

Abstract
BACKGROUND AND PURPOSE
Translational animal models are essential in the prediction of the efficacy and side effects of new chemical entities. We have carried out a thorough study of three distinct disease-modifying antirheumatic drugs (DMARDs) in an adjuvant-induced arthritis (AIA) model in the rat and critically appraised the results in the context of the reported clinical experience in rheumatoid arthritis (RA) patients.
EXPERIMENTAL APPROACH
Teriflunomide – a dihydroorotate dehydrogenase (DHODH) inhibitor; AL8697 – a selective p38 MAPK inhibitor; and tofacitinib – a Janus kinase (JAK) inhibitor; were selected as representatives of their class and dose-response studies carried out using a therapeutic 10-day administration scheme in arthritic rats. Paw swelling and body weight were periodically monitored, and joint radiology and histology, lymph organ weight and haematological and biochemical parameters evaluated at study completion.
KEY RESULTS
All three drugs demonstrated beneficial effects on paw swelling, bone lesions and splenomegalia, with p38 inhibition providing the best anti-inflammatory effect and JAK inhibition the best DMARD effect. Leukopenia, body weight loss and gastrointestinal toxicity were dose-dependently observed with teriflunomide treatment. p38 MAPK inhibition induced leukocytosis and increased total plasma cholesterol. JAK inhibition, normalized platelet, reticulocyte and neutrophil counts, and alanine aminotransferase (ALT) levels while inducing lymphopenia and cholesterolemia.
CONCLUSIONS AND IMPLICATIONS
This multiparametric approach can reveal specific drug properties and provide translational information. Whereas the complex profile for p38 inhibition in AIA is not observed in human RA, immunosuppressants such as DHODH and JAK inhibitors show DMARD properties and side effects seen in both AIA and RA.
Keywords: adjuvant-induced arthritis, rheumatoid arthritis, immunosuppressants, anti-inflammatory drugs, DMARD, translational research, DHODH, p38 MAPK, JAK
Introduction
Oral disease-modifying antirheumatic drugs (DMARDs) represent the standard therapy in rheumatoid arthritis (RA; Singer and Gibofsky, 2011) and the last approved oral DMARD was leflunomide in 1998. The mechanism of action of its active metabolite, teriflunomide, is the inhibition of dihydroorotate dehydrogenase (DHODH), a mitochondrial enzyme that is central in the de novo synthesis of pyrimidines (Breedveld and Dayer, 2000). This pathway is used by highly dividing cells when the supply of nucleotides through the salvage pathway becomes limiting. Thus, teriflunomide acts as a general antiproliferative molecule and most specifically as an immunosuppressant as it inhibits proliferation of T- and B-activated lymphocytes. The efficacy of leflunomide in RA is comparable with that of methotrexate (Singer and Gibofsky, 2011), whilst the most common adverse effects are gastrointestinal (diarrhoea, abdominal pain), along with alopecia, skin reactions and impaired liver function (van Riel et al., 2004).
Most recently, approved biological DMARDs such as the TNFα blockers have demonstrated greater effect and faster onset of action than the current standard therapies (Buch and Emery, 2011). Initially, p38 MAPK inhibitors were envisioned as orally bioavailable drugs with TNFα blocking activity given the central role of p38 MAPK in both the synthesis and the signalling of pro-inflammatory cytokines such as TNFα and IL-6 by monocyte/macrophages (Schett et al., 2008). Despite the clear efficacy of these agents in preclinical studies (Kumar et al., 2003), human clinical trials in RA carried out over the last 10 years have demonstrated limited efficacy and toxicity that have precluded further development (Genovese, 2009). Elevation of liver transaminases and a transient decrease in C-reactive protein (CRP) have been common findings across trials with different compounds (Cohen et al., 2009; Damjanov et al., 2009; Genovese et al., 2011). Other reported side effects include skin lesions, infections, gastrointestinal toxicity and dizziness.
Despite the discouraging results obtained with p38 MAPK inhibitors, another kinase inhibitor, tofacitinib, has been developed as a novel, orally-active DMARD (Kremer et al., 2009; Coombs et al., 2010). Tofacitinib is a potent inhibitor of the Janus kinases (JAK1, JAK2 and JAK3), which are involved in the signalling of a number of cytokines (Murray, 2007). In clinical trials the compound demonstrated both efficacy and a rapid onset of action. However, reported adverse effects include infections, anaemia, neutropenia, hypercholesterolemia, creatininemia and transaminase elevations (Kremer et al., 2009).
In the present report, we provide a comparison of three types of compounds, namely a DHODH inhibitor, a p38MAPK inhibitor and a JAK inhibitor in the rat adjuvant-induced arthritis (AIA) model.
Rat AIA is a robust animal model characterized by both local and systemic inflammation. Its resemblance to human RA, except for the absence of rheumatoid factor, has been well established (Rainsford, 1982; Whitehouse, 2007). A considerable amount of information is available on the articular (oedema, bone resorption) as well as extra-articular (haematological, biochemical, metabolic, organ-related) alterations induced in the adjuvant disease, which can be exploited in the combined analysis of the effects of new drugs. We have analysed the evidence of disease modification, and searched for mechanism of action-dependent effects for teriflunomide, tofacitinib and AL8697, a compound designed at Almirall as a p38 MAPK inhibitor (Vidal et al., 2008). Evaluation of various clinical, histological, haematological and biochemical parameters allows us to assign a primarily anti-inflammatory profile to AL8697, a broad anti-proliferative/immunosuppressant profile to teriflunomide and a particular immunosuppressant profile with strong DMARD properties to tofacitinib. These profiles have been compared with those reported in human studies. Broadly, this analysis indicates that the various effects of p38 inhibition in AIA are not reproducible in human disease, whereas the immunosuppressant modes of action and dependent side-effects of leflunomide and tofacitinib generally translate well from AIA into RA.
Materials and methods
Compounds
Teriflunomide, AL8697 and tofacitinib were synthesized in the Medicinal Chemistry Department of Almirall. AL8697 is a triazolopyridine, 3-(3-tert-butyl-6,8-difluoro[1,2,4]triazolo[4,3-a]pyridin-7-yl)-N-cyclopropyl-5-fluoro-4-methylbenzamide, designed through knowledge-based optimization and selected for the purpose of this study. Its chemical synthesis is described in Vidal et al. (2008).
Animals
All animal care and experimental procedures contained in this paper followed the European Community Directive 86/609/CEE and the Autonomous Catalan law (Decret 214/1997) for the use of laboratory animals and were approved by the Almirall Animal Experimentation Ethical Committee. Male Wistar rats weighing 150–175 g were purchased from Harlan Ibérica (St. Feliu de Codines, Spain). Animals were allowed to condition for 5 days in their new environment at 22°C ± 2°C, 40% to 70% relative humidity and 12 h:12 h light : dark cycles. Rats were housed in polycarbonate cages, with free access to water and non-purified stock diet from SAFE (Villemoisson-sur-Orge, France) during the full course of the studies. Procedures involving intraplantar injection or blood collection were performed under anesthesia induced by inhalation of 3–4% isofluorane/O2 mixture.
Induction of AIA, treatments and clinical evaluation
A total of 13 independent experiments were performed in the present study, including full dose-response studies for the selected compounds.
AIA was induced by a single intraplantar injection in the left hind foot paw of 0.1 mL of a 5 mg·mL−1Mycobacterium tuberculosis H37 RA (Difco, Detroit, MI, USA) suspension in paraffin oil (Merck, Darmstadt, Germany). Control rats received 0.1 mL of saline.
On day 11 post-induction, when signs of right paw oedema became obvious, and in order to ensure homogenous treatment groups, rats with right paw volumes around 2.0 mL, as measured by plethysmography (7140 UGO, Basile, Comerio, Italy) were selected and randomly divided into treatment groups of 6 rats each.
Dosing regimens were selected based on the available human equivalent doses and/or on oral rat pharmacokinetics data. Test compounds were freshly suspended in sterile 0.5% methylcellulose 0.1% Tween-80 solution (10 mL·kg−1 body weight). From day 11 to day 20 of protocol, rats were weighed each day and compounds administered by oral gavage according to the selected dosing and weight; control animals received an equivalent volume of vehicle.
Hind paw volumes were measured by plethysmography every other day, from day 11 (first day of treatment) to day 21 (study completion).
Sample collection and analysis
At study completion, animals were anaesthetized with isofluorane (Baxter, Deerfield, IL, USA) and 1 mL blood samples drawn from the retro-orbital plexus both in heparinized tubes and in EDTA tubes for plasma analysis and for blood cell counts respectively. Animals were killed, and the spleen, thymus and brain were removed and weighed.
Haemogram was determined using a XT-2000iv Sysmex haematological analyser (Sysmex, Kobe, Japan). Plasma α2-macroglobulin was assessed by elisa (Life Diagnostics, West Chester, PA, USA) according to the supplier's recommendations. Clinical biochemistry was analysed by means of an ABX Pentra 400 biochemical analyser (Horiba Diagnostics, Japan).
Hind paws were excised and X-rayed, or processed for histological evaluation, according to the study. X-ray image evaluation was performed by assessing the following parameters: soft tissue swelling, bone demineralization, periostitis, interarticular space reduction and bone cystic degeneration (adapted from Cai et al., 2006). An increasing score from 0 to 4, based on the lesion magnitude, was assigned to each parameter and the final score obtained by summing up all scores for each individual. Histology was evaluated by assessing the following parameters: inflammatory cell infiltration, synovial hyperplasia, cartilage damage, bone resorption and pannus formation (adapted from Yannaki et al., 2010). An increasing score from 0 to 3, based on the lesion magnitude, was assigned to each parameter and the final score obtained by summing up all scores for each individual.
Data analysis
Paw volume progression during the course of the treatment (days 11–21) was plotted and area under curve (AUC) calculated for each individual rat. The ratio between the right paw AUC mean value of the vehicle-treated induced rats and that of the vehicle-treated un-induced rats was used as a measure of arthritis induction, which generally was greater than 1.7. Within each study, the inhibition of oedema for each individual was calculated by referring its AUC value to the AUC mean value of the vehicle-treated induced group, using the AUC mean value of the un-induced rat control group as baseline. Inhibition of splenomegaly was calculated by using the individualized body weight-corrected spleen weights, and referred to the mean of the induced control group using the un-induced control group as baseline. Inhibition of thymus atrophy was calculated by using the individualized brain weight-corrected thymus weights, and referred to the mean of the induced control group using the un-induced control group as baseline.
Body weight change index was calculated as follows. Body weight progression from day 11 to 21 was plotted and AUC calculated for each individual rat. A ratio between the AUC value and the body weight recorded on the first day of treatment (day 11) was then calculated for each individual. In this protocol, a ratio of 10 indicates no net variation of body weight during treatment. Typically, vehicle-treated arthritic rats show values between 9.5–10 indicating weight loss, whereas vehicle-treated un-induced rats show values between 10–10.5 indicating weight gain. Each 0.1 units change equals a 2% weight gain or loss. Indexes were calculated for each rat versus the mean of the un-induced control group using the mean of the vehicle-treated induced group as baseline. Any positive value indicates body weight gain over the arthritic control group, a value of 1 representing the same percent weight gain as the non-arthritic control group, and a value of 0 meaning no change versus the arthritic control group. Negative values thus indicate additional weight loss beyond the arthritic control group. This calculation considers all variations of weight during treatment, not just the starting and ending weights.
Statistically significant differences were assessed by means of one-way anova test with Dunnett's post-test in relation to the vehicle-treated induced group, using GraphPad Prism version 5.00 (GraphPad Software, San Diego, CA, USA).
Results
In vitro and pharmacokinetic compound profiles
The compounds selected to represent each mechanism of action (inhibition of DHODH, p38 MAPK and JAKs) along with their chemical structure, in vitro and rat pharmacokinetic profiles are specified in Table 1.
Table 1.
Basic in vitro and pharmacokinetic profiles for teriflunomide, AL8697 and tofacitinib in Wistar rats
| In vitro profile | Rat pharmacokinetic parameterse | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Enzymatic data IC50 (nM) | t½ (h) | Cl (l (h·kg)-1 | Vss (l kg-1) | AUC p.o.(0–6 h) (ng·h) mL-1 | Cmax ng·mL−1 | F % | |||
| Teriflunomide | Human DHODHa | 1083 | 14 | 0.01 | 0.2 | 166437 | 35332 | 16 | |
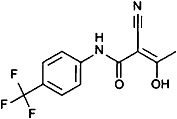 | |||||||||
| AL8697 | Human p38α | 6 | 3.3 | 0.135 | 0.58 | 16409 | 3471 | 50 | |
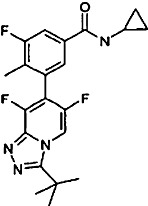 | |||||||||
| Tofacitinib | Human JAK1 | 12 | 0.9 | 2.6 | 1.69 | 3340 | 2579 | 87 | |
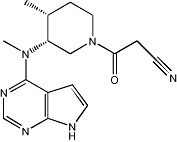 |
Human JAK2 | 1.2 | |||||||
| Human JAK3 | 1 | ||||||||
Inhibition of DHODH was measured as described by Phillips et al., (2008), using recombinant human DHODH enzyme.
Inhibition of p38 was measured as described by Lumeras et al., (2009) and selectivity of AL8697 is further shown in Supporting Information Table S1.
Inhibition of JAKs was assessed by time-resolved fluorescence resonance energy transfer, using the catalytic domains of human JAK1, JAK2, JAK3 (Carna Biosciences).
Pharmacokinetics were measured after an oral dose of 10 mg kg−1 and pharmacokinetic parameters calculated using WinNolin version 4.0.1.
Teriflunomide, a DHODH inhibitor, was used instead of leflunomide as the latter is almost completely converted into the former, the active metabolite, upon oral administration.
AL8697 is a specific p38α inhibitor, 14-fold less potent in p38β and at least 300-fold more selective in a panel of 91 kinases (Supporting Information Table S1). Despite not being a candidate molecule for human studies, its in vitro profile, comparable with the last generation p38 inhibitors (Goldstein et al., 2010), along with its pharmacokinetic properties in rats (see below), make it an adequate tool for in vivo studies.
Tofacitinib, also known as CP-690 550, is a JAK inhibitor currently in phase III clinical trials for RA. This compound inhibits human JAK1, JAK2 and JAK3 enzymes with a low nanomolar IC50 and is highly selective against a broad panel of human kinases (Changelian et al., 2003).
Pharmacokinetic analysis in the rat revealed that teriflunomide was the longest lasting compound with a 14 h plasma half-life, followed by the p38 inhibitor (3.3 h) and tofacitinib (0.9 h). Upon oral administration, teriflunomide showed the highest and longest sustained levels, as indicated by the Cmax and AUC values respectively. In contrast, tofacitinib, while attaining Cmax levels similar to those of AL8697, showed the shortest plasma half-life.
Evaluation of clinical parameters in AIA
Several independent dose-response studies were performed in AIA. Adjuvant disease was induced in male Wistar rats by intraplantar inoculation of complete Freund's adjuvant in the left hind paw. Establishment of arthritis was shown after 10 days by bilateral paw oedema, being more pronounced (about twofold) in the left paw. This is accompanied by a progressive decrease in body weight, an increase in spleen size and a boost in the synthesis of the rat acute phase response factor, α2-macroglobulin (α2M). This clinical course is indicative of systemic inflammatory disease.
All compounds and doses were administered once daily (qd) over the 10 day study period with the exception of tofacitinib for which, based on its PK profile, an additional control-matched twice daily (bid) dose-response study was performed.
Table 2 summarizes the findings of the arthritis studies in measurable efficacy parameters. Since the protocol records continuous paw volume and body weight measurements, we opted to use AUC rather than last time point measurements of these parameters for efficacy calculations (Figures 1, 2).
Table 2.
Efficacy results in adjuvant-induced arthritis
| Dose p.o. | Paw swelling | Radiology score | Splenomegaly | Thymus involution | Body weight change index | α2M | ||
|---|---|---|---|---|---|---|---|---|
| Compound | mg·kg−1 | R % Inhibition | L % Inhibition | R % Inhibition | % inhibition | % inhibition | % inhibition | |
| Teriflunomide | 10 qd | 86 ± 2ast;** | 47 ± 4ast;** | ND | 81 ± 4ast;** | ND | -0.1 ± 0.09 | ND |
| 3 qd | 61 ± 3ast;** | 28 ± 2ast;** | 52 ± 8ast;* | 64 ± 4ast;** | −32 ± 11 | 0 ± 0.06 | 63 ± 10ast;* | |
| 1 qd | 45 ± 9ast; | 18 ± 10 | ND | ND | ND | 0.21 ± 0.1 | ND | |
| AL8697 | 30 qd | 76 ± 3ast;** | 52 ± 3ast;** | ND | 73 ± 5ast;** | 72 ± 8ast;** | 0.49 ± 0.07ast;** | ND |
| 10 qd | 78 ± 2ast;** | 49 ± 2ast;** | 76 ± 4ast;** | 74 ± 3ast;** | 46 ± 12ast; | 0.57 ± 0.06ast;** | 95 ± 2ast;** | |
| 3 qd | 67 ± 4ast;** | 41 ± 3ast;** | 77 ± 6ast;** | 74 ± 3ast;** | ND | 0.31 ± 0.09ast; | 87 ± 3ast;** | |
| 1 qd | 59 ± 5ast;** | 36 ± 3ast;** | 20 ± 27 | 46 ± 10ast;** | ND | 0.14 ± 0.18 | 34 ± 28 | |
| Tofacitinib | 20 qd | 83 ± 2ast;** | 34 ± 3ast;** | 94 ± 4ast;** | 86 ± 4ast;** | ND | 0.38 ± 0.10ast; | 72 ± 3 ast;** |
| 10 qd | 79 ± 2ast;** | 34 ± 2ast;** | 92 ± 3ast;** | 77 ± 3ast;** | 8 ± 14 | 0.43 ± 0.07ast;** | 83 ± 5ast;** | |
| 3 qd | 51 ± 13ast;* | 17 ± 7ast; | ND | 25 ± 14 | ND | 0.19 ± 0.13 | ND | |
| 10 bid | 91 ± 1 ast;** | 48 ± 4 ast;** | 95 ± 6 ast;** | 98 ± 4 ast;** | ND | 1.08 ± 0.15ast;** | 96 ± 2 ast;** | |
| 3 bid | 84 ± 3 ast;** | 44 ± 3 ast;** | ND | 86 ± 4 ast;** | ND | 0.44 ± 0.14ast; | 90 ± 2 ast;** | |
| 1 bid | 75 ± 7 ast;** | 31 ± 4 ast;** | ND | 66 ± 7 ast;** | ND | 0.18 ± 0.15 | 57 ± 17 ast; | |
Data shown are means ± SEM. ***P < 0.001; **P < 0.01;*P < 0.05; significant inhibition, one-way anova.
ND, not determined; R, right hind paw; L, left hind paw.
Figure 1.
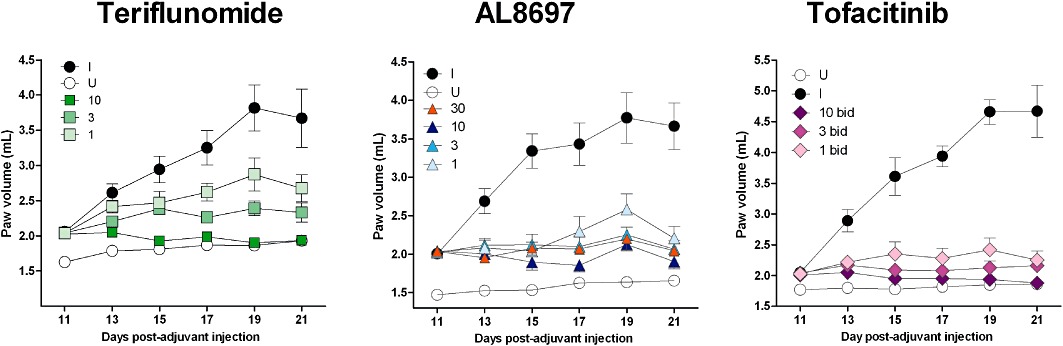
Progression of right paw volumes in full dose-response studies for teriflunomide (qd), AL8697 (qd) and tofacitinib (bid). Values represent mean of six animals ± SEM. U: vehicle-treated un-induced rats; I: vehicle-treated induced rats; 1, 3, 10, and 30: mg·kg−1 doses. For statistical analysis of these data see Table 2.
Figure 2.
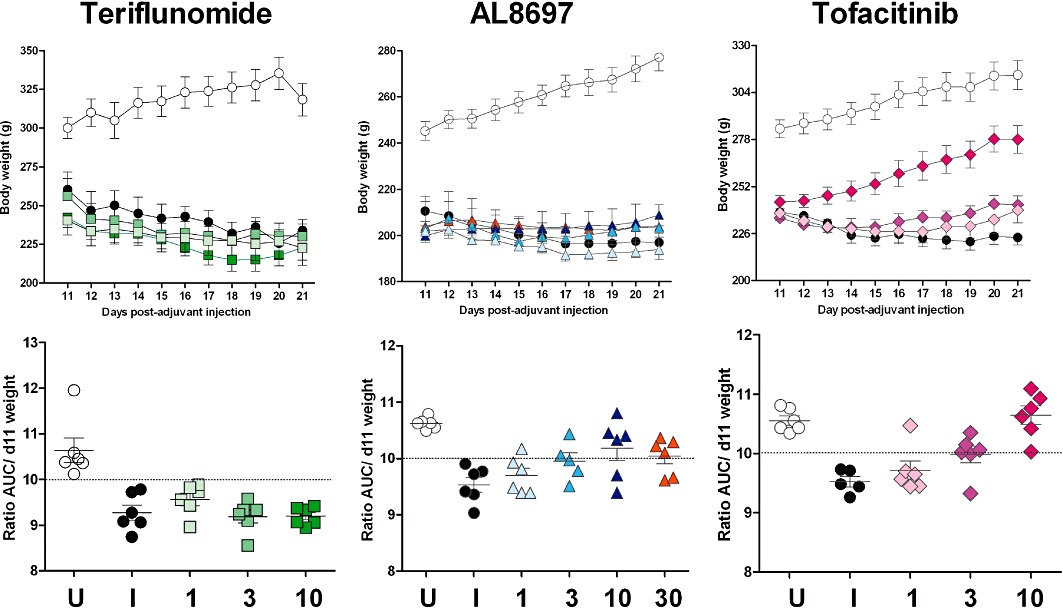
Progression of body weight in full dose-response studies for teriflunomide (qd), AL8697 (qd) and tofacitinib (bid; upper panel; same studies as in Figure 1). Values represent mean of six individuals ± SEM. Lower panel represents the ratio AUC/day 11 weight for each individual. Values above the dotted line indicate body weight gain, whereas values below the dotted line indicate body weight loss. U: vehicle-treated un-induced rats; I: vehicle-treated induced rats; 1, 3, 10 and 30: mg·kg−1 doses. For statistical analysis of these data see Table 2.
All three compounds dose-dependently decreased the oedema in right and left paws, causing a larger improvement in the contralateral un-injected paw. In this regard, results obtained in the qd dose-response studies were comparable among the compounds with the three mechanisms of action. AL8697 and tofacitinib reached an efficacy plateau around 80% inhibition at the highest two doses. In contrast, bid administration of tofacitinib provided higher efficacy in the right paw, as indicated by the 91% inhibition value obtained at 10 mg·kg−1 (Table 2).
Given that the injected paw is highly inflamed, it can be used as a measure of the anti-inflammatory activity. AL8697 was more efficacious at restoring the left paw volume than the other two compounds. Bid administration of the JAK inhibitor was not more effective than AL8697 in diminishing left paw oedema, even at the dose at which right paw volume was fully restored by tofacitinib treatment (Table 2). In addition, AL8697 showed an earlier onset of action than the other two treatments (see Supporting Information Figure S1).
Cachexia, as indicated by the loss of body cell mass, accompanies induction of arthritis (Roubenoff et al., 1997). We have determined that this represents an average body weight loss of approximately 10% during the last 10 days of the protocol. A positive effect on this parameter can thus be considered an indirect measure of efficacy, whereas a negative effect may indicate compound-induced toxicity or a mechanism-dependent effect. AL8697 and tofacitinib dose- dependently restored body weight in qd dosing (Table 2). Interestingly, bid dosing of tofacitinib provided complete restoration (i.e. same percent gain as the control rats) at 10 mg·kg−1 (Table 2 and Figure 2). In contrast, treatment with teriflunomide could not reverse the weight loss trend at any dose. In addition, the teriflunomide dose-response study was limited by gastrointestinal toxicity (diarrhoea) at 10 mg·kg−1.
In order to gain insight into the disease-modifying effects of the compounds, a radiographic analysis was made. Features of joint damage were clearly detected on arthritic rats on day 21 of the protocol. Because the contralateral paw presents the least severe lesions and has the highest potential to recover, only radiographic data for the contralateral paw have been included in Table 2. All compounds had an inhibitory effect on the radiological score. However, tofacitinib was consistently more effective than the other two compounds at normalizing the radiology of the right paw, even with the qd dosing (20 and 10 mg·kg−1 qd; Table 2 and Supporting Information Figure S2). To confirm these findings, right paws from rats treated with therapeutic doses of each compound were examined histologically for the degree of inflammatory cell infiltration, synovial hyperplasia, cartilage damage, bone resorption and pannus formation. As shown in Figure 3A and B, each treatment demonstrated a particular profile with tofacitinib obtaining the best overall average score. Interestingly, the three compounds had a similar inhibitory effect on bone resorption. However, the paws of rats treated with the p38 inhibitor showed a higher presence of inflammatory infiltrates, but less cartilage damage than with the other two therapies.
Figure 3.
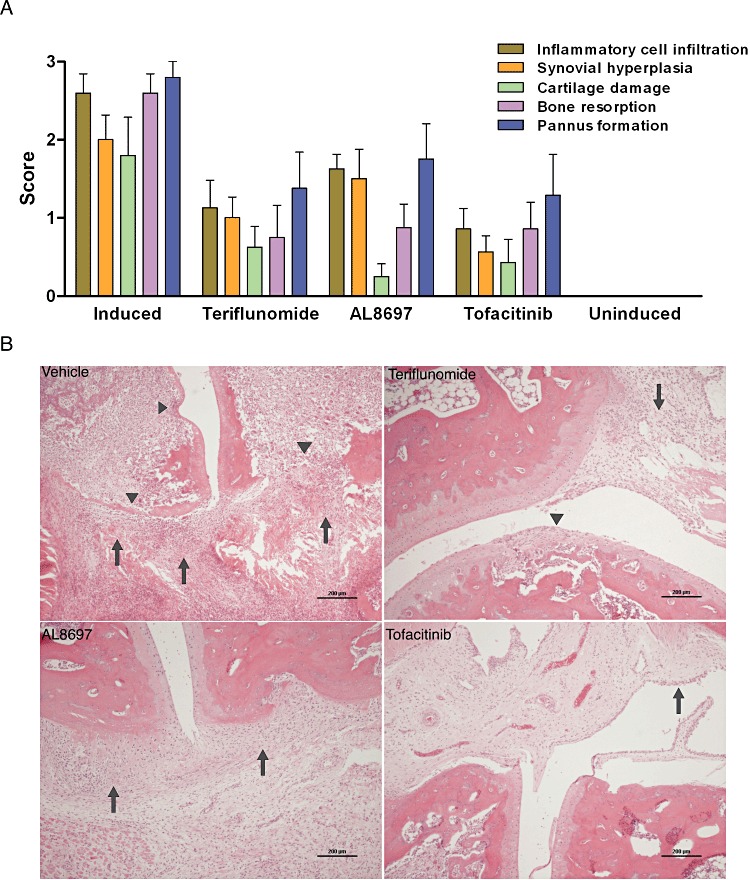
Histological assessment of contralateral paw joints of animals treated with therapeutic doses of teriflunomide (3 mg·kg−1 qd), AL8697 (10 mg·kg−1 qd) and tofacitinib (10 mg·kg−1 qd). (A) Histological score. Evaluation was done for each feature (represented by a different colour) on a scale of 0 (no effect) to 3 (marked effect). Results are the mean ± SEM of eight animals per group. (B) Representative micrographs of the groups analysed in panel A. Vehicle-treated induced control paw: severe thickening and swelling of the synovia and adjacent soft tissue due to severe inflammatory infiltration and fibrosis (arrows); extensive areas of cartilage degradation and bone destruction and erosion (arrowheads). Teriflunomide-treated paw: slight to moderate inflammation of the synovial membrane (arrow) and focal areas of cartilage degradation (arrowhead). AL8697-treated paw: slight to moderate inflammation of the synovial membrane (arrows) in the absence of cartilage damage. Tofacinib-treated paw: almost normal joint architecture with marked reduction of inflammatory cell infiltration and without cartilage and bone damage. Multilayered hyperplastic synovial villi are observed (arrow).
Spleen enlargement during adjuvant arthritis is a result of a combination of several factors including immune activation, granuloma formation secondary to Mycobacterium inoculation and extramedullary haematopoiesis (Billiau and Matthys, 2001). Histological examination on arthritic rat spleens revealed piogranulomatous serositis, increased cellularity in white and red pulps and multifocal granulomas (not shown). All three compounds effectively inhibited arthritis-induced splenomegaly indicating that they interfere with one or more processes involved in spleen enlargement (Table 2).
In addition to spleen enlargement, adjuvant arthritis induces thymus atrophy. The effect of compounds on thymus weight was studied in parallel at a therapeutic dose for each compound (Table 2). Arthritis caused a 1.8-fold decrease in normalized thymus weight and tofacitinib at 10 mg·kg−1 qd had no significant effect on thymus weight. In contrast, teriflunomide (3 mg·kg−1) caused further thymus weight loss (−31%) and interestingly, p38 inhibition reversed thymus atrophy with an average recovery of 46% at 10 mg·kg−1.
Finally, we evaluated α2M as the most abundant circulating acute phase protein in the rat (Heinrich et al., 1990). As shown in Table 2, all three inhibitors tested reduced α2M in plasma in parallel with the observed overall efficacy.
Evaluation of haematological and biochemical parameters in AIA
AIA is characterized by profound haematological changes that include leukocytosis; with extensive systemic neutrophilia, microcytic and hypochromic anaemia; with pronounced reticulocytosis of immature types, and thrombocytosis (Table 3 and Figure 4A). The effect of the test compounds on various haematological parameters was evaluated at therapeutic doses (summarized in Table 3). Teriflunomide at 3 mg·kg−1 caused a decrease in neutrophils, monocytes and reticulocytes (Figure 4A) relative to the arthritic rat counts, indicating restoration of the haematological normal values, as well as a decrease in lymphocytes. However, extensive pancytopenia (leukocytopenia, thrombocytopenia and erythropenia) relative to the un-induced rats was observed at 10 mg·kg−1 (not shown). This profile is due to the antiproliferative mechanism of action causing myelosuppression.
Table 3.
Haematological and biochemical changes observed in adjuvant arthritis studies
| Haematological changes | ||||||||
|---|---|---|---|---|---|---|---|---|
| Red lineage | White lineage | Biochemical changes | ||||||
| AIA | RBC/HTC | ↓ | Total leukocytes | ↑↑↑ | Glucose | ↓↓ | ALP | ↔ |
| Hgb | ↓ | Neutrophils | ↑↑↑↑ | TG | ↓↓↓ | ALT | ↓↓↓ | |
| Reticulocytes | ↑↑↑ | Lymphocytes | ↔ | CHOL | ↔ | AST | ↓ | |
| Platelets | ↑↑↑ | Monocytes | ↑↑↑ | |||||
| Teriflunomide | RBC/HTC | ↔ | Total leukocytes | ↓↓ | Glucose | ↔ | ALP | ↓↓ |
| Hgb | ↔ | Neutrophils | ↓↓↓ | TG | ↔ | ALT | ↔ | |
| Reticulocytes | ↓ | Lymphocytes | ↓↓ | CHOL | ↔ | AST | ↔ | |
| Platelets | ↔ | Monocytes | ↓↓ | |||||
| AL8697 | RBC/HTC | ↔ | Total leukocytes | ↑↑ | Glucose | ↑ | ALP | ↔ |
| Hgb | ↔ | Neutrophils | ↑↑ | TG | ↔ | ALT | ↔ | |
| Reticulocytes | ↔ | Lymphocytes | ↔ | CHOL | ↑↑ | AST | ↔ | |
| Platelets | ↓↓ | Monocytes | ↑↑ | |||||
| Tofacitinib | RBC/HTC | ↔ | Total leukocytes | ↓↓ | Glucose | ↔ | ALP | ↔ |
| Hgb | ↔ | Neutrophils | ↓↓↓ | TG | ↔ | ALT | ↑↑↑ | |
| Reticulocytes | ↓↓ | Lymphocytes | ↓↓ | CHOL | ↑↑ | AST | ↔ | |
| Platelets | ↓↓ | Monocytes | ↔ | |||||
Changes in AIA refer to the induced versus the un-induced control. Changes in treatment groups refer to each treatment group versus the induced control. Results are shown for teriflunomide (3 mg·kg−1 qd), AL8697 (10 mg·kg−1 qd) and tofacitinib (10 mg·kg−1 bid); except for biochemical changes for teriflunomide at 10 mg·kg−1. Only parameters with significant changes in any treatment group are shown. ↔: no change; ↓,↑: 5–20 % change; ↓↓,↑↑: 21–50% change; ↓↓↓,↑↑↑: 51–100% change; ↑↑↑↑: > 100%.
RBC, red blood cell count; HTC, hematocrit; Hgb, haemoglobin; TG, triglycerides; CHOL total cholesterol.
Figure 4.
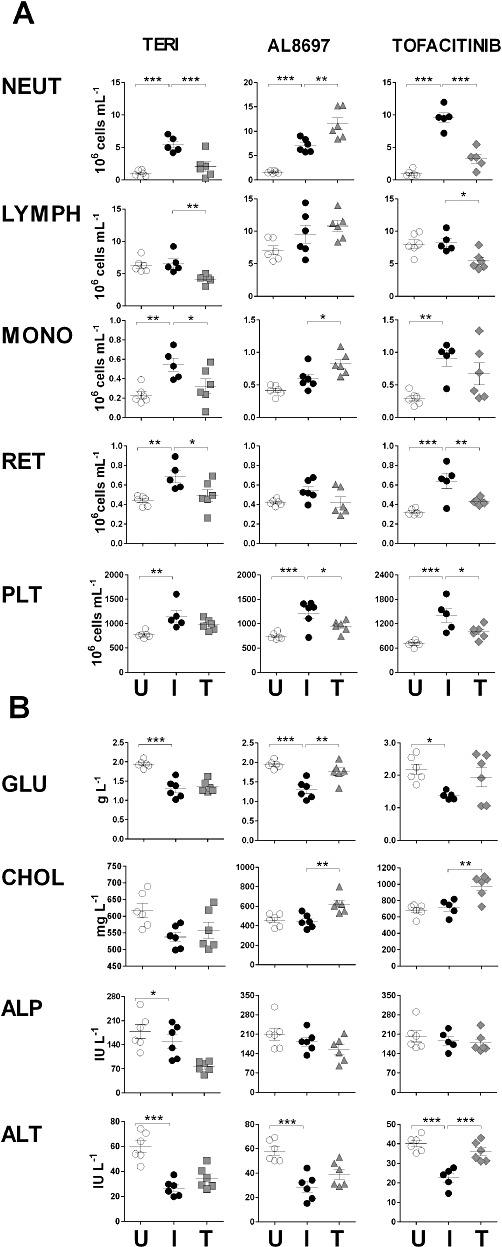
Haematological (A) and biochemical (B) changes in AIA. One independent representative study was selected for each mechanism of action. U: vehicle-treated un-induced control group; I: vehicle-treated induced control group; T: treatment group. Results are shown for teriflunomide, AL8697 and tofacitinib at 3 mg·kg−1 qd, 10 mg·kg−1 qd and 10 mg·kg−1 bid, respectively, except for biochemical changes for teriflunomide at 10 mg·kg−1 qd. ***P < 0.001, **P < 0.01, *P < 0.05, significantly different from the vehicle-treated induced control group(I). NEUT = neutrophils; LYMPH = lymphocytes; MONO = monocytes; RET = reticulocytes; PLT = platelets; GLU = glucose; CHOL = total cholesterol.
In contrast to teriflunomide, p38 inhibition caused a significant increase in neutrophils and monocytes (Figure 4A). This effect was clearly evident at 10 mg·kg−1 and occurred when using another p38 inhibitor of a different chemical series (not shown), suggesting that this may be a class effect. In addition, p38 inhibition partially restored the platelet count.
The haematological profile caused by JAK inhibition was distinctive in that it caused specific lymphocyte depletion in both qd and bid dosing regimens (mean 21% at 10 mg·kg−1 qd, 36% at 20 mg·kg−1 qd, 44% at 10 mg·kg−1 bid). Cytometric analysis of lymphocyte subsets in whole blood indicated that the most affected populations were NK cells and NK T-cells (>70% cell depletion) and CD8+ cells (50%; not shown), in accordance with other reports in rodents (Kudlacz et al., 2004). In addition, partial restoration of platelet and reticulocyte counts was also observed in both qd and bid regimens. In contrast, neutrophil counts showed a dose-dependent decrease towards normalization only with bid dosing (Figure 4A).
AIA is accompanied by profound metabolic alterations that affect different hepatic processes such as gluconeogenesis, glycogen synthesis, insulin response and lipogenesis (Fedatto-Júnior et al., 2002; Yassuda Filho et al., 2003). Arthritic rats show much lower glucose and triglyceride plasma levels than normal rats, whereas total cholesterol levels remain unaltered (Table 3 and Figure 4B). Restoration of glucose levels was observed upon treatment with the p38 inhibitor, with a similar trend showed by the JAK inhibitor (Table 3 and Figure 4B). Of note, AL8697 and tofacitinib in the bid dosing protocol caused an increase in total cholesterol over the levels in normal control rats (mean 39% and 26% respectively, at 10 mg·kg−1). These results suggest a role for p38 MAPK and JAK in cholesterol metabolism in the rat.
Plasma levels of the liver enzymes, alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP) and bilirubin (TBIL) are commonly employed as clinical disease indicators. We have found that arthritic rats have much lower levels of circulating AST and ALT than normal rats, while levels of ALP and TBIL are not altered. Because ALT and AST are biochemically involved in the synthesis of non-essential amino acids, this decrease may be a consequence of the hypermetabolic syndrome developed in AIA (Yassuda Filho et al., 2003). Tofacitinib, in both qd and bid regimens, caused a partial reversal in the levels of ALT (mean 70% increase), but not of AST, without obvious histological liver lesions (not shown). Similar to tofacitinib, the p38 inhibitor at 10 mg·kg−1 showed a trend to ALT recovery (Figure 4B) that became statistically significant at 30 mg·kg−1 (not shown). No other liver marker was altered. In contrast, a decrease in the plasma levels of ALP (mean 40%) was found only with teriflunomide at the 10 mg·kg−1 dose (Figure 4B).
Discussion
We have employed a multiparametric approach in a rat adjuvant arthritis model to profile drugs belonging to three different therapeutic classes (see Figure 5). By means of this approach, it is possible to reveal anti-inflammatory properties based on the compound's effect on the progression of the disease in both hind paws (especially the more inflamed paw); DMARD properties were characterised based on the effect on the radiological and histological alterations; immunosuppressive properties based on the effect on lymph organs and haematological counts; and anti-cachectic properties based on the improvement of body weight and metabolism normalization. In addition, side effects not directly related to the arthritic process can also be demonstrated using this model and used to characterise the compounds further. These other effects include the gastrointestinal toxicity observed with teriflunomide, or the cholesterol increase in the case of p38 and JAK inhibitors.
Figure 5.
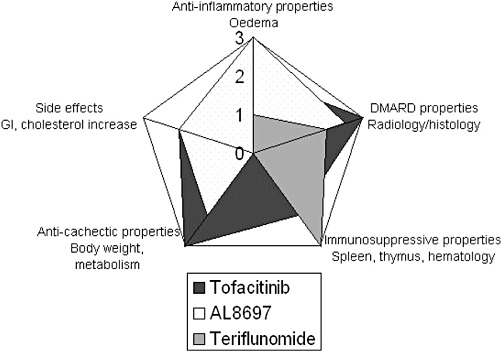
Radial diagram of the properties revealed in AIA for each drug studied, based on the parameters measured. The scale indicates a score assigned to each property and compound in which 0 = no effect; 1 = slight effect; 2 = moderate effect and 3 = marked effect (please note that the scale for side effects is actually reverse, so that marked side effects have the lowest score). The DHODH inhibitor shows a marked immunosuppressive effect with moderate DMARD and weak anti-cachectic properties, and with gastrointestinal toxicity that limits dosing. The p38 inhibitor shows marked anti-inflammatory properties, with moderate to marked DMARD, moderate anti-cachectic and slight pro-cholesterolemic effects, while being devoid of immunosuppressive effects. The JAK inhibitor shows marked DMARD and anti-cachectic properties, with moderate anti-inflammatory, immunosuppressive and slight pro-cholesterolemic effects.
It should be noted that drug-induced normalization of any altered haematological or biochemical value, when accompanied by disease amelioration, cannot be regarded unequivocally either as a drug-induced effect, a consequence of clinical improvement or both. Drug effects falling into this category include normalization of neutrophil, platelet and reticulocyte counts, as well as reversal of hypoglycaemia and ALT levels. Modification of parameters that are not altered by the disease, such as lymphocyte count, cholesterol or ALP levels, should be regarded as drug-induced effects. Our data also suggest that some AIA-induced changes may not be reversible, as attaining a maximal response in all efficacy parameters (tofacitinib 10 mg·kg−1 bid) is not accompanied by normalization of triglyceride or AST plasma levels (Table 3).
The results obtained with teriflunomide in AIA closely parallel the observed pharmacological effects reported in patients. Teriflunomide shows DMARD features as it reduces swelling and joint damage. In addition, the compound reduces spleen enlargement, thymus weight and leukocyte counts, attributable to its DHODH-dependent antiproliferative activity. These observations suggest that teriflunomide acts as a general immunosuppressant. Furthermore, as in humans, teriflunomide can cause gastrointestinal side effects secondary to its antiproliferative activity on the enteric epithelium. In this regard, given that intestinal ALP is the main circulating ALP isoform in the rat, the specific drop in plasma ALP observed at the 10 mg·kg−1 dose can be attributed to damage of the enteric epithelium along with a general state of malnutrition (Boone et al., 2005) and it would not be expected in humans. At the systemic level, body weight loss has been reported in arthritic patients treated with leflunomide (Alcorn et al., 2009). This effect is reproduced in AIA, where body weight recovery is clearly dissociated from an improvement in other efficacy parameters at all doses. Therefore, the profile of teriflunomide in AIA is that of an immunosuppressant, with DMARD properties. The compound has weak anti-cachectic activity and causes gastrointestinal toxicity, as seen in RA patients (Figure 5).
Based on its selectivity profile, AL8697 can be considered a selective p38α inhibitor. Because a common pattern has been observed for selective p38α inhibitors in preclinical and clinical studies, we believe that the results obtained with AL8697 are representative of its class. However, compound particularities cannot be excluded. The multiparametric approach used in this study demonstrated that AL8697 exhibits a complex profile. Inhibition of p38 produced a better anti-inflammatory effect on the ipsilateral-induced paw oedema than the other two compounds. This finding may be related to the known activity of p38 inhibitors on PGE2 production, through direct regulation of COX-2 mRNA stability (Lasa et al., 2000). AL8697 inhibits LPS-induced PGE2 production in human whole blood with an IC50 of 400 nM (unpublished results). Similarly, Hope et al. (2009) have reported inhibition of PGE2 production in IL-1β-challenged RA synovial fibroblasts using another p38 inhibitor.
In our studies, radiological and histological assessments revealed that AL8697 exhibits protective effects on joint destruction and cartilage tissue protection. In this regard, p38 MAPK inhibitors have been suggested to be chondroprotective based on the inhibition of IL-1β-induced chondrocyte expression of COX-2, MMP13 and inducible NOS (Joos et al., 2010). Moreover, AL8697 was less efficient at reducing the joint inflammatory infiltrates, possibly reflecting poorer immunosuppression. In fact, no sign of an immunosuppressive role for p38 inhibition was found. AL8697 did not diminish any circulating leukocyte subset at any dose. Conversely, there was an increase in circulating blood leukocytes in AIA, an effect which was also seen in a chronic study on normal rats at AIA therapeutic doses (unpublished results). These results could implicate p38 in the control of proliferation of leukocyte precursors. In fact, p38 MAPK has been shown to mediate the signalling of myelosuppressive cytokines in normal haematopoiesis in vitro and pharmacological inhibitors of p38 MAPK have been reported to reverse this modulation (Platanias, 2003). Furthermore, p38 inhibition prevented thymic atrophy suggesting a direct role of p38 in thymus homeostasis. In this regard, the p38 transduction pathway has been implicated in the control of thymocyte proliferation by apoptosis (Tanaka et al., 2002). Alternatively, an indirect effect through amelioration of clinical signs and decreased circulating cortisol levels cannot be excluded. In contrast to the increasing effect on thymus weight, p38 inhibition caused correction of AIA-induced splenomegaly. Given the role of TNFα and its signalling in secondary lymphoid follicle and granuloma formation in the spleen (Billiau and Matthys, 2001), we speculate that this apparent contradiction could be explained by the AL8697-mediated inhibition of TNFα. In this regard, AL8697 inhibits LPS-induced TNFα in human whole blood with an IC50 of 110 nM (unpublished results). In addition, p38 inhibition reversed the body weight loss induced by arthritis, possibly through the involvement of p38 in the signalling or production of pro-cachectic cytokines (i.e. TNFα; Roubenoff et al., 1997). Therefore, p38 inhibition in AIA shows the profile of an anti-inflammatory with moderate DMARD and anti-cachectic effects but devoid of immunosuppressive properties (Figure 5). This profile of activity if mimicked in RA patients would likely be that of an anti-inflammatory with potential anti-TNFα-mediated DMARD effects. However, efficacy reports for p38 inhibitors in the clinic showed a very modest effect on ACR20, resembling, at most, the efficacy of the non-steroidal anti-inflammatory drugs. An interesting clinical observation was an initial drop followed by a rebound in plasma levels of CRP (Cohen et al., 2009; Damjanov et al., 2009; Genovese et al., 2011). This observation suggested an unknown compensatory mechanism from p38 inhibition which occurs in humans. However, in AIA, reduction in α2M levels was clearly dose-dependent with no evidence of compensation, suggesting the existence of species-specific mechanisms. In addition, two human trials (Cohen et al., 2009; Damjanov et al., 2009) reported an increase in neutrophil counts in several patients. While several reasons could explain this finding, the leukocytosis observed in AIA is an indicator of potential haematological complications.
The efficacy of the JAK inhibitor tofacitinib in AIA clearly parallels the results reported in RA. Tofacitinib shows immunosuppressive properties and better DMARD properties than the other two compounds (Figure 5). In patients with RA, tofacitinib has been reported to affect steady-state neutrophil counts (Gupta et al., 2010) and to worsen anaemia (Kremer et al., 2009). Parallel findings in AIA, identified as a reversal of neutrophilia and normalization of reticulocyte counts, could be a consequence of the role of JAK signalling in emergency neutropoiesis and erythropoiesis, although the neutrophil count does not fall below the levels seen in un-induced rats. Alternatively, the effect could represent a consequence of continuous disease amelioration from the first day of administration. Similar conclusions have been suggested by others regarding neutrophil reduction in AIA (Meyer et al., 2010).
An interesting biochemical change is the total cholesterol increase induced by p38 MAPK and JAK inhibitors in AIA. A chronic study in normal rats at therapeutic doses of the p38 inhibitor revealed an increase in cholesterol (unpublished results), although no data in normal rats are available for tofacitinib. The AIA results resemble the increased cholesterolemia observed in tofacitinib-treated patients (Kremer et al., 2009) and, to our knowledge, has not been reported in any other animal model. Our results suggest that p38 MAPK and JAK may be acting on a common pathway. The fact that the anti-IL-6 antibody, tocilizumab, also alters cholesterol levels (Singh et al., 2011) suggests a central role for IL-6 in this effect. In fact, a link between IL-6 and cholesterol metabolism has been proposed (Hashizume et al., 2010), and it is well established that JAK proteins and p38 MAPK are key transducers in IL-6 signalling (Heinrich et al., 2003).
Hepatotoxicity, in the form of elevated transaminase levels, is a common finding for all three compound classes in RA. In general, rodents are known to be less sensitive to human hepatotoxins. Specifically in AIA, the adjuvant disease itself modifies the transaminase plasma levels as part of the general metabolic alteration. Therefore, it may be difficult to identify compound-induced changes in transaminase plasma levels which are a result of direct hepatotoxicity None of the compounds induced elevation of transaminases or bilirubin over the un-induced control. However, pan-JAK inhibition (10 mg·kg−1 qd and bid) and p38 inhibition (10 mg·kg−1 qd, 30 mg·kg−1) specifically induced a reversal of ALT, which was not paralleled by any particular histological liver lesion. These results, along with the trend to normalize glycaemia, may be related to the anti-cachectic effects observed for both compounds and suggest a direct or indirect role for JAK and p38 proteins in the regulation of metabolism in the rat.
In conclusion, our study demonstrates the usefulness of a multiparametric approach to reveal specific drug properties in AIA (Figure 5), and the valuable translational information obtained for immunosuppressors such as DHODH or JAK inhibitors. As for p38 inhibitors, based on the results obtained with our compound, we hypothesize that selective p38α inhibitors function primarily as anti-inflammatory molecules. In our view, the most likely explanations for their clinical failure lie in the pleiotropic functions of p38 MAPK with class-dependent side effects limiting the maximum tolerated dose for p38 inhibitors in humans, and in species-specific roles of p38 MAPK that could have prevented the prediction of important side effects (i.e. hepatotoxicity, skin lesions). In addition, cells have evolved mechanisms to counteract the inhibition of p38 MAPK (Cheung et al., 2003), which could have had a role in the rebound production of CRP. Likewise, diverse hypotheses have been put forward (Hammaker and Firestein, 2010), although further studies are warranted to explain the clinical results with the p38 inhibitors. In our view, JAK inhibitors appear to be the best candidates for new oral anti-rheumatic drugs.
Acknowledgments
The authors are indebted to the Almirall Medicinal Chemistry department for the synthesis of the compounds used in this study, the ADME and Screening departments for profiling data and Virginia Pes for excellent performance of arthritis-related animal work.
Glossary
- AIA
adjuvant-induced arthritis
- ALP
alkaline phosphatase
- ALT
alanine aminotransferase
- α2-M
α-2 macroglobulin
- AST
aspartate aminotransferase
- AUC
area under curve
- bid
twice a day
- CRP
C-reactive protein
- DHODH
dihydroorotate dehydrogenase
- DMARD
disease-modifying anti-rheumatic drug
- JAK
Janus kinase
- qd
once a day
- RA
rheumatoid arthritis
- TBIL
total bilirubin
Conflict of interest
All authors are current employees of Almirall.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Comparison of treatments on the progression of the left paw oedema in a single study. Black circles: induced vehicle-treated control group; Open circles: un-induced vehicle-treated control group; Dark squares: teriflunomide 3 mg·kg−1 qd; Light squares: Tofacitinib 10 mg·kg−1 qd; Triangles: AL8697 10 mg·kg−1 qd.
Figure S2 Representative X-ray pictorials of vehicle-treated and compound-treated right paws. A: induced vehicle-treated rat; B. un-induced vehicle-treated rat; C: teriflunomide 3 mg·kg−1; D: AL8697 10 mg·kg−1; E: tofacitinib 10mg·kg−1. Asterisks indicate areas of soft tissue swelling; large arrow-heads indicate bone demineralization areas with cystic degeneration (vehicle), and the small arrowhead indicates periostitis.
Table S1 Selectivity of AL8697 in a panel of 91 kinases. The compound was tested in duplicate at a single concentration of 10 μM. Values show percentage of remaining enzyme activity, or the value of IC50 for Ret, p38α (SAPK2a) and p38β (SAPK2b). Assays were performed by Upstate Ltd (Millipore Corp) except for p38α and p38β IC50 curves which were performed in-house
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alcorn N, Saunders S, Madhok R. Benefit-risk assessment of leflunomide: an appraisal of leflunomide in rheumatoid arthritis 10 years after licensing. Drug Saf. 2009;32:1123–1134. doi: 10.2165/11316650-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Billiau A, Matthys P. Modes of action of Freund's adjuvants in experimental models of autoimmune diseases. J Leukoc Biol. 2001;70:849–860. [PubMed] [Google Scholar]
- Boone L, Meyer D, Cusick P, Ennulat D, Bolliger AP, Everds N, et al. Selection and interpretation of clinical pathology indicators of hepatic injury in preclinical studies. Vet Clin Pathol. 2005;34:182–188. doi: 10.1111/j.1939-165x.2005.tb00041.x. [DOI] [PubMed] [Google Scholar]
- Breedveld FC, Dayer J-M. Leflunomide: mode of action in the treatment of rheumatoid arthritis. Ann Rheum Dis. 2000;59:841–849. doi: 10.1136/ard.59.11.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buch MH, Emery P. New therapies in the management of rheumatoid arthritis. Curr Opin Rheumatol. 2011;23:245–251. doi: 10.1097/BOR.0b013e3283454124. [DOI] [PubMed] [Google Scholar]
- Cai X, Wong YF, Zhou H, Xie Y, Liu ZQ, Jiang ZH, et al. The comparative study of Sprague-Dawley and Lewis rats in adjuvant-induced arthritis. Naunyn Schmiedebergs Arch Pharmacol. 2006;373:140–147. doi: 10.1007/s00210-006-0062-5. [DOI] [PubMed] [Google Scholar]
- Changelian PS, Flanagan ME, Ball DJ, Kent CR, Magnuson KS, Martin WH, et al. Prevention of organ allograft rejection by a specific Janus kinase 3 inhibitor. Science. 2003;302:875–878. doi: 10.1126/science.1087061. [DOI] [PubMed] [Google Scholar]
- Cheung PC, Campbell DG, Nebreda AR, Cohen P. Feedback control of the protein kinase TAK1 by SAPK2a/p38alpha. EMBO. 2003;22:5793–5805. doi: 10.1093/emboj/cdg552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SB, Cheng T, Chindalore V, Damjanov N, Burgos-Vargas R, DeLora P, et al. Evaluation of the efficacy and safety of pamapimod, a p38 MAP kinase inhibitor, in a double-blind, methotrexate-controlled study in patients with active rheumatoid arthritis. Arthritis Rheum. 2009;60:335–344. doi: 10.1002/art.24266. [DOI] [PubMed] [Google Scholar]
- Coombs JH, Bloom BJ, Breedveld FC, Fletcher MP, Gruben D, Kremer JM, et al. Improved pain, physical functioning and health status in patients with rheumatoid arthritis treated with tofacitinib, an orally active Janus kinase (JAK) inhibitor: results from a randomised, double-blind, placebo-controlled trial. Ann Rheum Dis. 2010;69:413–416. doi: 10.1136/ard.2009.108159. [DOI] [PubMed] [Google Scholar]
- Damjanov N, Kauffman RS, Spencer-Green GT. Efficacy, pharmacodynamics, and safety of VX-702, a novel p38 MAPK inhibitor, in rheumatoid arthritis. Results of two randomized, double-blind, placebo-controlled clinical studies. Arthritis Rheum. 2009;60:1232–1241. doi: 10.1002/art.24485. [DOI] [PubMed] [Google Scholar]
- Fedatto-Júnior Z, Ishii-Iwamoto EL, Caparroz-Assef SM, Vicentini GE, Bracht A, Kelmer-Bracht AM. Glycogen levels and glycogen catabolism in livers from arthritic rats. Mol Cell Biochem. 2002;229:1–7. doi: 10.1023/a:1017913124084. [DOI] [PubMed] [Google Scholar]
- Genovese MC. Inhibition of p38: has the fat lady sung? Arthritis Rheum. 2009;60:317–320. doi: 10.1002/art.24264. [DOI] [PubMed] [Google Scholar]
- Genovese MC, Cohen SB, Wofsy D, Weinblatt ME, Firestein GS, Brahn E, et al. A 24-week, randomized, double-blind, placebo-controlled, parallel group study of the efficacy of oral SCIO-469, a p38 mitogen-activated protein kinase inhibitor, in patients with active rheumatoid arthritis. J Rheumatol. 2011;38:846–854. doi: 10.3899/jrheum.100602. [DOI] [PubMed] [Google Scholar]
- Goldstein DM, Kuglstatter A, Lou Y, Soth MJ. Selective p38alpha inhibitors clinically evaluated for the treatment of chronic inflammatory disorders. J Med Chem. 2010;53:2345–2353. doi: 10.1021/jm9012906. [DOI] [PubMed] [Google Scholar]
- Gupta P, Friberg LE, Karlsson MO, Krishnaswami S, French J. A semi-mechanistic model of tofacitinib-induced reduction in neutrophil counts in patients with rheumatoid arthritis. J Clin Pharmacol. 2010;50:679–687. doi: 10.1177/0091270009346060. [DOI] [PubMed] [Google Scholar]
- Hammaker D, Firestein GS. ‘Go upstream, young man’: lessons learned from the p38 saga’. Ann Rheum Dis. 2010;69(Suppl. 1):i77–i82. doi: 10.1136/ard.2009.119479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashizume M, Yoshida H, Koike N, Suzuki M, Mihara M. Overproduced interleukin 6 decreases blood lipid levels via upregulation of very-low-density lipoprotein receptor. Ann Rheum Dis. 2010;69:741–746. doi: 10.1136/ard.2008.104844. [DOI] [PubMed] [Google Scholar]
- Heinrich PC, Castell JV, Andus T. Interleukin-6 and the acute phase response. Biochem J. 1990;265:621–636. doi: 10.1042/bj2650621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrich PC, Behrmann I, Haan S, Hermanns HM, Müller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hope HR, Anderson GD, Burnette BL, Compton RP, Devraj RV, Hirsch JL, et al. Anti-inflammatory properties of a novel N-phenyl pyridinone inhibitor of p38 mitogen-activated protein kinase: preclinical-to-clinical translation. J Pharmacol Exp Ther. 2009;331:882–895. doi: 10.1124/jpet.109.158329. [DOI] [PubMed] [Google Scholar]
- Joos H, Albrecht W, Laufer S, Brenner RE. Differential effects of p38MAP kinase inhibitors on the expression of inflammation-associated genes in primary, interleukin-1beta-stimulated human chondrocytes. Br J Pharmacol. 2010;160:1252–1262. doi: 10.1111/j.1476-5381.2010.00760.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer JM, Bloom BJ, Breedveld FC, Coombs JH, Fletcher MP, Gruben D, et al. The safety and efficacy of a JAK inhibitor in patients with active rheumatoid arthritis. Arthritis Rheum. 2009;7:1895–1905. doi: 10.1002/art.24567. [DOI] [PubMed] [Google Scholar]
- Kudlacz E, Perry B, Sawyer P, Conklyn M, McCurdy S, Brissette W, et al. The novel JAK-3 inhibitor CP-690550 is a potent immunosuppressive agent in various murine models. Am J Transplant. 2004;4:51–57. doi: 10.1046/j.1600-6143.2003.00281.x. [DOI] [PubMed] [Google Scholar]
- Kumar S, Boehm J, Lee JC. p38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nat Rev Drug Discov. 2003;2:717–726. doi: 10.1038/nrd1177. [DOI] [PubMed] [Google Scholar]
- Lasa M, Mahtani KR, Finch A, Brewer G, Saklatvala J, Clark AR. Regulation of cyclooxygenase 2 mRNA stability by the mitogen-activated protein kinase p38 signalling cascade. Mol Cell Biol. 2000;20:4265–4274. doi: 10.1128/mcb.20.12.4265-4274.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumeras W, Caturla F, Vidal L, Esteve C, Balagué C, Orellana A, et al. Design, synthesis, and structure-activity relationships of aminopyridine N-oxides, a novel scaffold for the potent and selective inhibition of p38 mitogen activated protein kinase. J Med Chem. 2009;52:5531–5545. doi: 10.1021/jm9008604. [DOI] [PubMed] [Google Scholar]
- Meyer DM, Jesson MI, Li X, Elrick MM, Funckes-Shippy CL, Warner JD, et al. Anti-inflammatory activity and neutrophil reductions mediated by the JAK1/JAK3 inhibitor, tofacitinib, in rat adjuvant-induced arthritis. J Inflamm. 2010;7:41. doi: 10.1186/1476-9255-7-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray PJ. The JAK-STAT signalling pathway: input and output integration. J Immunol. 2007;178:2623–2629. doi: 10.4049/jimmunol.178.5.2623. [DOI] [PubMed] [Google Scholar]
- Phillips MA, Gujjar R, Malmquist NA, White J, El Mazouni F, Baldwin J, et al. Triazolopyrimidine-based dihydroorotate dehydrogenase inhibitors with potent and selective activity against the malaria parasite Plasmodium falciparum. J Med Chem. 2008;51:3649–3653. doi: 10.1021/jm8001026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platanias LC. Map kinase signaling pathways and hematologic malignancies. Blood. 2003;12:3647. doi: 10.1182/blood-2002-12-3647. [DOI] [PubMed] [Google Scholar]
- Rainsford KD. Adjuvant polyarthritis in rats: is this a satisfactory model for screening anti-arthritic drugs? Agents Actions. 1982;12:452–458. doi: 10.1007/BF01965926. [DOI] [PubMed] [Google Scholar]
- van Riel PLCM, Smolen JS, Emery P, Kalden JR, Dougados M, Starnd CV, et al. Leflunomide: a manageable safety profile. J Rheumatol Suppl. 2004;71:21–24. [PubMed] [Google Scholar]
- Roubenoff R, Freeman LM, Smith DE, Abad LW, Dinarello CA, Kehayias JJ. Adjuvant arthritis as a model of inflammatory cachexia. Arthritis Rheum. 1997;40:534–539. doi: 10.1002/art.1780400320. [DOI] [PubMed] [Google Scholar]
- Schett G, Zwerina J, Firestein G. The p38 mitogen-activated protein kinase (MAPK) pathway in rheumatoid arthritis. Ann Rheum Dis. 2008;67:909–916. doi: 10.1136/ard.2007.074278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer O, Gibofsky A. Methotrexate versus leflunomide in rheumatoid arthritis: what is new in 2011? Curr Opin Rheumatol. 2011;23:288–292. doi: 10.1097/BOR.0b013e328344f2e4. [DOI] [PubMed] [Google Scholar]
- Singh JA, Beg S, Lopez-Olivo MA. Tocilizumab for rheumatoid arthritis: a cochrane systematic review. J Rheumatol. 2011;38:10–20. doi: 10.3899/jrheum.100717. [DOI] [PubMed] [Google Scholar]
- Tanaka N, Kamanaka M, Enslen H, Dong C, Wysk M, Davis RJ, et al. Differential involvement of p38 mitogen-activated protein kinase MKK3 and Mkk6 in T-cell apoptosis. EMBO Rep. 2002;3:785. doi: 10.1093/embo-reports/kvf153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal B, Eastwood PR, Gonzalez J, Esteve C. 2008. New 3-([1,2,4]Triazolo[4,3-A]Pyridin-7-4C) benzamide derivatives. Patent WO2008/107125.
- Whitehouse MW. Adjuvant arthritis 50 years on: the impact of the 1956 article by C.M. Pearson, ‘Development of arthritis, periarthritis and periostitis in rats given adjuvants. Inflamm Res. 2007;56:133–138. doi: 10.1007/s00011-006-6117-8. [DOI] [PubMed] [Google Scholar]
- Yannaki E, Papadopoulou A, Athanasiou E, Kaloyannidis P, Paraskeva A, Bougiouklis D, et al. The proteasome inhibitor bortezomib drastically affects inflammation and bone disease in adjuvant-induced arthritis in rats. Arthritis Rheum. 2010;62:3277–3288. doi: 10.1002/art.27690. [DOI] [PubMed] [Google Scholar]
- Yassuda Filho P, Bracht A, Ishii-Iwamoto EL, Lousano SH, Bracht L, Kelmer-Bracht AM. The urea cycle in the liver of arthritic rats. Mol Cell Biochem. 2003;243:97–106. doi: 10.1023/a:1021695625457. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.