

Abstract
BACKGROUND AND PURPOSE
PPARγ agonists, glitazones, have cardioprotective and anti-inflammatory actions associated with gene transcription interference. In this study, we determined whether chronic treatment of adult spontaneously hypertensive rats (SHR) with pioglitazone alters BP and vascular structure and function, and the possible mechanisms involved.
EXPERIMENTAL APPROACH
Mesenteric resistance arteries from untreated or pioglitazone-treated (2.5 mg·kg−1·day−1, 28 days) SHR and normotensive [Wistar Kyoto (WKY)] rats were used. Vascular structure was studied by pressure myography, vascular function by wire myography, protein expression by Western blot and immunohistochemistry, mRNA levels by RT-PCR, prostanoid levels by commercial kits and reactive oxygen species (ROS) production by dihydroethidium-emitted fluorescence.
KEY RESULTS
In SHR, pioglitazone did not modify either BP or vascular structural and mechanical alterations or phenylephrine-induced contraction, but it increased vascular COX-2 levels, prostacyclin (PGI2) production and the inhibitory effects of NS 398, SQ 29,548 and tranylcypromine on phenylephrine responses. The contractile phase of the iloprost response, which was reduced by SQ 29,548, was greater in pioglitazone-treated and pioglitazone-untreated SHR than WKY. In addition, pioglitazone abolished the increased vascular ROS production, NOX-1 levels and the inhibitory effect of apocynin and allopurinol on phenylephrine contraction, whereas it did not modify eNOS expression but restored the potentiating effect of N-nitro-L-arginine methyl ester on phenylephrine responses.
CONCLUSIONS AND IMPLICATIONS
Although pioglitazone did not reduce BP in SHR, it increased COX-2-derived PGI2 production, reduced oxidative stress, and increased NO bioavailability, which are all involved in vasoconstrictor responses in resistance arteries. These effects would contribute to the cardioprotective effect of glitazones reported in several pathologies.
Keywords: pioglitazone, PPARγ, hypertension, resistance arteries, prostacyclin, oxidative stress, NO
Introduction
PPARγ are ligand-activated transcription factors belonging to the nuclear hormone receptor superfamily that have an important role in adipocyte differentiation and carbohydrate homeostasis (Leff et al., 2004). PPARγ is expressed in all components of the vascular system including endothelial cells, vascular smooth muscle cells (VSMC) and monocytes/macrophages (Touyz and Schiffrin, 2006; Matsumoto et al., 2008), raising the possibility of direct effects of PPARγ activation on vascular tone and BP. PPARγ agonists such as pioglitazone and rosiglitazone have a BP lowering action in patients or animal models in which hypertension is associated with diabetes or with other factors of metabolic syndrome, such as obese Zucker fatty rats, high fat diet-induced obesity or fructose-fed rats (Sarafidis and Nilsson, 2006; Chen et al., 2008). However, results on BP in patients or animal models that do not have either diabetes or other symptoms of metabolic syndrome are controversial (Diep et al., 2002; Wakino et al., 2005; Llorens et al., 2007; Nakamura et al., 2007; Shinzato et al., 2007; Chan et al., 2010; Zhang et al., 2010). Interestingly, much evidence has been obtained indicating that these agents are able to interfere with the pathophysiology of target organ damage in hypertension (Dormandy et al., 2005; Nakamura et al., 2007; Shinzato et al., 2007). On the other hand, the correction of vascular structural abnormalities, associated with a reduction in BP, has also been described (Diep et al., 2002; Ledingham and Laverty, 2005; Zhang et al., 2010).
The mechanisms responsible for the beneficial effects of PPARγ agonists remain elusive. One attractive hypothesis proposed is that these drugs act as direct anti-inflammatory agents, regulating the production of immunomodulatory and inflammatory mediators. Thus, PPARγ is able to regulate gene expression in a DNA-independent fashion by interfering with pro-inflammatory transcription factors, such as NF-κB, activator protein-1 (AP-1) or signal transducer and activator of transcription (STAT) (Touyz and Schiffrin, 2006). Inflammation is a key feature in the initiation, progression and clinical implications of cardiovascular disorders, including essential hypertension. Thus, increased blood levels of pro-inflammatory cytokines, vascular COX-2 expression and oxidative stress together with reduced NO availability are well-established hallmarks of hypertension (Briones et al., 2002; Alvarez et al., 2005; Paravicini and Touyz, 2006; Savoia and Schiffrin, 2007; Virdis et al., 2009). PPARγ activation in the vascular wall inhibits among others, cytokine production, expression of adhesion molecules and metalloproteinases, and proliferation and migration of VSMC (Touyz and Schiffrin, 2006). In addition, PPARγ agonists inhibit the expression of several components of the NADPH oxidase, the main source of superoxide anion (O2•−) at the vascular level, and the subsequent production of reactive oxygen species (ROS), thus contributing to the anti-inflammatory and the vascular protective effects of these drugs (Hwang et al., 2005; Nakamura et al., 2007). In different inflammatory models, it has been shown that the anti-inflammatory effect of PPARγ agonists is associated with a reduction in the increased COX-2 expression (Sánchez-Hidalgo et al., 2005; Collino et al., 2006), although increased COX-2 expression has also been observed after PPARγ activation (Meade et al., 1999; Ye et al., 2006; Kang et al., 2008).
In the present study we investigated the effect of the treatment of spontaneously hypertensive rats (SHR) with pioglitazone on BP and on the structural, mechanical and functional properties of mesenteric resistance arteries (MRAs). In addition the role of COX-2-derived prostanoids, NO and ROS in phenylephrine responses was also examined.
Methods
Animals
All animal care and experimental procedures conformed to the current Spanish and European laws on animal use (RD 223/88 MAPA and 609/86). Six-month-old male normotensive Wistar Kyoto (WKY) rats (n= 30) and SHR rats (n= 83), obtained from colonies maintained at the Animal Quarters of the Facultad de Ciencias de la Salud of the Universidad Rey Juan Carlos (ES280920000023), were used. During the treatment, the rats were housed with constant room temperature, humidity and light cycle (12-h light/dark). Rats had free access to tap water and were fed with standard rat chow ad libitum.
The SHR rats were divided into two groups: control (received vehicle) and rats treated with the PPARγ agonist pioglitazone (2.5 mg·kg−1·day−1, for 28 days suspended in 0.5% methylcellulose and administered in drinking water). This dose has been reported to achieve a concentration equivalent to that reported in humans who were administered a 15 mg dose of pioglitazone (Ishibashi et al., 2002). Systolic BP was measured weekly using tail cuff plethysmography. Rats were killed by decapitation and the mesenteric arcade was removed and placed in Krebs Henseleit solution (KHS) of the following composition (in mM): NaCl 115.0; KCl 4.7; CaCl2 2.5; KH2PO4 1.2; MgSO4.7H2O 1.2; NaHCO3 25.0; glucose 11.1 and Na2EDTA 0.01, maintained at 4°C and continuously gassed with 95% O2 and 5% CO2. Segments of third-order branches of the mesenteric artery were dissected free of fat and connective tissue and used for vascular structure and function studies as well as O2•− production. The second- and third-order branches were used to analyse the production of prostanoids, mRNA levels and protein expression.
Blood samples were collected in tubes containing 15% K3EDTA as anticoagulant (BD Vacutainer Systems, Preanalytical Solutions, Plymouth, UK) and placed on ice. Blood samples were centrifuged at 1500× g for 15 min at 4°C. The plasma obtained was frozen at −20°C and kept at −70°C until used to determine malondialdehyde (MDA) concentration.
Pressure myography
The structural and mechanical properties of MRA were studied with a pressure myograph (Danish Myo Tech, Model P100, J.P. Trading I/S, Aarhus, Denmark). Vessels were placed on two glass microcannulae and secured with surgical nylon suture. After any small branches were tied off, vessel length was adjusted so that the vessel walls were parallel without stretch. Intraluminal pressure was then raised to 140 mmHg and the artery was unbuckled by adjusting the cannulae. The segment was then set to a pressure of 70 mmHg and allowed to equilibrate for 60 min at 37°C in calcium-free KHS (0 Ca2+; omitting calcium and adding 1 mM EGTA) intra- and extravascular perfused, gassed with a mixture of 95% O2 and 5% CO2. Intraluminal pressure was reduced to 3 mmHg. A pressure-diameter curve was obtained by increasing intraluminal pressure in 20 mmHg steps between 3 and 140 mmHg. Internal and external diameters were continuously measured under passive conditions (Di0Ca, De0Ca) for 3 min at each intraluminal pressure. The final value used was the mean of the measurements taken during the last 30 s when the measurements reached a steady state. Finally, the artery was set to 70 mmHg in 0 Ca2+-KHS, pressure-fixed with 4% paraformaldehyde (PFA; in 0.2 M phosphate buffer, pH 7.2–7.4) at 37°C for 60 min and kept in 4% PFA at 4°C for confocal microscopy studies.
Calculation of passive structural and mechanical parameters
From internal and external diameter measurements in passive conditions the following structural and mechanical parameters were calculated:
Incremental distensibility represents the percentage of change in the arterial internal diameter for each mmHg change in intraluminal pressure and was calculated according to the formula:
Circumferential wall strain (ε) = (Di0Ca− D00Ca)/D00Ca, where D00Ca is the internal diameter at 3 mmHg and Di0Ca is the observed internal diameter for a given intravascular pressure both measured in 0 Ca2+ medium.
Circumferential wall stress (σ) = (P × Di0Ca)/(2WT), where P is the intraluminal pressure (1 mmHg = 1.334 × 103 dynes·cm−2) and WT is wall thickness at each intraluminal pressure in 0 Ca2+-KHS.
Arterial stiffness independent of geometry is determined by the Young's elastic modulus (E = stress/strain). The stress-strain relationship is non-linear; therefore, it is more appropriate to obtain a tangential or incremental elastic modulus (Einc) by determining the slope of the stress-strain curve (Einc=δσ/δε). Einc was obtained by fitting the stress-strain data from each animal to an exponential curve using the equation: σ=σorigeβε, where σorig is the stress at the original diameter (diameter at 3 mmHg). Taking derivatives on the equation presented earlier, we see that Einc=βσ. For a given σ-value, Einc is directly proportional to β. An increase in β implies an increase in Einc, which means an increase in stiffness.
Confocal microscopy study of nuclei distribution
Pressure-fixed intact arteries were incubated with the nuclear dye Hoechst 33342 (Hoechst, Frankfurt, Germany) (0.01 mg·mL−1) for 15 min. After being washed, the arteries were mounted on slides with a well made of silicon spacers to avoid artery deformation. They were viewed using a Leica TCS SP5 confocal system (Leica Microsystems, Wetzlar, Germany) fitted with an inverted microscope and argon and helium-neon laser sources with oil immersion lens (×40) (Ex 351–364 nm and Em 400–500 nm). At least two serial optical sections (stacks of images) of 0.5 µm thick serial optical slices were taken from the adventitia to the lumen in different regions along the artery length. Thereafter, individual projections of the vessel were reconstructed with Metamorph image analysis software (Molecular Devices Corp., Downingtown, PA, USA) and a transversal view of the stack was obtained.
The total WT occupied by the different cell types was measured in the z-axis, as previously described (Arribas et al., 1997) with some modifications. Briefly, by using Metamorph software we measured the fluorescence intensity of stained nuclei. We then considered the first plane of the adventitia as the one with the maximum intensity on the first adventitial cell. Similarly, the last plane of the intima was considered to be that which showed the last endothelial nucleus at its maximum intensity.
Reactivity experiments
Ring segments, 2 mm in length, were mounted in a small vessel dual chamber myograph for measurement of isometric tension (Briones et al., 2002). Segment contractility was tested by an initial exposure to a high K+ solution (120 mM K+-KHS, which was identical to KHS except that NaCl was replaced by KCl on an equimolar basis). The response to K+-KHS was similar (P > 0.05) in arteries from the three groups (WKY: 3.3 ± 0.2 mN·mm−1, n= 30; SHR: 3.2 ± 0.1 mN·mm−1, n= 45; SHR pioglitazone: 3.1 ± 0.1 mN·mm−1, n= 38). The presence of endothelium was determined by the ability of 10 µM ACh to relax arteries precontracted with phenylephrine at a concentration that produce approximately 50% of the contraction induced by K+-KHS in each case. Thereafter, a cumulative concentration–response curve to phenylephrine (0.1–30 µM) was performed. The effects of NS 398, SQ 29,548, SC 19220, RO 1138452, SQ 29,548 plus RO 1138452, furegrelate, tranylcypromine, apocynin, allopurinol and N-nitro-L-arginine methyl ester (L-NAME) were analysed by their addition 30 min before the phenylephrine concentration–response curve. In some experiments, the endothelium was mechanically removed and the concentration–response curve to phenylephrine was performed in the absence and the presence of NS 398 and SQ 29,548. Endothelium removal was assessed by the inability of 10 µM ACh to produce vasodilatation. In another set of experiments, iloprost (1 µM) was added to arteries precontracted with phenylephrine at a concentration that produce approximately 50% of the contraction induced by K+-KHS in each case. The effects of SQ 29,548, SC 19220 and RO 1138452 on the iloprost-induced response were analysed by their addition 30 min before iloprost.
Vasoconstrictor responses were expressed as a percentage of the tone generated by K+-KHS. Vasodilator responses were expressed as a percentage of the previous tone in each case. To compare the effect of drugs on the response to phenylephrine in segments from different groups, results are expressed as differences of area under the concentration–response curves (dAUC) in control and experimental situations. AUCs were calculated from the individual concentration–response curve plots using a computer program (GraphPad Prism Software, San Diego, CA, USA); the differences were expressed as a percentage of the AUC of the corresponding control situation.
Protein expression determined by Western blot analysis
Protein expression was determined in homogenates from the mesenteric arteries obtained in ice-cold Tris-EDTA buffer (in mM: Tris-50, EDTA-1.0, pH = 7.4). Proteins [5 µg protein for Cu/Zn- and Mn-superoxide dismutase (SOD), 20 µg protein for EC-SOD and endothelial NOS (eNOS) and 40 µg protein for COX-2] were separated by 7.5% (eNOS), 10% (COX-2) or 12% (Cu/Zn-, Mn- and EC-SOD) SDS-PAGE. Proteins were transferred to polyvinyl difluoride membranes that were incubated with rabbit polyclonal antibodies for COX-2 (1:200; Cayman Chemical, Ann Arbor, MI, USA), Cu/Zn-SOD (0.1 µg·mL−1, StressGen Bioreagents Corp., Victoria, Canada), Mn-SOD (0.05 µg·mL−1, StressGen Bioreagents Corp.) or EC-SOD (10 µg·mL−1, StressGen Bioreagents Corp.) and with mouse monoclonal antibody (1:1000) for eNOS detection (Transduction Laboratories, Lexington, UK). After being washed, membranes were incubated with anti-rabbit (1:2000, Bio-Rad, Laboratories, Hercules, CA, USA) or antimouse (1:5000, StressGen Bioreagents Corp.) IgG antibody conjugated to horseradish peroxidase. The immunocomplexes were detected using an enhanced horseradish peroxidase-luminol chemiluminiscence system (ECL Plus, Amersham International, Little Chalfont, UK) and subjected to autoradiography (Minolta Film, Konica Minolta, Wayne, NJ, USA). Signals on the immunoblot were quantified using the NIH Image computer program (NIH, Bethesda, MD, USA, version 1.56). The same membrane was used to determine α-actin expression using a mouse monoclonal anti-α-actin-antibody (1:300 000, Sigma Chemical Co., St. Louis, MO, USA).
Data of protein expression are expressed as the ratio between signals on the immunoblot corresponding to the protein studied and that of α-actin. To compare the results for protein expression, we assigned a value of 1 to its expression in arteries from WKY rats.
mRNA levels determined by qRT-PCR assay
COX-2, IP receptor, TP receptor, NOX-1, NOX-2, p47phox, catalase and PPARγ mRNA levels were determined in mesenteric arteries. Total RNA was obtained by using TRIzol (Invitrogen Life Technologies, Carlsbad, CA, USA). A total of 1 µg of DNAse I treated RNA was reverse-transcribed into cDNA using the High Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA, USA) in a 50 µL reaction. PCR was performed in duplicate for each sample using 0.5 µL of cDNA as template, 1 × TaqMan Universal PCR Master Mix (Applied Biosystems) and 20 × of Taqman Gene Expression Assays (Applied Biosystems, COX-2: Rn00568225_m1; IP receptor: Rn01764022_m1; TP receptor: Rn00690601_m1; NOX-1: Rn00586652_m1; p47phox: Rn00586945_m1; catalase: Rn00560930_m1; PPARγ: Rn00440945_m1) or specific primers for NOX-2: (forward CCAGTGAAGATGTGTTCAGCT; reverse GCACAGCCAGTAGAAGTAGAT), purchased to Sigma-Aldrich, and Fast Start Universal SYBR Green Master (Rox) in a 20 µL reaction. For quantification, quantitative RT-PCR was carried out in an ABI PRISM 7000 Sequence Detection System (Applied Biosystems, from the CAT of Universidad Rey Juan Carlos) using the following conditions: 2 min 50°C, 10 min 95°C and 40 cycles: 15 s 95°C, 1 min 60°C. As a normalizing internal control we amplified β2 microglobulin (Rn00560865_m1). To calculate the relative index of gene expression, we used the 2-ΔΔCt method (Livak and Schmittgen, 2001) using WKY or untreated SHR as the control.
Measurement of PGF2α, prostacyclin and 8-isoprostane production
The levels of 13,14-dihydro-15-keto-PGF2α, the metabolite of PGF2α, 6-keto-PGF1α, the metabolite of prostacyclin (PGI2), and 8-isoprostane were measured in the incubation medium after completion of the phenylephrine concentration–response curves, using commercial enzyme immunoassay kits (Cayman Chemical). The medium was frozen in liquid nitrogen, kept at −70°C until analysis and processed following the manufacturer's instructions.
In situ detection of vascular O2•− production
The oxidative fluorescent dye dihydroethidium was used to evaluate O2•− production in situ, as described previously (García-Redondo et al., 2009). Fourteen-micrometre frozen cross-sections were incubated in a humidified chamber for 30 min in Krebs HEPES solution (in mM: 119 NaCl, 20 HEPES, 1 MgSO4, 0.15 Na2HPO4, 4.6 KCl, 0.4 KH2PO4, 5 NaHCO3, 1.2 CaCl2, 11.1 glucose, pH 7.4) at 37°C, and then incubated for 30 min in Krebs HEPES solution containing dihydroethidium (10 µM) in the dark. Each day of the experiment, images of the three different experimental conditions (WKY and pioglitazone-treated and untreated SHR) were taken with a fluorescent laser scanning confocal microscope (Leica TCS SP2) using a ×40oil objective with the 568 nm/600–700 nm excitation/emission filter always using the same imaging settings.
Measurement of MDA production
Plasma MDA levels were measured by a modified thiobarbituric acid (TBA) assay, as described previously (Alvarez et al., 2007).
Statistical analysis and drugs
All values are expressed as mean ± SEM; n represents the number of animals used in each experiment. The maximum response (Emax) and pD2 values were calculated by a non-linear regression analysis of each individual concentration–response curve using GraphPad Prism Software. Results were analysed by using Student's t-test or two-way anova followed by a Bonferroni's post hoc test. A probability value of less than 5% was considered significant.
Phenylephrine, ACh, SC 19220, furegrelate, tranylcypromine, L-NAME were obtained from Sigma Chemical, Co. NS 398 was obtained from Calbiochem-Novabiochem GmbH (Bad Soden, Germany), allopurinol from Research Biochemicals Incorporated (Natick, MA, USA), apocynin from Fluka-Sigma Chemical (Seelze, Germany) and SQ 29,548, RO 1138452 and iloprost from Cayman Chemical. Pioglitazone was generously supplied by Takeda-Lilly, Madrid, Spain.
Results
Pioglitazone treatment (2.5 mg·kg−1·day−1, 28 days) of SHR did not modify either systolic BP (before: 200.9 ± 3.2, after: 199.9 ± 1.7 mmHg, n= 17; P > 0.05) or body weight (results not shown).
Effect of pioglitazone treatment on vascular remodelling and stiffness
MRAs from hypertensive rats showed lower external (Figure 1A) and internal (Figure 1B) diameters compared with normotensive rats; pioglitazone treatment did not affect any of these parameters (Figure 1). Similar results were obtained when internal diameter was measured using the wire myograph; MRA from SHR showed a reduced internal diameter (WKY: 270.2 ± 4.8 µm, n= 30; SHR: 241.2 ± 2.9 µm, n= 45; P < 0.05), which was not modified by pioglitazone treatment (243.8 ± 1.2 µm, n= 38; P > 0.05). However, the wall : lumen ratio was greater in arteries from SHR and unaffected by pioglitazone (Figure 1C). Transversal confocal projections of the whole vascular wall of arteries also showed increased WT in the SHR vessels (WKY: 30.42 ± 1.47 µm, n= 5; SHR: 37.94 ± 1.47 µm, n= 5; P < 0.05) that was unaffected by pioglitazone treatment (39.72 ± 3.72 µm, n= 6; P > 0.05; Figure 1D).
Figure 1.
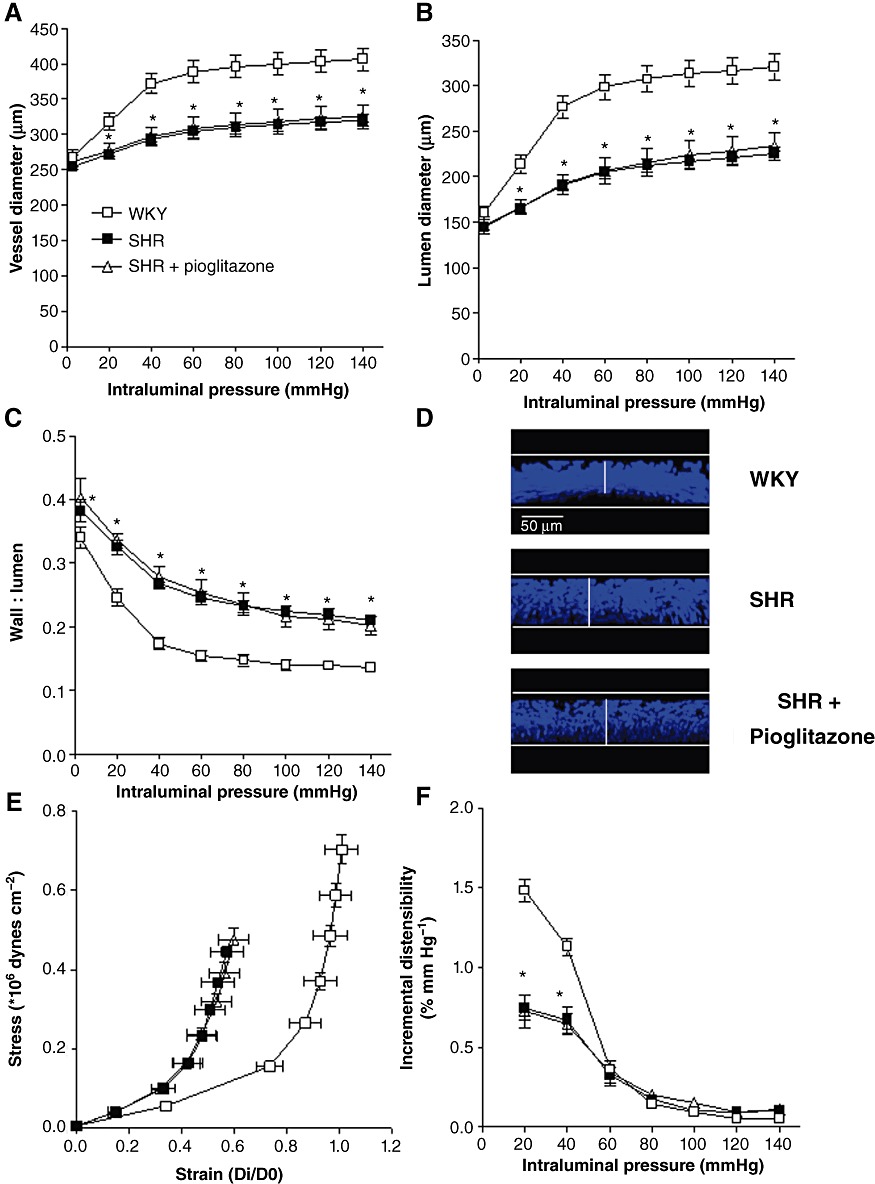
External and internal diameter-intraluminal pressure (A, B) and wall : lumen-intraluminal pressure (C) in MRA from WKY rats and SHR untreated or treated with pioglitazone incubated in 0 Ca2+-KHS. (D) Representative transversal confocal projections of the vascular wall of MRA from WKY, SHR and pioglitazone-treated SHR. Vessels were pressure-fixed at 70 mmHg, stained with Hoechst 33342 and mounted intact on a slide. Projections were obtained from serial optical sections captured with a fluorescence confocal microscope (x40oil immersion objective, zoomX2). Metamorph Image Analysis software was used to produce the transversal projection of the artery. Stress-strain relationship (E) and incremental distensibility-intraluminal pressure curves (F) in MRA from WKY, SHR and pioglitazone-treated SHR rats. *P < 0.05 versus WKY rats by two-way anova and Bonferroni post-test. n= 8–9 animals. Data are expressed as mean ± SEM.
Vessels from SHR rats showed decreased elasticity, as shown by the larger value of β (WKY: 4.75 ± 0.20, n= 8; SHR: 8.57 ± 1.02, n= 8; P < 0.05), and a leftward shift of the stress-strain relationship (Figure 1E). Incremental distensibility was also smaller in MRA from SHR (Figure 1F). Pioglitazone treatment did not affect any of the parameters studied (Figure 1E, F). Thus, the β value was similar in arteries from pioglitazone-treated (8.36 ± 0.71; n= 9; P > 0.05) and untreated SHR rats.
Effect of pioglitazone treatment on the participation of COX-2-derived prostanoids in phenylephrine contraction
The mRNA levels of PPARγ were lower in mesenteric arteries from hypertensive than normotensive rats; after pioglitazone treatment of SHR, these levels were greatly increased (Figure 2A). We had previously demonstrated that COX-2 protein expression is greater in MRA from SHR than WKY (Briones et al., 2002); these results were confirmed and we also observed increased COX-2 mRNA levels in SHR compared with WKY (Figure 2B). After pioglitazone treatment, both COX-2 protein and mRNA levels were increased (Figure 2B).
Figure 2.
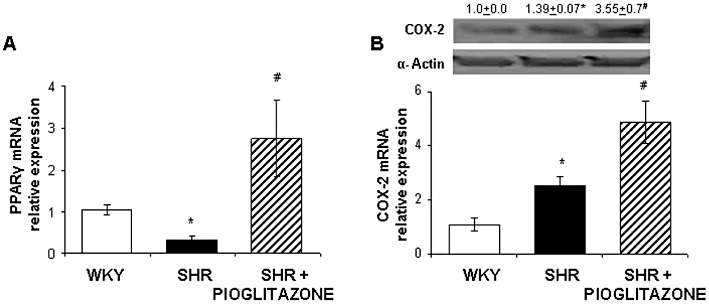
(A) Quantitative RT-PCR assessment of PPARγ mRNA levels in mesenteric arteries from WKY rats and SHR untreated and treated with pioglitazone. (B) Representative Western blot with densitometric analysis for the inducible isoform of COX-2 protein expression (upper panel) and quantitative RT-PCR assessment of COX-2 mRNA levels (lower panel) in arteries from WKY rats and SHR untreated and treated with pioglitazone. *P < 0.05 versus WKY rats, #P < 0.05 versus untreated SHR by Student's t test. n= 5–12 animals. Data are expressed as mean ± SEM.
The endothelium-dependent vasodilator response induced by ACh (10 µM) was reduced in MRA from SHR (WKY: 91.4 ± 0.9%, n= 30; SHR: 78.3 ± 1.1%, n= 45; P < 0.05); the treatment with pioglitazone improved this response, although it was still less than that observed in arteries from normotensive rats (83.1 ± 1.9%, n= 38, P < 0.05 vs. SHR and vs. WKY). Phenylephrine (0.1–30 µM) induced concentration-dependent contractile responses in MRA that were similar in arteries from normotensive and hypertensive rats; pioglitazone treatment did not modify the response to phenylephrine in SHR (Figure 3A). The selective COX-2 inhibitor NS 398 (1 µM) did not modify the phenylephrine-induced contraction in arteries from WKY and SHR (Figure 3B, C, Table 1). However, in endothelium-intact arteries from pioglitazone-treated SHR, NS 398 inhibited the phenylephrine response (Figure 3D, Table 1). These results suggest that pioglitazone treatment increases the participation of COX-2-derived contractile prostanoids in phenylephrine responses.
Figure 3.
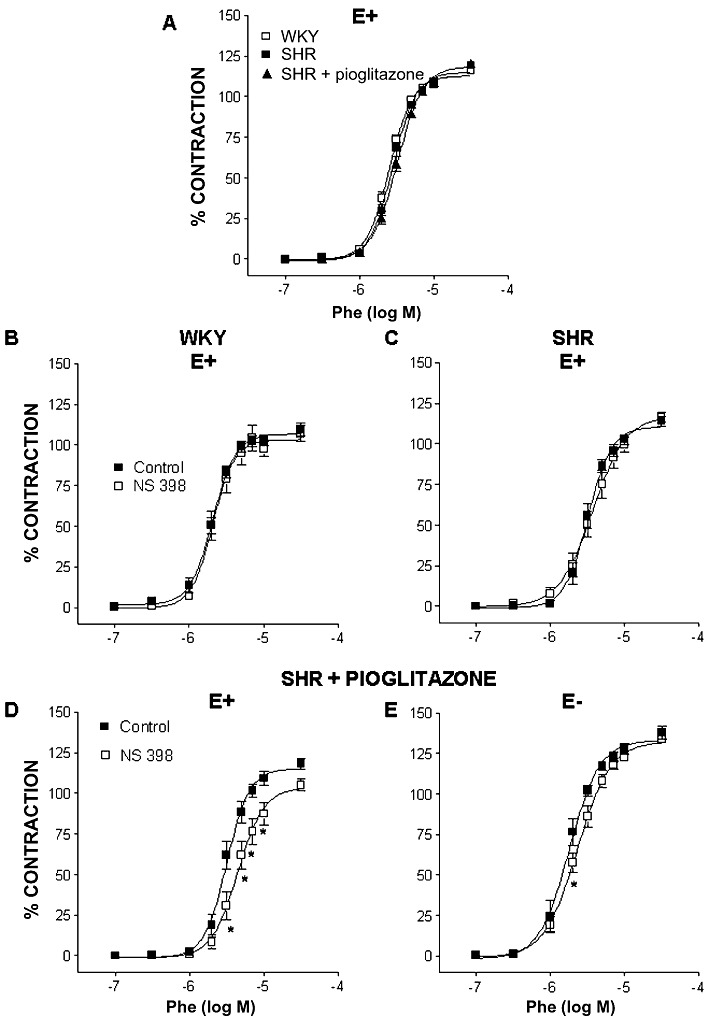
(A) Concentration–response curve to phenylephrine (Phe) in endothelium-intact (E+) MRA from WKY rats and SHR untreated or treated with pioglitazone. Effect of 1 µM NS 398 on the response to Phe in endothelium-intact (E+) MRA from WKY rats (B) and SHR untreated (C) or treated with pioglitazone (D) and in endothelium-denuded segments (E−) of MRA from pioglitazone-treated SHR (E). *P < 0.05 versus control by two-way anova and Bonferroni post-test. n= 6–10 animals. Data are expressed as mean ± SEM.
Table 1.
dAUC to phenylephrine response between control and experimental situations in endothelium-intact (E+) MRAs from WKY rats and SHR untreated or treated with pioglitazone and in endothelium-denuded segments (E−) of arteries from pioglitazone-treated SHR
| SHR + Pio | ||||
|---|---|---|---|---|
| WKY E+ | SHR E+ | E+ | E− | |
| NS 398 | ∼0 | ∼0 | 34.5 ± 6.9# | 13.2 ± 3.7+ |
| SQ 29,548 | ND | ∼0 | 19.5 ± 3.1# | ∼0+ |
| SC 19220 | ND | ∼0 | ∼0 | ND |
| RO 1138452 | ND | ND | ∼0 | ND |
| SQ 29,548 + RO 1138452 | ND | ND | 23.5 ± 5.8 | ND |
| Furegrelate | ND | ∼0 | ∼0 | ND |
| Tranylcypromine | ND | ∼0 | 16.0 ± 1.6# | ND |
| Apocynin | 14.2 ± 4.2 | 29.3 ± 4.3* | ∼0# | ND |
| Allopurinol | ∼0 | 15.0 ± 2.2* | ∼0# | ND |
| L-NAME | 51.9 ± 5.4 | 32.4 ± 4.9* | 53.6 ± 4.2# | ND |
AUCs were calculated from the individual concentration–response curve plots using a computer program (GraphPad Prism Software); the differences are expressed as a percentage of the AUC of the corresponding control situation. ND, not determined.
*P < 0.05 versus WKY rats, #P < 0.05 versus SHR untreated rats, +P < 0.05 versus endothelium-intact arteries by Student's t-test. n= 5–10 animals. Data are expressed as mean ± SEM.
After endothelium removal, the concentration–response curve to phenylephrine observed in segments from pioglitazone-treated rats was shifted leftward (Emax: 115.2 ± 2.1% vs. 130.9 ± 2.6 for E+, n= 38, and E−, n= 8, respectively, P < 0.05; pD2: 5.51 ± 0.02 vs. 5.74 ± 0.03 for E+ and E−, respectively, P < 0.05), to a similar extent to that observed in untreated SHR (data not shown). In endothelium-denuded segments from pioglitazone-treated rats, NS 398 (1 µM) inhibited the contractile response to phenylephrine (Figure 3E); however, this inhibitory effect was much smaller than that observed in endothelium-intact segments, as shown by the comparison of dAUC values (Table 1).
Involvement of prostacyclin in phenylephrine responses through TP receptor
The EP1/EP3 receptor antagonist SC 19220 (10 µM), the TP receptor antagonist SQ 29,548 (1 µM), the TXA2 synthase inhibitor furegrelate (1 µM) and the PGI2 synthase inhibitor tranylcypromine (10 µM) did not modify the concentration–response curve to phenylephrine in arteries from SHR (Table 1). In pioglitazone-treated rats, neither SC 19220 nor furegrelate affected the phenylephrine-induced contraction; this response was reduced by SQ 29,548 in endothelium-intact but not in endothelium-denuded segments (Figure 4). Tranylcypromine, but not the IP receptor antagonist RO 1138452 (1 µM), reduced the responses to phenylephrine. The combination of SQ 29,548 and RO 1138452 reduced the vasoconstrictor response (Figure 4) to a similar extent to that of SQ 29,548 alone, as shown by the comparison of dAUC values (Table 1). These results suggest that endothelium-derived prostanoids other than TXA2, probably PGI2, by acting on the TP receptor, contribute to the vasoconstrictor effect of phenylephrine on MRA from pioglitazone-treated SHR rats.
Figure 4.
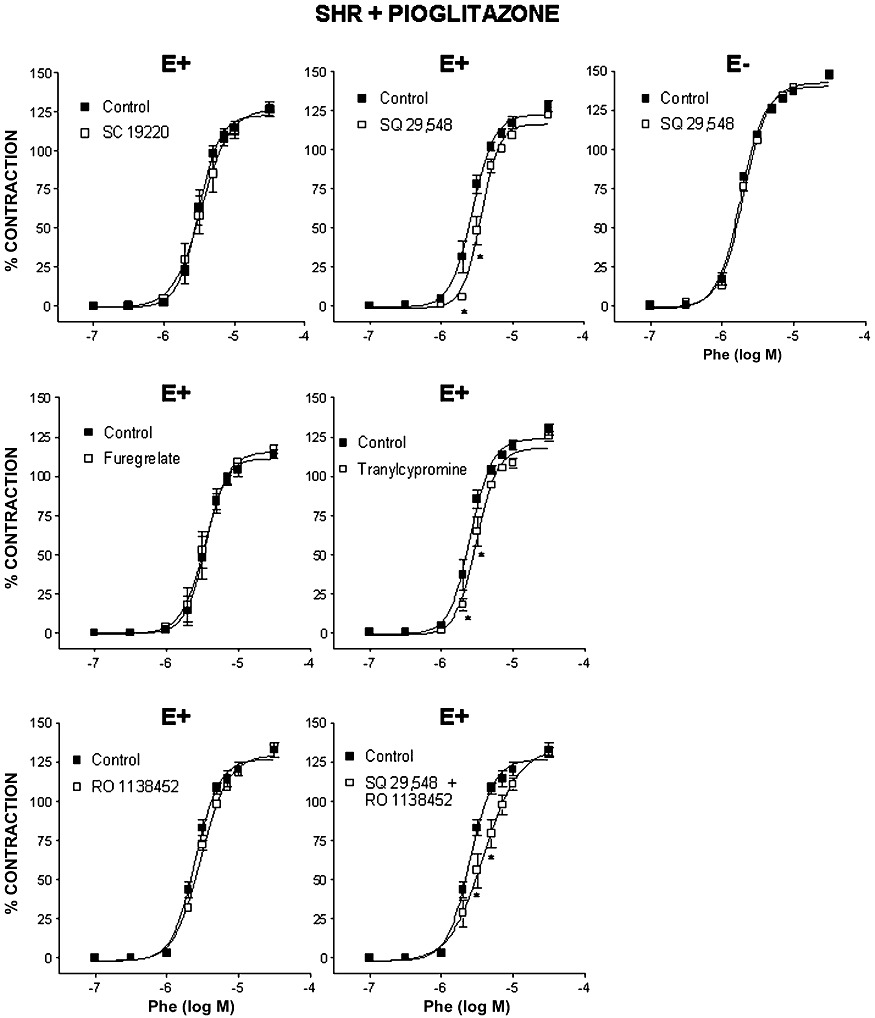
Effect of 10 µM SC 19220, 1 µM SQ 29,548, 1 µM furegrelate, 10 µM tranylcypromine, 1 µM RO 1138452 and the combination of SQ 29,548 and RO 1138452 on the concentration–response curve to phenylephrine (Phe) in endothelium-intact (E+) MRA from SHR treated with pioglitazone and effect of 1 µM SQ 29,548 on the response to Phe in endothelium-denuded segments (E−) of MRA from SHR treated with pioglitazone. *P < 0.05 versus control by two-way anova and Bonferroni post-test. n= 7–10 animals. Data are expressed as mean ± SEM.
The production of 6-keto-PGF1α was lower in the incubation medium of MRA from hypertensive than normotensive rats after stimulation with phenylephrine; these levels were greater in pioglitazone-treated rats (Figure 5A). 8-Isoprostane and PGF2α are also COX-derived products that may induce contraction by acting on TP receptors. The levels of 8-isoprostane were similar in arteries from WKY (0.062 ± 0.022 pg·µg−1 protein, n= 6) and SHR (0.061 ± 0.008 pg·µg−1 protein, n= 5, P > 0.05), and pioglitazone treatment did not modify these levels (0.046 ± 0.013 pg·µg−1 protein, n= 5, P > 0.05 vs. SHR). The levels of 13,14-dihydro-15-keto-PGF2α were similar in arteries from WKY (2.92 ± 0.59 pg·mg−1 protein, n= 6) and SHR (3.92 ± 1.19 pg·mg−1 protein, n= 5, P > 0.05 vs. WKY) and pioglitazone treatment did not modify these levels (5.92 ± 1.39 pg·mg−1 protein, n= 5, P > 0.05 vs. SHR).
Figure 5.
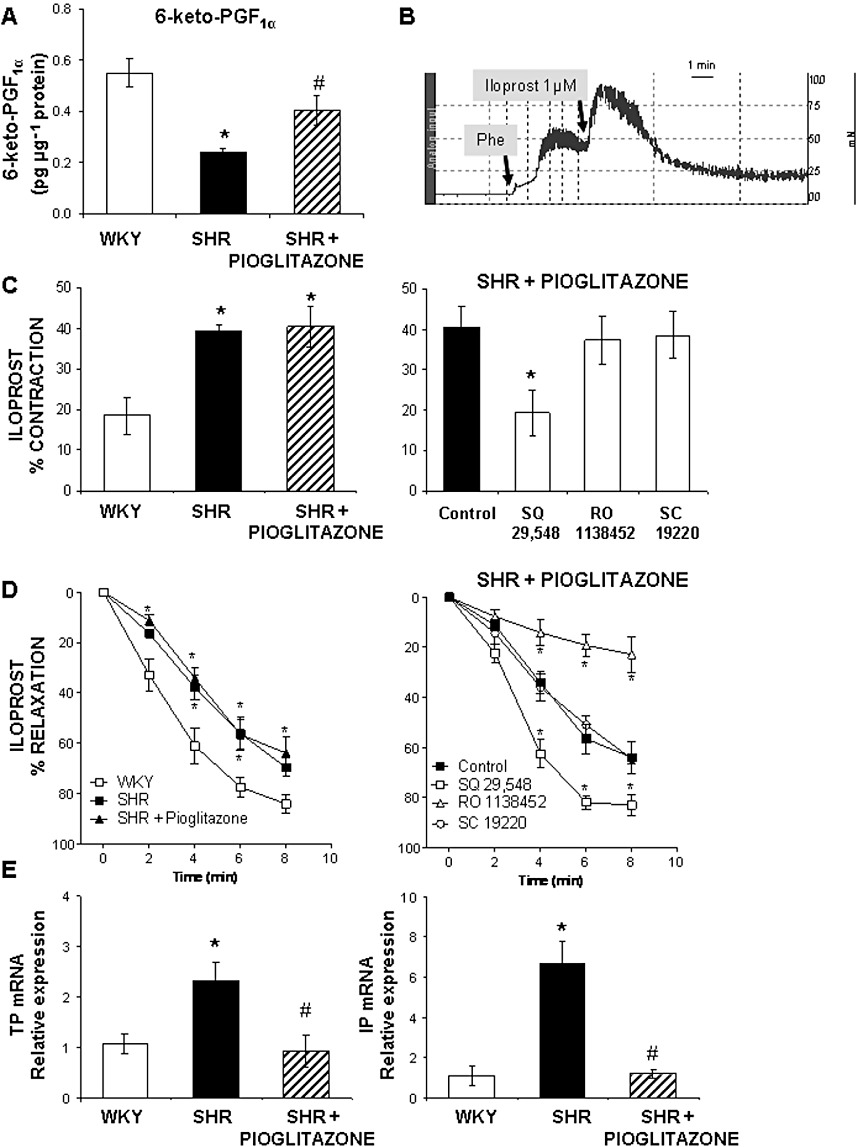
(A) Levels of 6-keto-PGF1α in the incubation medium after completion of the phenylephrine concentration–response curve in MRA from WKY rats and SHR untreated and treated with pioglitazone. (B) Representative record of the biphasic response elicited by iloprost (1 µM) in MRA precontracted with phenylephrine (Phe). (C) Contractile phase of the response to iloprost in MRA from WKY rats and SHR untreated or treated with pioglitazone and effect of 1 µM SQ 29,548, 1 µM RO 1138452 and 10 µM SC 19220 on the contraction induced by iloprost in pioglitazone-treated SHR. (D) Vasodilator phase of the response induced by iloprost in MRA from WKY rats and SHR untreated or treated with pioglitazone and effect of SQ 29,548, RO 1138452 and SC 19220 on the relaxation induced by iloprost in pioglitazone-treated SHR. (E) Quantitative RT-PCR assessment of TP and IP receptor mRNA levels in arteries from WKY rats and SHR untreated and treated with pioglitazone. *P < 0.05 versus WKY rats or versus control, #P < 0.05 versus SHR untreated rats by Student's t-test or by two-way anova and Bonferroni post-test. n= 5–7 animals. Data are expressed as mean ± SEM.
In segments precontracted with phenylephrine, the prostacyclin analogue iloprost (1 µM) induced a biphasic response consisting of a fast contraction followed by a slow relaxation (Figure 5B). The contractile phase was greater in arteries from SHR than WKY and remained unaltered after pioglitazone treatment (Figure 5C). In MRA from pioglitazone-treated rats, the contractile response elicited by iloprost was not modified by either RO 1138452 or SC 19220, but was reduced by SQ 29,548 (Figure 5C); similar results were obtained in untreated SHR (data not shown). The vasodilator phase of the response to iloprost was slower in arteries from SHR than WKY, as shown by the time–response curve (Figure 5D); pioglitazone treatment did not modify this response (Figure 5D). In MRA from pioglitazone-treated rats the relaxation induced by iloprost was almost abolished by RO 1138452 but unaffected by SC 19220; after incubation with SQ 29,548 this relaxing phase was faster (Figure 5D); in untreated SHR, similar results were observed (data not shown). All together, these results suggest that iloprost may act as a vasoconstrictor through TP receptors. The mRNA levels of both TP and IP receptors were greater in arteries from SHR than from WKY (Figure 5E); pioglitazone reduced these levels (Figure 5E).
Effect of pioglitazone treatment on the participation of ROS in phenylephrine contraction
The involvement of ROS was evaluated using the NADPH oxidase inhibitor apocynin and the xanthine oxidase inhibitor allopurinol. In MRA from both WKY and SHR rats, apocynin (0.3 mM) reduced the contractile response induced by phenylephrine (Figure 6A), although the inhibitory effect was greater in arteries from hypertensive rats, as shown by the analysis of dAUC (Table 1). Allopurinol (0.3 mM) reduced the response to phenylephrine only in segments from hypertensive rats; pioglitazone treatment abolished the effect of both apocynin and allopurinol (Figure 6A, Table 1).
Figure 6.
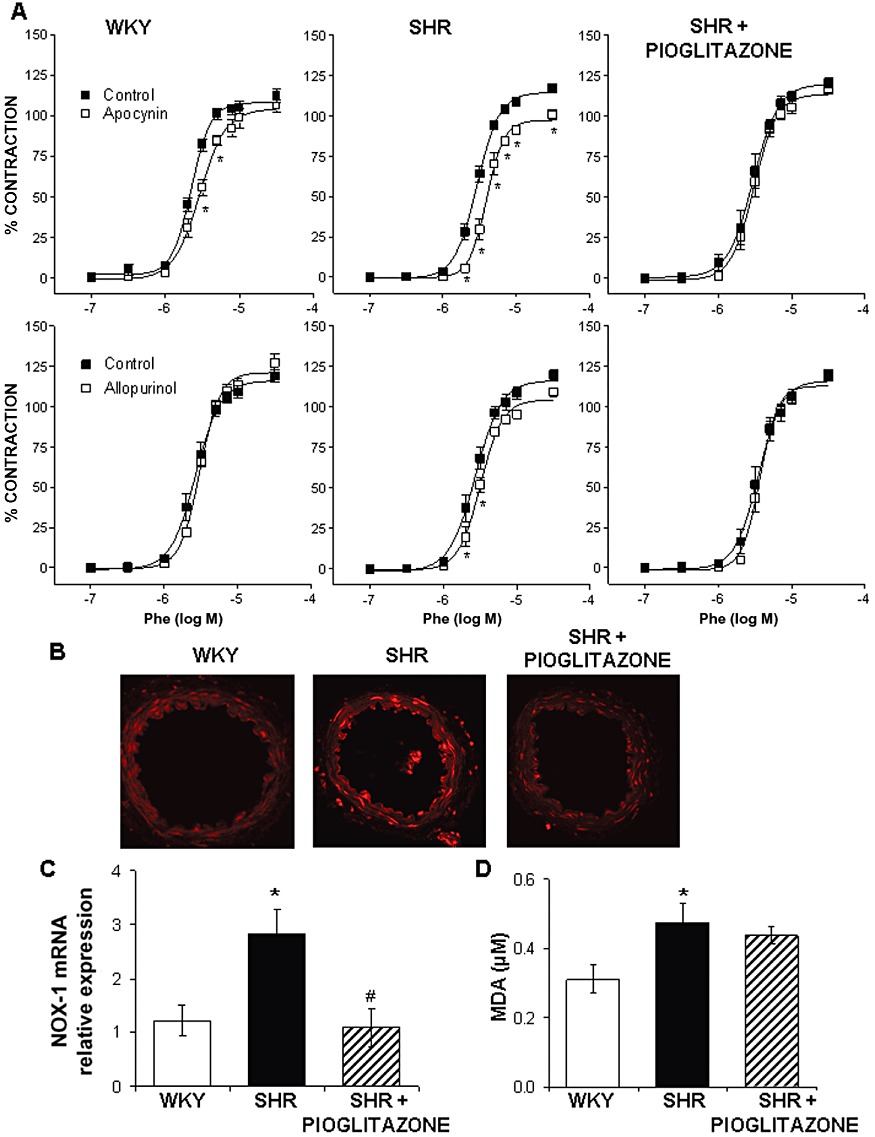
(A) Effect of 0.3 mM apocynin and 0.3 mM allopurinol on the concentration–response curve to phenylephrine (Phe) in MRA from WKY rats and SHR untreated and treated with pioglitazone. *P < 0.05 versus control by two-way anova and Bonferroni post-test. n= 7–12 animals. (B) Representative fluorescent photomicrographs of confocal microscopic sections of MRA from WKY rats and SHR untreated and treated with pioglitazone. Vessels were labelled with the oxidative dye dihydroethidium. Image size 375 × 375 µm. (C) Quantitative RT-PCR assessment of vascular NOX-1 mRNA levels and (D) plasma MDA levels in WKY rats and SHR untreated and treated with pioglitazone. *P < 0.05 versus WKY rats, #P < 0.05 versus SHR untreated rats by Student's t-test. n= 5–9 animals. Data are expressed as mean ± SEM.
Effect of pioglitazone on vascular O2•− production, mRNA levels of NADPH oxidase subunits and plasma MDA levels
As previously found (García-Redondo et al., 2009), O2•− production was greater in MRA from hypertensive than normotensive animals (Figure 6B). In agreement, vascular mRNA levels of NOX-1 were also greater in arteries from SHR than from WKY (Figure 6C). Pioglitazone treatment normalized the increased O2•− production and NOX-1 mRNA levels (Figure 6B, C). In addition, pioglitazone also reduced the mRNA levels of both NOX-2 (relative expression; SHR: 1.12 ± 0.25, n= 6; SHR-treated: 0.47 ± 0.10, n= 7, P < 0.05) and p47phox (relative expression; SHR: 1.14 ± 0.21, n= 8; SHR-treated: 0.25 ± 0.06, n= 7, P < 0.05).
Plasma MDA levels were also higher in SHR than in WKY rats, as previously reported (Alvarez et al., 2007). This difference was not affected by pioglitazone treatment of SHR (Figure 6D).
Effect of pioglitazone on vascular expression of detoxification enzymes
Western blot analysis revealed lower expression of Cu/Zn-, Mn- and EC-SOD in MRA from SHR than from WKY (Figure 7A). Pioglitazone-treatment did not modify the expression of Cu/Zn- and Mn-SOD, although it further reduced the EC-SOD expression (Figure 7A). The catalase mRNA levels were similar in arteries from normotensive and hypertensive rats (Figure 7B); after pioglitazone treatment, arteries from SHR show increased catalase mRNA levels (Figure 7B).
Figure 7.
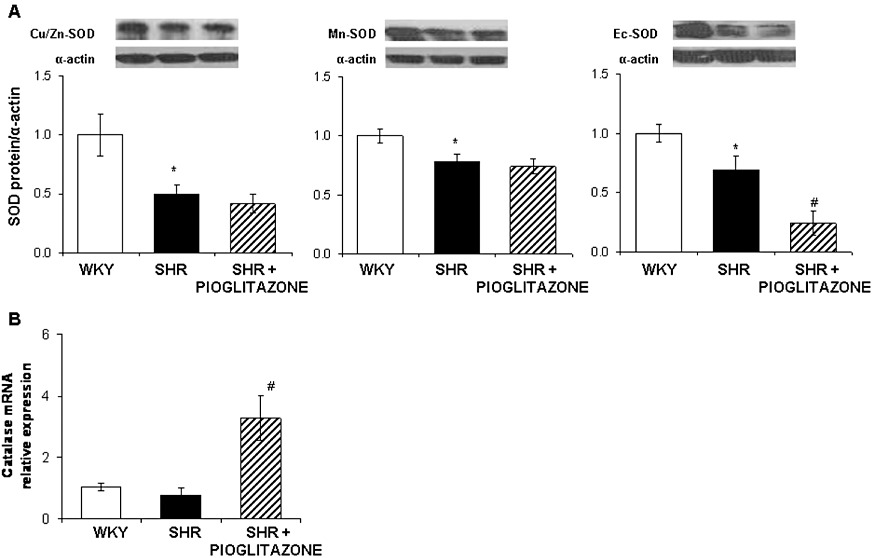
(A) Representative Western blot and densitometric analysis for Cu/Zn-, Mn- and EC-SOD protein expression and (B) quantitative RT-PCR assessment of catalase mRNA levels in mesenteric arteries from WKY rats and SHR untreated and treated with pioglitazone. *P < 0.05 versus WKY rats, #P < 0.05 versus SHR untreated rats by Student's t-test. n= 5–6 animals. Data are expressed as mean ± SEM.
Effect of pioglitazone treatment on the participation of NO in phenylephrine contraction
The NOS inhibitor L-NAME shifted the concentration–response curve to phenylephrine leftward, in segments from both WKY and SHR (Figure 8A); however, this effect was greater in normotensive rats, as shown by the dAUC analysis (Table 1). In pioglitazone-treated rats, L-NAME also increased the vasoconstrictor response to phenylephrine (Figure 8A) and this effect was greater than that observed in arteries from untreated SHR rats (Table 1).
Figure 8.
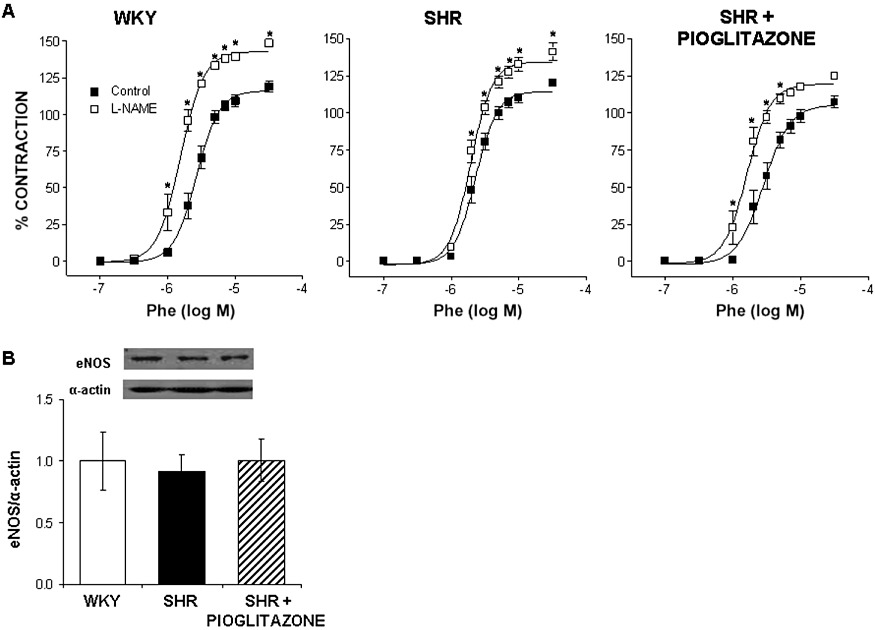
(A) Effect of 0.1 mM L-NAME on the concentration–response curve to phenylephrine (Phe) in WKY rats and SHR untreated and treated with pioglitazone. *P < 0.05 versus control by two-way anova and Bonferroni post-test. n= 6–7 animals. (B) Representative Western blot and densitometric analysis for eNOS protein expression in mesenteric arteries from SHR untreated and treated with pioglitazone. n= 5–6 animals. Data are expressed as mean ± SEM.
eNOS expression was similar in MRAs from normotensive and hypertensive rats; pioglitazone treatment did not modify the expression of this enzyme (Figure 8B).
Discussion
The present study shows the complex effect of PPARγ agonists on the modulation of vasoconstrictor responses. Thus, pioglitazone reduces ROS production and its contribution to phenylephrine contraction whereas it increases the contribution of NO to this response. Additionally, pioglitazone increases the production of COX-2-derived PGI2, which acts as a vasoconstrictor through TP receptors and is involved in the phenylephrine response. These effects, together with its lack of effect on vascular remodelling, might explain why pioglitazone does not affect BP in established hypertension, although it would contribute to the cardioprotective effect of glitazones widely reported.
The antihypertensive effects of PPARγ agonists are well-documented in patients and animal models of diabetes and/or metabolic syndrome (Sarafidis and Nilsson, 2006; Chen et al., 2008). However, when the hypertension is not associated with diabetes or with other factors of metabolic syndrome, the results are controversial. Glitazones prevent hypertension development (Sarafidis and Nilsson, 2006), but in established hypertension they have no effect on BP (Llorens et al., 2007; Nakamura et al., 2007; Shinzato et al., 2007), unless long-term treatment (Zhang et al., 2010) or high doses of glitazones (Wakino et al., 2005; Chan et al., 2010) are used. In adult SHR, with well-established hypertension, treatment for 28 days with 2.5 mg·kg−1·day−1 pioglitazone did not modify systolic BP.
The effects of glitazones on vascular structure are also debatable. In association with their preventative effect on the development of hypertension, glitazones also prevent vascular structural abnormalities (Diep et al., 2002; Ledingham and Laverty, 2005). In addition, long-term treatment with a high dose of rosiglitazone attenuated aortic remodelling and the rise in systolic BP in young SHR (Zhang et al., 2010). Furthermore, when glitazone treatment was started before or early after hypertension onset, only the attenuation of vascular remodelling was observed with no accompanying antihypertensive effect (Ishibashi et al., 2002; Nakamura et al., 2007; Cipolla et al., 2010). In contrast, in adult Zucker diabetic fatty rats, rosiglitazone did not modify the altered mechanical and structural properties despite the vascular function improvement (Lu et al., 2010). In MRA from 6 month-old SHR, which had been hypertensive for a long time, pioglitazone did not affect the reduced external and internal diameters, the increased wall : lumen ratio and vascular stiffness or the changes in distensibility. Differences in the time that rats have been hypertensive before treatment, which might result in remodelling variation, as well as in the duration and dosage of treatment, would explain such discrepancies.
Glitazones improve the impaired endothelium-dependent relaxation (Diep et al., 2002; Nakamura et al., 2007; Matsumoto et al., 2008) and reduce vascular contractility, although pioglitazone increased phenylephrine responses in SHR aorta (Llorens et al., 2007). In MRA from SHR, pioglitazone treatment slightly improved the impaired ACh-induced vasodilatation. Moreover, the phenylephrine contraction was similar in MRA from WKY and SHR, and remained unmodified after pioglitazone treatment; this result, which agrees with that found in isoprenaline-treated rats (Fukuda et al., 2008), apparently excludes an effect of the glitazone on vasoconstrictor responses.
A decrease in the expression of vascular PPARs might participate in the exacerbated proliferation, migration and inflammation observed in hypertension (Chan et al., 2010; Li et al., 2010; Zhang et al., 2010). Accordingly, MRA from SHR showed lower PPARγ mRNA levels than WKY. This reduction in PPARγ might be associated with the increased COX-2 expression in SHR, as COX-2 is regulated by transcription factors, the activation of which is reduced by PPARγ (Touyz and Schiffrin, 2006). Pioglitazone treatment increased MRA PPARγ levels, similarly to those found using rosiglitazone and pioglitazone in SHR aorta and rostral ventrolateral medulla (Chan et al., 2010; Zhang et al., 2010), suggesting that up-regulated PPARγ may contribute to the anti-inflammatory properties of glitazones. Despite this, pioglitazone treatment increased COX-2 expression in MRA from SHR. PPARγ agonists have been shown to decrease (Sánchez-Hidalgo et al., 2005; Collino et al., 2006) but also to increase COX-2 (Meade et al., 1999; Ye et al., 2006; Kang et al., 2008); the presence of a functional PPAR response element (PPRE) in the promoter region of COX-2 (Meade et al., 1999) would explain this increase. The augmented COX-2 expression after pioglitazone treatment had functional consequences, as a selective COX-2 inhibitor reduced phenylephrine responses, suggesting that COX-2-derived vasoconstrictor prostanoids are involved in the effects of pioglitazone treatment.
We next attempted to determine the prostanoid which is increased after pioglitazone treatment. The TP receptor antagonist reduced the phenylephrine responses, whereas the TXA2 synthase inhibitor furegrelate had no effect, suggesting that prostanoids other than TXA2, by acting on TP receptors, contribute to these responses. These prostanoids seem to be mainly of endothelial origin as endothelium removal greatly reduced the inhibitory effect of NS 398 and abolished that of SQ 29,548. PGF2α, isoprostanes and PGH2 would induce contraction by activating TP receptors (Félétou et al., 2009). However, the levels of PGF2α and 8-isoprostane were similar in samples from all groups, excluding the involvement of these compounds in the pioglitazone effect. Unfortunately, PGH2 levels are difficult to measure due to its short half-life and still need to be evaluated more precisely to determine their contribution to vascular responses (Félétou et al., 2009).
Prostacyclin, which acts on IP receptors, is the main vasodilator prostanoid generated by COX. However, there is growing evidence indicating that PGI2 acts as an endothelium-derived vasoconstrictor factor able to activate TP receptors in conditions such as hypertension or aging (Gluais et al., 2005; Gomez et al., 2008; Xavier et al., 2008; Félétou et al., 2009). Accordingly, the prostacyclin analogue iloprost induced a biphasic response with greater contractile effect in SHR than WKY, in agreement with reports using PGI2 (Gluais et al., 2005; Xavier et al., 2008). Additionally, the vasodilator phase was slower in SHR than WKY. Interestingly, pioglitazone treatment did not modify any of the two phases. The iloprost-induced vasoconstriction was reduced by the TP receptor antagonist in MRA from SHR. Furthermore, the relaxing phase was almost abolished by the IP receptor antagonist but increased by SQ 29,548 in MRA from both treated and untreated SHR, in agreement with the results of Gomez et al. (2008), suggesting that activation of TP receptors, even by a weak and partial agonist such as PGI2, can markedly blunt vascular relaxation. Whether changes in the expression of receptors would explain differences in the iloprost response between WKY and SHR remains elusive. Thus, Tang and Vanhoutte (2008) demonstrated that hypertension did not modify the genomic expression of either IP or TP receptors, whereas Numaguchi et al. (1999) found the expression of IP receptors was reduced in SHR. In MRA from SHR, we found that TP receptor levels were increased, in agreement with the higher contraction to iloprost observed and the increased response to the thromboxane analogue U46619 previously described (Xavier et al., 2008). Additionally, the IP receptor levels were greater in SHR, despite the slower iloprost-induced vasodilatation. Dysfunction of this receptor and possible alterations in the adenylate cyclase pathway has been described in SHR (Gluais et al., 2005; Gomez et al., 2008). Furthermore, pioglitazone treatment reduced TP and IP receptor levels. In this context, Sugawara et al. (2002) found that PPARγ activation reduced TP gene transcription. Despite this reduction in receptors expressed, the biphasic response to iloprost was similar in MRA from pioglitazone-treated and untreated rats. Similar contractile responses to U46619 were also observed (R. Hernanz, unpubl. results). Differences in the cellular mechanisms from the genomic expression to the presence of functional receptors in the membrane would explain such discrepancies.
Prostacyclin levels after phenylephrine stimulation were lower in samples from SHR than WKY, as demonstrated previously (Soma et al., 1985). Interestingly, pioglitazone treatment augmented these levels, in agreement with the findings of Ye et al. (2006), who established a relationship between these increased levels and the protective effect of pioglitazone in myocardial-reperfusion injury. Furthermore, the PGI2 synthesis inhibitor, but not the IP receptor antagonist, reduced the phenylephrine response only in segments from pioglitazone-treated rats. Overall, these results suggest that pioglitazone increases prostacyclin levels, which by acting on TP receptors are involved in the phenylephrine response.
Increased O2•− production from different origins, including NADPH oxidase, participates in contractile responses in hypertension (Alvarez et al., 2008; García-Redondo et al., 2009). Accordingly, increased expression of NOX-1, one of the catalytic subunits of NADPH oxidase, has been demonstrated in SHR (Briones et al., 2011). Additionally, reduced antioxidant defences might contribute to the increased oxidative stress described in this pathology (Redón et al., 2003). This increased oxidative stress would contribute to the altered vascular responses by reducing NO availability (Virdis et al., 2009). Our results support this hypothesis because compared with WKY, SHR showed: (i) greater inhibitory effect of apocynin and allopurinol on phenylephrine responses; (ii) increased O2•− production and MDA plasma levels; (iii) increased NOX-1 mRNA levels; (iv) reduced Cu/Zn-, Mn- and EC-SOD expression, whereas the catalase levels were similar in both strains; and (v) lower leftward shift of the phenylephrine concentration–response curve after NO inhibition. Glitazones reduce oxidative stress by inhibition of NADPH oxidase activity (Bagi et al., 2004; Hwang et al., 2005); this contributes to their anti-inflammatory effect and protective action against hypertension-induced cerebrovascular injury (Nakamura et al., 2007). In our conditions pioglitazone treatment also displayed antioxidant properties as: (i) it abolished the apocynin and allopurinol effects on the phenylephrine response; (ii) normalized the increased O2•− production; and (iii) reduced the NOX-1, NOX-2 and p47phox mRNA levels. Moreover, despite the lack of an effect on Cu/Zn- and Mn-SOD and the decreased EC-SOD, catalase mRNA levels were increased by pioglitazone treatment, in agreement with the results of Bagi et al. (2004); this would be explained by the existence of a PPRE in the catalase promoter region (Girnun et al., 2002). Apocynin might reduce ROS availability through its antioxidant properties independent of NADPH oxidase inhibition (Heumüller et al., 2008). Interestingly, we found that the increased ROS production in SHR was reduced after pioglitazone treatment, although we do not know the exact mechanism of action of apocynin in our conditions.
The antioxidant effect of glitazones might have a very important functional consequence, that is increased NO availability. The effects of glitazone on NO generation are unclear. Although some studies have found that glitazones increase NO release and/or NO synthase activity (Calnek et al., 2003; Wakino et al., 2005; Llorens et al., 2007), others have found they have no effect (Ye et al., 2006; Li et al., 2010). In our study, pioglitazone treatment improved the impaired ACh-induced vasodilatation in SHR and L-NAME greatly potentiated the phenylephrine responses without affecting eNOS protein expression. Similarly, Calnek et al. (2003) showed increased NO release by endothelial cells without increased eNOS mRNA or protein levels.
In conclusion, in resistance arteries several factors contribute to the phenylephrine response and, although pioglitazone does not have antihypertensive effects, it modifies the levels of different mediators involved in this response. Thus, pioglitazone increased PGI2 production, probably by increasing COX-2 activity. Although in our experimental conditions PGI2 seems to act as a vasoconstrictor, increased levels of this prostanoid would be beneficial; through its inhibition of platelet aggregation and pleiotropic effects on vascular smooth muscle, PGI2 has an important cardioprotective role. Further investigations are needed to address this issue. In addition, the reduction in oxidative stress, which can increase NO bioavailability, would also contribute to the protective effect of glitazones widely reported in several pathologies.
Acknowledgments
This study was supported by Fundación Mutua Madrileña, Ministerio de Ciencia e Innovación (SAF2009-07201), Instituto de Salud Carlos III (Red RECAVA, RD06/0014/0011) and Sociedad Española de Farmacología-Almirall Prodesfarma. AMB is supported by the Ramon y Cajal program (RYC-2010-06473) from MICINN.
Glossary
- dAUC
differences of area under the concentration–response curves
- KHS
Krebs Henseleit solution
- L-NAME
N-nitro-L-arginine methyl ester
- MDA
malondialdehyde
- MRA
mesenteric resistance artery
- NS 398
N-(2-cyclohexyloxy-4-nitrophenyl)-methanesulfonamide
- O2•−
superoxide anion
- PGI2
prostacyclin
- RO 1138452
1H-imidazol-2-amine,4,5-dihydro-N-[4-{[4-(1-methylethoxy)phenyl]methyl}phenyl]
- ROS
reactive oxygen species
- SC 19220
8-chloro-dibenz[b,f][1,4]oxazepine-10(11H)-carboxy-(2-acetyl)hydrazide
- SHR
spontaneously hypertensive rats
- SOD
superoxide dismutase
- SQ 29,548
{1S-[1α,2α(Z),3α,4α]}-7-{3-[{2-[(phenylamino)carbonyl]hydrazino]methyl}-7-oxabicyclo[2.2.1]hept-2-yl}-5 heptenoic acid
- VSMC
vascular smooth muscle cells
- WKY
Wistar Kyoto
Conflict of interest
None.
References
- Alvarez Y, Briones AM, Balfagón G, Alonso MJ, Salaices M. Hypertension increases the participation of vasoconstrictor prostanoids from cyclooxygenase-2 in phenylephrine responses. J Hypertens. 2005;23:767–777. doi: 10.1097/01.hjh.0000163145.12707.63. [DOI] [PubMed] [Google Scholar]
- Alvarez Y, Pérez-Girón JV, Hernanz R, Briones AM, García-Redondo A, Beltrán A, et al. Losartan reduces the increased participation of cyclooxygenase-2-derived products in vascular responses of hypertensive rats. J Pharmacol Exp Ther. 2007;321:381–388. doi: 10.1124/jpet.106.115287. [DOI] [PubMed] [Google Scholar]
- Alvarez Y, Briones AM, Hernanz R, Pérez-Girón JV, Alonso MJ, Salaices M. Role of NADPH oxidase and iNOS in vasoconstrictor responses of vessels from hypertensive and normotensive rats. Br J Pharmacol. 2008;153:926–935. doi: 10.1038/sj.bjp.0707575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arribas SM, Hillier C, González C, McGrory S, Dominiczak AF, McGrath JC. Cellular aspects of vascular remodeling in hypertension revealed by confocal microscopy. Hypertension. 1997;30:1455–1464. doi: 10.1161/01.hyp.30.6.1455. [DOI] [PubMed] [Google Scholar]
- Bagi Z, Koller A, Kaley G. PPARγ activation, by reducing oxidative stress, increases NO bioavailability in coronary arterioles of mice with Type 2 diabetes. Am J Physiol Heart Circ Physiol. 2004;286:H742–H748. doi: 10.1152/ajpheart.00718.2003. [DOI] [PubMed] [Google Scholar]
- Briones AM, Alonso MJ, Hernanz R, Tovar S, Vila E, Salaices M. Hypertension alters the participation of contractile prostanoids and superoxide anions in lipopolysaccharide effects on small mesenteric arteries. Life Sci. 2002;71:1997–2014. doi: 10.1016/s0024-3205(02)01967-7. [DOI] [PubMed] [Google Scholar]
- Briones AM, Tabet F, Callera GE, Montezano AC, Yogi A, He Y, et al. Differential regulation of Nox1, Nox2 and Nox4 in vascular smooth muscle cells from WKY and SHR. J Am Soc Hypertens. 2011;5:137–153. doi: 10.1016/j.jash.2011.02.001. [DOI] [PubMed] [Google Scholar]
- Calnek DS, Mazzella L, Roser S, Roman J, Hart CM. Peroxisome proliferator-activated receptor γ ligands increase release of nitric oxide from endothelial cells. Arterioscler Thromb Vasc Biol. 2003;23:52–57. doi: 10.1161/01.atv.0000044461.01844.c9. [DOI] [PubMed] [Google Scholar]
- Chan SH, Wu KL, Kung PS, Chan JY. Oral intake of rosiglitazone promotes a central antihypertensive effect via upregulation of peroxisome proliferator-activated receptor-γ and alleviation of oxidative stress in rostral ventrolateral medulla of spontaneously hypertensive rats. Hypertension. 2010;55:1444–1453. doi: 10.1161/HYPERTENSIONAHA.109.149146. [DOI] [PubMed] [Google Scholar]
- Chen R, Liang F, Moriya J, Yamakawa J, Takahashi T, Shen L, et al. Peroxisome proliferator-activated receptors (PPARs) and their agonists for hypertension and heart failure: are the reagents beneficial or harmful? Int J Cardiol. 2008;130:131–139. doi: 10.1016/j.ijcard.2008.03.080. [DOI] [PubMed] [Google Scholar]
- Cipolla MJ, Bishop N, Vinke RS, Godfrey JA. PPARγ activation prevents hypertensive remodeling of cerebral arteries and improves vascular function in female rats. Stroke. 2010;41:1266–1270. doi: 10.1161/STROKEAHA.109.576942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collino M, Aragno M, Mastrocola R, Gallicchio M, Rosa AC, Dianzani C, et al. Modulation of the oxidative stress and inflammatory response by PPAR-γ agonists in the hippocampus of rats exposed to cerebral ischemia/reperfusion. Eur J Pharmacol. 2006;530:70–80. doi: 10.1016/j.ejphar.2005.11.049. [DOI] [PubMed] [Google Scholar]
- Diep QN, El Mabrouk M, Cohn JS, Endemann D, Amiri F, Virdis A, et al. Structure, endothelial function, cell growth, and inflammation in blood vessels of angiotensin II-infused rats: role of peroxisome proliferator-activated receptor-γ. Circulation. 2002;105:2296–2302. doi: 10.1161/01.cir.0000016049.86468.23. [DOI] [PubMed] [Google Scholar]
- Dormandy JA, Charbonnel B, Eckland DJ, Erdmann E, Massi-Benedetti M, Moules IK, et al. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet. 2005;366:1279–1289. doi: 10.1016/S0140-6736(05)67528-9. [DOI] [PubMed] [Google Scholar]
- Félétou M, Verbeuren TJ, Vanhoutte PM. Endothelium-dependent contractions in SHR: a tale of prostanoid TP and IP receptors. Br J Pharmacol. 2009;156:563–574. doi: 10.1111/j.1476-5381.2008.00060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda LE, Davel AP, Verissimo-Filho S, Lopes LR, Cachofeiro V, Lahera V, et al. Fenofibrate and pioglitazone do not ameliorate the altered vascular reactivity in aorta of isoproterenol-treated rats. J Cardiovasc Pharmacol. 2008;52:413–421. doi: 10.1097/FJC.0b013e31818a8927. [DOI] [PubMed] [Google Scholar]
- García-Redondo AB, Briones AM, Avendaño MS, Hernanz R, Alonso MJ, Salaices M. Losartan and tempol treatments normalize the increased response to hydrogen peroxide in resistance arteries from hypertensive rats. J Hypertens. 2009;27:1814–1822. doi: 10.1097/HJH.0b013e32832d23e6. [DOI] [PubMed] [Google Scholar]
- Girnun GD, Domann FE, Moore SA, Robbins ME. Identification of a functional peroxisome proliferator-activated receptor response element in the rat catalase promoter. Mol Endocrinol. 2002;16:2793–2801. doi: 10.1210/me.2002-0020. [DOI] [PubMed] [Google Scholar]
- Gluais P, Lonchampt M, Morrow JD, Vanhoutte PM, Feletou M. Acetylcholine-induced endothelium-dependent contractions in the SHR aorta: the Janus face of prostacyclin. Br J Pharmacol. 2005;146:834–845. doi: 10.1038/sj.bjp.0706390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez E, Schwendemann C, Roger S, Simonet S, Paysant J, Courchay C, et al. Aging and prostacyclin responses in aorta and platelets from WKY and SHR rats. Am J Physiol Heart Circ Physiol. 2008;295:H2198–H2211. doi: 10.1152/ajpheart.00507.2008. [DOI] [PubMed] [Google Scholar]
- Heumüller S, Wind S, Barbosa-Sicard E, Schmidt HH, Busse R, Schröder K, et al. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension. 2008;51:211–217. doi: 10.1161/HYPERTENSIONAHA.107.100214. [DOI] [PubMed] [Google Scholar]
- Hwang J, Kleinhenz DJ, Lassègue B, Griendling KK, Dikalov S, Hart CM. Peroxisome proliferator-activated receptor-γ ligands regulate endothelial membrane superoxide production. Am J Physiol Cell Physiol. 2005;288:C899–C905. doi: 10.1152/ajpcell.00474.2004. [DOI] [PubMed] [Google Scholar]
- Ishibashi M, Egashira K, Hiasa K, Inoue S, Ni W, Zhao Q, et al. Antiinflammatory and antiarteriosclerotic effects of pioglitazone. Hypertension. 2002;40:687–693. doi: 10.1161/01.hyp.0000036396.64769.c2. [DOI] [PubMed] [Google Scholar]
- Kang YJ, Kim HS, Choi HC. Troglitazone increases IL-1β induced cyclooxygenase-2 and inducible nitric oxide synthase expression via enhanced phosphorylation of IκBα in vascular smooth muscle cells from Wistar-Kyoto rats and spontaneously hypertensive rats. Biol Pharm Bull. 2008;31:1955–1958. doi: 10.1248/bpb.31.1955. [DOI] [PubMed] [Google Scholar]
- Ledingham JM, Laverty R. Effects of glitazones on blood pressure and vascular structure in mesenteric resistance arteries and basilar artery from genetically hypertensive rats. Clin Exp Pharmacol Physiol. 2005;32:919–925. doi: 10.1111/j.1440-1681.2005.04285.x. [DOI] [PubMed] [Google Scholar]
- Leff T, Mathews ST, Camp HS. Review: peroxisome proliferator-activated receptor-γ and its role in the development and treatment of diabetes. Exp Diabesity Res. 2004;5:99–109. doi: 10.1080/15438600490451668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Zhang H, Wang W, Wang X, Huang Y, Huang C, et al. Vascular insulin resistance in prehypertensive rats: role of PI3-kinase/Akt/eNOS signaling. Eur J Pharmacol. 2010;628:140–147. doi: 10.1016/j.ejphar.2009.11.038. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Llorens S, Mendizabal Y, Nava E. Effects of pioglitazone and rosiglitazone on aortic vascular function in rat genetic hypertension. Eur J Pharmacol. 2007;575:105–112. doi: 10.1016/j.ejphar.2007.08.002. [DOI] [PubMed] [Google Scholar]
- Lu X, Guo X, Karathanasis SK, Zimmerman KM, Onyia JE, Peterson RG, et al. Rosiglitazone reverses endothelial dysfunction but not remodeling of femoral artery in Zucker diabetic fatty rats. Cardiovasc Diabetol. 2010;9:19. doi: 10.1186/1475-2840-9-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto T, Kobayashi T, Kamata K. Relationships among ET-1, PPARγ, oxidative stress and endothelial dysfunction in diabetic animals. J Smooth Muscle Res. 2008;44:41–55. doi: 10.1540/jsmr.44.41. [DOI] [PubMed] [Google Scholar]
- Meade EA, McIntyre TM, Zimmerman GA, Prescott SM. Peroxisome proliferators enhance cyclooxygenase-2 expression in epithelial cells. J Biol Chem. 1999;274:8328–8334. doi: 10.1074/jbc.274.12.8328. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Yamamoto E, Kataoka K, Yamashita T, Tokutomi Y, Dong YF, et al. Pioglitazone exerts protective effects against stroke in stroke-prone spontaneously hypertensive rats, independently of blood pressure. Stroke. 2007;38:3016–3022. doi: 10.1161/STROKEAHA.107.486522. [DOI] [PubMed] [Google Scholar]
- Numaguchi Y, Harada M, Osanai H, Hayashi K, Toki Y, Okumura K, et al. Altered gene expression of prostacyclin synthase and prostacyclin receptor in the thoracic aorta of spontaneously hypertensive rats. Cardiovasc Res. 1999;41:682–688. doi: 10.1016/s0008-6363(98)00239-9. [DOI] [PubMed] [Google Scholar]
- Paravicini TM, Touyz RM. Redox signaling in hypertension. Cardiovasc Res. 2006;71:247–258. doi: 10.1016/j.cardiores.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Redón J, Oliva MR, Tormos C, Giner V, Chaves J, Iradi A, et al. Antioxidant activities and oxidative stress byproducts in human hypertension. Hypertension. 2003;41:1096–1101. doi: 10.1161/01.HYP.0000068370.21009.38. [DOI] [PubMed] [Google Scholar]
- Sánchez-Hidalgo M, Martín AR, Villegas I, Alarcón De La Lastra C. Rosiglitazone, an agonist of peroxisome proliferator-activated receptor gamma, reduces chronic colonic inflammation in rats. Biochem Pharmacol. 2005;69:1733–1744. doi: 10.1016/j.bcp.2005.03.024. [DOI] [PubMed] [Google Scholar]
- Sarafidis PA, Nilsson PM. The effects of thiazolidinediones on blood pressure levels – a systematic review. Blood Press. 2006;15:135–150. doi: 10.1080/08037050600853720. [DOI] [PubMed] [Google Scholar]
- Savoia C, Schiffrin EL. Vascular inflammation in hypertension and diabetes: molecular mechanisms and therapeutic interventions. Clin Sci (Lond) 2007;112:375–384. doi: 10.1042/CS20060247. [DOI] [PubMed] [Google Scholar]
- Shinzato T, Ohya Y, Nakamoto M, Ishida A, Takishita S. Beneficial effects of pioglitazone on left ventricular hypertrophy in genetically hypertensive rats. Hypertens Res. 2007;30:863–873. doi: 10.1291/hypres.30.863. [DOI] [PubMed] [Google Scholar]
- Soma M, Manku MS, Jenkins DK, Horrobin DF. Prostaglandins and thromboxane outflow from the perfused mesenteric vascular bed in spontaneously hypertensive rats. Prostaglandins. 1985;29:323–333. doi: 10.1016/0090-6980(85)90212-6. [DOI] [PubMed] [Google Scholar]
- Sugawara A, Uruno A, Kudo M, Ikeda Y, Sato K, Taniyama Y, et al. Transcription suppression of thromboxane receptor gene by peroxisome proliferator-activated receptor-gamma via an interaction with Sp1 in vascular smooth muscle cells. J Biol Chem. 2002;277:9676–9683. doi: 10.1074/jbc.M104560200. [DOI] [PubMed] [Google Scholar]
- Tang EH, Vanhoutte PM. Gene expression changes of prostanoid synthases in endothelial cells and prostanoid receptors in vascular smooth muscle cells caused by aging and hypertension. Physiol Genomics. 2008;32:409–418. doi: 10.1152/physiolgenomics.00136.2007. [DOI] [PubMed] [Google Scholar]
- Touyz RM, Schiffrin EL. Peroxisome proliferator-activated receptors in vascular biology-molecular mechanisms and clinical implications. Vascul Pharmacol. 2006;45:19–28. doi: 10.1016/j.vph.2005.11.014. [DOI] [PubMed] [Google Scholar]
- Virdis A, Colucci R, Versari D, Ghisu N, Fornai M, Antonioli L, et al. Atorvastatin prevents endothelial dysfunction in mesenteric arteries from spontaneously hypertensive rats: role of cyclooxygenase 2-derived contracting prostanoids. Hypertension. 2009;53:1008–1016. doi: 10.1161/HYPERTENSIONAHA.109.132258. [DOI] [PubMed] [Google Scholar]
- Wakino S, Hayashi K, Tatematsu S, Hasegawa K, Takamatsu I, Kanda T, et al. Pioglitazone lowers systemic asymmetric dimethylarginine by inducing dimethylarginine dimethylaminohydrolase in rats. Hypertens Res. 2005;28:255–262. doi: 10.1291/hypres.28.255. [DOI] [PubMed] [Google Scholar]
- Xavier FE, Aras-López R, Arroyo-Villa I, del Campo L, Salaices M, Rossoni LV, et al. Aldosterone induces endothelial dysfunction in resistance arteries from normotensive and hypertensive rats by increasing thromboxane A2 and prostacyclin. Br J Pharmacol. 2008;154:1225–1235. doi: 10.1038/bjp.2008.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Lin Y, Atar S, Huang MH, Perez-Polo JR, Uretsky BF, et al. Myocardial protection by pioglitazone, atorvastatin, and their combination: mechanisms and possible interactions. Am J Physiol Heart Circ Physiol. 2006;291:H1158–H1169. doi: 10.1152/ajpheart.00096.2006. [DOI] [PubMed] [Google Scholar]
- Zhang L, Xie P, Wang J, Yang Q, Fang C, Zhou S, et al. Impaired peroxisome proliferator-activated receptor-γ contributes to phenotypic modulation of vascular smooth muscle cells during hypertension. J Biol Chem. 2010;285:13666–13677. doi: 10.1074/jbc.M109.087718. [DOI] [PMC free article] [PubMed] [Google Scholar]