

Abstract
BACKGROUND AND PURPOSE
Endostatin (ES) is a c-terminal proteolytic fragment of collagen XVIII with promising antitumour properties in several tumour models, including human glioblastoma. We hypothesized that this peptide could interact with plasma membrane ion channels and modulate their functions.
EXPERIMENTAL APPROACH
Using cell proliferation and migration assays, patch clamp and Western blot analysis, we studied the effects of ES on the proliferation and migration of human glioblastoma U87 cells, mediated by T-type Ca2+ channels.
KEY RESULTS
Extracellular application of ES reversibly inhibited T-type Ca2+ channel currents (T-currents) in U87 cells, whereas L-type Ca2+ currents were not affected. This inhibitory effect was associated with a hyperpolarizing shift in the voltage-dependence of inactivation but was independent of G-protein and protein tyrosine kinase-mediated pathways. All three α1 subunits of T-type Ca2+ channels (CaV3), α1G (CaV3.1), α1H (CaV3.2) and α1I (CaV3.3), were endogenously expressed in U87 cells. Using transfected HEK293 or CHO cells, we showed that only CaV3.1 and CaV3.2, but not CaV3.3 or CaV1.2 (L-type), channel currents were significantly inhibited. More interestingly, ES inhibited the proliferation and migration of U87 cells in a dose-dependent manner. Pretreatment of the cells with the specific T-type Ca2+ channel blocker mibefradil occluded these inhibitory effects of ES.
CONCLUSION AND IMPLICATIONS
This study provides the first evidence that the antitumour effects of ES on glioblastoma cells is through direct inhibition of T-type Ca2+ channels and gives new insights into the future development of a new class of antiglioblastoma agents that target the proliferation and migration of these cells.
LINKED ARTICLE
This article is commented on by Santoni et al., pp. 1244–1246 of this issue. To view this commentary visit http://dx.doi.org/10.1111/j.1476-5381.2012.01908.x
Keywords: endostatin, T-type Ca2+ channels, glioblastoma cells, proliferation
Introduction
Glioblastoma is the most common and most aggressive malignant primary brain tumour in humans. This glia-derived brain tumour is highly invasive, rapidly proliferates and neurologically destructive, and responds poorly to chemotherapy (Walker and Kaye, 2001). The treatment of glioblastoma remains palliative and no significant advancements in the treatment have developed in the past years. Recently, several tumour-derived, circulating angiogenesis inhibitors generated in vivo by proteolytic degradation have been identified (Cao, 2001; Skovseth et al., 2005). In particular, a 20-kDa C-terminal proteolytic fragment of collagen XVIII, termed endostatin (ES), inhibits tumour growth in several in vivo tumour models, including human glioblastoma (Boehm et al., 1997; Sorensen et al., 2002). For example, the endogenous expression of ES by C6 glioma cells results in a reduced tumour growth rate in vivo (Peroulis et al., 2002). Also, local intracerebral microinfusion of ES improves treatment efficiency and survival in an orthotopic human glioblastoma model (Schmidt et al., 2004). The antitumour effect of ES is probably through the inhibition of angiogenesis (Sorensen et al., 2002; Schmidt et al., 2004). Interestingly, there is growing evidence that ES can elicit a direct effect in tumour cells (Yang et al., 2011). It has been reported recently that peptide 30 derived from ES suppresses the proliferation and migration of HepG2 cells in vitro (Li et al., 2011). However, whether and how ES directly affects tumour cells, especially glioblastoma cells, is less clear.
T-type Ca2+ channels are a class of calcium-permeable low-voltage-activated (LVA) channels that open after small depolarizations of the membrane. Through conducting Ca2+ entry and changing [Ca2+]i, T-type Ca2+ channel is crucial for the orderly progression of the cell cycle and plays a vital role in the regulation of cell proliferation, growth and gene expression (Ciapa et al., 1994). In mammals, three α1-subunit genes have been described that encode distinct T-type Ca2+ channels with unique biophysical and pharmacological properties: Cav3.1 (α1G), Cav3.2 (α1H) and Cav3.3 (α1I) (Perez-Reyes, 2003). It has long been hypothesized that there exists a link between T-type Ca2+ channels and cancer incidence and progression (Lory et al., 2006). Recent studies also show that α1 subunits of T-type Ca2+ channels are expressed in cancerous cells and participate in tumour pathophysiology; however, the investigations of their functions have just begun (Gray and Macdonald, 2006). For example, aberrant up-regulation of the gene encoding T-type Ca2+ channel α1G subunit was detected in various human primary tumours, suggesting that T-type Ca2+ channels may play a role in cancer development by modulating Ca2+ signalling (Toyota et al., 1999). Indeed, the T-type Ca2+ channel has been implicated in proliferation in several tumours (Panner and Wurster, 2006). There is a growing body of evidence suggesting that tumour cell proliferation could be halted by the use of T-type Ca2+ channel blockers. Furthermore, knocking down the expression of T-type Ca2+ channels with siRNA targeting both α1G and α1H resulted in growth inhibition in MCF-7 cells, a human breast cancer cell line (Taylor et al., 2008). Since T-type Ca2+ channels regulate cell proliferation, which is a key feature of tumour cells, we hypothesize that the manipulation of T-type Ca2+ channels could have promising clinical potential for treating highly proliferative tumours, such as glioblastoma.
In this study, we identified a novel function of ES in modulating cell proliferation and migration by targeting T-type Ca2+ channels in U87 human glioblastoma cells, in which all three α1 subunits of CaV3 were endogenously expressed. By using heterologously HEK293 or CHO cell expressing system, we found that only CaV3.1 and CaV3.2, but not CaV3.3 or CaV1.2, channel currents were inhibited by ES. Our results highlight the novel mechanism and therapeutic potential of ES via targeting T-type Ca2+ channels for the treatment of human glioblastoma.
Methods
Cell culture and transfection
All reagents were obtained from Sigma (St. Louis, MO, USA) unless otherwise stated. The drug/molecular target nomenclature (e.g. receptors, ion channels and so on) used in the present study conforms to BJP's Guide to Receptors and Channels (Alexander et al., 2011). The U87 human glioblastoma cell lines were obtained from American Type Culture Collection (ATCC, Rockville, MD, USA). U87, HEK-293 and CHO cells were cultured using standard techniques as described in our previous reports (Tao et al., 2008; 2009a). To measure the U87 cell membrane currents, the cells from the stock culture were plated onto glass coverslips and used for experiments 2–3 days after plating. Transient transfection in HEK293 or CHO was performed using the standard calcium phosphate transfection method (Tao et al., 2008) with a DNA mix containing 1:9 ratios (by weight) of GFP plasmid and constructs encoding for human CaV3.1, CaV3.2 and CaV3.3 isoforms. The full-length human CaV3.1, CaV3.2 and CaV3.3 α1-subunits (kindly provided by Dr Terry P Snutch, University of British Columbia, Canada) were cloned in the pcDNA3 vectors.
Reverse transcription-PCR (RT-PCR)
Total RNA was extracted from U87 cells as described previously (Tao et al., 2009a; Wang et al., 2011). Reverse transcription was carried out with SuperScriptTMII (Invitrogen, Carlsbad, CA, USA). The sequences of the primers employed in this study are summarized in Table 1. The PCR protocol includes a denaturation step at 95°C for 2 min, denaturation, annealing and elongation were carried out at 94°C for 30 s, at 65°C for 20 s and at 72°C for 1 min. PCR was carried out for 33 cycles. PCR analysis was repeated at least twice with the same samples to confirm reproducibility of the results.
Table 1.
Sequence of primers for three α1 subunits (α1G, α1H, α1I) of T-type Ca2+ channels
Western blot analysis
Western blotting was performed by following the procedures as described in our previous studies (Wang et al., 2011; Zhang et al., 2011). For antibody detection, after blocking with 5% non-fat milk in TBST for 1 h at room temperature, membranes were incubated with diluted primary polyclonal goat antihuman CaV3.1, CaV3.2 or CaV3.3 antibodies (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) (1:500) and incubated at 4°C for overnight. After five washes with TBST, membranes were incubated for 2 h with 1:5000 diluted donkey antigoat secondary antibody (Sigma, St. Louis, MO, USA). After five washes, the specific binding of the primary antibody was detected with SuperSignal Ultra chemiluminescent substrate (Pierce, Rockford, IL, USA).
Whole-cell patch clamp recording
Recordings were made using standard whole-cell techniques at room temperature as previously described (Tao et al., 2008; 2009b; Wang et al., 2011; Zhang et al., 2011). Electrodes were pulled from borosilicate glass microcapillary tubes (World Precision Instruments, Sarasota, FL, USA). They had resistances from 2 to 3 MΩ when filled with internal solution. We made recordings using a MultiClamp 700B amplifier (Molecular Devices, Union City, CA, USA) and controlled voltage commands and digitization of membrane currents using a Digidata 1440A interfaced with Clampex 10.2 of the pClamp software package (Molecular Devices), running on a personal computer. Currents were low-pass filtered at 2–5 kHz. Series resistance (Rs) and capacitance (Cm) values were taken directly from readings of the amplifier after electronic subtraction of the capacitive transients. Series resistance was compensated to the maximum extent possible (at least 80%). Current traces were corrected for linear capacitive leak with online P/6 trace subtraction. The external solution was composed of (in mM): 10 BaCl2, 125 tetraethylammonium chloride (TEA-Cl), 10 HEPES and 10 glucose (pH 7.3, adjusted with TEA-OH). The pipette solution contained (in mM): 120 CsCl, 2 MgCl2, 11 EGTA, 15 HEPES, 4 Mg-ATP and 10 glucose (pH 7.3, adjusted with CsOH). To isolate T-currents, we blocked the L-type Ca2+ channels with application of 10 µM nifedipine in the external solution (Zhang et al., 2011). Stock solutions of ES, NNC 55-0396, mibefradil, GDP-β-S and ATP-γ-S were prepared in distilled deionized water. Stock solutions of nifedipine, Bay K8644 and lavendustin C were prepared in dimethyl sulfoxide (DMSO). The concentration of DMSO in the bath solution is expected to be less than 0.01% and had no functional effects on T-type Ca2+ channels (Tao et al., 2009a). The stock solutions were diluted in the external or pipette solution just before use. Unless otherwise indicated, ES was bath applied with an air-pressure injector (PicoPump PV820, World Precision Instruments). The micropipette was located at distances ranging from 100 to 150 µm from the recorded cells. In experiments in which ES was intracellularly applied with the pipette solution, current measurements were started at least 5 min after breaking the patch.
[3H]-thymidine incorporation assay
Proliferation of U87 cells was determined by quantitating the incorporation of [3H]-thymidine as an indicator of DNA synthesis. To determine the effects of ES on the proliferation of U87 cells, 4 × 105 cells per well treated with different concentrations of ES were cultured in flat bottom 96-well plates for 3 days. Cells were pulsed with 1 µCi per well of [3H]-thymidine for the last 18 h of the culture period. Following incubation, cells were rinsed three times with ice-cold PBS and 5% TCA and lysed with 0.5 M NaOH. Subsequently, the cells were transferred into liquid scintillator in scintillation vials, and the radioactivity was measured by a liquid scintillation spectrometer. Data on [3H]-thymidine uptake into U87 cells are presented as % of controls.
Small interfering RNA (siRNA) transfection
U87 cells were seeded (4 × 105 cells per well) onto laminin-polyornithine-coated coverslips. siRNA (chemically synthesized) targeting both α1G (CaV3.1) and α1H (CaV3.2) T-type Ca2+ channels (sense, 5′-GCCAUCUUCCAGGUCAUCACATT-3′, antisense, 5′-UGUGAUGACCUGGAAGAUGGCTT-3′) was purchased from Qiagen (Valencia, CA, USA). The negative control siRNA (5′-UAGUGAAGGGAGUCGGAUCUC-3′) was used as control. Cells were transfected with 0.6 µg of α1G/H siRNAs or control siRNA at a final concentration of 100 nM by using Oligofectamine (Invitrogen, Karlsruhe, Germany). Then 48 h post transfection, U87 cells were subjected to the [3H]-thymidine incorporation assay.
Cell migration
In vitro tumour cell migration was assessed using a Biocoat™ Matrigel chamber (BD Biosciences, Bedford, MA, USA) with cell culture inserts containing an 8-µm pore size membrane with a thin Matrigel (40 µL of 1 mg·mL−1) basement membrane matrix. Half a millilitre of cells (5 × 104 cells ml-1) in serum-free DMEM was added to the cell culture insert of a Biocoat™ Matrigel-coated chamber. To avoid gradients (Kim et al., 2001), ES was added to both the upper and lower compartments before the measurement of migration/invasion. Fibronectin plays an important role both as a substrate adhesion molecule as well as a chemokinetic agent (Ohnishi et al., 1997) and was added in the bottom chamber as a chemoattractant (25 µg·mL−1) (Huang et al., 2004a; Li et al., 2007). The cells were then incubated at 37°C in humidified 5% CO2 conditions for 10 h. To quantify tumour cell migration, non-invading cells were removed from the top surface of the membrane by scrubbing gently with a cotton-tipped swab. The cells on the bottom surface of the membrane were fixed with Diff-Quik stain set (Dade Behring, Deerfield, IL) and counted to determine the number of cells that passed through the Matrigel and membrane layers.
Data analysis
All data are expressed as mean ± SEM, and Prism 5.0 (GraphPad Software Inc., La Jolla, CA, USA) was used for data plotting. Student's t-tests or one-way anova with post hoc Bonferroni were used to compare the different values and were considered significant at P < 0.05. Dose–response curves were fitted by non-linear regression Y= 1/(1 + 10(log IC50 −X)nH), where X is the decadic logarithm of the concentration used, IC50 is the concentration at which the half-maximum effect occurs and nH is the Hill coefficient. I–V curves were fitted by IBa=Gmax(V−Erev) / {1 + exp[(V−V1/2)/k]}, where Gmax is maximum conductance, Erev is reversal potential, V1/2 is half voltage activation and k is slope factor. The activation data were fitted with a Boltzmann equation: G/Gmax= 1/{1 + exp[(V1/2−V)/k]}, where G/Gmax is the relative conductance normalized by the maximal conductance, V1/2 is the potential required for half-activation of the current and k is the Boltzmann coefficient. Steady-state inactivation data were fitted with the Boltzmann equation: I/Imax= 1/{1 + exp[(V1/2−V)/k]}, where V1/2 and k are the half-maximum inactivation potential and the slope factor, respectively.
Results
Characterization of voltage-gated Ca2+ channels in human glioblastoma U87 cells
Voltage-gated Ca2+ channels (VGCC) fall into two categories: high-voltage activated (HVA), including L-, N-, P/Q- and R-type, and low-voltage activated (LVA) T-type. To test whether ES regulates VGCC, we first determined the subtypes of VGCC in U87 human glioblastoma cells. Whole-cell currents were recorded using 10 mM Ba2+ as charge carrier. The HVA channel currents were elicited by a depolarization step from −60 to 0 mV (Figure 1A). Application of nifedipine (10 µM), a specific L-type Ca2+ channel blocker, completely abolished the HVA channel currents (n= 7, P < 0.001, Figure 1A), indicating that only L-type HVA channels are functional in U87 cells. To further confirm our hypothesis, Bay K8644, a specific L-type Ca2+ channel activator, significantly increased the current density from 12.8 ± 3.9 pA·pF−1 to 24.7 ± 4.2 pA·pF−1 (n= 5, P < 0.001, Figure 1B). In addition, LVA T-type Ca2+ channel currents (T-currents) in U87 cells were further characterized. As shown in Figure 1C, a current trace was recorded when a U87 cell was given the designated depolarizing test pulse from −90 to −30 mV with application of 10 µM nifedipine in the external solution. There were few steady-state components in the current (Figure 1C). Addition of NiCl2 (100 µM), a specific T-type Ca2+ channel blocker (Todorovic and Lingle, 1998; Lee et al., 1999), inhibited the inward currents by ∼83.5% (n= 6, P < 0.01, Figure 1C). The remaining ∼16.5% T-current could be attributed to the different sensitivity of three subtypes of T-type Ca2+ channels (CaV3.1, CaV3.2 and CaV3.3) to Ni2+ (Perez-Reyes, 2003). Furthermore, application of mibefradil (100 µM), another T-type Ca2+ channel blocker, almost completely blocked the inward currents (inhibition = 93.6 ± 6.9%, n= 5). The low voltage-activated, fast inactivating, steady-state component-free and Ni2+-sensitive current showed typical properties of the T-currents.
Figure 1.
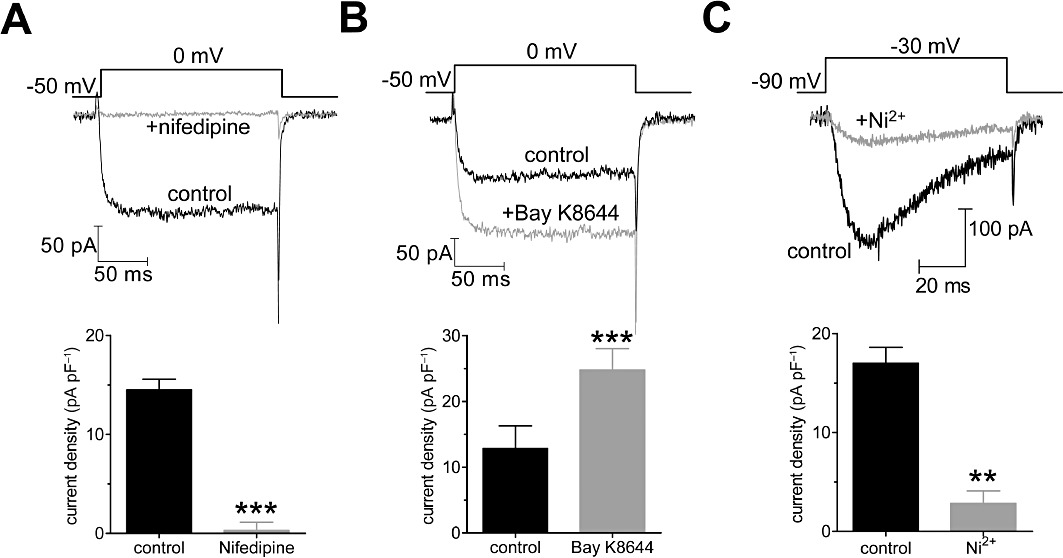
Characterization of voltage-gated Ca2+ channel currents in U87 human glioblastoma cells. Examples of traces and pooled data showing the effects of nifedipine (10 µM, A) or Bay K8644 (1 µM, B) on barium currents elicited by a 150-ms long depolarizing step pulse from the holding potential of −60 to 0 mV. (C) Examples of traces and pooled data showing the effects of NiCl2 (100 µM) on T-currents. Currents with 10 mM barium as a charge carrier were elicited by a 80-ms-long depolarization step pulse from the holding potential of −90 to −30 mV. **P < 0.01 versus control, ***P < 0.001 versus control.
ES selectively inhibited T-currents
After having identified that U87 cells express both L- and T-type Ca2+ channels, we further investigated which type of Ca2+ channel was affected by ES. We first determined the effects of ES on L-type Ca2+ channels and found that bath application of ES at 0.1 µM did not significantly affect the L-type Ca2+ channel currents (n= 7, Figure 2A). However, bath application of 0.1 µM ES significantly inhibited the amplitude of the basal T-type Ca2+ channel currents (T-currents) by 23.7 ± 2.9% (n= 9, P < 0.01, Figure 2B,C) in U87 human glioblastoma cells, whereas intracellular application of ES elicited no such effects (n= 8, Figure 2D). Upon washout of ES, the amplitude of T-currents partially returned within 3 min (Figure 2C), which indicated that the effect of ES on T-currents in U87 cells was not due to rundown. A current–voltage (I–V) curve was evoked by a series of depolarizing pulses from a holding potential of −90 mV to test potentials between −80 and 0 mV. Population data showed that at a higher depolarizing voltage, above −60 mV, 0.1 µM ES significantly up-shifted the I–V curve, and at −30 mV the current density declined from 16.4 ± 0.9 pA·pF−1 to 12.8 ± 1.5 pA·pF−1 (n= 8, P < 0.01, Figure 2E). From the size of the effect of ES on currents elicited by depolarization to −30 mV, it is clear that ES inhibited T-currents in a dose-dependent manner (Figure 2F). The relationship between the concentration of ES used and the degree of T-current inhibition observed is described by a logistic equation where the concentration of ES producing half-maximal inhibition (IC50) is 0.32 µM, the apparent Hill coefficient is 0.92 and the maximal inhibitory effect is 46.6 ± 2.7% (n= 8, P < 0.01; Figure 2F). To further confirm the selectivity of ES on T-currents, we pretreated U87 cells with NNC 55-0396, a mibefradil nonhydrolysable analogue without L-type channel efficacy (Huang et al., 2004b) and found that in the cells pretreated with NNC 55-0396 (8 µM) the ES-induced T-current inhibition was completely abolished (inhibition = 2.3 ± 0.3%, n= 7; Figure 2G,H).
Figure 2.
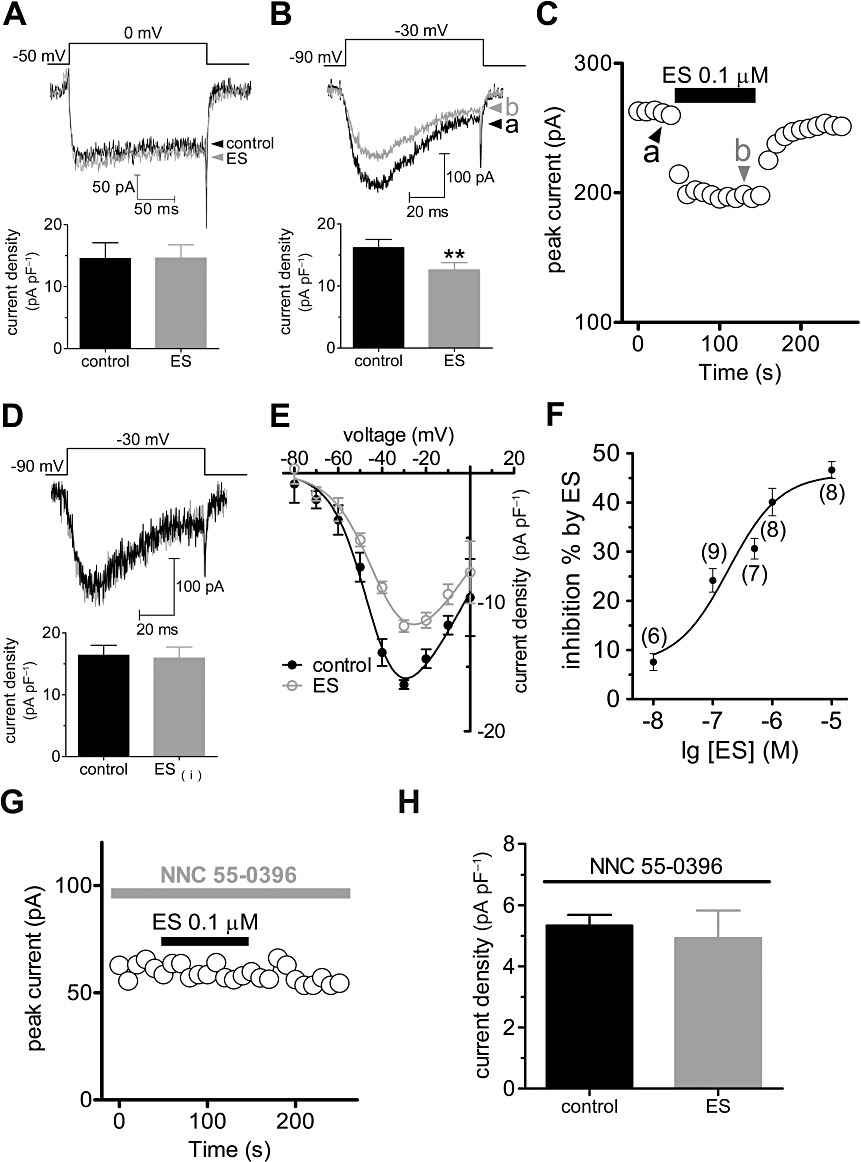
ES selectively inhibited T-currents. Examples of traces and pooled data of HVA L-type Ca2+ currents (A) or LVA T-currents (B) recorded under the control conditions and during exposure to 0.1 µM ES. (C) Time course of changes in amplitude of T-currents in control conditions, during exposure to 0.1 µM ES and washout. (D) Examples of traces and pooled data showing no inhibition of T-currents by intracellular application of ES. (E) Current–voltage (I–V) curve (evoked by a series of depolarizing pulses from a holding potential of −90 mV to test potentials between −80 and 0 mV, in 10-mV increments) for the inhibitory effects of 0.1 µM ES on T-currents. (F) Dose–response curve for the inhibitory effects of ES on T-currents. The line represents the best fit of the data points to the sigmoidal Hill equation (see Methods). Number of cells tested at each concentration of ES is indicated in parentheses. Time course (G) and pooled data (H) showing that pretreatment of cells with NNC 55-0396 (8 µM) completely abolished the inhibitory effect of ES on the T current; **P < 0.01 versus control.
ES leftward shifted steady-state inactivation curve
Next, we further investigated whether the electrophysiological properties of T-type Ca2+ channels were affected by ES; steady-state activation and inactivation potentials of T-type Ca2+ channels were studied (Figure 3A,B). ES did not significantly shift, in the hyperpolarized direction, the activation potential (V1/2 from −34.9 ± 1.7 mV to −36.1 ± 1.3 mV, and k-value from 6.8 ± 0.6 to 6.9 ± 0.9, n= 9) (Figure 3C,D). However, ES 0.1 µM leftward shifted the steady-state inactivation potentials of T-type Ca2+ channels by −15 mV (V1/2 from −47.9 ± 1.6 mV to −63.5 ± 2.7 mV, P < 0.01, and k-value from −6.5 ± 0.5 to −8.7 ± 0.3, n= 8, P < 0.01) (Figure 3C,D). These results suggest that the reduced T-currents observed upon application of ES could be due to more channels remaining in the inactivated state after activation.
Figure 3.
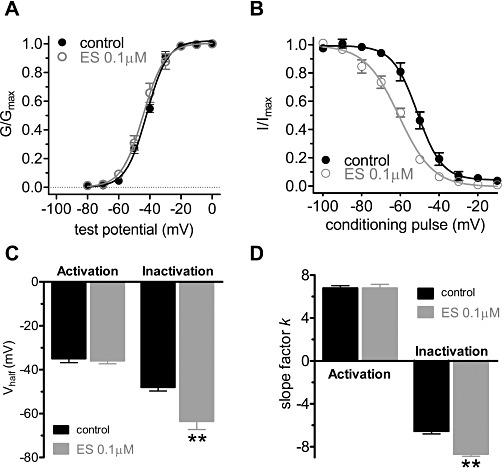
ES shifted the steady-state inactivation curve in a hyperpolarizing direction. (A) The steady-state activation of T-type calcium channels was not altered by 0.1 ES. Tail currents were elicited by repolarization to −110 mV after 40 ms test pulses from −80 to 0 mV in increments of 10 mV. (B) ES shifted steady-state inactivation curve of T-type calcium channels to the hyperpolarizing direction. Steady-state inactivation curves were obtained by 40 ms test pulse to −30 mV after the 3 s conditioning pulses ranging from −110 to +10 mV with 10 mV increments. (C,D) Pooled data showing the changes in Vhalf and k (slope factor) indicated in (A) and (B), respectively.
G-protein and tyrosine kinases were not involved in ES induced T-current inhibition
Next we tried to find out the mechanisms underlying ES-induced T-current inhibition. To determine whether G-proteins are involved in ES-mediated inhibition of T-type Ca2+ channels, we dialysed cells with guanosine-5′-O-(2-thiodiphosphate) (GDP-β-S, 1 mM), a non-hydrolysable GDP analogue. Our results showed that ES (0.1 µM) still dramatically inhibited the T-currents in the presence of GDP-β-S (inhibition% = 23.7 ± 4.3, n= 7, Figure 4A,D), indicating that ES-induced T-current inhibition was not via GPCRs. ES was reported to interact with VEGFR-1 that coupled to a family of receptor protein tyrosine kinase (PTK) (Kim et al., 2002). To determine whether inhibition of T-currents by ES is TK-dependent, we pretreated the cells with lavendustin C, a potent PTK inhibitor, and found that ES still robustly inhibited the T-currents in these pretreated cells (Figure 4B); in the presence of 5 µM lavendustin C, 0.1 µM ES reduced the peak T-currents by 24.2 ± 1.6% (n= 9, P < 0.05, Figure 4D), which was not significantly different from ∼23.7% inhibition observed under control conditions (Figure 4B,D). Lavendustin C alone had no effect on the basal T-currents (data not shown). Evidence supporting the finding that the inhibition of T-currents by ES in U87 cells is not dependent on PTK was obtained by dialysing the cells with 5 mM ATP-γ-S, a non-hydrolysable analogue of ATP. If basal TK activity was responsible for the inhibitory effect of ES, then the dialysis of cells with a pipette solution containing ATP-γ-S (5 mM) would be expected to produce irreversible, tyrosine thiophosphorylation of the Ca2+ channel protein (or other auxiliary proteins) and attenuated the ES-induced inhibition. However, in the presence of ATP-γ-S, 0.1 µM ES inhibited the amplitude of the T-currents by 27.2 ± 3.5% (n= 8; Figure 4C,D), which is not significantly different from the level of inhibition exhibited by cells dialysed with the control pipette solution (P > 0.05). It should be noted that ATP-γ-S alone produced a stimulant effect on the basal T-currents (∼46%) (n= 7). Taken together, these results strongly suggest that other mechanisms such as direct inhibition, rather than G-protein or PTK-mediated pathway, are involved in ES-induced T-current inhibition.
Figure 4.
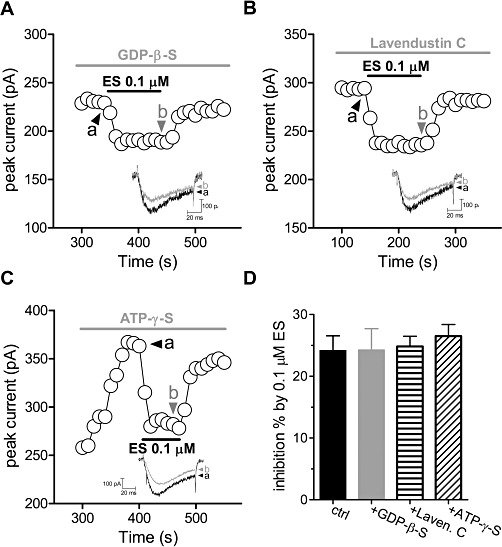
G-protein and PTK are not involved in ES-induced T-current inhibition. Time course showing the effects ES (0.1 µM) on T-currents in the presence of GDP-β-S (1 mM, A) or lavendustin C (5 µM, B). Inset: an example of the current traces indicated, respectively, in (A) and (B). Numbers on plot indicate which points were used for sample traces. (C) Intracellular ATP-γ-S has no effect on ES-induced T-current inhibition. Time course of changes in amplitude of T-currents in U87 cells dialysed with a pipette solution containing 5 mM ATP-γ-S in the absence (a) or presence of 0.1 µM ES (b). (D) Pooled data showing the effects of ES (0.1 µM) on T-currents in the presence of GDP-β-S (1 mM), lavendustin C (5 µM) or ATP-γ-S (5 mM), respectively.
Expression of α1 subunits of CaV3 in U87 human glioblastoma cells
It has been reported that CaV3.1, CaV3.2, and CaV3.3 are differentially expressed in the brain and various peripheral tissues (Perez-Reyes, 2003). To determine which subtypes of CaV3 are endogenously expressed in U87 human glioblastoma cells, we first examined the expression of the three T-type Ca2+ channel α1 subunits at transcription level by RT-PCR. For each α1 subunit, the corresponding cDNA clone was used as a positive control in amplification. Our results showed that the transcripts for the α1G (CaV3.1, predicted size of amplicon is 197 bp), α1H (CaV3.2, predicted size of amplicon is 193 bp) and α1I (CaV3.3, predicted size of amplicon is 203 bp) subunits were present in cultured U87 cells. Negative control reactions, in which reverse transcriptase was not added during the reverse transcription step, showed no PCR products (Figure 5A). We further determined the expression of the three α1 subunits at the protein level by immunoblotting with subunit-specific antibodies. HEK293 cells transfected with α1G, α1I, or α1H cDNA served as corresponding positive controls. Western blotting results showed that U87 cells express all of three α1 subunits (α1G, α1H and α1I) of CaV3 (Figure 5B). Each band had a molecular weight above 200 kDa, consistent with the predicted sizes of the α1 subunits obtained from human sequences.
Figure 5.
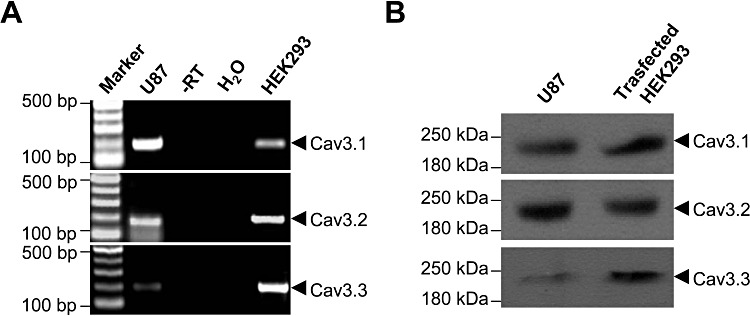
Expression analysis of α1 subunits of T-type Ca2+ channels in U87 human glioblastoma cells. (A) RT-PCR analysis of voltage-gated Ca2+ channel α1 subunit transcripts. RNA was isolated from cultured U87 cells and RT-PCR was performed using primers as described in Table 1. The PCR template from reverse transcription without RTase (–RT), or from water (H2O) served as negative controls. HEK293 cells transfected with CaV3.1, CaV3.2 or CaV3.3 cDNA were used as corresponding positive controls. (B) Western blot analysis of α1 subunits of CaV3 channels. Proteins extracted from cultured U87 cells were probed with goat polyclonal antibodies against the different CaV3 α1 subunits. Proteins extracted from HEK293 cells transfected with CaV3.1, CaV3.2 or CaV3.3 were used as corresponding positive controls. The molecular weight is shown in the left lane.
ES selectively inhibits CaV3.1 and CaV3.2, but not CaV3.3, T-type Ca2+ channels
To further determine which subtype of CaV3 is inhibited by ES, we examined the inhibitory effects of ES on the three clones of HEK293 cells that had been transfected with corresponding CaV3 subunits. HEK293 is a human embryonic kidney cell line, and does not express CaV3 subunits endogenously. Our results show that application of 0.1 µM ES robustly inhibited both CaV3.1 and CaV3.2 channel currents by 24.1% (I/Icontrol= 0.76 ± 0.05, n= 9, P < 0.01; Figure 6A) and 28.4% (I/Icontrol= 0.72 ± 0.07, n= 11, P < 0.01; Figure 6B), respectively, while the CaV3.3 channel currents were not affected (I/Icontrol= 0.98 ± 0.05, n= 9, P > 0.05; Figure 6C). Similar inhibitions of CaV3.1 and CaV3.2 channel currents were observed at all test potentials examined (Figure 6A–C). Consistent with the inhibition in U87 cells, this current inhibition in transfected HEK293 cells was selective to T-channels since no effect was observed on CaV1.2 L-type currents (Figure 6D), even when the ES concentration was increased to 10 µM (n= 6; data not shown). A similar inhibitory effect was observed in CHO cells (Figure 6E), indicating that the effect was not restricted to cell type. Overall, the direct inhibition of T-type Ca2+ channels in U87 human glioblastoma cells was mediated through only CaV3.1 and CaV3.2, but not CaV3.3.
Figure 6.
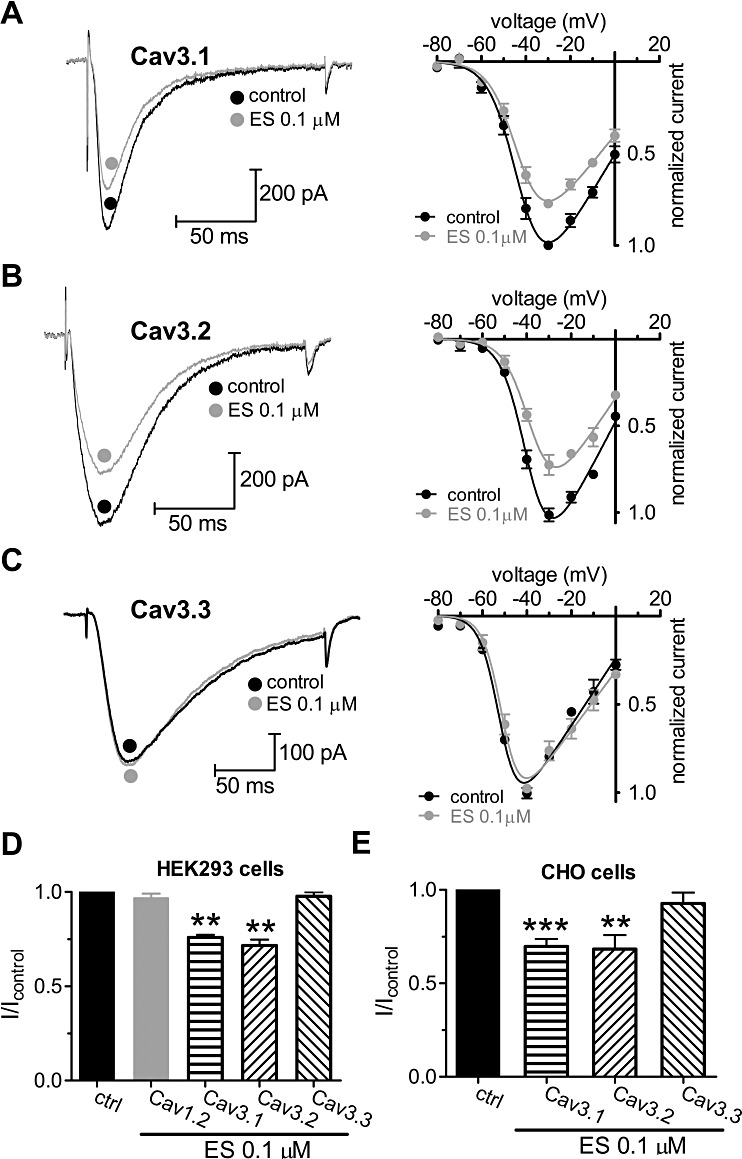
ES selectively inhibits CaV3.1 and CaV3.2, but not CaV3.3 T-type Ca2+ channels. (A–C) Left panels: representative traces showing the effect of 0.1 µM ES on cloned CaV3 channel currents elicited by a −30 mV test pulse. The holding potential (HP) was −80 mV. Right panels: current–voltage (I–V) profiles (evoked by a series of depolarizing pulses from a holding potential of −90 mV to test potentials between −80 and 0 mV, in 10-mV increments) obtained for cloned human α1G (CaV3.1) (A, n= 11), α1H (CaV3.1) (B, n= 13) and α1I (CaV3.1) subunits (C, n= 9). (D) Pooled data showing the effect of 0.1 µM ES on cloned human CaV3.1, CaV3.2 and CaV3.3 channel currents indicated in (A), (B) and (C), respectively. ES blocked T-currents but not L-type CaV1.2 (α1C) channel currents. CaV1.2 channel currents were elicited by a +10 mV test pulse applied from a HP of −80 mV. (E) ES at 0.1 µM produced a similar block of Ba2+ (10 mM) currents when α1 subunits of CaV3 were expressed in CHO cells. **P < 0.01 versus control, ***P < 0.001 versus control.
ES attenuated U87 cell proliferation via T-type Ca2+ channels
Previous studies have indicated that functional T-type Ca2+ channels endogenously expressed in many tumour cells play important roles in cell proliferation (Panner et al., 2005; Panner and Wurster, 2006). As we showed that ES robustly inhibits the CaV3.1 and CaV3.2 channel currents, we determined whether ES also attenuates the proliferation of U87 human glioblastoma cells. In six experiments, it was shown that serum-induced proliferation of U87 cells was inhibited by ES (0.01–10 µM) in a dose-dependent manner (Figure 7A). Mibefradil (100 µM), an antagonist of T-type Ca2+ channels, inhibits T-type Ca2+ channel currents in U87 cells by 93.6%. In this study, the effect of mibefradil on the cell proliferation was also examined. Mibefradil (100 µM) significantly inhibited the cell proliferation by 38.7 ± 3.5% (n= 6, P < 0.01, Figure 7B), implying the involvement of T-type Ca2+ channels in U87 cell proliferation. In contrast, the L-type Ca2+ channel blocker nifedipine had no such effects on cell proliferation at pharmacologically appropriate doses (10 µM) (Figure 7B). Similar results were observed with another L-type Ca2+ channel blocker nimodipine (10 µM) (Figure 7B). Application of ES (0.1 µM) in U87 cells pretreated with mibefradil (100 µM) failed to produce any further inhibition (Figure 7B), which indicates that T-type Ca2+ channels are involved in ES-induced inhibition of U87 cell proliferation. To further determine the role of CaV3.1 and CaV3.2 T-type Ca2+ channels in ES-induced cell proliferation, we transfected U87 cells with siRNA targeting both α1G and α1H (α1G/H). Western blot analysis showed that the expression of either α1G or α1H was significantly reduced in U87 cells transfected with α1G/H siRNA compared with the counterparts transfected with control siRNA (Figure 7C). As shown in Figure 7D, knockdown of α1G/H in U87 cells resulted in near complete abolishment of the inhibitory effect of ES on cell proliferation (n= 6, Figure 7D), whereas ES still significantly inhibited the cell proliferation in the control siRNA-transfected cells (n= 6, Figure 7D)(94.5 ± 5.7%, n= 6). These results strongly indicate that T-type Ca2+ channels play a critical role in the proliferation of U87 cells, and the effect of ES on cell proliferation is due to the blockade of CaV3.1/CaV3.2 T-type Ca2+ channels in U87 human glioblastoma cells.
Figure 7.
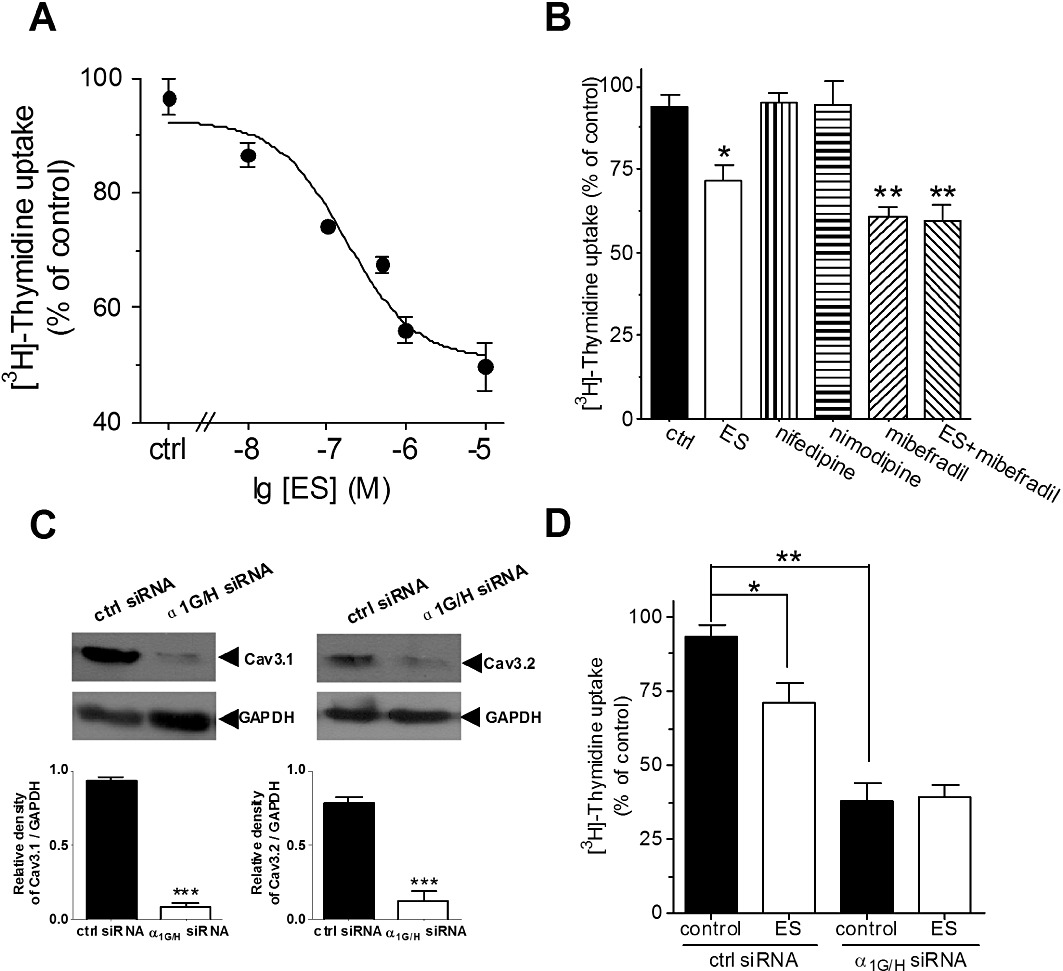
Inhibition of T-type Ca2+ channels by ES attenuated U87 cell proliferation. (A) ES inhibited cell proliferation of U87 cells in a dose-dependent manner. U87 cells, 2 × 106 per well, were seeded in 96-well plates and treated with different concentrations of ES for 3 days and were pulsed with 1 µCi [3H]-thymidine for the last 18 h. Cell proliferation values are expressed relative to those wells where no ES was added (100% control value). (B) Mibefradil (100 µM), the T-type Ca2+ channel blocker, but not nifedipine, a L-type Ca2+ channel blocker, inhibited U87 cell proliferation. Mibefradil at 100 µM occluded the inhibitory effects of cell proliferation induced by ES. (C) Western blot analysis of α1G and α1H expression in negative-control siRNA (ctrl siRNA) and α1G/H siRNA-treated U87 cells. GAPDH was used as a positive control. (D) Pooled data showed the effect of α1G/H siRNA on the inhibitory effects of 0.1 µM ES on cell proliferation in U87 cells. Values represent the mean ± SEM of six experiments. *P < 0.05 versus control, **P < 0.01 versus control.
ES inhibits U87 cell migration via T-type Ca2+ channels
Because T-type Ca2+ channels also play important roles in cell migration, we tested the ability of ES to inhibit U87 cell migration in vitro. Cell suspensions were loaded to the top chamber with or without the addition of ES. After 10 h incubation, transmigrated cells on the underside of the inserts were fixed, stained and counted. ES significantly inhibited U87 cell transmigration in a dose-dependent fashion (Figure 8A). However, the L-type Ca2+ channel blocker nifedipine (10 µM) had no such effects on cell migration at pharmacologically appropriate doses (10 µM) (Figure 8B). In contrast, the T-type Ca2+ channel blocker mibefradil significantly inhibited the cell transmigration by 29.2 ± 5.1% (P < 0.01; Figure 8B) at the concentration of 100 µM. To further test whether ES inhibits cell migration via T-type Ca2+ channels, we investigated whether mibefradil would occlude ES-mediated effects. Indeed, similat to ES, application of 100 µM mibefradil inhibited U87 cell transmigration (Figure 8B). Notably, application of ES during the maximum mibefradil-induced response failed to produce any further inhibition (Figure 8B). Therefore, these results suggest that inhibition of T-type Ca2+ channels by ES attenuates cell migration in U87 human glioblastoma cells.
Figure 8.
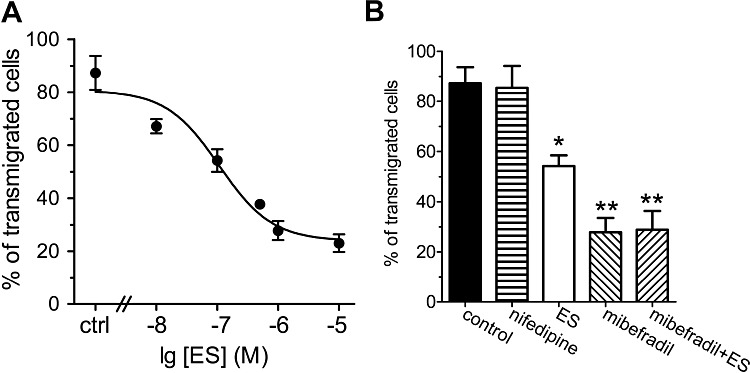
Effects of ES on U87 cell migration. (A) ES inhibited U87 cell migration in a dose-dependent manner. U87 cells were seeded into the top Matrigel chamber with or without the addition of ES or mibefradil at various concentrations. A final concentration of 25 µg·mL−1 fibronectin was added to the bottom chamber medium as a chemoattractant. (B) Mibefradil occluded the ES-induced inhibition of cell migration. Mibefradil (100 µM), but not nifedipine (10 µM), inhibited U87 cell migration (n= 5). Results represent means ± SEM of six experiments. *P < 0.05 versus control, **P < 0.01 versus control.
Discussion and conclusions
Previous in vitro and in vivo studies have suggested that T-type Ca2+ channels are important in tumour cell proliferation and migration (Huang et al., 2004a; Panner and Wurster, 2006; Li and Xiong, 2011). The current study revealed that ES directly inhibits U87 human glioblastoma cell proliferation and migration by targeting T-type Ca2+ channels. Several lines of evidence support this conclusion. Firstly, ES selectively inhibited T-type Ca2+ channels in U87 cells, whereas L-type Ca2+ channels were not affected. Secondly, this inhibition was shown to be independent of either G-protein or protein tyrosine kinases. Using heterologously expressing CaV3 in HEK293 or CHO cells, we found that only CaV3.1 and CaV3.2, but not CaV3.3 or CaV1.2, were significantly inhibited. Thirdly, ES suppressed cell proliferation and migration in a dose-dependent manner. These ES induced inhibitory effects were occluded by the T-type Ca2+ channel blocker, mibefradil, which itself also inhibited U87 cell proliferation and migration. Our results thus suggest a novel mechanism for the antiproliferation effects of ES via T-type Ca2+ channels that might be a potential target for human glioblastoma therapy.
Although the mechanisms of in vivo antitumour action are less understood and remain controversial, ES was previously considered to interact with several endothelial cell-surface receptors including tropomyosin, caveolin-1 and VEGFR-1 that are involved in angiogenesis (Kim et al., 2002; Wickstrom et al., 2002; Skovseth et al., 2005). For example, Peroulis et al. (2002) has reported that the endogenous expression of ES by C6 glioma cells results in a reduced tumour growth rate in vivo. Furthermore, Schmidt et al. (2004) showed that local intracerebral microinfusion of ES improved treatment efficiency and survival in an orthotopic human glioblastoma model. These results suggested that antiangiogenesis effect was critically required for ES in vivo in the growth and expansion of tumours including glioblastoma. However, several studies later failed to observe an antitumour effect of ES (Marshall, 2002). In contrast, growing evidences have recently shown that ES could play a direct role in inhibiting tumour cells (Yang et al., 2011). Also, it has been reported recently that peptide 30 derived from ES suppresses the proliferation and migration of HepG2 cells in vitro (Li et al., 2011). In the present study, we revealed that, besides its antiangiogenesis effects, ES played a direct role in inhibition of U87 human glioblastoma cell proliferation and migration in vitro by targeting T-type Ca2+ channels.
T-channels are present in several cancerous cell lines, such as neuroblastoma, retinoblastoma and glioma cells (Chemin et al., 2002; Latour et al., 2004). Recent studies have shown that α1G (Cav3.1), one of the T-channel subunits, has been shown in various human tumours, including colon cancer, pancreatic tumour and glioblastoma, as well as in acute myelogenous leukaemia (Toyota et al., 1999; Lory et al., 2006). Abnormal up-regulation of the gene encoding T-type Ca2+ channel α1G (Cav3.1) subunit was detected in various human primary tumours (Toyota et al., 1999), suggesting that T-type Ca2+ channels play a role in cancer development. In the present study, we reported that all three subtypes of Cav3.1, Cav3.2, and Cav3.3 were shown to be endogenously expressed in U87 human glioblastoma cells. There has long been hypothesized that a link exists between regulation of these channels and cancer progression (Lory et al., 2006). For example, silencing of Cav3.1 (α1G) channels by methylation of CpG islands (GC-rich regions of DNA, mainly in promoter regions) was found in number of primary tumours (Lory et al., 2006), which indicated that Cav3.1 may be a putative tumour suppressor gene (Toyota et al., 1999). In addition, Cav3.2 T-channel (α1H) may also be involved in cancer growth since neuroendocrine differentiation of cancer epithelial cancer cells was associated with an increase in their functional expression (Mariot et al., 2002). Furthermore, the impairment of T-channels showed an inhibitory role in cancer development and progression (Lory et al., 2006). Consistent with these results, we found in the present study that blockade of T-channels inhibited U87 cell proliferation and migration, whereas inhibition of L-channels elicited no such effects. Recent studies have shown that T-type Ca2+ channels are expressed in several cancerous cells (Gray and Macdonald, 2006), although their functional role has only begun to be investigated. At low voltages, T-type Ca2+ channels are known to mediate a phenomenon known as ‘window current’ (Panner and Wurster, 2006; Vassort et al., 2006). The term ‘window’ refers to the voltage overlap between the activation and steady-state inactivation at low or resting membrane potentials. As a result, there is a sustained inward current carried by a small portion of channels that are not completely inactivated. Window current allows T-type Ca2+ channels to regulate Ca2+ homeostasis under nonstimulated or resting membrane conditions (Perez-Reyes, 2003). In the present study, we observed that inhibition of T-currents by ES is highly dependent on the inactivation state of the channels. ES hyperpolarized induced an approximately −15 mV shift of the steady-state inactivation curve in U87 cells, whereas activation of curve was not affected. Although it is unclear whether the hyperpolarizing shift of the steady-state inactivation curve would produce a significantly modification in the T-type ‘window current’, our results suggested that the reduced T-currents observed upon application of ES could be due to more channels remaining in the inactivated state after activation. Further studies will be needed to address how making less T-type Ca2+ channels available for opening mechanistically contributes to the inhibitory effect of ES in U87 human glioblastoma cells.
It is well established that the pathology of tumour mainly consists of proliferation and migration (Kunzelmann, 2005). Particularly, proliferation was thought to be a key factor in the development of tumour (Kunzelmann, 2005; Lory et al., 2006). A number of prior studies suggested a potential role of T-type Ca2+ channels in controlling cell proliferation and migration. T-type Ca2+ channels played the key role in the regulation of intracellular Ca2+ during the tumour development (Kunzelmann, 2005; Panner and Wurster, 2006; Gray and Macdonald, 2006), which was further confirmed by the fact that T-type Ca2+ channel blockers inhibited the tumour growth (Wang et al., 2004). The pharmacological inhibitors of T-type Ca2+ channels, such as mibefradil and pimozide, have been demonstrated to be effective in decreasing cell proliferation in glioma cells (Panner et al., 2005) and breast cancer cells (Strobl et al., 1998). Previous studies have revealed the antiproliferative effect of mibefradil in various cell types (Lory et al., 2006). Among them a few have succeed in demonstrating that the effect was in fact due to the T-type Ca2+ channel blockade in rat neonatal cardiomyocytes (Li et al., 2005). Furthermore, inhibition of T-type Ca2+ channels reduces cell proliferation in human esophageal carcinomas via a p53-denendent pathway (Lu et al., 2008). In contrast, nonspecific antiproliferative effects of mibefradil in pancreatic beta-cells may be due to accumulation inside cells and hydrolysis to metabolites which exert L-type Ca2+ channel inhibition other than blocking T-type Ca2+ channels (Wu et al., 2000). However, in our present study, L-type Ca2+ channels were not involved in U87 cell proliferation because blockade of L-type Ca2+ channels by nifedipine or nimodipine did not affect the U87 cell proliferation and migration. In addition, pretreatment of cells with NNC 55-0396, a mibefradil nonhydrolysable analogue without L-type Ca2+ channel efficacy, completely abolished the ES-induced T-current inhibition (Figure 2G,H), which further confirm with the absence of any effects of ES on L-type currents. In addition, T-type Ca2+ channel could also inhibit the new vascularization in the tumour (Lory et al., 2006). Our results showed that ES could directly inhibit T-currents in U87 cells independent of GPCRs and PKs. Instead, this inhibitory effect of ES on inward currents occurred via a direct inhibition of T-type Ca2+ channels. In addition, our results showed that ES dose-dependently inhibited the proliferation of U87 cells. Therefore, it is reasonable to infer that in addition to its anti-angiogenesis effects ES could play a direct antitumour role via the inhibition of the cell proliferation via T-type VGCC. To clarify the effects of ES on Ca2+ channels, we used the whole-cell patch clamp technique to detect Ca2+ channel currents in U87 cells, which express both L- and T-type calcium channels. Ba2+ was used as the charge carrier in the present study. Typically, T-type Ca2+ channels have been characterized by their distinct permeability to divalent ions. CaV3.1 had significantly larger currents in calcium than in barium (Serrano et al., 2000; McRory et al., 2001; Adams and Snutch, 2007), whereas CaV3.2 had significantly larger currents in barium than in calcium (McRory et al., 2001). Interestingly, there were no major changes in the kinetics of CaV3.1 and CaV3.2 channel currents using the different charge carriers. Rundown of ionic currents is always a concern in whole-cell voltage clamp recording. We minimized time-dependent changes in barium currents by using high-resistance pipettes filled with Mg-ATP 4 µM (Wang et al., 2011) and beginning the experiments within 10 min after membrane rupture. In addition, we examined the inhibitory effects of ES on the three human CaV3 channel clones by heterologously expressing them in the HEK293 or CHO cells and found that ES selectively inhibited both CaV3.1 and CaV3.2 channel currents, whereas CaV1.2 or CaV3.3 was not affected. Together, these findings show that direct blockade of T-currents via CaV3.1 and CaV3.2 channels contributes to the ES-induced antiglioblastoma effects. However, our results are inconsistent with some previous reports. Overexpression of T-channels (CaV3.1 and CaV3.2 subunits) in HEK-293 cells did not affect the proliferation rate of these cells (Chemin et al., 2000). In NIE-115 cells, there is a decrease in T-currents that correlates with an increase in proliferation (Panner et al., 2005), leading the authors to conclude that proliferation of these cell lines is regulated by the expression of T-channels. The reasons for these differences remain to be explored, but they could be attributed to differences in T-channels expressed and/or the different cell types/passages used.
In conclusion, our present studies characterized a novel functional role of ES in modulating cell proliferation and migration by targeting T-type Ca2+ channels in U87 human glioblastoma cells, in which all three subunits of CaV3 were endogenously expressed. Using a heterologously expressing system, we demonstrated that only CaV3.1 and CaV3.2, but not CaV3.3 or CaV1.2 channel currents, were directly inhibited. Our results highlight the novel mechanism and therapeutic potential of ES via targeting T-type calcium channels for the treatment of human glioblastoma.
Acknowledgments
This work was supported by National Natural Science Foundation of China (No. 30900437 and 81171056), Natural Science Funding of Jiangsu Province (No. BK2009118 and BK2011440), Natural Science Funding for Colleges and Universities in Jiangsu Province (No. 09KJB180008), Scientific Research Foundation for the Returned Overseas Chinese Scholars of State Education Ministry (JT), Start-up Funding of Soochow University (No. Q4134901) and Dong-Wu Scholar Funding of Soochow University (JT).
Glossary
- ES
endostatin
- GDP-β-S
guanosine-5′-O-(2-thiodiphosphate)
- HVA
high-voltage activated
- LVA
low-voltage activated
- PTK
protein tyrosine kinase
- T-currents
T-type Ca2+ channel currents
- VGCC
voltage-gated Ca2+ channels
Conflicts of interest
The authors declare that they have no conflicts of interests.
References
- Adams PJ, Snutch TP. Calcium channelopathies: voltage-gated calcium channels. Subcell Biochem. 2007;45:215–251. doi: 10.1007/978-1-4020-6191-2_8. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th Edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm T, Folkman J, Browder T, O'Reilly MS. Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature. 1997;390:404–407. doi: 10.1038/37126. [DOI] [PubMed] [Google Scholar]
- Cao Y. Endogenous angiogenesis inhibitors and their therapeutic implications. Int J Biochem Cell Biol. 2001;33:357–369. doi: 10.1016/s1357-2725(01)00023-1. [DOI] [PubMed] [Google Scholar]
- Chemin J, Monteil A, Briquaire C, Richard S, Perez-Reyes E, Nargeot J, et al. Overexpression of T-type calcium channels in HEK-293 cells increases intracellular calcium without affecting cellular proliferation. FEBS Lett. 2000;478:166–172. doi: 10.1016/s0014-5793(00)01832-9. [DOI] [PubMed] [Google Scholar]
- Chemin J, Nargeot J, Lory P. Neuronal T-type alpha 1H calcium channels induce neuritogenesis and expression of high-voltageactivated calcium channels in the NG108-15 cell line. J Neurosci. 2002;22:6856–6862. doi: 10.1523/JNEUROSCI.22-16-06856.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciapa B, Pesando D, Wilding M, Whitaker M. Cell-cycle calcium transients driven by cyclic changes in inositol trisphosphate levels. Nature. 1994;368:875–878. doi: 10.1038/368875a0. [DOI] [PubMed] [Google Scholar]
- Gray LS, Macdonald TL. The pharmacology and regulation of T type calcium channels: new opportunities for unique therapeutics for cancer. Cell Calcium. 2006;40:115–120. doi: 10.1016/j.ceca.2006.04.014. [DOI] [PubMed] [Google Scholar]
- Huang JB, Kindzelskii AL, Clark AJ, Petty HR. Identification of channels promoting calcium spikes and waves in HT1080 tumor cells: their apparent roles in cell motility and invasion. Cancer Res. 2004a;64:2482–2489. doi: 10.1158/0008-5472.can-03-3501. [DOI] [PubMed] [Google Scholar]
- Huang L, Keyser BM, Tagmose TM, Hansen JB, Taylor JT, Zhuang H, et al. NNC 55-0396 [(1S,2S)-2-(2-(N-[(3-benzimidazol-2-yl)propyl]-N-methylamino)ethyl)-6-fluoro- 1,2,3,4-tetrahydro-1-isopropyl-2-naphtyl cyclopropanecarboxylate dihydrochloride]: a new selective inhibitor of T-type calcium channels. J Pharmacol Exp Ther. 2004b;309:193–199. doi: 10.1124/jpet.103.060814. [DOI] [PubMed] [Google Scholar]
- Kim D, Kim S, Koh H, Yoon SO, Chung AS, Cho KS, et al. Akt/PKB promotes cancer cell invasion via increased motility and metalloproteinase production. FASEB J. 2001;15:1953–1962. doi: 10.1096/fj.01-0198com. [DOI] [PubMed] [Google Scholar]
- Kim YM, Hwang S, Kim YM, Pyun BJ, Kim TY, Lee ST, et al. Endostatin blocks vascular endothelial growth factor-mediated signaling via direct interaction with KDR/Flk-1. J Biol Chem. 2002;277:27872–27879. doi: 10.1074/jbc.M202771200. [DOI] [PubMed] [Google Scholar]
- Kunzelmann K. Ion channels and cancer. J Membr Biol. 2005;205:159–173. doi: 10.1007/s00232-005-0781-4. [DOI] [PubMed] [Google Scholar]
- Latour I, Louw DF, Beedle AM, Hamid J, Sutherland GR, Zamponi GW. Expression of T-type calcium channel splice variants in human glioma. Glia. 2004;48:112–119. doi: 10.1002/glia.20063. [DOI] [PubMed] [Google Scholar]
- Lee JH, Gomora JC, Cribbs LL, Perez-Reyes E. Nickel block of three cloned T-type calcium channels: low concentrations selectively block alpha1H. Biophys J. 1999;77:3034–3042. doi: 10.1016/S0006-3495(99)77134-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Gondi CS, Dinh DH, Olivero WC, Gujrati M, Rao JS. Transfection with anti-p65 intrabody suppresses invasion and angiogenesis in glioma cells by blocking NF-κB transcriptional activity. Clin Cancer Res. 2007;13:2178–2190. doi: 10.1158/1078-0432.CCR-06-1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Xiong ZG. Ion channels as targets for cancer therapy. Int J Physiol Pathophysiol Pharmacol. 2011;3:156–166. [PMC free article] [PubMed] [Google Scholar]
- Li M, Zhang M, Huang L, Zhou J, Zhuang H, Taylor JT, et al. T-type Ca2+ channels are involved in high glucose-induced rat neonatal cardiomyocyte proliferation. Pediatr Res. 2005;57:550–556. doi: 10.1203/01.PDR.0000155756.89681.3C. [DOI] [PubMed] [Google Scholar]
- Li S, Wei J, Yuan L, Sun H, Liu Y, Zhang Y, et al. RGD-Modified Endostatin Peptide 30 Derived from Endostatin Suppresses Invasion and Migration of HepG2 Cells Through the αvβ3 Pathway. Cancer Biother Radiopharm. 2011;26:529–538. doi: 10.1089/cbr.2011.0978. [DOI] [PubMed] [Google Scholar]
- Lory P, Bidaud I, Chemin J. T-type calcium channels in differentiation and proliferation. Cell Calcium. 2006;40:135–146. doi: 10.1016/j.ceca.2006.04.017. [DOI] [PubMed] [Google Scholar]
- Lu F, Chen H, Zhou C, Liu S, Guo M, Chen P, et al. T-type Ca2+ channel expression in human esophageal carcinomas: a functional role in proliferation. Cell Calcium. 2008;43:49–58. doi: 10.1016/j.ceca.2007.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariot P, Vanoverberghe K, Lalevee N, Rossier MF, Prevarskaya N. Overexpression of an alpha 1H (CaV3.2) T-type calcium channel during neuroendocrine differentiation of human prostate cancer cells. J Biol Chem. 2002;277:10824–10833. doi: 10.1074/jbc.M108754200. [DOI] [PubMed] [Google Scholar]
- Marshall E. Cancer therapy, setbacks for endostatin. Science. 2002;295:2198–2199. doi: 10.1126/science.295.5563.2198. [DOI] [PubMed] [Google Scholar]
- McRory J, Santi CM, Hamming KSC, Mezeyova J, Sutton KG, Baillie DL, et al. Molecular and functional characterization of a family of rat brain T-type calcium channels. J Biol Chem. 2001;276:3999–4011. doi: 10.1074/jbc.M008215200. [DOI] [PubMed] [Google Scholar]
- Ohnishi T, Arita N, Hiraga S, Taki T, Izumoto S, Fukushima Y, et al. Fibronectin-mediated cell migration promotes glioma cell invasion through chemokinetic activity. Clin Exp Metastasis. 1997;15:538–546. doi: 10.1023/a:1018422926361. [DOI] [PubMed] [Google Scholar]
- Panner A, Wurster RD. T-type calcium channels and tumor proliferation. Cell Calcium. 2006;40:253–259. doi: 10.1016/j.ceca.2006.04.029. [DOI] [PubMed] [Google Scholar]
- Panner A, Cribbs LL, Zainelli GM, Origitano TC, Singh S, Wurster RD. Variation of T-type calcium channel protein expression affects cell division of cultured tumor cells. Cell Calcium. 2005;37:105–119. doi: 10.1016/j.ceca.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Perez-Reyes E. Molecular physiology of low-voltage-activated T-type calcium channels. Physiol Rev. 2003;83:117–161. doi: 10.1152/physrev.00018.2002. [DOI] [PubMed] [Google Scholar]
- Peroulis I, Jonas N, Saleh M. Antiangiogenic activity of endostatin inhibits C6 glioma growth. Int J Cancer. 2002;97:839–845. doi: 10.1002/ijc.10115. [DOI] [PubMed] [Google Scholar]
- Schmidt NO, Ziu M, Carrabba G, Giussani C, Bello L, Sun Y, et al. Antiangiogenic therapy by local intracerebral microinfusion improves treatment efficiency and survival in an orthotopic human glioblastoma model. Clin Cancer Res. 2004;10:1255–1262. doi: 10.1158/1078-0432.ccr-03-0052. [DOI] [PubMed] [Google Scholar]
- Serrano JR, Dashti SR, Perez-Reyes E, Jones SW. Mg2+ block unmasks Ca2+/Ba2+ selectivity of α1G T-type calcium channels. Biophys J. 2000;79:3052–3062. doi: 10.1016/S0006-3495(00)76540-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skovseth DK, Veuger MJ, Sorensen DR, De Angelis PM, Haraldsen G. Endostatin dramatically inhibits endothelial cell migration, vascular morphogenesis, and perivascular cell recruitment in vivo. Blood. 2005;105:1044–1051. doi: 10.1182/blood-2004-03-1164. [DOI] [PubMed] [Google Scholar]
- Sorensen DR, Read TA, Porwol T, Olsen BR, Timpl R, Sasaki T, et al. Endostatin reduces vascularization, blood flow, and growth in a rat gliosarcoma. Neuro-Oncol. 2002;4:1–8. doi: 10.1215/15228517-4-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strobl JS, Melkoumian Z, Peterson VA, Hylton H. The cell death response to gamma-radiation in MCF-7 cells is enhanced by a neuroleptic drug, pimozide. Breast Cancer Res Treat. 1998;51:83–95. doi: 10.1023/a:1006046604062. [DOI] [PubMed] [Google Scholar]
- Tao J, Hildebrand ME, Liao P, Liang MC, Tan G, Li S, et al. Activation of corticotropin-releasing factor receptor 1 selectively inhibits CaV3.2 T-type calcium channels. Mol Pharmacol. 2008;73:1596–1609. doi: 10.1124/mol.107.043612. [DOI] [PubMed] [Google Scholar]
- Tao J, Zhang Y, Li S, Sun W, Soong TW. Tyrosine kinase-independent inhibition by genistein on spermatogenic T-type calcium channels attenuates mouse sperm motility and acrosome reaction. Cell Calcium. 2009a;45:133–143. doi: 10.1016/j.ceca.2008.07.004. [DOI] [PubMed] [Google Scholar]
- Tao J, Zhang Y, Huang H, Jiang X. Activation of corticotropin-releasing factor 2 receptor inhibits Purkinje neuron P-type calcium currents via G(o)alpha-dependent PKC epsilon pathway. Cell Signal. 2009b;21:1436–1443. doi: 10.1016/j.cellsig.2009.05.002. [DOI] [PubMed] [Google Scholar]
- Taylor JT, Huang L, Pottle JE, Liu K, Yang Y, Zeng X, et al. Selective blockade of T-type Ca2+ channels suppresses human breast cancer cell proliferation. Cancer Lett. 2008;267:116–124. doi: 10.1016/j.canlet.2008.03.032. [DOI] [PubMed] [Google Scholar]
- Todorovic SM, Lingle CJ. Pharmacological properties of T-type Ca2+ current in adult rat sensory neurons: effects of anticonvulsant and anesthetic agents. J Neurophysiol. 1998;79:240–252. doi: 10.1152/jn.1998.79.1.240. [DOI] [PubMed] [Google Scholar]
- Toyota M, Ho C, Ohe-Toyota M, Baylin SB, Issa JP. Inactivation of CACNA1G, a T-type calcium channel gene, by aberrant methylation of its 5′ CpG island in human tumors. Cancer Res. 1999;59:4535–4541. [PubMed] [Google Scholar]
- Vassort G, Talavera K, Alvarez JL. Role of T-type Ca2+ channels in the heart. Cell Calcium. 2006;40:205–220. doi: 10.1016/j.ceca.2006.04.025. [DOI] [PubMed] [Google Scholar]
- Walker DG, Kaye AH. Diagnosis and management of astrocytomas, oligodendrogliomas and mixed gliomas: a review. Australas Radiol. 2001;45:472–482. doi: 10.1046/j.1440-1673.2001.00959.x. [DOI] [PubMed] [Google Scholar]
- Wang F, Zhang Y, Jiang X, Zhang Y, Zhang L, Gong S, et al. Neuromedin U inhibits T-type Ca2+ channel currents and decreases membrane excitability in small dorsal root ganglia neurons in mice. Cell Calcium. 2011;49:12–22. doi: 10.1016/j.ceca.2010.11.002. [DOI] [PubMed] [Google Scholar]
- Wang G, Lemosm JR, Iadecolam C. Herbal alkaloid tetrandrine: fron an ion channel blocker to inhibitor of tumor proliferation. Trends Pharmacol Sci. 2004;25:120–123. doi: 10.1016/j.tips.2004.01.009. [DOI] [PubMed] [Google Scholar]
- Wickstrom SA, Alitalo K, Keski-Oja J. Endostatin associates with integrin alpha5beta1 and caveolin-1, and activates Src via a tyrosyl phosphatase-dependent pathway in human endothelial cells. Cancer Res. 2002;62:5580–5589. [PubMed] [Google Scholar]
- Wu S, Zhang M, Vest PA, Bhattacharjee A, Liu L, Li M. A mibefradil metabolite is a potent intracellular blocker of L-type Ca2+ currents in pancreatic beta-cells. J Pharmacol Exp Ther. 2000;292:939–943. [PubMed] [Google Scholar]
- Yang L, Lin Z, Lin J, Weng S, Huang Q, Zhang P, et al. Antitumor effect of endostatin overexpressed in C6 glioma cells is associated with the down-regulation of VEGF. Int J Oncol. 2011;38:465–471. doi: 10.3892/ijo.2010.871. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Zhang L, Wang F, Zhang Y, Wang J, Qin Z, et al. Activation of M3 muscarinic receptors inhibits T-type Ca2+ channel currents via pertussis toxin-sensitive novel protein kinase C pathway in small dorsal root ganglion neurons. Cell Signal. 2011;23:1057–1067. doi: 10.1016/j.cellsig.2011.02.001. [DOI] [PubMed] [Google Scholar]