). Extracellular solution (ECS) contained the following (in mmol·L−1): 160 NaCl; 2.5 KCl; 10 HEPES; 10 glucose; 0.2 EDTA; and 0.7 CaCl2 (pH-adjusted to 7.3 with NaOH). Glycine (10 µmol·L−1), a NMDA receptor co-agonist, was present in the control and test solutions. All steroid solutions were made from freshly prepared 20 mmol·L−1 stock in DMSO. The same concentration of DMSO was added to all ECSs.
Drug applications were made with a microprocessor-controlled multibarrel fast-perfusion system. The estimated solution exchange times were 11 ± 3 ms (n= 6) (10–90% rise), measured by the change in the whole-cell current recorded from HEK293 cells expressing recombinant NMDA receptors and induced by stepping from glutamate-containing control ECS to the same solution with reduced sodium (50 mmol·L−1). Experiments were performed at room temperature.
Microscopy of live cells
Fluorescence measurements were performed using inverted microscope Olympus IX81 at 20× magnification. The Polychromator V (Till Photonics, Munich, Germany) was used for the excitation of specimen at specific wavelength (7 nm excitation bandwidth). The emission was detected by CCD camera Hamamatsu Orca ER (16 bits per pixel, 2 × 2 binning). Exposure time was 100 ms at a repetition rate of 300 ms or 1 s, depending on the type of experiment. For 7-nitrobenz-2-oxa-1,3-diazol-4-yl (NBD) fluorescent-labelled steroid we used the GFP cube (excitation 460–490 nm, DM 505 nm, emission 515 nm), and for Alexa-labelled steroid we used the Rhodamine cube (excitation 530–550 nm, DM 570 nm, emission 575 nm). The experiment was controlled by Cell^R software (Olympus, Hamburg, Germany), and for further data analysis ImageJ software (NIH, Bethesda, MD, USA) was used. Application of the fluorescently-labelled steroid was performed by blowing the solution from a patch clamp pipette (tip resistance ∼5 MΩ) placed next to the cell.
Molecular modelling
Molecular modelling studies were performed using program Hyperchem 8 (Hypercube, Gainesville, FL, USA). The molecular geometry and partial charges were optimized by semiempirical method PM3 considering appropriate total charge of molecule and singlet state. Consequently, the general protocol for obtaining the lowest energy conformers by simulated annealing was used: optimized starting structure was subjected to a dynamic run (0.2 ps heating from 300 to 1000°K), 1 ps equilibration, and 2 ps cooling to 200°K, followed by energy minimization. Every subsequent run started from the previously minimized structure. A set of 100 structures was obtained for each compound, from which the lowest energy conformation was taken. The Polak-Ribiere conjugate gradient method for energy minimizations was used for convergence (less than 0.04 kJ·mol−1 RMS force). Force field MM+ was used for all computations. All calculations were done in vacuo.
Calculations of log P were made using the on-line program ALOGPS (http://www.vcclab.org/lab/alogps/) (Tetko, 2005; Tetko et al., 2005).
Data analysis
The results are presented as mean ± SD, with n equal to the number of cells studied. Statistical comparisons were made using Student's t-test or a one-way anova with post hoc Tukey's test. P < 0.05 was used to determine the significance.
Materials
All drugs, unless otherwise stated, were purchased from Sigma (St. Louis, MO, USA). 5β-pregnane analogues were synthesized as described previously (Stastna et al., 2009) and the details are given in the Supplemental text. The structures of all synthesized steroids were confirmed by IR, NMR, MS and HR-MS spectra.
Results
Structural-functional relation of steroid action at NMDA receptors
Currents elicited by 100 µmol·L−1 glutamate were recorded from HEK293 cells, voltage-clamped at a holding potential of −60 mV, which expressed recombinant GluN1/GluN2B receptors. In accordance with previous results, pregnanolone sulphate (PA-6; see structure in Figure 1A) diminished the amplitude of GluN1/GluN2B receptor responses (Table 1). To determine what structural requirements were important for the steroid molecule to inhibit NMDA receptors, we prepared a series of 18 C3-substituted analogues of PA-6 differing by residue charge and its distance from the steroid skeleton (Figure 1A). We then tested these analogues at a single concentration (10–200 µmol·L−1) and calculated their inhibition of responses to fast application of 100 µmol·L−1 glutamate at GluN1/GluN2B receptors (Figure 1B) and the corresponding value of IC50 (Table 1). The IC50 was determined from a single dose of steroid using the formula 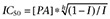 , where I is the relative degree of inhibition, [PA] is the steroid concentration used, and h (fixed at 1.2) is the apparent Hill coefficient (Petrovic et al., 2005). For two compounds (PA-6, PA-27), the IC50 was also calculated from a full concentration-response curve, fitted to a logistic equation (see Figure 3). Calculations of IC50 were made assuming 100% inhibition at saturating steroid concentration. Our results showed that all steroids with a negatively charged C3 residue had an inhibitory effect at responses mediated by GluN1/GluN2B receptors, with the IC50 differing by more than 10-fold (Table 1).
, where I is the relative degree of inhibition, [PA] is the steroid concentration used, and h (fixed at 1.2) is the apparent Hill coefficient (Petrovic et al., 2005). For two compounds (PA-6, PA-27), the IC50 was also calculated from a full concentration-response curve, fitted to a logistic equation (see Figure 3). Calculations of IC50 were made assuming 100% inhibition at saturating steroid concentration. Our results showed that all steroids with a negatively charged C3 residue had an inhibitory effect at responses mediated by GluN1/GluN2B receptors, with the IC50 differing by more than 10-fold (Table 1).
Figure 1.
Chemical structures of steroids tested for biological activity at NMDA receptors. (A) Structure of 5β-20-oxo-pregnane (PA) and residues substituted in the position of carbon C3 (A-S). (B) Examples of traces obtained from HEK293 cells transfected with cDNAs encoding NR1/NR2B receptor subunits. PA-6 (100 µmol·L−1), PA-27 (10 µmol·L−1) and PA-5 (200 µmol·L−1) were applied simultaneously with 100 µmol·L−1 glutamate (duration of steroid and glutamate application is indicated by filled and open bars respectively).
Table 1.
Effects of PA-6 and its synthetic analogues on responses of GluN1/GluN2B receptors in HEK293 cells to glutamate
| NS | Steroid concentration (µmol·L−1) | Mean % change ±SD (n) | IC50 (µmol·L−1) |
|---|---|---|---|
| PA-4 | 200 | +1.9 ± 6.2 (4) | ND |
| PA-5 | 200 | +14.1 ± 7.5 (5) | ND |
| PA-6 | 100 | −84.4 ± 3.4 (5) | 24.6 ± 5.3 |
| 3–300 | (5) | 31.1 ± 5.9 (DR) | |
| PA-11 | 200 | −71.5 ± 13.3 (4) | 97.3 ± 48.3 |
| PA-12 | 200 | −84.7 ± 0.6 (4) | 48.1 ± 1.8 |
| PA-13 | 100 | −69.2 ± 7.0 (6) | 51.9 ± 14.2 |
| PA-14 | 100 | −80.9 ± 3.6 (4) | 30.1 ± 5.9 |
| PA-15 | 50 | −81.6 ± 6.1 (6) | 14.4 ± 4.9 |
| PA-18 | 200 | −83.6 ± 2.7 (5) | 51.7 ± 8.6 |
| PA-21 | 50 | −63.7 ± 2.9 (5) | 31.4 ± 3.3 |
| PA-22 | 200 | −87.3 ± 8.9 (4) | 40.8 ± 27.8 |
| PA-25 | 200 | −77.1 ± 5.2 (5) | 73.4 ± 18.0 |
| PA-27 | 10 | −69.2 ± 9.6 (7) | 5.3 ± 2.1 |
| 0.3–30 | (7) | 6.8 ± 1.3 (DR) | |
| PA-33 | 50 | −31.5 ± 2.5 (4) | 95.9 ± 8.8 |
| PA-34 | 200 | −61.5 ± 10.6 (5) | 141.1 ± 54.3 |
| PA-35 | 10 | −65.4 ± 3.4 (5) | 5.9 ± 0.7 |
| PA-36 | 200 | −54.0 ± 10.8 (5) | 184.0 ± 70.2 (S) |
| PA-17 | 100 | −54.1 ± 8.4 (5) | 89.7 ± 23.8 |
The structures of PA-6 and its C3-substituted analogues are shown in Figure 1. The effect of the steroids on the current induced by fast application of 100µmol·L−1 glutamate is expressed as % change (means ± SD), with n indicating the number of cells studied. The IC50 values (means ± SD) were calculated from the single concentrations shown and, for PA-6 and PA-27, also from a full concentration-response curve (marked DR), as described in the text.
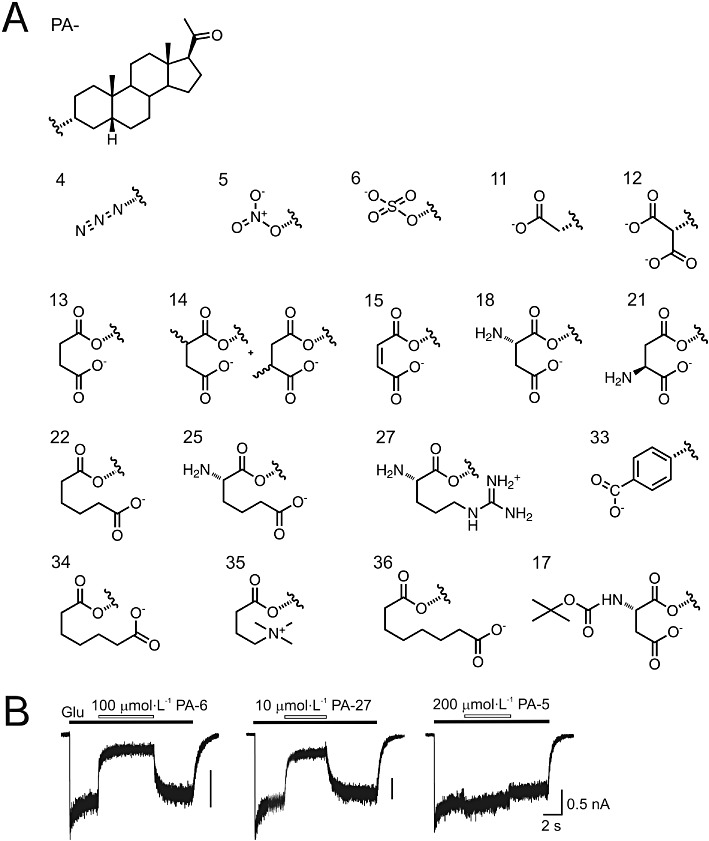
Figure 3.
Concentration-dependent inhibition by PA-6 and PA-27 at NR1/NR2B receptors. Examples of traces obtained from HEK293 cells expressing recombinant NMDA receptors activated by 100 µmol·L−1 glutamate and its co-application with 3 and 30 µmol·L−1 PA-6 (A) and 3 and 30 µmol·L−1 PA-27) (B) (duration of glutamate and steroid is indicated by an open and filled bars respectively). (C) Concentration-response curves for the PA-6 and PA-27 effect at NR1/NR2B receptors. Steroid-induced inhibition was fitted to the following logistic equation: I= 1/(1([PA]/IC50)h), where IC50 is the concentration of steroid that produces a 50% inhibition of agonist-evoked current, [PA] is the PA-6 or PA-27 concentration, and h is the apparent Hill coefficient. Smooth curves are calculated from the mean values (PA-6 IC50= 31.1 µmol·L−1, Hill coefficient = 1.1, n= 5; PA-27 IC50= 6.8 µmol·L−1, Hill coefficient = 1.9, n= 7). Data shown are means ± SD.
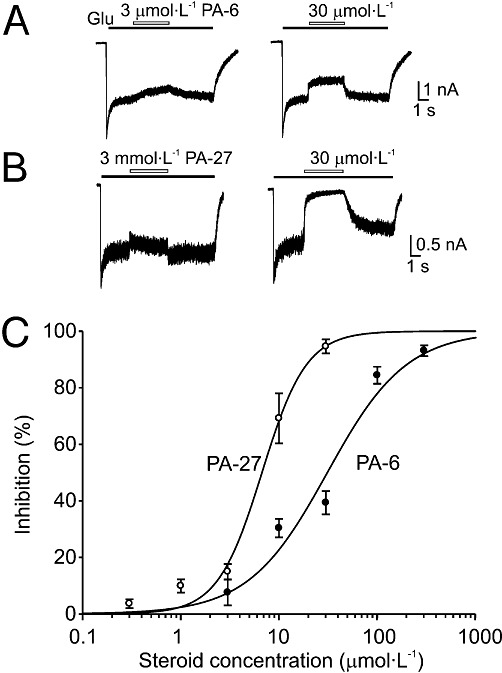
We also tested the effects of C3-substituted steroid analogues bearing a positive charge. Two positively charged PA-6 analogues – sulphate replaced by arginine (PA-27) and permethylated γ-amino butyrate (PA-35) (Figure 1) – had profound inhibitory effects at low micromolar concentrations at recombinant GluN1/GluN2B receptors (Table 1). As positively charged compounds can produce a voltage-dependent block of NMDA receptor channels (Mayer et al., 1984; Nowak et al., 1984), we calculated the inhibition by PA-27 of NMDA receptor-mediated currents at various negative and positive holding potentials (Figure 2). An I/V plot of the PA-27-induced inhibition showed that the inhibition was the same at positive and negative membrane potentials and was therefore unlikely to be a result of to binding of the positively charged steroid inside the ion channel pore.
Figure 2.
PA-27 is a voltage-independent inhibitor of NMDA receptors. (A) Examples of responses induced by glutamate (1 mmol·L−1) in HEK293 cells expressing NR1/NR2B receptors, recorded at −60 and +60 mV. The glutamate-evoked currents recorded at each of the membrane potentials indicated are reversibly inhibited by co-application of PA-27 (10 µM) at the point indicated by the open bar. (B) Plot of the mean inhibition induced by PA-27 versus holding potential. Note that PA-27-induced inhibition was not affected by membrane potential.
To test the importance of the presence of the charged residue at the C3 of the steroid skeleton for its inhibitory effect at NMDA receptors, we prepared a series of compounds substituted at C3 with an uncharged residue. Out of six compounds, only 3α-azido-5β-pregnan-20-one (PA-4) and 5β-pregnan-20-one-3α-yl nitrate (PA-5) did not precipitate when added to the ECS. At a concentration of 200 µmol·L−1, both PA-4 and PA-5 were devoid of any inhibitory action at responses induced by 100 µmol·L−1 glutamate in GluN1/GluN2B receptors; in fact, both had small potentiating effects (Table 1), indicating that the presence of the charged residue at the C3 position was critical for the PA-6 inhibitory effect at NMDA receptors.
To further explore the differences in the effects of positively and negatively charged steroids at NMDA receptors, we analysed the concentration–response relationships of PA-6 and PA-27. Analysis of the inhibitory effect of PA-6 at a range of concentrations (3–300 µmol·L−1) on the responses of GluN1/GluN2B receptors induced by glutamate (100 µmol·L−1) yielded a values for IC50= 31.1 ± 5.9 µmol·L−1 and the Hill coefficient = 1.1 ± 0.1 (n= 5) (see Figure 3A,C). These data agree well with the IC50 determined earlier (Park-Chung et al., 1994; Petrovic et al., 2005). Figure 3B,C also shows the results of experiments in which PA-27 (0.1–30 µmol·L−1) was tested under similar conditions. Analysis of these data provided IC50= 6.8 ± 1.3 µmol·L−1 and a Hill coefficient = 1.9 ± 0.1 (n= 7). These observations showed that PA-27 was a more potent NMDA receptor inhibitor than PA-6.
To assess the structure activity relationships of steroid action at NMDA receptors, we used molecular modelling to determine the steroid 3D structure (see Methods). Supporting Information Figure S1 shows a plot in which the value of IC50 is plotted versus the distance of terminal charge from the C3 of the steroid molecule. Poor correlation was found for the value of IC50 and the distance of the most distal charge moiety to C3 versus IC50 (Pearson correlation coefficient, r = 0.60, P= 0.0142), indicating that, in addition to the charge distance from the steroid molecule, there are other structural characteristics of the residue that underlie the differences in the potency of steroids at NMDA receptors.
Use-dependent and use-independent steroid inhibition
We have shown previously that the inhibitory action of PA-6 at NMDA receptors depends on receptor activation and is therefore use dependent (Petrovic et al., 2005). To test whether the action of PA-27 is similar to that of PA-6 at NMDA receptors, we used two protocols of the steroid and glutamate applications: (i) a co-application of steroid and glutamate made after the onset of the response to glutamate; and (ii) a sequential application of glutamate after pre-application of steroid.
Figure 4A shows the responses of GluN1/GluN2B receptors to a co-application of PA-6 (300 µmol·L−1) with glutamate (1 mmol·L−1) made after the onset of the response. The steroid induced almost complete inhibition (96.8 ± 1.1%; n= 5) of the responses of the NMDA receptors. After cessation of the co-application of the steroid, the response to glutamate slowly recovered (τ(off)= 714 ± 96 ms; n= 5). On the other hand, the responses to glutamate made immediately after PA-6 (300 µmol·L−1) pre-application for 10 s were characterized by fast onset (τ(off)= 30 ± 7 ms; n= 5). This shows that the steroid binding requires NMDA receptor activation and is therefore use-dependent, as described previously (Petrovic et al., 2005). Onset of the control glutamate response (without the steroid) [τ(on)= 7 ± 3 ms, n= 5] was even faster than that made after PA-6 pre-application. Even though the kinetics of the control glutamate response is likely to be affected by the rate of solution exchange around the studied cell (see Methods), the difference in the onset of control glutamate response and that made after steroid pre-application may indicate relatively fast steroid unbinding from the non-activated NMDA receptors or, more likely, that during the switch between the steroid and glutamate solution, the mixing of both solutions for a short period results in partial receptor inhibition.
Figure 4.

Use-dependent and use-independent inhibition of NMDA receptor channels by steroids. The Figure shows examples of records obtained from HEK293 cells transfected with NR1 and NR2B subunits. (A) On the left, response to co-application of 300 µmol·L−1 PA-6 and 1 mmol·L−1 glutamate (Glu) made after the onset of the response to the agonist was inhibited by 97% and recovered from the inhibition on a slow timescale. In the middle, the onset of the response to application of 1 mmol·L−1 glutamate made immediately after pre-application of 300 µmol·L−1 PA-6 for 10 s was rapid, similar to the control glutamate response. (B) On the left, response to co-application of 30 µmol·L−1 PA-27 and 1 mmol·L−1 glutamate (Glu) made after the onset of the response to the agonist was inhibited by 98% and recovered from the inhibition on a slow timescale. In the middle, the onset of the response to application of 1 mmol·L−1 glutamate made immediately after pre-application of 30 µmol·L−1 PA-27 for 10 s was slow, similar to the recovery after PA-27 and glutamate co-application. (C) On the left, response to co-application of 200 µmol·L−1 PA-22 and 1 mmol·L−1 glutamate (Glu) made after the onset of the response to the agonist was inhibited by 95% and recovered from the inhibition on a slow timescale. In the middle, the onset of the response to application of 1 mmol·L−1 glutamate made immediately after pre-application of 200 µmol·L−1 PA-22 for 10 s was complex characterized by biphasic response – the first being fast and the second slow. (A-C) On the right, recovery from inhibition induced by steroid and glutamate co-application (in red) and steroid pre-application (in black) are displayed overlaid to show the difference in the rate of recovery. Note that the recovery from inhibition was similar for all three steroids; in the case of co-application, there were differences in the recovery after the steroid application.
To study the nature of the action of positively charged steroids with the NMDA receptors, we used simultaneous and sequential applications of glutamate and PA-27. Figure 4B shows that the time-course of the onset of the glutamate response made after PA-27 (30 µmol·L−1) pre-application for 10 s (τ(on)= 458 ± 253 ms; n= 5) was similar to the time-course of the onset after glutamate and PA-27 co-application (τ(on)= 503 ± 253 ms; paired t-test P= 0.613). Our data show that, in contrast to the use-dependent action of PA-6, PA-27 binding does not require NMDA receptor activation and is therefore use-independent.
Next, experiments were designed to test whether the difference in the use-dependent and use-independent action of PA-6 and PA-27 is due to differences in the charge or length of C3-conjugated residue. Structural analysis of the newly synthesized C3-substituted steroids (Figure 1) allowed us to select a steroid – PA-22 – in which the distance between the end terminal charge and C3 (6.3 Å) is similar to that between N+ and C3 (5.6 Å) in PA-27; however, in contrast to PA-27, PA-22 is negatively charged. Figure 4C shows typical NMDA receptor responses induced by glutamate after PA-22 pre-application and its co-application with glutamate. The onset of PA-22 (30 µmol·L−1)-induced inhibition was best fit by a double exponential function (τ1= 236 ± 124 ms; τ2= 1514 ± 713 ms; A1= 58 ± 24%) (τw= 668 ± 210 ms; n= 5) and the recovery from inhibition by a single exponential function (τ= 1797 ± 417 ms; n= 5). The response to glutamate made after PA-22 (30 µmol·L−1) pre-application for 10 s was characterized by complex kinetics. First, the initial peak response reached an amplitude similar to that of the control response made without the steroid pre-application. Following the peak, the current diminished (τ= 58 ± 26 ms; n= 5) to a minimum level, from which it slowly recovered (τ= 2147 ± 621 ms; n= 5; not significantly different from recovery after steroid and glutamate co-application, P= 0.202). The results suggest that, similar to PA-6, PA-22 is also a use-dependent inhibitor of NMDA receptors and, further, that the complex kinetics of PA-22 inhibition after its pre-application is due to steroid accumulation in a pool from which it can easily access the NMDA receptor but can slowly diffuse to the extracellular space. In addition, these results indicate that the onset of inhibition during simultaneous steroid and glutamate application is not diffusion limited and reflects the slow accumulation of steroid in a compartment(s) from which it can then reach the receptor.
Complex kinetics of steroid onset and offset of inhibition
The electrophysiological records shown in Figure 3 indicate that the rates of recovery after PA-6- and PA-27-induced inhibition differ, according to the dose of steroid used. This may indicate a more complex mechanism of action of these steroids. Figure 5 illustrates the results of the experiments in which we studied the kinetics of the onset and offset of the PA-6-induced inhibition of responses mediated by GluN1/GluN2B receptors. The onset of PA-6 inhibition was, in all studied steroid concentrations (10–300 µmol·L−1), best fitted by a double exponential function (Figure 5A). The plot of the time constants of PA-6-induced inhibition as a function of steroid concentration indicates significant differences in the time course of steroid-induced inhibition at 30 µmol·L−1 PA-6 versus that assessed at 10 and 300 µmol·L−1 PA-6 (Figure 5B). The complex kinetics of the onset of inhibition was observed for the fast and the slow component and both components weighted [τ(on)1, τ(on)2, τ(on)w]; however, the relative amplitude (A) of both the fast and slow components remained unchanged.
Figure 5.
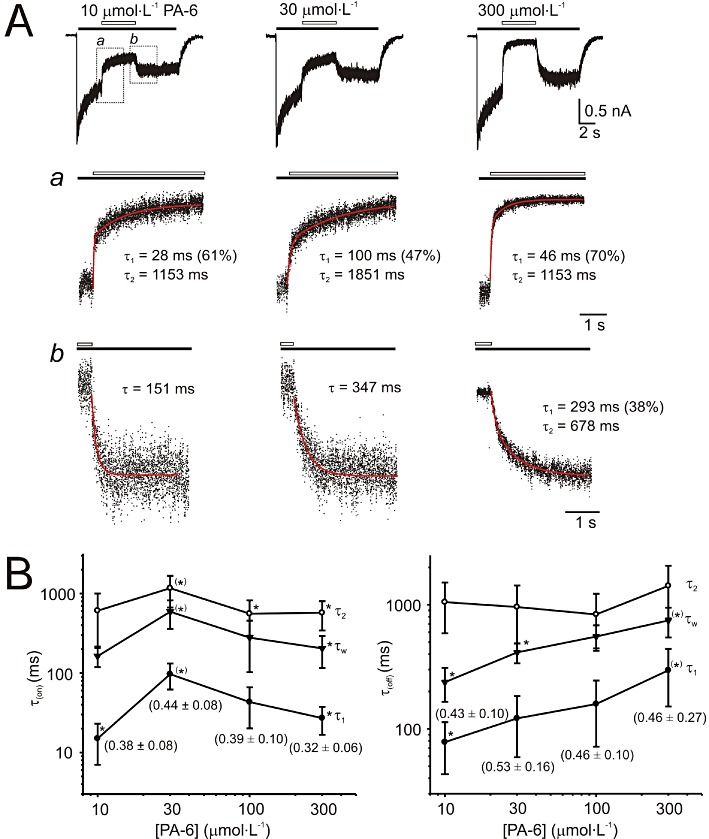
Analysis of the time course of the onset and offset of PA-6-induced inhibition. (A) Whole-cell responses to 12 s application of 1 mmol·L−1 glutamate (indicated by filled bar) recorded from HEK293 cells expressing NR1/NR2B receptors and its co-application with 10, 30, and 300 µmol·L−1 PA-6 (indicated by open bar). Normalized onset of responses to PA-6 (represented by dots) is shown in (a) on expanded time scale and the fit by a double exponential function [indicated by superimposed red line; values of the fast component (τ1) with its relative contribution indicated in parentheses and the slow component (τ2)] are attached. Normalized offset of responses to PA-6 is shown in (b) on expanded time scale and the fit by a double exponential function [indicated in red; values of the fast component (τ1) with its relative contribution indicated in parenthesis and the slow component (τ2)] are attached. Plot of the concentration dependence of the mean time constants ± SD (τ1, τ2 and both components weighted τw) describing onset and offset (B) of PA-6-induced inhibition for 6 cells analysed. Relative contribution of the slow component is attached (mean ± SD). *P < 0.05, significantly different from values marked †; one-way anova with post hoc Tukey's test. No differences were found in the relative amplitude of τ1 and τ2 describing onset and offset of PA-6-induced inhibition. Note bell-shape of the dependence of time constants describing the onset of inhibition and slowdown of recovery of steroid-induced inhibition as a function of PA-6 concentration.
Similar analysis performed on the offset of 30 µmol·L−1 PA-6 inhibition indicated that the recovery after steroid inhibition was best fitted in four out of six cells by a double exponential function and in the remaining two cells by a single exponential function (Figure 5A,B). The plot of the time constants of recovery from PA-6-induced inhibition as a function of steroid concentration indicates significant differences in the time course of recovery: it is slower at higher steroid concentrations for the fast component and for both components weighted (τ(off)1, τ(off)2, τ(off)w) (Figure 5B). The slow component and the relative amplitude of both the fast and slow components remained unchanged. Similarly, analysis of the offset of PA-27 inhibition of responses to glutamate (1 mmol·L−1) showed that the recovery from inhibition is dependent on the concentration of the steroid used [at 10 µmol·L−1 PA-27, τ(off)= 301 ± 87 ms (n= 6); at 30 µmol·L−1 PA-27, τ(off)= 674 ± 182 ms (n= 6); significant difference at P= 0.007]. The complex kinetics of the onset and offset of PA-6 and PA-27 inhibition of NMDA receptors is inconsistent with a simple drug receptor interaction in the aqueous environment.
Complex kinetics of the onset and offset of PA-6 inhibition of NMDA receptor responses (Figure 5) and its use-dependent mechanism of action (Figure 4) may suggest that the steroid-induced inhibition is affected by the degree of receptor activation. Figure 6 shows the results of experiments in which the inhibition of PA-6 (a use-dependent inhibitor) and PA-27 (use-independent inhibitor) was studied at low (1 µmol·L−1) and high (1 mmol·L−1) glutamate concentrations. Figure 6 shows that inhibition induced by both steroids was influenced by glutamate concentration. Responses induced by 1 µmol·L−1 glutamate were inhibited more by PA-6 (20 µmol·L−1) and PA-27 (5 µmol·L−1) than those induced by high glutamate concentration (1 mmol·L−1). It is, however, likely that the dependence of steroid potency on the agonist concentration is complex, as responses induced by high glutamate concentration (1 mmol·L−1) were inhibited more by high steroid concentration (100 µmol·L−1 PA-6). Onset of PA-6 (20 µmol·L−1) inhibition of responses to glutamate was similar for responses induced by 1 µmol·L−1 and 1 mmol·L−1[τ(on)w= 320 ± 195 ms at 1 µmol·L−1 glutamate and τ(on)w= 262 ± 68 ms at 1 mmol·L−1 glutamate; n= 5, P= 0.706]. In contrast, offset of inhibition differed at both glutamate concentrations [τ(off)= 221 ± 127 ms at 1 µmol·L−1 glutamate, τ(off)= 524 ± 188 ms at 1 mmol·L−1 glutamate; n= 5, P= 0.023]. Surprisingly, analysis of the kinetics of inhibition induced by PA-27 (5 µmol·L−1) showed that the onset of the inhibition of responses to glutamate was different for those induced by 1 µmol·L−1 and 1 mmol·L−1[τ(on)w= 508 ± 120 ms at 1 µmol·L−1 glutamate and τ(on)w= 711 ± 65 ms at 1 mmol·L−1 glutamate, n= 7, P= 0.003]. In contrast, offset of inhibition differed at both glutamate concentrations [τ(off)= 595 ± 238 ms (at 1 µmol·L−1 glutamate), τ(off)= 746 ± 173 ms (at 1 mmol·L−1 glutamate), n= 7, P= 0.133].
Figure 6.
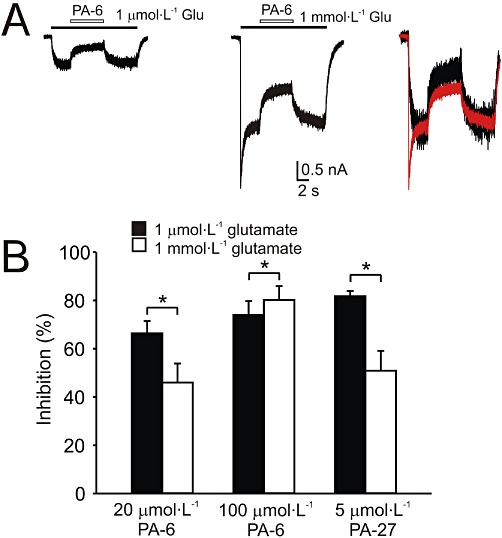
The effect of steroids on responses induced by low and high concentration of glutamate. (A) Example of responses induced by 1 µmol·L−1 and 1 mmol·L−1 glutamate in HEK293 cells expressing NR1/NR2B receptors. The responses diminished during co-application with 20 µmol·L−1 PA-6 by 64 and 42% respectively. On the right, both responses are displayed overlaid and normalized to show the difference in the degree of steroid-induced inhibition. (B) Bar graph represents mean ± SD (n= 5–7 cells) inhibition by PA-6 (20 µmol·L−1, 100 µmol·L−1) or PA-27 (5 µmol·L−1) of responses induced by glutamate (1 µmol·L−1, 1 mmol·L−1). *P < 0.001, significantly different from 1 µmol·L−1 glutamate; paired t-test. Note that 20 µmol·L−1 PA-6 was a more powerful inhibitor of responses induced by 1 µmol·L−1 glutamate, whereas at 100 µmol·L−1 it was a more powerful inhibitor of responses induced by 1 mmol·L−1 glutamate.
Our results suggest that the mechanism of inhibitory steroid action at NMDA receptor is complex. More powerful PA-6 (100 µmol·L−1) inhibition of responses to 1 mmol·L−1 glutamate than those induced by 1 µmol·L−1 glutamate is compatible with use-dependent action of this steroid at NMDA receptors. However, similar relative difference in the inhibition induced by steroids with the use-dependent and use-independent action (relative steroid inhibition of responses induced by 1 µmol·L−1 glutamate/relative inhibition of responses induced by 1 mmol·L−1 glutamate was 1.44 for 20 µmol·L−1 PA-6, the use-dependent inhibitor, and 1.61 for 5 µmol·L−1 PA-27, the use-independent inhibitor) suggest that at intermediate steroid concentrations additional mechanism(s) may account for the use-independent effect. This effect has some characteristics similar to that described earlier for lysophospholipids (Casado and Ascher, 1998), however, at this stage it is not clear whether inhibition induced by intermediate steroid concentrations and lysophospholipids are mediated by the similar or different mechanisms.
Membrane-delimited route of steroid access to the receptor
We considered the possibility that the kinetics of the steroid onset and offset inhibition governs its accumulation and release from a non-aqueous reservoir (perhaps the plasma membrane) from which the steroid reaches the binding site(s) at the NMDA receptor. To explore the possible involvement of the cytoplasmic membrane as an important reservoir for inhibitory steroids acting at NMDA receptors, we used cyclodextrins. These cyclic sugar molecules are capable, due to their inner hydrophobic structure, of accommodating lipophilic compounds such as cholesterol and of manipulating the content of these compounds in the cytoplasmic membrane (Yancey et al., 1996; Szejtli, 1998; Shu et al., 2004). We expected that, if the rate of recovery after the steroid-induced NMDA receptor inhibition is limited by steroid dissociation from the membrane, it would be accelerated by cyclodextrins, but if the rate of recovery is limited by the steroid unbinding from the receptor to the aqueous solution, then it would probably not be affected by the cyclodextrins.
Figure 7 shows the results of our experiments in which we studied the effects of three cyclodextrins (α-, β- and γ-), which differ by the substrate preference on the rate of recovery after PA-27 inhibition of responses to 1 mmol·L−1 glutamate. The results showed that the rate of recovery after 10 µmol·L−1 PA-27 and 1 mmol·L−1 glutamate co-application [τ(off)= 807 ± 226 ms; n= 5] was not significantly affected by 10 mmol·L−1α-cyclodextrin [τ(off)= 608 ± 65; n= 5; P= 0.143] (see superimposed responses on the right in Figure 7A). In the case of two other cyclodextrins, the recovery from PA-27 inhibition was significantly shortened, with τ(off)1= 133 ± 45 ms (A1= 68 ± 22%) and τ(off)2= 599 ± 104 ms (n= 5) in the presence of β-cyclodextrin (10 mmol·L−1) and τ(off)1= 68 ± 11 ms (A1= 63 ± 6%) and τ(off)2= 413 ± 108 ms (n= 5) in the presence of γ-cyclodextrin (10 mmol·L−1) (Figure 7A,B). We also studied the effects of cyclodextrins on the rate of recovery after the PA-6-induced inhibition of responses of recombinant GluN1/GluN2B receptors. Similarly, the rate of recovery of responses to 1 mmol·L−1 glutamate after 100 µmol·L−1 PA-6 co-application was accelerated by β-cyclodextrin (10 mmol·L−1) and γ-cyclodextrin (10 mmol·L−1), whereas α-cyclodextrin (10 mmol·L−1) had no effect (data not shown).
Figure 7.
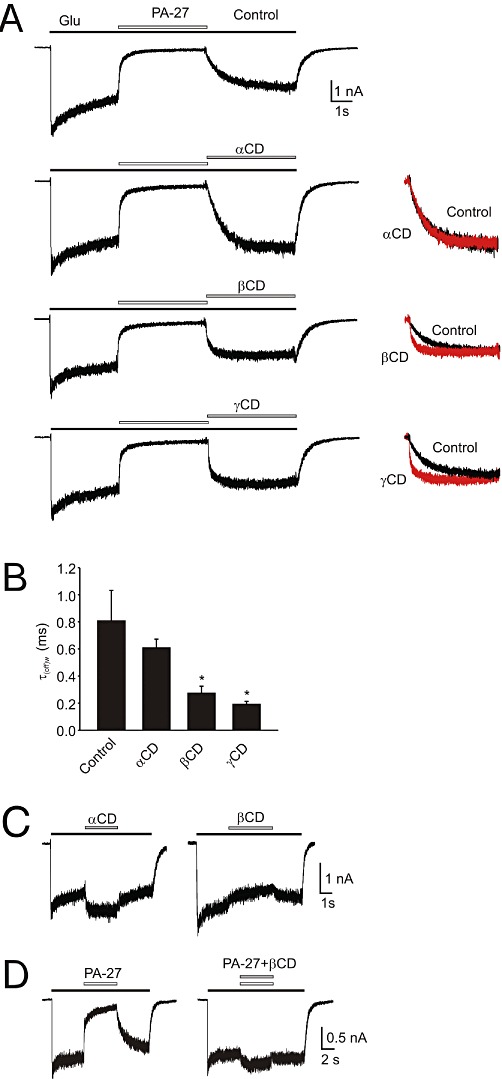
Cyclodextrin effects on the rate of recovery from PA-27-induced inhibition. (A) Example of a response to 1 mmol·L−1 glutamate and its co-application with 10 µmol·L−1 PA-27 (Control), and that recorded in the presence of 10 mmol·L−1α-cyclodextrin (αCD), 10 mmol·L−1β-cyclodextrin (βCD) or 10 mmol·L−1γ-cyclodextrin (γCD). On the left, recovery recorded in the absence of cyclodextrin (Control) and that recorded in the presence of cyclodextrin (in red) are shown normalized and superimposed. Note that the rate of recovery from PA-27-induced inhibition was accelerated in the presence of βCD and γCD. All records are from the same cell. (B) Summary of the effects of α-, β- and γ-cyclodextrin on the rate of recovery from inhibition induced by PA-27. Abscissa shows mean ± SD of the weighted single or double exponential fit to the recovery after steroid application (n= 5). *P < 0.05, significantly different from Control, paired t-test. (C) Effect of 10 mmol·L−1α- and β-cyclodextrin on 1 mmol·L−1 glutamate-induced responses. (D) Example of a response to 1 mmol·L−1 glutamate and its co-application with 10 µmol·L−1 PA-27 (Control), and that recorded in the presence of mixture of 10 mmol·L−1β-CD and 10 µmol·L−1 PA-27.
We subsequently performed control experiments in which we analysed the effect of α-, β- and γ-cyclodextrin on NMDA receptor responses (Figure 7C). In all studied cells 10 mmol·L−1α-cyclodextrin potentiated (+18 ± 2%; n= 4) responses induced by 1 mmol·L−1 glutamate, whereas β-cyclodextrin (10 mmol·L−1) and γ-cyclodextrin (10 mmol·L−1) inhibited the responses by −11 ± 2% and −8 ± 1% (n= 4) respectively. In addition, control experiments were performed in which the effectiveness of β-cyclodextrin to accommodate PA-27 was tested. Figure 7D shows that the inhibitory effect of PA-27 (10 µmol·L−1) on response mediated by NMDA receptors was abolished when PA-27 was co-applied with β-cyclodextrin (10 mmol·L−1). This indicates that β-cyclodextrin effectively reduces the free PA-27 concentration. Taken together, these data suggest that the slow recovery after steroid application is not a result of the slow rate of steroid unbinding from the receptor but rather of the slow emptying from the membrane.
To further support our hypothesis that the steroids accessed the receptor from cell membranes, we synthesized (see Methods for details) fluorescent analogues of PA-18 tagged with 7-nitrobenzo-2-oxa-1,3-diazole (NBD) (PA-38) and Alexa (PA-37) (Figure 8A). As shown in Figure 8B,C, both fluorescent steroids retained their activity at NMDA receptors. At 30 µmol·L−1 PA-38, the responses induced by 1 mmol·L−1 glutamate in HEK293 cell-expressing GluN1/GluN2B receptors were inhibited by 82.0 ± 4.8% (IC50= 8.5 ± 2.3 µmol·L−1, n= 6). The kinetics of the onset of inhibition was fit by double exponential function (τ(on)w= 885 ± 345 ms (n= 6). The recovery from inhibition was also slow, and was different for PA-38 (30 µmol·L−1) application for 3 s (τ(off)= 2.33 ± 0.55 s) and for 30 s (τ(off)= 3.84 ± 0.93 s; n= 6; t-test, P= 0.009). As observed with PA-38, PA-37 (60 µmol·L−1) also inhibited the responses to 1 mmol·L−1 glutamate by 77.9 ± 3.7% (IC50= 21.1 ± 3.7 µmol·L−1; n= 7), with slow on kinetics (τ(on)w= 743 ± 343 ms) and off kinetics (τ(off)= 1561 ± 389 ms; n= 7). These results showed that tagging PA-18 with a large fluorescent substitute (NBD or Alexa) did not substantially affect its inhibitory activity at NMDA receptors.
Figure 8.
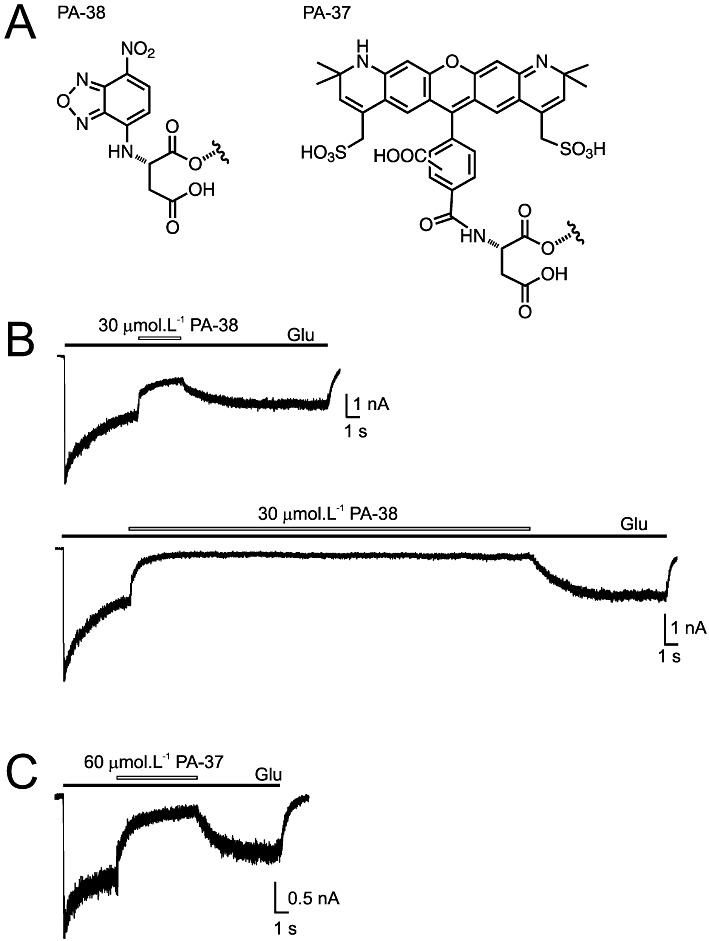
Fluorescent 3α5β analogues of PA retain their activity at NMDA receptors. (A) Chemical structures of fluorescent steroids used in electrophysiological and imaging experiments. (B) Inhibition of responses to 1 mmol·L−1 glutamate by co-application with 30 µmol·L−1 PA-38 applied for 3 s and 30 s (indicated by open bar) in HEK293 cells expressing NR1/NR2B receptors. (C) Response to 1 mmol·L−1 glutamate recorded from HEK293 cells expressing NR1/NR2B receptors and its inhibition by 60 µmol·L−1 PA-37.
Next, we aimed to assess whether fluorescently labelled steroids are retained in the membranes, as expected from our electrophysiological experiments. Using microscopy on live cells, we studied fluorescence labelling by PA-38 (30 µmol·L−1) of HEK293 cells. The fluorescence intensity increased linearly during PA-38 application, lasting seconds to a minute (see inset in Figure 9B). The fluorescence intensity was confined to the perinuclear region and reduced in the nuclear region (Figure 9A), indicating that PA-38 is a membrane-permeable dye that accumulates intracellularly. Following PA-38 application for 8 s and subsequent washing of the cell by control ECS, the fluorescence intensity diminished with time constant (τ= 1.6–2 min, depending on the cell region measured: the cell edge or the perinuclear region). For PA-38 applications lasting 1 min, the fluorescence intensity diminished quite slowly, with a half decay time of ∼5 min. Differences in the on and off kinetics of PA-38 fluorescence intensity and inhibition of NMDA receptor responses (more than 10-fold; Figures 8B and 9B) suggested that there are multiple membrane pools in which PA-38 concentrated; however, only some pools may be important for direct access of the steroid to the NMDA receptor.
Figure 9.
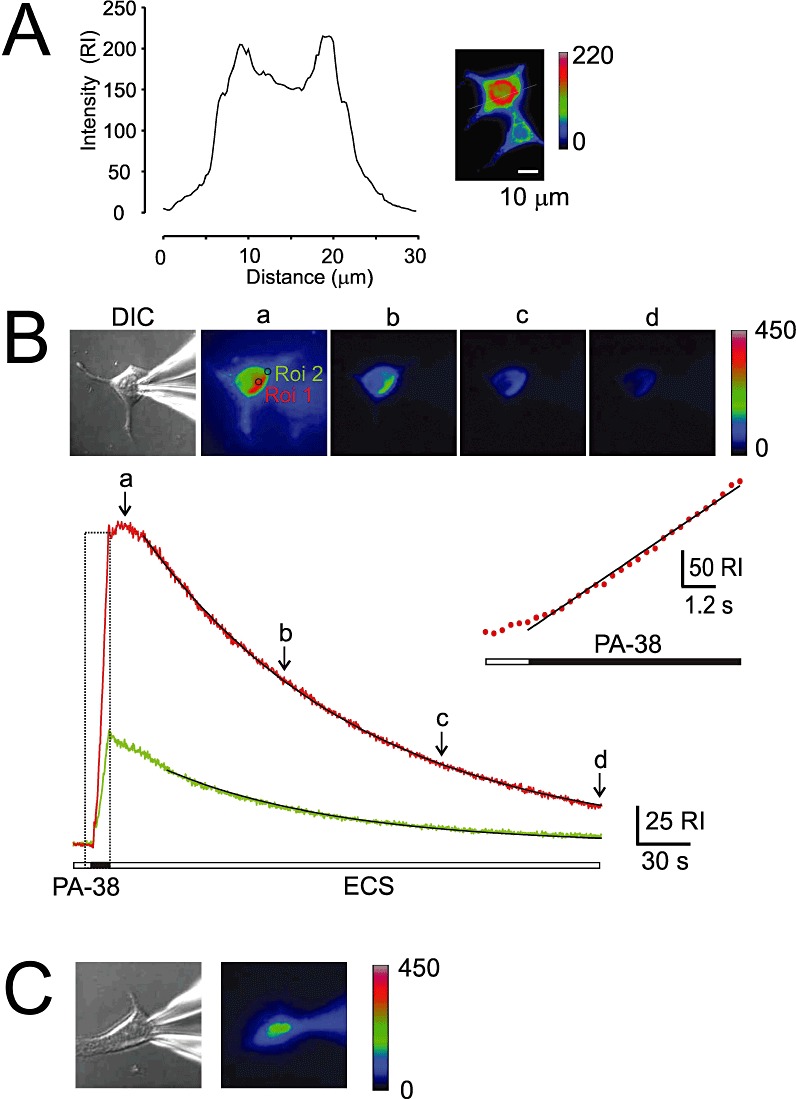
Imaging analysis of fluorescent steroid analogues. (A) HEK293 cells were stained with 100 µmol·L−1 PA-38 extracellularly applied for 5 s and subsequently washed with ECS for 3 s before the image was acquired. Fluorescence intensity is indicated in pseudocolours. Graph shows relative fluorescence intensity (relative intensity, RI, indicates fluorescence intensity measured outside the cell subtracted from that in the cell) in a cross-section (indicated by a line). (B) HEK293 cell was stained with PA-38 (30 µmol·L−1) extracellularly applied for 8 s. The images of the cell were acquired using DIC and fluorescence microscopy at 300 ms intervals. Analysis of the fluorescence accumulation in two cell regions (R1, perinuclear in red, and R2, cell edge in green) as a function of time. Images were acquired at time intervals indicated (a-d). Time-course of the fluorescence onset (boxed region) was fitted to a linear regression (slope 1632 ± 24 RI·min−1) and is shown on expanded time scale in inset. Time-course of the fluorescence offset was fitted by a single exponential function (2 min perinuclear region; 1.6 min for cell edge region). Note that the fluorescence intensity increased rapidly during steroid application; however, it slowly continued for 10 s after the steroid was removed from the ECS. (C) DIC and fluorescence image of a HEK293 cell acquired 15 min after the start of whole-cell recording and intracellular dialysis with ICS containing 100 µM PA-37.
The results of imaging experiments with PA-38 are compatible with the idea that intracellularly accumulated steroid is not directly necessary for receptor inhibition, and they indirectly strengthen the hypothesis that cytoplasmic membrane accumulation is how the steroid accesses the receptor. To test this hypothesis further, we used another fluorescent steroid PA-37 (Figure 8). This compound is highly charged and therefore likely to be less membrane permeant (Akk et al., 2005; Revankar et al., 2005). We found that extracellularly applied PA-37 (100 µmol·L−1) failed to stain live cells even when applied for minutes (data not shown). In contrast, imaging of patch-clamped HEK293 cells intracellularly dialysed with 100 µmol·L−1 PA-37 revealed a gradual increase in fluorescence intensity during the period of whole-cell recording lasting for 15 min (Figure 9C). Lack of staining of live cells after extracellular PA-37 application observed in our experiments was also described for Alexa-labelled steroid acting at GABAA receptors (Akk et al., 2005). The results of our experiments have shown that fluorescence intensity of PA-37 dissolved in protic solvent (aqueous solution) was markedly reduced in aprotic solvents (sixfold in DMSO and 23-fold in acetone). This is the opposite of what was determined for NBD-labelled compounds, which are almost nonfluorescent in aqueous solvents (Chattopadhyay, 1990). It is therefore likely that both Alexa and NBD fluorescent steroids concentrate in the cell membrane to modulate the activity of NMDA receptors; however, in contrast to PA-38, PA-37 is almost non-fluorescent when placed at the lipid–water interface.
Discussion
The studies presented here engaged electrophysiological and optical techniques to characterize the mechanism by which PA-6 and its synthetic analogues inhibit NMDA receptors. Our results indicate that steroids can reach the binding site(s) at the NMDA receptor via a membrane-delimited route rather than directly from the aqueous milieu. Second, we show that a charged residue attached to steroid C3 is required for inhibition of NMDA receptors. Third, we show that the onset of inhibition induced by different C3-substituted analogues can be use-dependent or use-independent. Finally, we show that the onset and offset of cell staining by fluorescent steroid analogues are slower than the onset and offset of inhibition induced by these steroids.
There has been considerable interest in understanding the molecular mechanisms of the pharmacological regulation of NMDA receptor function. Several binding sites providing allosteric regulation of agonist binding and channel opening and closing have been identified on the extracellular domains of the NMDA receptor (Traynelis et al., 2010). The membrane region of the receptor has been investigated, particularly for substances such as small cations that block the open channel; however, surprisingly little is known about possible modulation involving the membrane domain at the lipid-protein interface (Salous et al., 2009; Ogden and Traynelis, 2011).
Kinetic experiments in which we studied the time-course of steroid-induced inhibition indicate that onset of inhibition had a complex dependency on the steroid concentration; this is in contrast to the recovery from inhibition, which was dependent on the concentration of the steroid applied (Figure 4). Theoretical assumptions require ligand binding in the aqueous solution kon[Drug] > koff. The experimentally obtained estimates of kon and koff for steroid acting at NMDA receptors (see Figure 4) provided either that kon[Drug] < koff (4/6 cells when studied at 10 µmol·L−1 PA-6; 5/6 cells at 30 µmol·L−1 PA-6; 1/6 cells at 100 µmol·L−1 PA-6) or that the affinity (calculated as Kd= koff/kon) was different from the IC50 (Kd range 7–307 µmol·L−1). Several factors may account for this atypical drug-receptor interaction (Popescu, 2005); for instance, the kinetics of the steroid-induced inhibition may not be determined by the rate constants of steroid binding and unbinding, or the free steroid concentration at the site of action may be different from that applied in aqueous solution. Indeed, the results of our experiments in which cyclodextrins were used, which showed that the rate of recovery of responses after steroid inhibition of NMDA receptors was accelerated in the presence of cyclodextrins, strongly indicate that at least the rate of recovery from steroid inhibition is not strictly determined by steroid koff (Figure 7).
Cyclodextrins are known to remove various amphipathic drugs from cell membranes, including cholesterol and neurosteroids (Adam et al., 2002; Shu et al., 2004; Zidovetzki and Levitan, 2007). On the basis of these results, we propose that neuroactive steroids may target the NMDA receptor through a site that is accessible through lateral membrane diffusion rather than by aqueous access. The slow kinetics of the onset and offset of inhibition reflect the time required for their diffusion from the ECS into the membrane and back. The suggestion that inhibitory steroids access the binding site(s) at the NMDA receptor via a membrane-delimited route is further supported by recent results from Farb's group showing that replacement of alanine 651 at the third membrane region of GluN2A with threonine diminished inhibition by PA-6 at GluN1/GluN2A receptors expressed in oocytes (Kostakis et al., 2011).
To further test the hypothesis of membrane-delimited action of inhibitory steroids at NMDA receptors, we made an attempt to calculate the partition coefficient {log P; = log [(steroid)octanol/(steroid)water} of studied steroids. It is generally difficult to predict log P of amphipathic compounds as the lipophilic moiety preferentially partitions into the lipid environment and the charged group preferentially partitions into the aqueous environment. Therefore, for the first approximation, we calculated log P for steroids in which the part of the carbon skeleton after the first ionized residue of the C3 subsistent was substituted for methyl (e.g. aspartate residue of PA-21 was substituted for propionate; Figure 10). Even though estimates of log P must be interpreted cautiously, the plot of the IC50 of steroid inhibition of NMDA receptors as a function of log P exhibited positive correlation (r = 0.804 for plot of steroids derived from those bearing positive and negative charge; r = 0.819 for only negative charged steroids).
Figure 10.
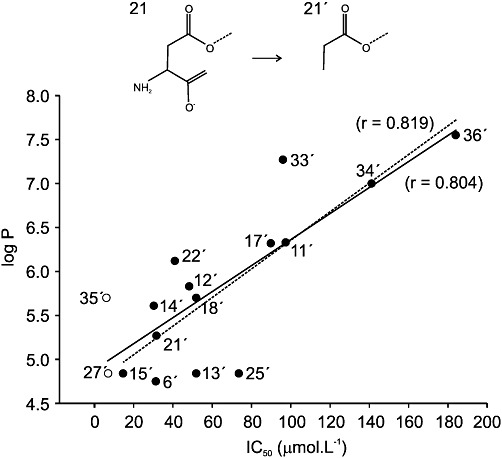
Correlation between inhibitory effects of steroid analogues and their lipophilicity. The ordinate gives the IC50 for inhibition (see Table 1), and the abscissa shows the lipophilicity index (log P). The lipophilicity index was estimated for steroids in which the part of the carbon skeleton after the first ionized residue of the C3 subsistent was substituted for methyl (e.g. aspartate residue of PA-21 was substituted for propionate). The solid line shows the regression of IC50 on log P (Pearson correlation coefficient, r = 0.804, P = 0.00017) for structures derived from negatively charged (closed symbols) and all charged (open symbols) steroids, whereas the dashed line is the regression (Pearson correlation coefficient r = 0.819, P= 0.00034) for structures derived from only negatively charged steroids.
Surprisingly, the relation between steroid IC50 and its lipophilicity indicates that more powerful inhibitors are less lipophilic compounds. To understand this apparent paradox, one has to bear in mind that IC50 is an empirical term that relates to free aqueous concentration and the effect (affinity and efficacy). As the drug's lipophilicity and its affinity are not directly related, it is likely that, on the one hand, the structural alteration of the steroid molecule can increase lipophilicity – resulting in diminution of the value of IC50– whereas, on the other hand, the same structural change may result in a decrease of the drug's affinity and therefore increase the value of IC50. The resultant inhibition will therefore be dependent on both the lipophilicity and the affinity/efficacy. A similar paradox was observed for the potentiation of GABAA responses by steroid analogues – when the efficacy of four selected steroids was compared, the efficacy and log P were inversely correlated, but for a larger group of 68 compounds there was a positive correlation (Chisari et al., 2009).
In addition, the plot of the steroid IC50 versus its lipophilicity indicates that positively charged steroids are distributed along the linear regression of negatively charged steroids, indicating that high-affinity steroid action at NMDA receptors (PA-27 and PA-35) may not be attributed to their positive charge but rather to their low lipophilicity (Figure 10).
Arguments supporting the hypothesis of membrane-delimited action of inhibitory steroids at NMDA receptors might be hampered by the fact that the rate of recovery of NMDA receptor currents after steroid inhibition was different from that determined for the diminution of fluorescence after PA-38 application (Figures 8 and 9). There could be several reasons for this difference. The site of steroid action at NMDA receptors is likely to be spatially restricted, and perhaps only steroid access from the exterior leaf of the cell membrane is important for receptor inhibition. In optical measurements, the slow rate of fluorescence diminution after PA-38 application reflects steroid diffusion from cell membranes, including the intracellular compartment, which comprises most of the cell membrane content (Blouin et al., 1977). In addition, the difference in the recovery rate after steroid application observed in electrophysiological and optical experiments may be caused by differences in the rate of steroid diffusion from the membrane. The cytoplasmic membrane is thought to contain both raft (defined as cholesterol- and sphingolipid-enriched membrane microdomains) and non-raft domains, with NMDA receptors preferentially associated with the lipid rafts (Besshoh et al., 2007; Korade and Kenworthy, 2008). As for different compounds, including lipids, which preferentially concentrate in different membrane domains, it is quite likely that the steroid concentration in raft and non-raft domains is different.
We have shown previously that PA-6 is a use-dependent allosteric inhibitor of NMDA receptors (Petrovic et al., 2005). Results of experiments presented here (Figure 5) indicate that NMDA receptor responses made immediately after steroid pre-application in the absence of agonist have three different onsets. First, typical for PA-6, indicating that this steroid does not bind to the non-activated receptor and, further, that at the time of NMDA receptor activation there is no spare pool of the steroid that could inhibit the receptor after its activation. Second, typical of PA-22, indicating that this steroid also does not bind to the non-activated receptor; however, at the time of NMDA receptor activation there is presumably a spare pool of the steroid that could inhibit the receptor almost completely after its activation. Finally, typical of PA-27, indicating that this steroid binding to the receptor is use-independent. It is difficult to hypothesize on the molecular mechanism of the use-dependent and use-independent action of steroids at NMDA receptors without detailed knowledge of the biophysics of steroid interaction with the membrane and the receptor structure. Although our results seem to be compatible with the hypothesis of steroid accumulation in the membrane and inhibition of the receptor after its activation, some results are difficult to accommodate in this mode of action. For instance, it is not clear why PA-22, which has a log P similar to that of PA-6, induces subsequent inhibition of NMDA receptor responses after receptor activation, in contrast to PA-6. This invites speculation on the structural requirements of the steroid action at the NMDA receptor and whether there is only one pathway involving membrane to receptor interaction or whether the same site can be reached from the receptor vestibule after receptor activation. In any case the crystal structure of the AMPA receptor provides space that the receptor vestibule can adopt for the steroid molecule (Sobolevsky et al., 2009).
In summary, on the basis of the results of kinetic and pharmacological experiments, we propose that NMDA receptor activity can be inhibited by the membrane-delimited action of neuroactive steroids. This may help to identify the steroid binding sites at the receptor and the molecular mechanism of their action. In addition, the results of our experiments may improve the drug design of new pharmaceutical compounds for treating symptoms and diseases in which the pathology of NMDA receptors is involved (Endele et al., 2010).
Acknowledgments
This work was supported by the Grant Agency of the Czech Republic (309/07/0271; 203/08/1498; 309/08/H079; P303/11/0075; P303/12/1464); ED0007/01/01, Research Project of the AS CR AV0Z 50110509, and Ministry of Education, Youth and Sports of the Czech Republic (1 M0517 and LC554). Work of E. S, V. K, B. S., H. C. was supported by Research Project Z4 055 0506. We thank M. Kuntosova and D. Hybsova for excellent technical assistance.
Glossary
- ECS
extracellular solution
- GFP
green fluorescent protein
- HR-MS
high-resolution MS
- ICS
intracellular solution
- NBD
7-nitrobenz-2-oxa-1,3-diazol-4-yl
- PA-4
3α-azido-5β-pregnan-20-one
- PA-5
5β-pregnan-20-one-3α-yl nitrate
- PA-6
pyridinium 20-oxo-5β-pregnan-3α-yl sulphate
- PA-11
2-(20-oxo-5β-pregnan-3α-yl)-acetic acid
- PA-12
2-(20-oxo-5β-pregnan-3α-yl)-propanedioic acid
- PA-13
20-oxo-5β-pregnan-3α-yl hemisuccinate, PA-14, 20-oxo-5β-pregnan-3α-yl 3-carboxy-ξ-methyl-propanoate
- PA-15
20-oxo-5β-pregnan-3α-yl (Z)-3-carboxyprop-2-enoate
- PA-17
20-oxo-5β-pregnan-3α-yl N-(tert-butoxycarbonyl)-L-aspartyl 1-ester
- PA-18
20-oxo-5β-pregnan-3α-yl L-aspartyl 1-ester
- PA-21
20-oxo-5β-pregnan-3α-yl L-aspartyl 4-ester
- PA-22
20-oxo-5β-pregnan-3α-yl adipyl 1-ester
- PA-25
20-oxo-5β-pregnan-3α-yl L-glutamyl 1-ester, PA-27, 20-oxo-5β-pregnan-3α-yl L-argininate dihydrochloride salt
- PA-33
4-(20-oxo-5β-pregnan-3α-yl)-benzoic acid
- PA-34
20-oxo-5β-pregnan-3α-yl 6-carboxyhexanoate
- PA-35
20-oxo-5β-pregnan-3α-yl 4-(trimethylammonio)-butanoate chloride
- PA-36
20-oxo-5β-pregnan-3α-yl 7-carboxyheptanoate
Conflict of interest
No conflict of interests exists.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Correlation between inhibitory effects of steroid analogues and their structure. Shown is a scatter plot of data for sixteen C3-substituted steroid analogues (see Figure 1 for their structure). The ordinate gives the IC50 for inhibition (see Table 1), and the abscissa shows the distance between the charged moiety and the C3 of the steroid skeleton (distance) (see Methods for details). The solid line shows the regression of IC50 on distance (correlation coefficient r = 0.60). Inset shows an example of a 3D structure of PA-25 as determined from 3D modelling (see Methods).
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Adam JM, Bennett DJ, Bom A, Clark JK, Feilden H, Hutchinson EJ, et al. Cyclodextrin-derived host molecules as reversal agents for the neuromuscular blocker rocuronium bromide: synthesis and structure-activity relationships. J Med Chem. 2002;45:1806–1816. doi: 10.1021/jm011107f. [DOI] [PubMed] [Google Scholar]
- Akk G, Shu HJ, Wang C, Steinbach JH, Zorumski CF, Covey DF, et al. Neurosteroid access to the GABAA receptor. J Neurosci. 2005;25:11605–11613. doi: 10.1523/JNEUROSCI.4173-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th Edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besshoh S, Chen S, Brown IR, Gurd JW. Developmental changes in the association of NMDA receptors with lipid rafts. J Neurosci Res. 2007;85:1876–1883. doi: 10.1002/jnr.21336. [DOI] [PubMed] [Google Scholar]
- Blouin A, Bolender RP, Weibel ER. Distribution of organelles and membranes between hepatocytes and nonhepatocytes in the rat liver parenchyma. A stereological study. J Cell Biol. 1977;72:441–455. doi: 10.1083/jcb.72.2.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock AE, Clark AL, Grady SR, Robinson SF, Slobe BS, Marks MJ, et al. Neurosteroids modulate nicotinic receptor function in mouse striatal and thalamic synaptosomes. J Neurochem. 1997;68:2412–2423. doi: 10.1046/j.1471-4159.1997.68062412.x. [DOI] [PubMed] [Google Scholar]
- Cais O, Sedlacek M, Horak M, Dittert I, Vyklicky L., Jr Temperature dependence of NR1/NR2B NMDA receptor channels. Neuroscience. 2008;151:428–438. doi: 10.1016/j.neuroscience.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Casado M, Ascher P. Opposite modulation of NMDA receptors by lysophospholipids and arachidonic acid: common features with mechanosensitivity. J Physiol (Lond) 1998;513:317–330. doi: 10.1111/j.1469-7793.1998.317bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay A. Chemistry and biology of N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)-labeled lipids: fluorescent probes of biological and model membranes. Chem Phys Lipids. 1990;53:1–15. doi: 10.1016/0009-3084(90)90128-e. [DOI] [PubMed] [Google Scholar]
- Chisari M, Eisenman LN, Krishnan K, Bandyopadhyaya AK, Wang C, Taylor A, et al. The influence of neuroactive steroid lipophilicity on GABAA receptor modulation: evidence for a low-affinity interaction. J Neurophysiol. 2009;102:1254–1264. doi: 10.1152/jn.00346.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corpechot C, Robel P, Axelson M, Sjovall J, Baulieu EE. Characterization and measurement of dehydroepiandrosterone sulfate in rat brain. Proc Natl Acad Sci U S A. 1981;78:4704–4707. doi: 10.1073/pnas.78.8.4704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endele S, Rosenberger G, Geider K, Popp B, Tamer C, Stefanova I, et al. Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nat Genet. 2010;42:1021–1026. doi: 10.1038/ng.677. [DOI] [PubMed] [Google Scholar]
- Flood JF, Roberts E. Dehydroepiandrosterone sulfate improves memory in aging mice. Brain Res. 1988;448:178–181. doi: 10.1016/0006-8993(88)91116-x. [DOI] [PubMed] [Google Scholar]
- Grobin AC, Roth RH, Deutch AY. Regulation of the prefrontal cortical dopamine system by the neuroactive steroid 3a,21-dihydroxy-5a-pregnane-20-one. Brain Res. 1992;578:351–356. doi: 10.1016/0006-8993(92)90270-j. [DOI] [PubMed] [Google Scholar]
- Korade Z, Kenworthy AK. Lipid rafts, cholesterol, and the brain. Neuropharmacology. 2008;55:1265–1273. doi: 10.1016/j.neuropharm.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostakis E, Jang MK, Russek SJ, Gibbs TT, Farb DH. A steroid modulatory domain in NR2A collaborates with NR1 exon-5 to control NMDAR modulation by pregnenolone sulfate and protons. J Neurochem. 2011;119:486–496. doi: 10.1111/j.1471-4159.2011.07442.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kussius CL, Kaur N, Popescu GK. Pregnanolone sulfate promotes desensitization of activated NMDA receptors. J Neurosci. 2009;29:6819–6827. doi: 10.1523/JNEUROSCI.0281-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapchak PA. The neuroactive steroid 3-alpha-ol-5-beta-pregnan-20-one hemisuccinate, a selective NMDA receptor antagonist improves behavioral performance following spinal cord ischemia. Brain Res. 2004;997:152–158. doi: 10.1016/j.brainres.2003.10.047. [DOI] [PubMed] [Google Scholar]
- Mayer ML, Westbrook GL, Guthrie PB. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature. 1984;309:261–263. doi: 10.1038/309261a0. [DOI] [PubMed] [Google Scholar]
- Nasman B, Olsson T, Backstrom T, Eriksson S, Grankvist K, Viitanen M, et al. Serum dehydroepiandrosterone sulfate in Alzheimer's disease and in multi-infarct dementia. Biol Psychiatry. 1991;30:684–690. doi: 10.1016/0006-3223(91)90013-c. [DOI] [PubMed] [Google Scholar]
- Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A. Magnesium gates glutamate-activated channels in mouse central neurones. Nature. 1984;307:462–465. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- Ogden KK, Traynelis SF. New advances in NMDA receptor pharmacology. Trends Pharmacol Sci. 2011;32:726–733. doi: 10.1016/j.tips.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park-Chung M, Wu FS, Farb DH. 3 alpha-Hydroxy-5 beta-pregnan-20-one sulfate: a negative modulator of the NMDA-induced current in cultured neurons. Mol Pharmacol. 1994;46:146–150. [PubMed] [Google Scholar]
- Petrovic M, Sedlacek M, Horak M, Chodounska H, Vyklicky L., Jr 20-oxo-5beta-pregnan-3alpha-yl sulfate is a use-dependent NMDA receptor inhibitor. J Neurosci. 2005;25:8439–8450. doi: 10.1523/JNEUROSCI.1407-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popescu G. Mechanism-based targeting of NMDA receptor functions. Cell Mol Life Sci. 2005;62:2100–2111. doi: 10.1007/s00018-005-5227-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy DS. Neurosteroids: endogenous role in the human brain and therapeutic potentials. Prog Brain Res. 2010;186:113–137. doi: 10.1016/B978-0-444-53630-3.00008-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307:1625–1630. doi: 10.1126/science.1106943. [DOI] [PubMed] [Google Scholar]
- Rupprecht R, Holsboer F. Neuroactive steroids: mechanisms of action and neuropsychopharmacological perspectives. Trends Neurosci. 1999;22:410–416. doi: 10.1016/s0166-2236(99)01399-5. [DOI] [PubMed] [Google Scholar]
- Salous AK, Ren H, Lamb KA, Hu XQ, Lipsky RH, Peoples RW. Differential actions of ethanol and trichloroethanol at sites in the M3 and M4 domains of the NMDA receptor GluN2A (NR2A) subunit. Br J Pharmacol. 2009;158:1395–1404. doi: 10.1111/j.1476-5381.2009.00397.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedlacek M, Korinek M, Petrovic M, Cais O, Adamusova E, Chodounska H, et al. Neurosteroid modulation of ionotropic glutamate receptors and excitatory synaptic transmission. Physiol Res. 2008;57(Suppl. 3):S49–S57. doi: 10.33549/physiolres.931600. [DOI] [PubMed] [Google Scholar]
- Shu HJ, Eisenman LN, Jinadasa D, Covey DF, Zorumski CF, Mennerick S. Slow actions of neuroactive steroids at GABAA receptors. J Neurosci. 2004;24:6667–6675. doi: 10.1523/JNEUROSCI.1399-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobolevsky AI, Rosconi MP, Gouaux E. X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature. 2009;462:745–756. doi: 10.1038/nature08624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stastna E, Chodounska H, Pouzar V, Kapras V, Borovska J, Cais O, et al. Synthesis of C3, C5, and C7 pregnane derivatives and their effect on NMDA receptor responses in cultured rat hippocampal neurons. Steroids. 2009;74:256–263. doi: 10.1016/j.steroids.2008.11.011. [DOI] [PubMed] [Google Scholar]
- Szejtli J. Introduction and General Overview of Cyclodextrin Chemistry. Chem Rev. 1998;98:1743–1754. doi: 10.1021/cr970022c. [DOI] [PubMed] [Google Scholar]
- Tetko IV. Computing chemistry on the web. Drug Discov Today. 2005;10:1497–1500. doi: 10.1016/S1359-6446(05)03584-1. [DOI] [PubMed] [Google Scholar]
- Tetko IV, Gasteiger J, Todeschini R, Mauri A, Livingstone D, Ertl P, et al. Virtual computational chemistry laboratory–design and description. J Comput Aided Mol Des. 2005;19:453–463. doi: 10.1007/s10822-005-8694-y. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, et al. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev. 2010;62:405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallee M, Mayo W, Darnaudery M, Corpechot C, Young J, Koehl M, et al. Neurosteroids: deficient cognitive performance in aged rats depends on low pregnenolone sulfate levels in the hippocampus. Proc Natl Acad Sci U S A. 1997;94:14865–14870. doi: 10.1073/pnas.94.26.14865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver CE, Jr, Marek P, Park-Chung M, Tam SW, Farb DH. Neuroprotective activity of a new class of steroidal inhibitors of the N-methyl-D-aspartate receptor. Proc Natl Acad Sci U S A. 1997;94:10450–10454. doi: 10.1073/pnas.94.19.10450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu FS, Gibbs TT, Farb DH. Inverse modulation of gamma-aminobutyric acid- and glycine-induced currents by progesterone. Mol Pharmacol. 1990;37:597–602. [PubMed] [Google Scholar]
- Wu FS, Gibbs TT, Farb DH. Pregnenolone sulfate: a positive allosteric modulator at the N-methyl-D- aspartate receptor. Mol Pharmacol. 1991;40:333–336. [PubMed] [Google Scholar]
- Yancey PG, Rodrigueza WV, Kilsdonk EP, Stoudt GW, Johnson WJ, Phillips MC, et al. Cellular cholesterol efflux mediated by cyclodextrins. Demonstration Of kinetic pools and mechanism of efflux. J Biol Chem. 1996;271:16026–16034. doi: 10.1074/jbc.271.27.16026. [DOI] [PubMed] [Google Scholar]
- Zidovetzki R, Levitan I. Use of cyclodextrins to manipulate plasma membrane cholesterol content: evidence, misconceptions and control strategies. Biochim Biophys Acta. 2007;1768:1311–1324. doi: 10.1016/j.bbamem.2007.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.