

Abstract
BACKGROUND AND PURPOSE
Lipoxidation-derived reactive carbonyl species (RCS) such as 4-hydroxy-2-nonenal (HNE) react with proteins to form advanced lipoxidation end products (ALEs), which have been implicated in both atherosclerosis and renal disease. L-carnosine acts as an endogenous HNE scavenger, but it is rapidly inactivated by carnosinase. This study aimed at assessing the effect of the carnosinase-resistant, D-carnosine, on HNE-induced cellular injury and of its bioavailable prodrug D-carnosine octylester on experimental atherosclerosis and renal disease.
EXPERIMENTAL APPROACH
Vascular smooth muscle cells (VSMCs) were exposed to HNE or H2O2 plus D-carnosine. ApoE null mice fed a Western, pro-atherogenic diet were treated with D-carnosine octylester for 12 weeks.
KEY RESULTS
In vitro, D-carnosine attenuated the effect of HNE, but not of H2O2, on VSMCs. In vivo, D-carnosine octylester-treated mice showed reduced lesion area and a more stable plaque phenotype compared with untreated animals, with reduced foam cell accumulation, inflammation and apoptosis and increased clearance of apoptotic bodies and collagen deposition, resulting in decreased necrotic core formation. Likewise, renal lesions were attenuated in D-carnosine octylester-treated versus untreated mice, with lower inflammation, apoptosis and fibrosis. This was associated with increased urinary levels of HNE-carnosine adducts and reduced protein carbonylation, circulating and tissue ALEs, expression of receptors for these products, and systemic and tissue oxidative stress.
CONCLUSIONS AND IMPLICATIONS
These data indicate RCS quenching with a D-carnosine ester was highly effective in attenuating experimental atherosclerosis and renal disease by reducing carbonyl stress and inflammation and that this may represent a promising therapeutic strategy in humans.
Keywords: reactive carbonyl species, 4-hydroxy-2-nonenal, advanced lipoxidation end products, carnosine, atherosclerosis, renal disease
Introduction
Reactive carbonyl species (RCS) are highly reactive electrophilic compounds derived from oxidation of lipids and carbohydrates, but also from non-oxidative metabolism (Negre-Salvayre et al., 2008). RCS, which include the aldehydes acrolein, malondialdehyde, 4-hydroxy-2-nonenal (HNE) and 4-hydroxy-2-hexanal and the α-oxoaldehydes glyoxal and methylglyoxal, react with proteins to generate stable adducts called advanced lipoxidation end products (ALEs) or advanced glycosylation end products (AGEs), depending on whether they derive from lipids or sugars respectively. ALEs/AGEs exert both direct, physico-chemical and indirect, biological effects, the latter mediated by pattern recognition receptors of innate immunity, including the scavenger receptors (SRs), the toll-like receptors (TLRs) and the classical AGE receptors galectin-3 and receptor for AGEs (RAGE), which mediate ligand internalization and removal, as well as initiation of pro-inflammatory signal cascades. Both direct (Uchida, 2000) and receptor-mediated (Go and Cooper, 2008) effects of ALEs/AGEs, particularly those contained in oxidized LDLs (oxLDLs), have been implicated in the pathogenesis of atherosclerosis (Lusis, 2000), by promoting vascular tissue inflammation and remodelling; similar mechanisms seem to be operating also in renal disease (Heeringa and Cohen Tervaert, 2000). In particular, we have previously shown that ablation of galectin-3, a receptor favouring ALE/AGE removal, accelerates progression from fatty streaks to complex lesions in the aorta (Iacobini et al., 2009a) and aggravates renal disease (Iacobini et al., 2009b) in mice on high-fat intake. This was associated with marked inflammatory changes, increased accumulation of ALEs/AGEs, and up-regulation of other receptors for these compounds, including the pro-inflammatory RAGE (Iacobini et al., 2009a,b).
Thus, therapeutic strategies using RCS scavengers represent a very promising approach to atherosclerosis and renal disease, as shown in experimental animal models (Ellis, 2007). In fact, these agents are very efficient in reacting with RCS during their generation or chemical reactions involved in adduct formation, thus intervening at an early step in ALE/AGE formation and disease progression (Aldini et al., 2006). Some of these agents have been used in preclinical studies, mainly targeting AGE accumulation in diabetic subjects, with inconclusive results, except for pyridoxamine, due to safety concerns and inconsistent efficacy (Go and Cooper, 2008), at least partly attributable to promiscuous activity and lack of selectivity due to cross-reactivity with physiological aldehydes, such as pyridoxal (Aldini et al., 2006). In addition, though most of these agents act also as inhibitors of ALE formation, there is increasing interest in the development of compounds effectively sequestering lipoxidation-derived aldehydes such as HNE, which appears to play a major role in the progression of atherosclerosis (Leonarduzzi et al., 2005).
L-carnosine (β-alanyl-L-histidine) is a histidine-containing dipeptide, particularly abundant in the nervous system and skeletal muscle, which serves as a major endogenous quencher of RCS and particularly of HNE, via intra-molecular Michael addition (Aldini et al., 2005). L-carnosine was found to be effective in several disease models in which lipid peroxidation is thought to play a central pathogenic role, such as ischaemic and/or toxic injury of the kidney (Kurata et al., 2006), lung (Cuzzocrea et al., 2007) and brain (Rajanikant et al., 2007). Moreover, the prodrug N-acetyl-L-carnosine was shown to prevent and reverse age-related cataract, another condition involving glyco/lipoxidation, in humans and dogs (Babizhayev et al., 2004). However, previous reports suggested other protective mechanisms of L-CAR action in addition to RCS quenching, including inhibition of glycation (Price et al., 2001) and oxidative stress (Mozdzan et al., 2005) and endoplasmic reticulum stress (Oh et al., 2009).
Unfortunately, in humans, L-carnosine has a short half-life, due to its rapid inactivation by serum and tissue carnosinase. The shortest allelic variant of the serum carnosinase CNDP1 gene determining reduced enzyme levels was shown to be associated with protection against diabetic nephropathy (Janssen et al., 2005) and end-stage renal disease (Freedman et al., 2007), but not with longevity and protection against coronary heart disease (Zschocke et al., 2008). The carnosinase-resistant compound D-carnosine was shown to be as effective as L-carnosine as a quencher of HNE and other aldehydes and also in preventing renal disease and metabolic dysfunction in Zucker rats, but renal carnosine level were much higher with D-carnosine than with L-carnosine (Aldini et al., 2011). However, unlike L-carnosine, D-carnosine is poorly recognized by the intestinal hPepT1 transporter due to its chiral configuration. Thus, we developed a drug discovery approach aimed at designing, synthesizing and evaluating novel D-carnosine derivatives, which are efficiently absorbed at the intestinal level (Vistoli et al., 2009). D-carnosine octylester (Canevotti et al., 2011) was chosen for further study because of its rapid hydrolysis by esterase enzymes to the bioactive metabolite D-carnosine in vitro and an oral bioavailability of D-carnosine, which was 2.6-fold higher with D-carnosine octylester than with D-carnosine (Orioli et al., 2011).
This study was aimed at assessing whether (1) incubation of cultured vascular smooth muscle cells (VSMCs) and other cell types with the RCS scavenger D-carnosine attenuated HNE-induced cytotoxicity; and (2) administration of its bioavailable prodrug D-carnosine octylester to ApoE null mice fed with a pro-atherogenic diet conferred protection from atherosclerosis and renal disease by interrupting the vicious cycle of increased carbonyl stress leading to tissue inflammation, which ultimately drives lesion progression.
Methods
Design
In the in vitro studies, VSMCs, the main target cells of RCS action together with macrophages (Leonarduzzi et al., 2005), were isolated from female C57BL6 mice (Charles River, Calco, LC, Italy) using standard technique, then incubated with HNE (Cayman, Ann Harbor, MI, USA) or saline, in the presence or absence of D-carnosine (20 mM, Flamma, Chignolo d'Isola, BG, Italy), based on previous reports showing that this carnosine concentration is within the physiological range in brain and muscle (Kohen et al., 1988) and that it is effective in affording neuroprotection in vitro (Cheng et al., 2010). Monolayers were also incubated with H2O2 in order to verify whether D-carnosine has also direct antioxidant properties. Data with 10 µM HNE for up to 4 h (Leonarduzzi et al., 2005) were compared with those obtained with 300 µM H2O2 for 1 h, since the latter dose and incubation time produced the same extent of reduction in cell viability and increase in cell apoptosis and reactive oxygen species (ROS) levels as with HNE. Moreover, such a brief exposure to H2O2 reduced the possibility that the effect of this agent could be mediated via RCS generation. The experiments with HNE with or without D-carnosine were replicated in mouse mesangial cells isolated from female C57BL6 mice using standard technique and in the murine macrophage cell line J774 (Sigma-Aldrich, St. Louis, MO, USA).
For in vivo studies, all animal care and study protocols were approved by the ethical committee of the Catholic University of Rome, Italy (n. 355/2008). The animals were housed and cared in accordance with the ‘the International Guiding Principles for Biomedical Research Involving Animals’ and received water and food ad libitum. Adult (aged 6 weeks) female ApoE null mice (Charles River) were fed with a Western, high-fat diet (HFD, Teklad TD 88137: 42% fat, 0.2% cholesterol; Harlan, Indianapolis, IN, USA) or a standard, normal-fat diet (NFD: 4% fat; Charles River) and were treated with D-carnosine octylester (60 mg·kg−1 body weight in the drinking water; Flamma) or vehicle for 12 weeks. The ApoE null mouse lacks the apoprotein involved in the receptor-mediated endocytosis and catabolism of chylomicron and very low-density lipoprotein remnants by the liver, thus developing type III hyperlipoproteinemia, in which both cholesterol and triglyceride levels are increased (mixed dyslipidemia), as well as lipid-induced atherosclerosis (Plump et al., 1992) and renal disease (Wen et al., 2002). C57BL/6J wild-type mice served as control. Each group consisted of 20 mice. At the end of the 12-week period, the animals were placed into metabolic cages to collect urine. The next day, body weights were recorded, then mice were anaesthetized with intraperitoneal ketamine (Imalgene, 60 mg·kg−1) and xylazine (Rompum®, 7.5 mg·kg−1); a blood specimen was obtained and the heart with attached aorta and both kidneys were removed, fixed in 4% formaldehyde and embedded in paraffin. In selected mice, death was preceded by measurement of BP by the tail-cuff method, using the CODA System (Kent Scientific, Torrington, CT, USA), then the vasculature was perfused with 4% formaldehyde before removing the aorta, in order to prepare en face aorta specimens.
Cell culture measurements
Cell proliferation and viability were assessed using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)-based colorimetric kit (Roche Applied Science, Mannheim, Germany). Cells grown in 96-well plates were incubated with MTT for 4 h at room temperature and the formazan salt crystal formed were solubilized overnight and quantified spectrophotometrically at 540 nm using an elisa plate reader. Results were expressed as percent viability versus control. Cell apoptosis rate and ROS levels were assessed using the Annexin-V-FLUOS staining kit (Roche Applied Science) and by evaluating formation of the intracellular trapped fluorescent compound resulting from the oxidation of 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA; Molecular Probes, Eugene, OR, USA) respectively. Cells grown in multi-chamber glass slides were incubated with Annexin-V-FLUOS solution for 10 min at room temperature or with 5 µmol·L−1 CM-H2DCFDA in serum-free medium for 30 min at 37°C, and analysed at a fluorescence microscope (Zeiss-Axioplan 2, Carl Zeiss Italy, Arese, MI, Italy). For detection of Annexin-V-FLUOS, 450 nm excitation and 515 nm emissions were used, whereas CM-H2DCFDA fluorescence was analysed after fixation in 2% paraformaldehyde at 488 nm excitation and 530 nm emissions. Annexin-V-positive cells were counted and the intensity of DCF fluorescence generated by CM-H2DCFDA oxidation was measured, respectively, by developing two image analysis routines with the Optimas 6.5® image analyser (Bioscan, Washington DC, USA), according to previously reported procedures (Pläsier et al., 1999; Kubota et al., 2010). Results were expressed as percent of apoptotic versus total cells per field and percent of fluorescence intensity versus control respectively.
Blood and urine measurements
Blood glucose was measured with an automated colorimetric instrument (Glucocard G meter®, Menarini, Florence, Italy), and serum cholesterol and triglycerides by enzymatic colorimetric methods (Roche Diagnostics, Milan, Italy). Serum levels of insulin and isoprostane 8-epi-PGF2α were assessed using elisa kits from Mercodia AB (Uppsala, Sweden) and Cayman respectively. The Homeostasis Model of Assessment – Insulin Resistance (HOMA-IR) index was calculated from glucose and insulin levels. Total carbonylated proteins (PCOs) were assessed by slot blot immunoassay using an anti-dinitrophenylhydrazone antibody, after carbonyl derivatization with dinitrophenylhydrazine (Vistoli et al., 2009; Orioli et al., 2011); serum ALE/AGE levels by a competitive elisa technique (Iacobini et al., 2005); serum pentosidine levels by HPLC (Odetti et al., 1992). Total carnosine, D-carnosine octylester and the Michael adduct of HNE to carnosine (CAR-HNE) by LC-ESI-MS/MS (MRM mode; Vistoli et al., 2009; Orioli et al., 2011). Briefly, to each sample, 10 µL of a H-Tyr-His-OH solution. (500 µM solution in 1 mM PBS, pH 7.4) was added as an internal standard and the samples then deproteinized by adding 110 µL of 700 mM perchloric acid solution. After 15 min at 4°C, the samples were centrifuged (26 000× g, 10 min), and the supernatants were diluted (1:1 v/v) with the mobile phase A (water : acetonitrile : heptafluorobutyric acid; 90:10:0.1, v/v/v), filtered and analysed by LC-MS/MS. The following parent ion → product ions transitions were used for quantitative analysis: total carnosine: 227.1 m/z → 110.1 m/z + 156.6 m/z (collision energy, 38 eV); D-carnosine octylester: 339.2 m/z → 110.2 m/z + 164.2 m/z (collision energy 27eV); CAR-HNE: 383.1 m/z → 110.1 m/z + 266.2 m/z (collision energy 40 eV). Serum and urine creatinine levels were measured by HPLC, whereas proteinuria was assessed using the Bradford dye-binding protein assay kit (Pierce, Rockford, IL, USA), and results were normalized by urine creatinine (Iacobini et al., 2009b).
Morphology/morphometry
For the assessment of atherosclerotic lesions (Iacobini et al., 2009a), 5 µm sections obtained from the aortic sinus and the brachiocephalic artery were stained with haematoxylin and eosin for histological analysis and with the Weigert-van Gieson method for elastic and collagen fibres for morphometric analysis. Measurement of lesion area, necrotic core area and collagen content in the aortic sinus, and extent of luminal occlusion in the brachiocephalic artery was performed with the aid of a computer-assisted image analysis system (Optimas 6.5®, Bioscan). Elastin degradation in the aortic sinus was assessed as number of elastic lamina ruptures, whereas pseudoaneurysm formation was evaluated as disruption of all of the medial elastic laminae by a plaque extending abluminally beyond the external elastic lamina. For en face aorta preparation, after perfusion, aortas were removed, opened longitudinally in situ, layered on ovalbumin-coated glass slides, allowed to dry for 1 h and fixed in formalin for 24 h. Cryosections of the aortic sinus and en face aortic preparations were evaluated for lipid accumulation after staining with oil red O, with the aid of the Optimas 6.5®, Bioscan) image analysis system. For the assessment of renal lesions (Iacobini et al., 2009b), 4 µm sections were stained with periodic acid-Schiff (PAS) and Masson's trichrome to assess glomerular and tubulointerstitial damage by light microscopy. Briefly, the areas of at least 60 glomerular tuft profiles per sample were measured, the harmonic mean of the profile area (mean glomerular area, mGA) was obtained and the mean glomerular volume (mGV) was estimated from it. Percentage of glomerular area occupied by matrix and percentage of glomerular tufts presenting foam cells and microaneurysms (ballooning dilation of glomerular capillaries) were also evaluated, together with extent of tubulo-interstitial fibrosis.
Immunohistochemistry
Immunohistochemical analysis was performed as previously reported (Iacobini et al., 2009a,b) to assess the aortic and kidney content and distribution of markers of apoptotic cells (active caspase-3), murine macrophage activation (F4/80) and in aortas only, activated Th1 lymphocytes (chemokine receptor CXCR3), and VSMCs (α-smooth muscle actin, α-SMA); the lipoxidation products HNE adducts and oxLDLs; and the marker of oxidative stress nitrotyrosine.
RT-PCR
Total RNA was extracted from the aortic root and the kidneys by the guanidine thiocyanate-phenol-chloroform method using TRIzol® Reagent (Invitrogen Italia, San Giuliano Milanese, MI, Italy). Competitive RT-PCR (Iacobini et al., 2009a,b) or real-time PCR were used for measuring the following transcripts in the aorta: the inflammatory and lesion progression markers vascular cell adhesion molecule-1 (VCAM-1), CCL2 (monocyte chemoattractant protein-1, MCP-1), F4/80, CXCR3, TNF-α, IFN-γ, IL-1β, 4 and 10, and matrix metalloproteinase (MMP)-2 and 9; the endoplasmic reticulum stress CCAAT/enhancer binding protein (C/EBP) homologous protein (CHOP); the regulators of clearance of apoptotic cells (i.e. efferocytosis) annexin, transglutaminase-2 (TG-2) and milk fat globule-EGF factor 8 protein (MGFE8)/lactadherin; and the SRs and AGE receptors CD36, TLR-2 and 4, galectin-3 and RAGE. Real-time PCR was used for measuring the following transcripts in the kidney: fibronectin, collagen IV, TGF-β, connective tissue growth factor (CTGF), CCL2, TNF-α, CHOP, CD36, TLR-2 and 4, RAGE, and galectin-3.
Statistical analysis
Results are expressed as mean ± SD. Statistical significance was evaluated by one-way anova followed by the Student-Newman-Keuls test for multiple comparisons. A P-value < 0.05 was considered significant. All statistical tests were performed on raw data.
Results
In vitro studies
HNE-induced reduction in cell viability was significantly improved by D-carnosine, whereas the effect of this agent in monolayers exposed to H2O2 was negligible (Figure 1A). Likewise, D-carnosine blunted significantly the increase in apoptosis rate and ROS generation induced by HNE, but not that induced by H2O2, though there was a trend toward reduction also the latter (Figure 1B, C). D-carnosine was also effective in preventing cytotoxicity in mesangial and J774 cells (not shown).
Figure 1.
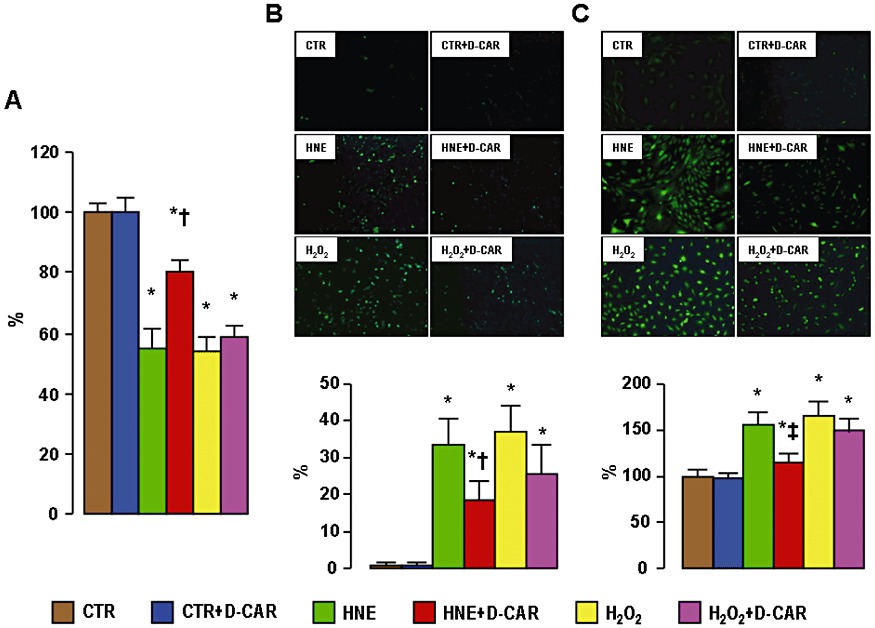
In vitro studies. Quantification of cell viability (panel A), Annexin-V-FLUOS fluorescence from representative monolayers and quantification of apoptosis rate (panel B) and DCF fluorescence from representative monolayers and quantification of ROS levels (panel C) in VSMCs incubated with saline CTR, HNE (10 µM for 4 h) or H2O2 (300 µM for 1 h), in the presence or absence of D-carnosine 20 mM. *P < 0.001 versus CTR monolayers; †P < 0.001 or ‡P < 0.01 versus monolayers incubated with HNE without D-carnosine. CTR = control.
In vivo studies
No aortic and renal lesions were detected in control C57BL/6J mice on HFD, though these animals, as compared with NFD-fed mice, showed increased serum lipid levels and changes in the gene expression of several inflammatory markers, which were generally attenuated by treatment with D-carnosine octylester (not shown). Here, we present results obtained in the ApoE null mice.
Metabolic and haemodynamic parameters
Body weight, blood glucose, serum insulin, cholesterol and triglycerides, and HOMA-IR increased significantly in animals fed with a HFD and were not affected by treatment with D-carnosine octylester. Serum PCO, ALE/AGE, pentosidine and isoprostane-8-epi-PGF2αlevels also increased significantly in mice on HFD, with significantly lower increments in D-carnosine octylester-treated animals. BP did not differ among groups (Table 1). Total carnosine levels increased markedly in plasma and urine of D-carnosine octylester-treated versus untreated mice, whereas D-carnosine octylester was detected only in urines of D-carnosine octylester-treated mice and in a negligible amount as compared with total carnosine (<1%), thus indicating an efficient prodrug conversion to the bioactive form (Figure 2A–C). CAR-HNE levels in urine samples were significantly increased in HFD- compared with NFD-fed mice and in animals treated with D-carnosine octylester, compared with untreated controls (Figure 2D).
Table 1.
Metabolic, haemodynamic and renal function parameters
| Untreated | Treated | Untreated | Treated | |
|---|---|---|---|---|
| NFD-fed | NFD-fed | HFD-fed | HFD-fed | |
| Body weight g | 21.6 ± 1.6 | 21.7 ± 2.4 | 27.3 ± 2.7* | 27.2 ± 1.8* |
| Glucose mM | 4.87 ± 0.54 | 4.83 ± 0.31 | 5.67 ± 0.62† | 5.54 ± 0.55‡ |
| Insulin pM | 54.6 ± 11.5 | 52.1 ± 9.6 | 85.2 ± 14.4 | 82.6 ± 8.8 |
| Cholesterol mM | 9.76 ± 1.64 | 8.05 ± 2.99 | 37.94 ± 4.19 | 35.28 ± 2.84 |
| Triglycerides mM | 0.76 ± 0.37 | 0.79 ± 0.23 | 1.29 ± 0.20 | 1.42 ± 0.16 |
| HOMA-IR | 1.59 ± 0.26 | 1.60 ± 0.43 | 2.75 ± 0.48* | 2.82 ± 0.30* |
| PCOs pmol mg protein-1 | 1.79 ± 0.47 | 1.58 ± 0.38 | 3.76 ± 0.58* | 2.07 ± 0.58§ |
| ALEs/AGEs AU·mL−1 | 3.91 ± 0.96 | 3.41 ± 1.48 | 9.61 ± 2.12* | 5.89 ± 0.78*§ |
| Pentosidine pg·mL−1 | 77.3 ± 6.7 | 65.7 ± 12.7 | 135.8 ± 12.8* | 88.5 ± 11.3‡§ |
| 8-epi-PGF2α pg·mL−1 | 67.8 ± 14.6 | 72.0 ± 12.4 | 148.4 ± 15.7* | 107.5 ± 13.7*§ |
| Systolic BP mmHg | 111.3 ± 8.5 | 107.5 ± 9.6 | 110.0 ± 7.1 | 110.0 ± 7.1 |
| Diastolic BP mmHg | 72.5 ± 6.5 | 75.0 ± 7.1 | 77.5 ± 2.9 | 75.0 ± 4.1 |
| Mean BP mmHg | 85.4 ± 6.9 | 85.8 ± 7.4 | 88.3 ± 4.1 | 86.7 ± 4.9 |
| Creatinine µM | 28.6 ± 1.9 | 28.9 ± 1.9 | 29.0 ± 0.7 | 29.1 ± 0.9 |
| Urinary protein : creatinine ratio mg·g−1 | 1.80 ± 0.75 | 1.82 ± 0.84 | 3.48 ± 0.98* | 2.49 ± 0.47§ |
*P < 0.001, †P < 0.01 or ‡P < 0.05 versus NFD-fed mice; §P < 0.001 or #P < 0.01 versus untreated mice.
Comparison of untreated and D-carnosine octylester-treated ApoE null mice fed a NFD or a HFD.
Figure 2.
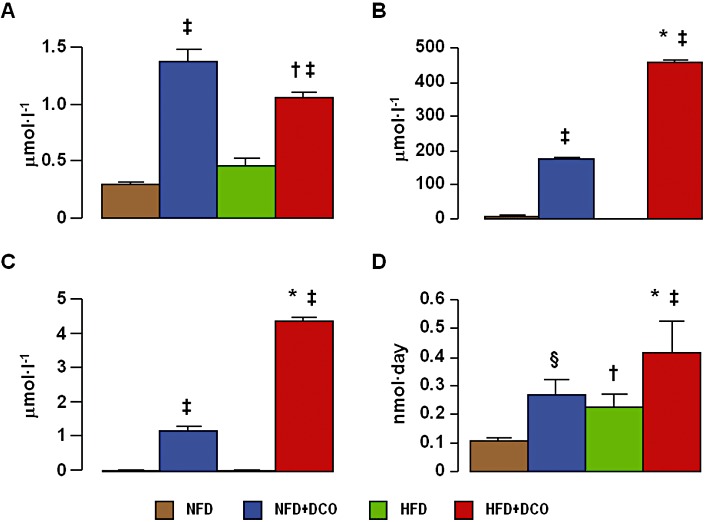
Carnosine levels. Plasma (panel A) and urinary (panel B) total carnosine, urinary D-carnosine octylester (panel C) and urine CAR-HNE (panel D) levels, in untreated and D-carnosine octylester-treated ApoE null mice fed a NFD or a HFD (mean ± SD; n= 4 per group). *P < 0.001 or †P < 0.05 versus NFD-fed mice; ‡P < 0.001 or §P < 0.01 versus untreated mice. DCO = D-carnosine octylester.
Aorta
Oil red O staining of en face aorta preparations (Supporting Information Figure S1) showed a significant reduction of lipid accumulation in D-carnosine octylester-treated versus untreated mice on a HFD. Morphometric evaluation of aortic lesions in sections stained with oil red O and the Weigert-Van Gieson method showed a significant reduction (by 18%) of lesion area in D-carnosine octylester-treated versus untreated mice fed with a HFD at the level of the aortic sinus (Figure 3A, B). D-carnosine octylester treatment also produced a more stable plaque phenotype, with less foam cell accumulation, inflammation and apoptosis, resulting in reduced necrotic core formation (−54%, Figure 3C) and more fibrosis (+48%, Figure 3D). Disruption of vessel wall architecture was also reduced, with less elastin degradation and pseudo-aneurysm formation (Supporting Information Figure S2A–C). Lesion extent was reduced more markedly in the brachiocephalic artery, with a 48% decrease of luminal occlusion (Supporting Information Figure S2D, E). Staining for active caspase-3 was significantly lower in D-carnosine octylester-treated mice, which showed less apoptotic bodies, mainly localized at the luminal side of fatty streaks, as compared with untreated animals, showing apoptotic cells surrounding the necrotic core and localized at the abluminal side of lesions. Apoptosis of cells with morphological and topographical features of VSMCs was also lower in D-carnosine octylester-treated versus untreated mice (Figure 4A). Increases of protein content of F4/80 and CXCR3 (Figure 4B, C) as well as of mRNA expression of F4/80, CXCR3, VCAM-1, CCL2, TNF-α, IFN-γ, IL-1β, MMP-9 and CHOP were significantly lower (or even normalized) and those of anti-inflammatory IL-4 and Il-10 significantly higher in D-carnosine octylester-treated versus untreated mice (Table 2), with the inverse changes in Il-1β and IL-4 mRNA levels suggesting a shift in macrophage phenotype from M1 to M2. Conversely, expression of the VSMC-specific marker α- smooth muscle actin (α-SMA) was conserved in D-carnosine octylester-treated HFD-fed mice, which also showed more VSMC-rich fibrous caps, compared with untreated mice (Figure 4D). The regulator of efferocytosis annexin increased significantly more in D-carnosine octylester-treated than in untreated HFD-fed mice, whereas TG-2 and MGFE8/lactadherin did not differ between these two groups (Table 2); however, MGFE8, which is expressed preferentially by macrophages, was up-regulated more markedly in D-carnosine octylester-treated versus untreated HFD-fed mice when mRNA levels were normalized to macrophage content (not shown). Aortic content of HNE adducts, oxLDLs and nitrotyrosine (Figure 5) and mRNA expression of CD36, TLR-2 and 4, RAGE and galectin-3 (Table 2) were significantly lower in D-carnosine octylester-treated versus untreated HFD-fed mice, with no difference versus NFD-fed animals for some of these parameters.
Figure 3.
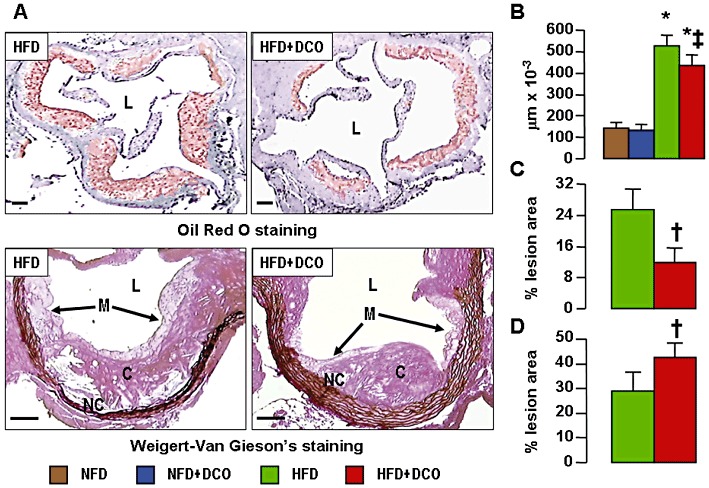
Aortic lesions. Oil Red O and Weigert-Van Gieson's staining of aortic sinus from representative untreated and D-carnosine octylester-treated ApoE null mice fed a HFD (panel A); and quantification of lesion area (panel B), necrotic core (panel C) and collagen content (panel D) in untreated and D-carnosine octylester-treated ApoE null mice fed a NFD (only for lesion area) or a HFD (mean ± SD; n= 10 per group); scale bar = 100 µm. *P < 0.001 versus NFD-fed mice; †P < 0.001 or ‡P < 0.01 versus untreated mice. L = lumen; M = macrophages; NC = necrotic core; C = collagen; DCO = D-carnosine octylester.
Figure 4.
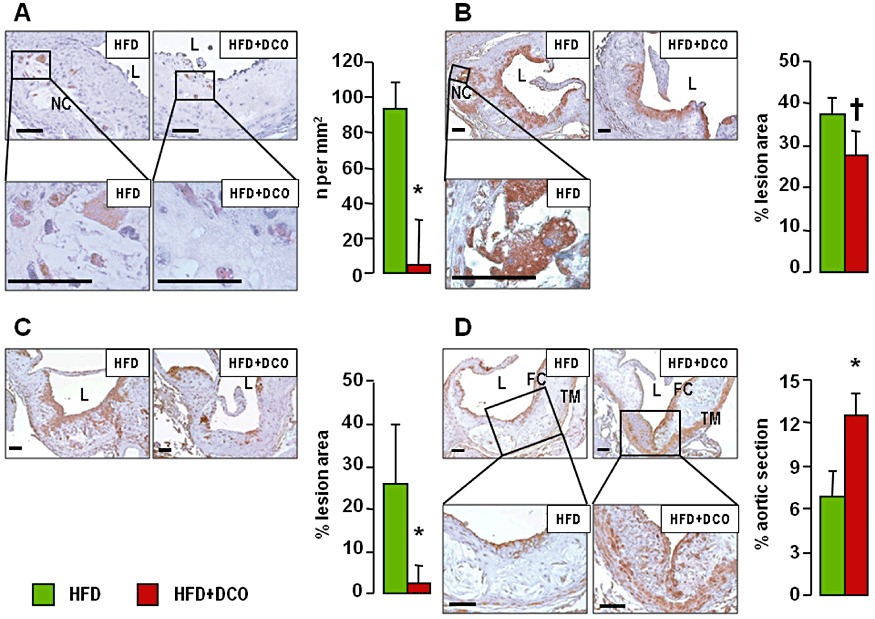
Aortic apoptosis, inflammation and VSMCs. Immunohistochemistry of aortic sinus from representative untreated and D-carnosine octylester-treated ApoE null mice fed a HFD and quantification of expression (mean ± SD; n= 5 per group) for active caspase-3 (panel A), F4/80 (panel B), CXCR3 (panel C) and α-SMA (panel D) in untreated and D-carnosine octylester-treated ApoE null mice fed a HFD; scale bar = 50 µm. *P < 0.001 or †P < 0.01 versus untreated mice. CXCR3 = chemokine receptor; α-SMA =α- smooth muscle actin; L = lumen; NC = necrotic core; TM = tunica media; FC = fibrous cap; DCO = D-carnosine octylester.
Table 2.
Aortic gene expression
| Untreated | Treated | Untreated | Treated | |
|---|---|---|---|---|
| NFD-fed | NFD-fed | HFD-fed | HFD-fed | |
| F4/80 | 0.95 ± 0.07 | 0.54 ± 0.08 | 5.65 ± 0.28* | 0.57 ± 0.14‡ |
| CXCR3 | 1.25 ± 0.07 | 1.10 ± 0.01 | 2.55 ± 0.07* | 1.40 ± 0.01*‡ |
| VCAM-1 | 2.40 ± 0.57 | 2.28 ± 0.04 | 6.10 ± 0.14* | 3.65 ± 0.21*‡ |
| CCL2 | 1.05 ± 0.07 | 1.05 ± 0.07 | 1.78 ± 0.04* | 1.05 ± 0.07‡ |
| TNF-α | 1.10 ± 0.01 | 1.00 ± 0.01 | 1.63 ± 0.04* | 1.10 ± 0.14‡ |
| IFN-γ | 1.40 ± 0.14 | 1.08 ± 0.11 | 4.12 ± 0.26* | 1.92 ± 0.12*‡ |
| IL-1β | 0.32 ± 0.02 | 0.32 ± 0.03 | 0.58 ± 0.01* | 0.27 ± 0.02 |
| IL-4 | 0.48 ± 0.04 | 0.54 ± 0.04 | 0.31 ± 0.02* | 0.44 ± 0.04†‡ |
| IL-10 | 0.12 ± 0.02 | 0.13 ± 0.02 | 0.14 ± 0.04 | 0.19 ± 0.01†§ |
| MMP-2 | 0.79 ± 0.04 | 0.80 ± 0.03 | 0.83 ± 0.03 | 0.75 ± 0.07 |
| MMP-9 | 1.01 ± 0.04 | 0.96 ± 0.05 | 2.29 ± 0.09* | 1.43 ± 0.06*‡ |
| CHOP | 2.88 ± 0.16 | 2.75 ± 0.13 | 3.72 ± 0.10* | 3.13 ± 0.06†‡ |
| Annexin | 0.58 ± 0.11 | 0.58 ± 0.03 | 0.84 ± 0.02† | 1.02 ± 0.05*§ |
| TG-2 | 0.62 ± 0.02 | 0.64 ± 0.06 | 0.63 ± 0.04 | 0.66 ± 0.04 |
| MGFE8 | 0.46 ± 0.03 | 0.46 ± 0.01 | 0.55 ± 0.03 | 0.51 ± 0.02 |
| CD36 | 1.96 ± 0.07 | 1.96 ± 0.05 | 7.47 ± 0.15* | 2.08 ± 0.10‡ |
| TLR-2 | 0.73 ± 0.06 | 0.73 ± 0.07 | 1.04 ± 0.05* | 0.79 ± 0.04‡ |
| TLR-4 | 0.64 ± 0.04 | 0.58 ± 0.08 | 0.78 ± 0.03* | 0.52 ± 0.03‡ |
| RAGE | 1.18 ± 0.04 | 1.16 ± 0.03 | 2.05 ± 0.07* | 1.50 ± 0.01*‡ |
| Galectin-3 | 1.19 ± 0.01 | 1.19 ± 0.04 | 5.53 ± 0.25* | 1.58 ± 0.08†‡ |
*P < 0.001 or †P < 0.01 versus NFD-fed mice; ‡P < 0.001 or §P < 0.05 versus untreated mice.
Comparison of untreated and D-carnosine octylester-treated ApoE null mice fed a NFD or a HFD.
Figure 5.
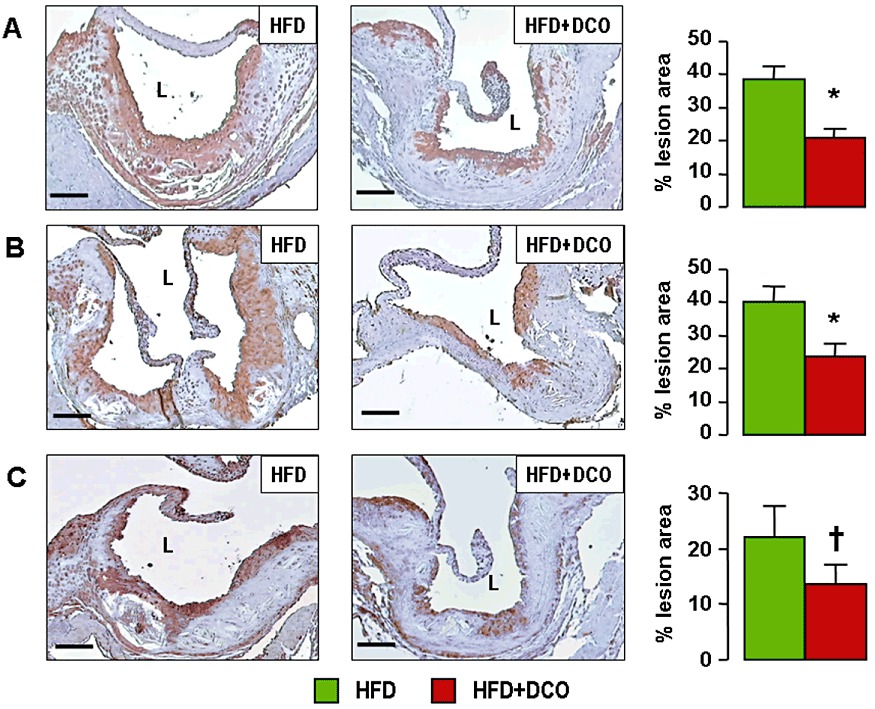
Aortic ALE levels and oxidative stress. Immunohistochemistry of aortic sinus from representative untreated and D-carnosine octylester-treated ApoE null mice fed a HFD and quantification of expression (mean ± SD; n= 5 per group) for HNE adducts (panel A), oxLDLs (panel B) and nitrotyrosine (panel C); scale bar = 100 µm. *P < 0.001 or †P < 0.01 versus the untreated mice. L = lumen; DCO = D-carnosine octylester.
Kidney
Serum creatinine levels did not differ among groups, whereas the increase in proteinuria in HFD-fed mice was significantly reduced by 28% by D-carnosine octylester treatment (Table 1). Glomerulopathy was detected in HFD-fed mice, with higher mGA, MGV, percentage of glomeruli presenting foam cells and microaneurysms, and glomerular and interstitial fibrosis without significant glomerulosclerosis or tubulo-interstitial damage. All these structural parameters were significantly reduced by D-carnosine octylester treatment (Figure 6). Also, the renal content of active caspase-3, F4/80, HNE adducts, oxLDL and nitrotyrosine (Supporting Information Figure S3) and the gene expression level of fibronectin, collagen IV, TGF-β, CTGF, CCL2, TNF-α, TLR-2 and RAGE (Supporting Information Table S1) were significantly reduced or even normalized in D-carnosine octylester-treated mice on a HFD.
Figure 6.
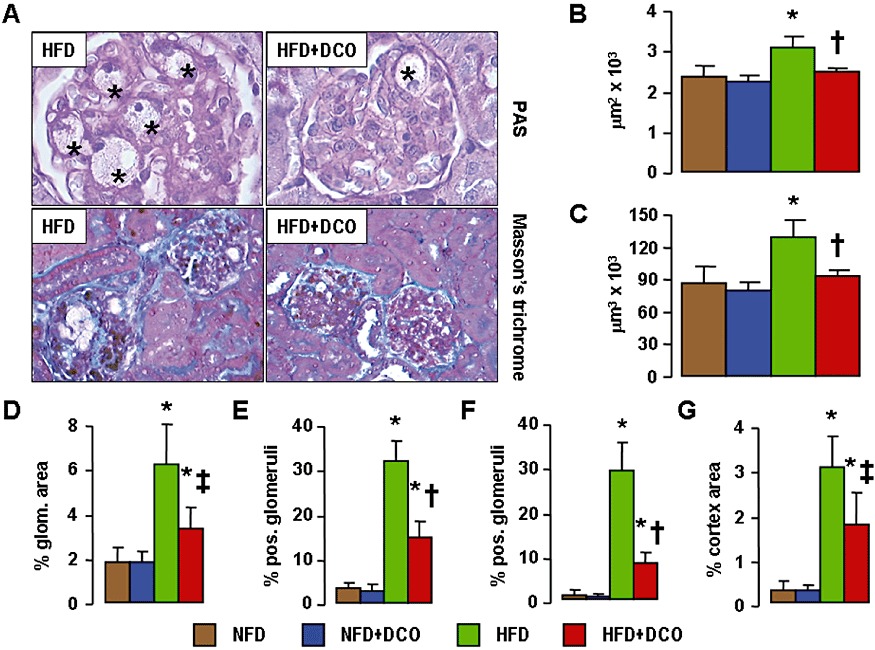
Kidney lesions. PAS and Masson's trichrome staining of kidneys from representative untreated and D-carnosine octylester-treated ApoE null mice fed a HFD (panel A; *= microaneurysms containing foam cells); and quantification of mGA (panel B), mGV (panel C), glomerular matrix (panel D), foam cells (panel E), microaneurysms (panel F) and tubulo-interstitial fibrosis (panel G) in untreated and D-carnosine octylester-treated ApoE null mice fed a NFD or a HFD (mean ± SD; n= 10 per group); scale bar = 50 µm. *P < 0.001 versus NFD-fed mice; †P < 0.001 versus untreated mice. DCO = D-carnosine octylester.
Discussion and conclusions
This study demonstrates the efficacy of D-carnosine octylester, a prodrug of the carnosinase-resistant D-carnosine, in attenuating atherosclerosis and renal disease in ApoE null mice fed with a Western, HFD diet, by efficiently scavenging RCS and particularly HNE formed under these conditions, thus reducing carbonylation of proteins, generation of ALEs/AGEs and promotion of inflammation driving lesion progression.
The negligible plasma levels of D-carnosine-octylester are consistent with a rapid hydrolysis by esterases. These enzymes are ubiquitous, though they are expressed at the highest levels in liver microsomes; in several mammalian species including mice and humans, butyrylcholinesterase and paraoxonase hydrolyse compounds having ester and amide bonds (Li et al., 2005). Moreover, the resistance of D-carnosine to carnosinase translated into much higher carnosine levels not only in plasma but also in the kidney, as compared with treatment with L-carnosine (Aldini et al., 2011), thus resulting in enhanced carnosine quenching activity towards circulating and tissue RCS. That attenuation of both atherosclerosis and renal disease induced by D-carnosine octylester is mainly attributable to the efficacy of this agent as an RCS and particularly HNE scavenger (Vistoli et al., 2009; Orioli et al., 2011) is shown by the marked reduction of protein carbonylation and ALE/AGE levels, and more importantly by the increase of urinary CAR-HNE levels. The adducts of HNE to carnosine were in fact enhanced both in mice receiving D-carnosine octylester, thus providing the experimental evidence of the scavenging efficacy of this compound, and in untreated animals on HFD, this finding supporting a relevant activity of L-carnosine as an endogenous RCS scavenger. This concept is further supported by the attenuation of HNE-induced injury in cultured VSMCs. Moreover, based on previous reports (Price et al., 2001; Mozdzan et al., 2005; Oh et al., 2009), the effect of carnosine could also be related to inhibition of glycation and oxidative and endoplasmic reticulum stress. However, a direct antioxidant activity of carnosine has been recently ruled out by the demonstration that the metal ion chelating (Decker et al., 2000) and radical scavenging (Velez et al., 2008) activities of this compound are much lower than those of histidine and α-tocopherol respectively. Moreover, the unchanged rate of cell death and ROS generation that we observed in D-carnosine-treated VSMCs in response to short-term incubation with H2O2 argues against a direct antioxidant activity of this compound. Thus, the in vivo antioxidant activity of D-carnosine octylester could also be attributed to the RCS scavenging activity, causing reduced formation of ALEs/AGEs, which are known to induce pro-oxidant species (Lusis, 2000), that in turn would be implicated in the induction of endoplasmic reticulum stress (and vice versa). Nonetheless, other pleiotropic actions of D-carnosine octylester cannot be ruled out and require further investigation.
Treatment with D-carnosine octylester resulted in qualitative more than quantitative changes in aortic lesions. The 30% decrease of fat accumulation in the whole aorta and the 18% reduction of lesion area at the level of the aortic sinus are consistent with persistence of very high serum lipid levels, particularly cholesterol, despite D-carnosine octylester treatment. In fact, by scavenging RCS, this therapy was aimed at reducing ALE generation and tissue injury induced by these compounds, thus influencing progression towards advanced lesions more than early changes such as fatty streak formation, which depends mainly on hyperlipidaemia. However, at the level of the brachiocephalic artery, lesion extent was markedly reduced in D-carnosine octylester-treated mice, a finding which may be very relevant, as this is the best characterized anatomical site in mice, in terms of production of unstable lesions (Jackson et al., 2007).
Independent of site, plaque phenotype was dramatically modified by D-carnosine octylester treatment, with reduced signs of lesion progression and increased features of stability, as compared with untreated mice. In particular, there was a significant decrease of content and media invasion by lipid-laden macrophages, which also showed a reduced rate of apoptosis. Macrophage depletion by apoptosis was shown to promote plaque stability in the early but not later stages of lesion development. This has been related to clearance of apoptotic cells, which initially appears to be efficient, thus leading to diminished lesion cellularity and reduced production of pro-inflammatory cytokines. Conversely, in advanced lesions, efferocytosis is defective, possibly due to accumulation of oxLDLs, thus resulting in secondary necrosis of apoptotic cells and consequent pro-inflammatory response, necrotic core generation and promotion of further inflammation, plaque instability, and ultimately thrombosis (Tabas, 2005). Treatment with D-carnosine octylester, possibly by reducing oxLDL content, produced a significant amelioration of efferocytosis, as indicated by both reduced morphological evidence of apoptotic bodies and increased expression of regulators of this process, such as annexin and MGFE8. Conversely, TG-2 expression was unaffected by D-carnosine octylester treatment; however, a recent report indicates that TG-2 deficiency does not influence plaque stability (Williams et al., 2010). Features of macrophage shift towards a M2 phenotype, that is down-regulation of IL-1β and up-regulation of IL-4, also indicated reduced inflammation. Furthermore, collagen deposition was increased and apoptosis of VSMCs, expression of MMP-9, extent of elastin degradation and number of pseudo-aneurysms were decreased in D-carnosine octylester-treated mice, consistent with a more stable plaque phenotype. This favourable impact on plaque stability suggests that D-carnosine octylester might be very effective in the prevention and treatment of atherosclerosis by preventing increased necrotic core area and reduced fibrous cap thickness, leading to plaque erosion/rupture and consequent thrombosis.
In addition to atherosclerosis, renal lesions were also significantly attenuated in D-carnosine octylester-treated mice, with reduction of proteinuria, glomerular hypertrophy and glomerular and tubulo-interstitial fibrosis. This was associated with lower inflammation and apoptosis as well as with reduction of lesions characteristic of lipid-induced renal disease occurring in ApoE null mice, such as accumulation of foam cells and formation of microaneurysms due to mesangiolysis (Wen et al., 2002). These data are consistent with our previous study in the Zucker rat (Aldini et al., 2011) and also with the recent reports that L-carnosine reduced proteinuria in the db/db mouse by preventing alterations of renal carnosine metabolism (Peters et al., 2011) and podocyte and pericyte loss in the streptozotocin-diabetic rat by preventing apoptosis (Pfister et al., 2011; Riedl et al., 2011).
In conclusion, our data, in addition to supporting a central role for lipoxidation-derived carbonyl stress in atherosclerosis and renal disease, show that D-carnosine octylester is highly effective in attenuating HNE-induced cellular injury as well as in reducing progression of atherosclerotic plaque and kidney lesions in an experimental model of lipid-induced injury. This is attributable to an efficient scavenging of RCS and particularly of HNE, formed under these conditions, which interrupts the vicious cycle fuelling tissue inflammation. Thus, the use of this novel compound may represent a promising preventive and therapeutic strategy in humans.
Acknowledgments
This work was supported by a research grant of the Research Foundation of the Italian Society of Diabetology (Fo.Ri.SID) and by an unconditional grant from Flamma S.p.A., Chignolo d'Isola, BG, Italy. The sponsors had no role in study design; in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the article for publication.
Glossary
- AGEs
advanced glycation end products
- ALEs
advanced lipoxidation end products
- α-SMA
α- smooth muscle actin
- BP
blood pressure
- CAR-HNE
HNE-carnosine adducts
- CHOP
CCAAT/enhancer binding protein (C/EBP) homologous protein
- CM-H2DCFDA
5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate
- CTGF
connective tissue growth factor; HFD, high-fat diet
- HNE
4-hydroxy-2-nonenal
- HOMA-IR
Homeostasis Model of Assessment – Insulin Resistance
- MCP-1
monocyte chemoattractant protein-1
- mGA
mean glomerular area
- MGFE8
milk fat globule-EGF factor 8 protein
- mGV
mean glomerular volume
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NFD
normal-fat diet
- oxLDLs
oxidized LDLs
- PAS
periodic acid-Schiff
- PCOs
protein carbonyl derivatives
- RAGE
receptor for AGEs
- RCS
reactive carbonyl species
- ROS
reactive oxygen species
- SRs
scavenger receptors
- TG-2
transglutaminase-2
- TGF-β
transforming growth factor β
- TLRs
toll-like receptors
- VCAM-1
vascular cell adhesion molecole-1
- VSMCs
vascular smooth muscle cells
Conflicts of interest
The authors declare no relevant conflict of interest to disclose, except for Dr. Canevotti, who is an employee of Flamma S.p.A. the manufacturers of D-carnosine-octylester.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Aortic lipid accumulation. Oil red Ostaining of en-face aorta preparations from representative untreated and D-carnosine octylester-treated ApoE null mice fed a NFD or a HFD (Panel A); and quantification of staining (Panel B) in untreated and D-carnosine octylester-treated ApoE null mice fed a NFD or a HFD (mean ± SD; n = 10 per group); scale bar = 5 mm. *P < 0.001 vs. NFD-fed mice; †P < 0.001 vs. untreated mice. CAR = carnosine; NFD = normal fat diet; HFD = high fat diet; DCO = D-carnosine octylester.
Figure S2 Aortic lesions. Weigert-Van Gieson's staining of aortic sinus from representative untreated and D-carnosine octylester-treated ApoE null mice fed a HFD (Panel A), and quantification of elastin degradation (Panel B), and number of pseudo aneurysms (Panel C) in untreated and D-carnosine octylester-treated ApoE null mice feda HFD (mean ± SD; n = 10 per group). Weigert-Van Gieson's staining of brachiocephalic artery from representative of untreated and D-carnosine octylester-treated ApoE null mice fed a HFD (Panel D), and quantification of luminal occlusion (Panel E) in untreated and D-carnosine octylester-treated ApoE null mice fed a HFD (mean ± SD;n = 10 per group); scale bar = 100 μm. *P < 0.001 vs. untreated mice. CAR = carnosine; NFD = normal fat diet; HFD = high fat diet; L = lumen; IEL = internal elastic lamina; D = disruption of elastic laminae; E = ectasia; TM = tunica media; NC = necrotic core; BFC = buried fibrous caps; DCO = D-carnosine octylester.
Figure S3 Kidney apoptosis, inflammation, ALEs and oxidative stress. Immunohistochemistry of kidneys from representative untreated and D-carnosine octylester-treated ApoE null mice fed a HFD and quantification of expression in untreated and D-carnosine octylester-treated ApoE null mice fed a NFD or a HFD (mean ± SD; n = 5 per group) for active caspase-3 (Panel A), F4/80 (Panel B; → = F4/80 positive cells, showing a foam cell phenotype in the untreated HFD-fed mouse), HNE adducts (Panel c), oxLDLs (Panel D) and nitrotyrosine (Panel E); scale bar = 50 μm. *P< 0.001 or †P < 0.01 vs. NFD-fed mice; ‡P < 0.001 or §P < 0.01 vs. untreated mice. CAR = carnosine; HFD = high fat diet; NFD = normal fat diet; HNE = 4-hydroxy-2-nonenal; oxLDLs = oxidized LDLs; DCO = D-carnosine octylester.
Table S1 Kidney gene expression. Comparison of untreated and D-carnosine octylester-treated Apo E null mice fed a NFD or a HFD
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Aldini G, Maffei Facino R, Beretta G, Carini M. Carnosine and related dipeptides as quenchers of reactive carbonyl species: from structural studies to therapeutic perspectives. Biofactors. 2005;24:77–87. doi: 10.1002/biof.5520240109. [DOI] [PubMed] [Google Scholar]
- Aldini G, Dalle-Donne I, Colombo R, Maffei Facino R, Milzani A, Carini M. Lipoxidation-derived reactive carbonyl species as potential drug targets in preventing protein carbonylation and related cellular dysfunction. ChemMedChem. 2006;1:1045–1058. doi: 10.1002/cmdc.200600075. [DOI] [PubMed] [Google Scholar]
- Aldini G, Orioli M, Rossoni G, Savi F, Braidotti P, Vistoli G, et al. The carbonyl scavenger carnosine ameliorates dyslipidaemia and renal function in Zucker obese rats. J Cell Mol Med. 2011;15:1339–1354. doi: 10.1111/j.1582-4934.2010.01101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babizhayev MA, Deyev AI, Yermakova VN, Brikman IV, Bours J. Lipid peroxidation and cataracts: N-acetylcarnosine as a therapeutic tool to manage age-related cataracts in human and in canine eyes. Drugs R D. 2004;5:125–139. doi: 10.2165/00126839-200405030-00001. [DOI] [PubMed] [Google Scholar]
- Canevotti R, Negrisoli G, Previtali M. 2008. Dipeptide compounds containing D-histidine. WO2008001175(A2)
- Cheng J, Wang F, Yu DF, Wu PF, Chen JG. The cytotoxic mechanism of malondialdehyde and protective effect of carnosine via protein cross-linking/mitochondrial dysfunction/reactive oxygen species/MAPK pathway in neurons. Eur J Pharmacol. 2010;650:184–194. doi: 10.1016/j.ejphar.2010.09.033. [DOI] [PubMed] [Google Scholar]
- Cuzzocrea S, Genovese T, Failla M, Vecchio G, Fruciano M, Mazzon E, et al. Protective effect of orally administered carnosine on bleomycin-induced lung injury. Am J Physiol. 2007;292:L1095–L1104. doi: 10.1152/ajplung.00283.2006. [DOI] [PubMed] [Google Scholar]
- Decker EA, Livisay SA, Zhou S. A re-evaluation of the antioxidant activity of purified carnosine. Biochemistry (Mosc) 2000;65:766–770. [PubMed] [Google Scholar]
- Ellis EM. Reactive carbonyls and oxidative stress: potential for therapeutic intervention. Pharmacol Ther. 2007;115:13–24. doi: 10.1016/j.pharmthera.2007.03.015. [DOI] [PubMed] [Google Scholar]
- Freedman BI, Hicks PJ, Sale MM, Pierson ED, Langefeld CD, Rich SS, et al. A leucine repeat in the carnosinase gene CNDP1 is associated with diabetic end-stage renal disease in European Americans. Nephrol Dial Transplant. 2007;22:1131–1135. doi: 10.1093/ndt/gfl717. [DOI] [PubMed] [Google Scholar]
- Go S-Y, Cooper ME. The role of advanced glycation end products in progression and complications of diabetes. J Clin Endocrinol Metab. 2008;93:1143–1152. doi: 10.1210/jc.2007-1817. [DOI] [PubMed] [Google Scholar]
- Heeringa P, Cohen Tervaert JW. Role of oxidized low-density lipoprotein in renal disease. Curr Opin Nephrol Hypertens. 2000;11:287–293. doi: 10.1097/00041552-200205000-00004. [DOI] [PubMed] [Google Scholar]
- Iacobini C, Oddi G, Menini S, Amadio L, Ricci C, Di Pippo C, et al. Development of age-dependent glomerular lesions in galectin-3/AGE-receptor- 3 knockout mice. Am J Physiol. 2005;289:F611–F621. doi: 10.1152/ajprenal.00435.2004. [DOI] [PubMed] [Google Scholar]
- Iacobini C, Menini S, Ricci C, Scipioni A, Sansoni V, Cordone S, et al. Accelerated lipid-induced atherogenesis in galectin-3 deficient mice: role of lipoxidation via receptor-mediated mechanisms. Arterioscler Thromb Vasc Biol. 2009a;29:831–836. doi: 10.1161/ATVBAHA.109.186791. [DOI] [PubMed] [Google Scholar]
- Iacobini C, Menini S, Ricci C, Scipioni A, Sansoni V, Mazzitelli G, et al. Advanced lipoxidation end-products mediate lipid-induced glomerular injury: role of receptor-mediated mechanisms. J Pathol. 2009b;218:360–369. doi: 10.1002/path.2536. [DOI] [PubMed] [Google Scholar]
- Jackson CL, Bennett MR, Biessen EA, Johnson JL, Krams R. Assessment of unstable atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2007;27:714–720. doi: 10.1161/01.ATV.0000261873.86623.e1. [DOI] [PubMed] [Google Scholar]
- Janssen B, Hohenadel D, Brinkkoetter P, Peters V, Rind N, Fischer C, et al. Carnosine as a protective factor in diabetic nephropathy: association with a leucine repeat of the carnosinase gene CNDP1. Diabetes. 2005;54:2320–2327. doi: 10.2337/diabetes.54.8.2320. [DOI] [PubMed] [Google Scholar]
- Kohen R, Yamamoto Y, Cundy KC, Ames BN. Antioxidant activity of carnosine, homocarnosine, and anserine present in muscle and brain. Proc Natl Acad Sci USA. 1988;85:3175–3179. doi: 10.1073/pnas.85.9.3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota C, Torii S, Hou N, Saito N, Yoshimoto Y, Imai H, et al. Constitutive reactive oxygen species generation from autophagosome/lysosome in neuronal oxidative toxicity. J Biol Chem. 2010;285:667–674. doi: 10.1074/jbc.M109.053058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurata H, Fujii T, Tsutsui H, Katayama T, Ohkita M, Takaoka M, et al. Renoprotective effects of l-carnosine on ischemia/reperfusion-induced renal injury in rats. J Pharmacol Exp Ther. 2006;319:640–647. doi: 10.1124/jpet.106.110122. [DOI] [PubMed] [Google Scholar]
- Leonarduzzi G, Chiarpotto E, Biasi F, Poli G. 4-Hydroxynonenal and cholesterol oxidation products in atherosclerosis. Mol Nutr Food Res. 2005;49:1044–1049. doi: 10.1002/mnfr.200500090. [DOI] [PubMed] [Google Scholar]
- Li B, Sedlacek M, Manoharan I, Boopathy R, Duysen EG, Masson P, et al. Butyrylcholinesterase, paraoxonase, and albumin esterase, but not carboxylesterase, are present in human plasma. Biochem Pharmacol. 2005;70:1673–1684. doi: 10.1016/j.bcp.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Lusis AJ. Atherosclerosis. Nature. 2000;407:233–241. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozdzan M, Szemraj J, Rysz J, Nowak D. Antioxidant properties of carnosine re-evaluated with oxidizing systems involving iron and copper ions. Basic Clin Pharmacol Toxicol. 2005;96:352–360. doi: 10.1111/j.1742-7843.2005.pto_03.x. [DOI] [PubMed] [Google Scholar]
- Negre-Salvayre A, Coatrieux C, Ingueneau C, Salvayre R. Advanced lipid peroxidation end products in oxidative damage to proteins. Potential role in diseases and therapeutic prospects for the inhibitors. Br J Pharmacol. 2008;153:6–20. doi: 10.1038/sj.bjp.0707395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odetti P, Fogarthy J, Sell D, Monnier VM. Chromatographic quantitation of plasma and erythrocyte pentosidine in diabetic and uremic subjects. Diabetes. 1992;41:153–159. doi: 10.2337/diab.41.2.153. [DOI] [PubMed] [Google Scholar]
- Oh YM, Jang EH, Ko JH, Kang JH, Park CS, Han SB, et al. Inhibition of 6-hydroxydopamine-induced endoplasmic reticulum stress by L-carnosine in SH-SY5Y cells. Neurosci Lett. 2009;459:7–10. doi: 10.1016/j.neulet.2009.04.047. [DOI] [PubMed] [Google Scholar]
- Orioli M, Vistoli G, Regazzoni L, Pedretti A, Lapolla A, Rossoni G, et al. Design, synthesis, ADME Properties, and pharmacological activities of β-alanyl-D-histidine (D-Carnosine) prodrugs with improved bioavailability. ChemMedChem. 2011;6:1269–1282. doi: 10.1002/cmdc.201100042. [DOI] [PubMed] [Google Scholar]
- Peters V, Schmitt CP, Zschocke J, Gross M-L, Brismar K, Forsberg E. Carnosine treatment largely prevents alterations of renal carnosine metabolism in diabetic mice. Amino Acids. 2011 doi: 10.1007/s00726-011-1046-4. e-pub Aug 11. [DOI] [PubMed] [Google Scholar]
- Pfister F, Riedl E, Wang Q, vom Hagen F, Deinzer M, Harmsen MC, et al. Oral carnosine supplementation prevents vascular damage in experimental diabetic retinopathy. Cell Physiol Biochem. 2011;28:125–136. doi: 10.1159/000331721. [DOI] [PubMed] [Google Scholar]
- Pläsier B, Lloyd DR, Paul GC, Thomas CR, Al-Rubeai M. Automatic image analysis for quantification of apoptosis in animal cell culture by annexin-V affinity assay. J Immunol Methods. 1999;229:81–95. doi: 10.1016/s0022-1759(99)00107-6. [DOI] [PubMed] [Google Scholar]
- Plump AS, Smith JD, Hayek T, Aalto-Setala K, Walsh A, Verstuyft JG, et al. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell. 1992;71:343–353. doi: 10.1016/0092-8674(92)90362-g. [DOI] [PubMed] [Google Scholar]
- Price DL, Rhett PM, Thorpe SR, Baynes JW. Chelating activity of advanced glycation end-product inhibitors. J Biol Chem. 2001;276:48967–48972. doi: 10.1074/jbc.M108196200. [DOI] [PubMed] [Google Scholar]
- Rajanikant GK, Zemke D, Senut MC, Frenkel MB, Chen AF, Gupta R, et al. Carnosine is neuroprotective against permanent focal cerebral ischemia in mice. Stroke. 2007;38:3023–3031. doi: 10.1161/STROKEAHA.107.488502. [DOI] [PubMed] [Google Scholar]
- Riedl E, Pfister F, Braunagel M, Brinkkötter P, Sternik P, Deinzer M, et al. Carnosine prevents apoptosis of glomerular cells and podocyte loss in STZ diabetic rats. Cell Physiol Biochem. 2011;28:279–288. doi: 10.1159/000331740. [DOI] [PubMed] [Google Scholar]
- Tabas I. Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: the importance of lesion stage and phagocytic efficiency. Arterioscler Thromb Vasc Biol. 2005;25:2255–2264. doi: 10.1161/01.ATV.0000184783.04864.9f. [DOI] [PubMed] [Google Scholar]
- Uchida K. Role of reactive aldehyde in cardiovascular diseases. Free Radic Biol Med. 2000;28:1685–1696. doi: 10.1016/s0891-5849(00)00226-4. [DOI] [PubMed] [Google Scholar]
- Velez S, Nair NG, Reddy VP. Transition metal ion binding studies of carnosine and histidine: biologically relevant antioxidants. Colloids Surf B Biointerfaces. 2008;66:291–294. doi: 10.1016/j.colsurfb.2008.06.012. [DOI] [PubMed] [Google Scholar]
- Vistoli G, Orioli M, Pedretti A, Regazzoni L, Canevotti R, Negrisoli G, et al. Design, synthesis, and evaluation of carnosine derivatives as selective and efficient sequestering agents of cytotoxic reactive carbonyl species. ChemMedChem. 2009;4:967–975. doi: 10.1002/cmdc.200800433. [DOI] [PubMed] [Google Scholar]
- Wen M, Segerer S, Dantas M, Brown PA, Hudkins KL, Goodpaster T, et al. Renal injury in apolipoprotein E-deficient mice. Lab Invest. 2002;82:999–1006. doi: 10.1097/01.lab.0000022222.03120.d4. [DOI] [PubMed] [Google Scholar]
- Williams H, Pease RJ, Newell LM, Cordell PA, Graham RM, Kearney MT, et al. Effect of transglutaminase 2 (TG2) deficiency on atherosclerotic plaque stability in the apolipoprotein E deficient mouse. Atherosclerosis. 2010;210:94–99. doi: 10.1016/j.atherosclerosis.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zschocke J, Nebel A, Wicks K, Peters V, El Mokhtari NE, Krawczak M, et al. Allelic variation in the CNDP1 gene and its lack of association with longevity and coronary heart disease. Mech Ageing Dev. 2008;127:817–820. doi: 10.1016/j.mad.2006.08.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.