

Abstract
BACKGROUND AND PURPOSE
Transient receptor potential cation channel subfamily M member 7 (TRPM7) is a bifunctional protein comprising a TRP ion channel segment linked to an α-type protein kinase domain. TRPM7 is essential for proliferation and cell growth. Up-regulation of TRPM7 function is involved in anoxic neuronal death, cardiac fibrosis and tumour cell proliferation. The goal of this work was to identify non-toxic inhibitors of the TRPM7 channel and to assess the effect of blocking endogenous TRPM7 currents on the phenotype of living cells.
EXPERIMENTAL APPROACH
We developed an aequorin bioluminescence-based assay of TRPM7 channel activity and performed a hypothesis-driven screen for inhibitors of the channel. The candidates identified were further assessed electrophysiologically and in cell biological experiments.
KEY RESULTS
TRPM7 currents were inhibited by modulators of small conductance Ca2+-activated K+ channels (KCa2.1–2.3; SK) channels, including the antimalarial plant alkaloid quinine, CyPPA, dequalinium, NS8593, SKA31 and UCL 1684. The most potent compound NS8593 (IC50 1.6 µM) specifically targeted TRPM7 as compared with other TRP channels, interfered with Mg2+-dependent regulation of TRPM7 channel and inhibited the motility of cultured cells. NS8593 exhibited full and reversible block of native TRPM7-like currents in HEK 293 cells, freshly isolated smooth muscle cells, primary podocytes and ventricular myocytes.
CONCLUSIONS AND IMPLICATIONS
This study reveals a tight overlap in the pharmacological profiles of TRPM7 and KCa2.1–2.3 channels. NS8593 acts as a negative gating modulator of TRPM7 and is well-suited to study functional features and cellular roles of endogenous TRPM7.
Keywords: transient receptor potential, TRPM7, SK channels, KCa2.1–2.3 channels, magnesium, cell motility
Introduction
TRPM7 (transient receptor potential cation channel, subfamily M, member 7) is a bifunctional protein comprising two distinct functional moieties: an ion channel segment and a protein kinase domain. The channel segment of TRPM7 displays high homology to other melastatin-related TRP (TRPM) channels (reviewed in Chubanov et al., 2005), whereas the kinase domain is a member of the family of α-type serine/threonine protein kinases with structural similarity to PKA (Ryazanov et al., 1999; Yamaguchi et al., 2001). Although TRPM channels are highly conserved throughout the animal kingdom (Mederos y Schnitzler et al., 2008; Hofmann et al., 2010), only in TRPM7 and a genetically related protein, TRPM6, the ion pore-forming subunits are covalently linked to α-kinase domains.
In heterologous expression systems, TRPM7 behaves as a constitutively active cation channel that is highly permeable to a broad range of divalent cations, including Ca2+ and Mg2+ (Nadler et al., 2001; Runnels et al., 2001; Monteilh-Zoller et al., 2003). Intracellular and extracellular Mg2+ tightly regulates TRPM7 channel activity. External Mg2+ is a permeant blocker of TRPM7 (Nadler et al., 2001; Monteilh-Zoller et al., 2003; Kozak et al., 2005). Internal Mg2+ (as well as Mg2+-ATP) inhibits TRPM7 via a nucleotide-binding site in the kinase domain synergistically with another as yet unidentified Mg2+ binding site extrinsic to the kinase domain (Nadler et al., 2001; Demeuse et al., 2006). Native TRPM7-like currents have been detected in all cell types examined so far (Kozak et al., 2002; Aarts et al., 2003; Gwanyanya et al., 2004; Kim et al., 2007; Numata et al., 2007b; Mishra et al., 2009), indicating that this channel type plays a role indispensable for cell function. It has been suggested that TRPM7-mediated cation entry regulates many essential cellular functions such as cellular Mg2+ homeostasis (Schmitz et al., 2003; Chubanov et al., 2004; Sahni and Scharenberg, 2008; Ryazanova et al., 2010), cell spreading (Clark et al., 2006; Su et al., 2006; Wei et al., 2009), proliferation (Nadler et al., 2001; Schmitz et al., 2003; Sahni and Scharenberg, 2008), mechanosensitivity (Oancea et al., 2006; Numata et al., 2007a; Wei et al., 2009) and exocytosis (Brauchi et al., 2008). TRPM7 was found to play a role in anoxic neuronal death (Aarts et al., 2003), hypertension (Touyz, 2008), neurodegenerative disorders (Hermosura et al., 2005; Tseveleki et al., 2010), cardiac fibrosis (Du et al., 2010) and tumour cell proliferation (Hanano et al., 2004; Jiang et al., 2007; Kim et al., 2008; Guilbert et al., 2009)
Despite the physiological and clinical importance of TRPM7, only a few modulators of TRPM7 channel are currently available (Table 1), including a subset of non-specific ion channel blockers like spermine and 2-aminoethoxydiphenyl borate (2-APB) (Kerschbaum et al., 2003; Monteilh-Zoller et al., 2003; Jiang et al., 2005; Parnas et al., 2009; Chen et al., 2010a,b). However, as TRPM7 null mice are embryonic lethal (Jin et al., 2008; Ryazanova et al., 2010), an indiscriminate pharmacological inactivation of TRPM7 will be most probably toxic and, therefore, is not a preferable option. In contrast, similar to the situation with voltage-gated sodium channel inhibitors, drugs acting as ‘use-dependent’ or ‘state-dependent’ TRPM7 channel inhibitors may be particularly valuable. Accordingly, organic compounds that would interfere with Mg2+-dependent gating of TRPM7 are of particular interest since they might be instrumental in developing this type of TRPM7 drugs.
Table 1.
Organic compounds inhibiting TRPM7 currents
| Compound | IC50 (µM)* | Description of the block | Reference |
|---|---|---|---|
| 2-APB | 178 | Reversible | Prakriya and Lewis, 2002, Li et al., 2006 |
| Spermine | 2.3† | Reversible, voltage dependent | Kozak et al., 2002 |
| Carvacrol | 306 | Reversible | Parnas et al., 2009 |
| NDGA | n.d. | Tested only at 10 and 20 µM | Chen et al., 2010b |
| AA861 | n.d. | Tested only at 10 and 40 µM | Chen et al., 2010b |
| MK886 | n.d. | Tested only at 20 µM | Chen et al., 2010b |
| Nafamostat mesylate | 617 | Reversible, voltage and extracellular divalents dependent | Chen et al., 2010a |
| Waixenicin A | 7.0 | Irreversible, [Mg2+]i dependent | Zierler et al., 2011 |
| NS8593 | 1.6 | Reversible, [Mg2+]i dependent | Present study |
IC50 values were determined for recombinant TRPM7 in patch-clamp measurements in the absence of internal Mg2+.
The dose-dependent effect of spermine was evaluated on endogenous MIC currents in divalent-free external solution.
n.d., not determined.
The gating mechanisms of TRPM7 as well as its high sensitivity to internal Mg2+ are poorly understood and any relevant results are controversial (Nadler et al., 2001; Runnels et al., 2001; 2002; Kozak and Cahalan, 2003; Kozak et al., 2005). It is possible that intracellular Mg2+ regulates TRPM7 via one of the mechanisms described for a subset of K+ channels. For instance, intracellular Mg2+ blocks small conductance Ca2+-activated K+ (SK1-3 or KCa2.1–2.3 according to Alexander et al., 2011) channels in a voltage-dependent manner, resulting in a rectified current–voltage relationship (Soh and Park, 2002). A corresponding divalent cation-binding site in KCa2.1–2.3 channels was identified in a pore-forming loop similar to that in TRPM7 (Figure 1A). The phenomenon of inward rectification in inward rectifier K+ channels type 2.1–2.4 (Kir2.1–2.4) channels is a result of voltage-dependent block by internal polyamines and Mg2+ acting via a site located in one of the transmembane helices and in the C-terminus of the ion channel (Figure 1A) (Stanfield et al., 1994; Wible et al., 1994; Taglialatela et al., 1995). High-conductance Ca2+-activated K+ (BK or KCa1.1) channels are also sensitive to intracellular Mg2+ (Shi and Cui, 2001; Yang et al., 2008; Wu et al. 2010). Unlike KCa2.1–2.3 and KIR2.1–2.4, Mg2+ binds to a C-terminal domain to positively modulate KCa1.1 channels (Figure 1A).
Figure 1.
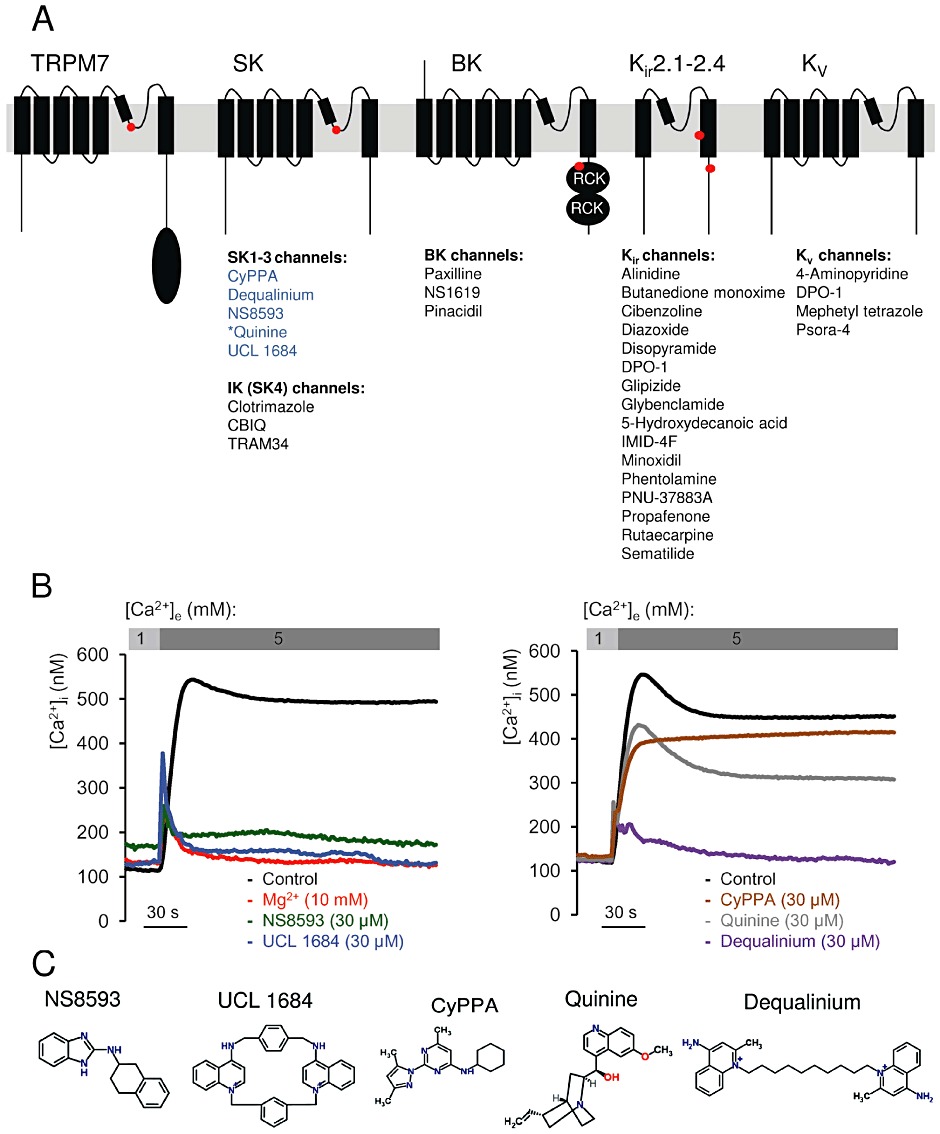
Identification of TRPM7 channel inhibitors among known modulators of Mg2+ sensitive K+ channels. (A) Domain architecture of TRPM7, KCa2.1–2.3 (SK), KCa3.1 (IK, SK4), KCa1.1 (BK), KIR and KV channels. Locations of Mg2+-binding sites in TRPM7 (formed by E1047 and Y1049 in mouse TRPM7 (Mederos y Schnitzler et al., 2008), KCa2.1–2.3 channels [S359 in rat KCa2.2 (Soh and Park, 2002)], KCa1.1 channels (formed by E374 and E399 in human BK1) (Wu et al. 2010; Yang et al., 2008), KIR channels (D172 and E224 in mouse KIR2.1) (Stanfield et al., 1994; Wible et al., 1994; Taglialatela et al., 1995) are indicated by red dots. KD, Ser/Thr kinase domain in TRPM7; RCK, ‘regulating the conductance of K+’ domain in KCa1.1 channels. The modulators of K+ channels tested in a primary screen for TRPM7 inhibitors are listed below their known targets. Compounds labelled in blue were found to be inhibitors of TRPM7, while modulators in black showed no inhibitory effect on TRPM7. *Quinine blocks KCa1.1 and KCa3.1 channels. (B) Primary assessment of modulators using a bioluminescence-based assay of TRPM7 channel activity. Representative traces are shown from two independent experiments with similar results. The indicated compounds (30 µM) or Mg2+ (10 mM) were applied to ponasterone A-induced HEK 293 cells, and the measurements were performed in the presence of 1 or 5 mM external Ca2+ ([Ca2+]e) as indicated. (C) Chemical structures of TRPM7 inhibitors identified.
In a hypothesis-driven chemical screen to identify novel modulators of TRPM7 channel activity, we discovered that TRPM7 is the target of commonly used activators and inhibitors of KCa2.1–2.3 channels. We noted that the antimalarial plant alkaloid quinine as well as synthetic KCa2.1–2.3 modulators, such as CyPPA, dequalinium, NS8593, SKA31 and UCL 1684 (reviewed in Weatherall et al., 2010), are potent inhibitors of TRPM7. The most efficient compound, NS8593, acts as a gating modulator of the TRPM7 channel, similar to the situation with KCa2.1–2.3 channels (Strobaek et al., 2006; Jenkins et al., 2011). These findings suggest that genetically unrelated TRPM7 and KCa2.1–2.3 channels harbour a similar drug-binding site functionally linked to divalent cation-dependent gating of the channels. Accordingly, existing drugs targeting KCa2.1–2.3 channels may be applied to test their potential towards TRPM7.
Methods
Compounds
Alinidine, 2-aminobenzimidazole (ABI), 4-aminopyridine, butanedione monoxime, benzimidazol-1-methylethyl-benzimidazole (BMB), CBIQ, 2-chlorobenzimidazole (CBI), clotrimazole, cibenzoline, dequalinium, diazoxide, disopyramide, DPO-1, glipizide, glybenclamide, 5-hydroxydecanoic acid, IMID-4F, nimodipin, mephetyl tetrazole, minoxidil, phentolamine, pinacidil, PNU-37883A, pregnenolone sulfate (PS), propafenone, rutaecarpine, sematilide, psora-4, paxilline, NS1619, NS8593, quinine, quinidine and 1,2,3,4-tetrahydro-1-naphthylamine (THT) were purchased from Sigma-Aldrich (Taufkirchen, Germany). Synthetic apamin, CyPPA, icilin, NS309, SKA31, TRAM 34, UCL 1684 and tetrodotoxin (TTX) were procured from Tocris Bioscience (Bristol, UK). Synthetic tamapin was from Alomone Labs (Jerusalem, Israel). The molecular target nomenclature was according to Alexander et al. (2011).
Molecular biology, cell culture and immunofluorescence staining
Mouse TRPM7, in pIRES-eGFP vector (Clontech Laboratories Mountain View, CA, USA), was generated as described previously (Chubanov et al., 2004; 2007; Mederos y Schnitzler et al., 2008). Mouse TRPM3 cDNA (JF706722) was cloned from kidney mRNA by an RT-PCR approach and inserted into the pcDNA3.1-TOPO vector (Invitrogen, Darmstadt, Germany).
HEK 293 cells were maintained at 37°C and 5% CO2 in Earl's minimal essential medium supplemented with 10% fetal calf serum, 100 µg·mL−1 streptomycin and 100 U·mL−1 penicillin (Invitrogen). Cells were transiently transfected using the Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's instructions.
A stable HEK 293 cell line expressing mouse TRPM7 was generated using an ecdysone-inducible expression system (Invitrogen) as reported previously (Chubanov et al., 2007). The cell line was maintained at 37°C and 5% CO2 in Dulbecco's modified Earl's medium (DMEM, Invitrogen) supplemented with 10% fetal calf serum, 100 µg·mL−1 streptomycin and 100 U·mL−1 penicillin, 400 µg·mL−1 hygromycin and 400 µg·mL−1 zeocin (Invitrogen). To induce the expression of recombinant mouse TRPM7, 10 µM ponasterone A (Invitrogen) was added to the cell culture medium.
A stable HEK 293 cell line expressing human TRPM7 was obtained from Carsten Schmitz (Schmitz et al., 2003). The cell line was maintained at 37°C and 5% CO2 in DMEM (Invitrogen) supplemented with 10% fetal calf serum, 100 µg·mL−1 streptomycin and 100 U·mL−1 penicillin, 400 µg·mL−1 zeocin and 10 µg·mL−1 blasticidin (Invitrogen). To confirm tetracyclin-dependent expression of recombinant TRPM7 protein, cells were examined by immunofluorescence microscopy using a TRPM7-specific antibody as described previously (Chubanov et al., 2007).
Smooth muscle cells were enzymatically isolated from cerebellar and basilar arteries of C57Bl/J mice as described previously (Dietrich et al., 2005) and were used immediately for patch-clamp experiments. Primary human ventricular cardiomyocytes were purchased (Promocell, Heidelberg, Germany) and cultured according to the manufacturer's instructions. The experiments with cardiomyocytes were performed with the fourth passage of cells.
Primary podocytes were isolated from the kidneys of 4–8-week-old C57Bl/J mice using a conventional sieving approach (Schlondorff, 1990), with some modifications. Kidneys (four organs per batch) were mechanically disrupted in 200 mL Ham's F12 medium containing 10% FCS and 100 U·mL−1 penicillin (Invitrogen) using a set of steel sieves with a pore size of 100, 75, 50 and 36 µm (Retsch, Haan, Germany) coated by Ham's F12 medium containing 50% FCS (Invitrogen) at room temperature. Freshly isolated cells from the final fraction were centrifuged for 5 min at 200×g and cultured in Ham's F12 medium supplemented with 10% FCS, 2 mM glutamine, 100 U·mL−1 penicillin, 100 µg·mL−1 streptomycin, 100 µg·mL−1 normocin, 5 µg·mL−1 transferrin, 5 ng·mL−1 natriumselenite, 100 nM hydrocortisone and 5 µg·mL−1 recombinant human insulin (Sigma, Taufkirchen, Germany). Enrichment of podocytes was over 95% as assessed by cell morphology and staining with an anti-podocin antibody (Sigma). Primary podocytes were analysed after the second cell passage.
Imaging of living cells, wound healing and viability assay
Wild-type or tetracycline-inducible HEK 293 cells were grown in 35 mm dishes (1 × 104 cells per dish) in the absence or presence of NS8593 for 24–36 h as indicated in the figure legends. Images from living cells were acquired using an Axiovert 40 CFL inverted microscope equipped with an LD-A-plan 20×/0.30PhI objective and an AxioCam ICc 1 CCD camera (Carl Zeiss, Göttingen, Germany). To evaluate effects of NS8593 on cellular viability, cells were washed twice with PBS and incubated at 37°C and 5% CO2 in 1 mL of 0.05% trypsin/EDTA solution (Invitrogen). The incubation was terminated by addition of 1 mL of the corresponding cell culture medium. Cells were vigorously resuspended and counted using a Neubauer chamber.
For the wound healing assay, confluent wild-type HEK 293 cells grown in 35 mm dishes were scratched by a yellow pipette tip, washed three times with PBS and supplemented with fresh culture medium with or without NS8593. The wound areas (∼600 µm) were imaged immediately or after 24 h as described above. To quantify the effect of NS8593 on cell motility, the ImageJ software (http://rsbweb.nih.gov/ij/index.html) was used to outline the wound area and calculate the percentage of closure.
Aequorin-based [Ca2+]i measurements
In initial screening experiments, the HEK 293 cell line stably expressing mouse TRPM7 was used. Cells cultured in 100 mm dishes were transfected with 2 µg·per dish of plasmid DNAs containing a pG5A construct encoding enhanced green fluorescent protein fused in-frame to Aequorea victoria apo-aequorin (Baubet et al., 2000). Four hours after transfection, the cell culture medium was replaced by fresh medium containing 10 µM ponasterone A. Then 24 h after induction, cells were washed twice with PBS and incubated with 0.05% trypsin, 1 mM EDTA in PBS for 1 min at room temperature. Cell suspensions were centrifuged twice at 600×g for 3 min and resuspended in Mg2+-free HEPES-buffered saline (HBS; Mg2+-free HBS: 140 mM NaCl, 6 mM KCl, 1 mM CaCl2, 10 mM HEPES, 5 mM glucose and 0.1% BSA, pH 7.4). For reconstitution of aequorin, cell suspensions were incubated with 5 µM coelenterazin (Biaffin GmbH, Kassel, Germany) in Mg2+-free HBS for 30 min at room temperature. Cells were washed twice by centrifugation at 200×g for 3 min followed by resuspension of the pellet in Mg2+-free HBS and placed in 96-well plates (1 × 105 cells per well). Luminescence was detected using a FLUOstar OPTIMA microplate reader at 37°C (BMG LABTECH GmbH, Ortenberg, Germany). To monitor TRPM7-mediated Ca2+ influx, extracellular Ca2+ was increased by injection of Mg2+-free HBS containing 5 mM CaCl2 (final concentration). Experiments were terminated by lysing cells with 0.1% (v v-1) Triton X-100 in HBS to record total bioluminescence. Bioluminescence rates (counts·s−1) were analysed at 1 s intervals and calibrated as [Ca2+]i values using the following equation:
where k represents the rate of aequorin consumption (i.e. counts·s−1 divided by total number of counts).
Determination of Ca2+ and Ba2+ influx in HEK 293 cells with Fura-2
HEK 293 cells grown on 25 mm glass coverslips in 35 mm dishes were transiently transfected with 2 µg per dish mTRPM7 cDNA in pIRES2-eGFP vector. Ba2+ entry was measured 18–24 h after transfection. Cells were loaded with 5 µM fura-2 acetoxymethyl ester (Sigma) in HBS (140 mM NaCl, 6 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 10 mM HEPES, 5 mM glucose, 0.1% BSA, pH 7.4) at room temperature for 45 min. After an additional 10 min incubation in HBS, coverslips were placed in a perfusion chamber, washed three times by Ca2+ and Mg2+ free HBS (140 mM NaCl, 6 mM KCl, 10 mM HEPES, 5 mM glucose, pH 7.4) and immediately used for measurements at room temperature. Fura-2 fluorescence was recorded from eGFP-positive cells at an 340/380 nm excitation wavelength with 1 s intervals using a Polychrome V monochromator (TILL Photonics, Gräfelfing, Germany) and 14-Bit EMCCD; XON3 885 Andor camera (Andor Technology, South Windsor, CT, USA) coupled to an inverted IX71 microscope equipped with an UApo 340 40×/1.35 oil objective (Olympus, Hamburg, Germany).
Electrophysiological techniques
HEK 293 cells grown in 35 mm dishes to about 70% confluence were transfected with mouse TRPM7 cDNA in pIRES2-eGFP vector (Clontech Laboratories) (1–2 µg per dish) 20–30 h before analysis. Whole-cell patch-clamp recordings were carried out at room temperature (22°C).
Unless stated otherwise, cells were superfused with a standard physiological solution: 140 mM NaCl, 5 mM CsCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM glucose, 10 mM HEPES, pH 7.4 with NaOH. The Cs+-based external bath solution contained 130 mM CsCl, 50 mM mannitol, 10 mM HEPES, pH 7.4 with CsOH. In experiments with primary human ventricular cardiomyocytes, the external solution contained 10 µM TTX und 10 µM nimodipin in order to fully inhibit voltage-gated Na+ and Ca2+ currents, respectively. Whole-cell patch-clamp recordings were performed with an EPC10 patch-clamp amplifier (HEKA, Lambrecht, Germany) using the PatchMaster v2x52 software (HEKA). Patch pipettes were made of borosilicate glass (Science Products, Hofheim, Germany) and had resistances between 1.2 to 2.3 MΩ when filled with the standard Mg2+ free intracellular solution: 130 mM CsCl, 0.635 mM CaCl2[5.5 nM calculated free [Ca2+] calculated with CaBuf programme (http://ftp.cc.kuleuven.ac.be/pub/droogmans/cabuf.zip)], 10 mM BAPTA, 1 mM HEDTA, 10 mM HEPES, pH 7.2. The Mg2+-containing pipette solution was adjusted to 300 µM calculated free [Mg2+]. The liquid junction potential was +5.1 or +4.7 mV, respectively, and corrected by the PatchMaster software. Series resistance compensation of 80% was used to reduce voltage errors in all experiments. Data were acquired at a frequency of 5 kHz after filtering at 1.67 kHz. The osmolality of all solutions was controlled by the vapour osmometer Vapro 5520 (Wescor Inc., Logan, UT, USA) and was 298 ± 3 mOsm·kg−1.
To determine IC50 values, data were fitted with the Hill equation:
with E being the effect/current at a given concentration c of activator/inhibitor, Emax the maximally achievable effect, IC50 the half-maximal concentration and n the Hill factor, being negative in the case of inhibitory action.
For cell-attached recordings of TRPM7 currents, HEK 293 cells were transfected with TRPM7 in pIRES-eGFP for 24 h. Prior to recording, cells were perfused 3–20 min with bath solution containing 140 mM NaCl, 5 mM CsCl, 1 mM MgCl2, 2 mM CaCl2 10 mM HEPES (pH 7.4), 10 mM glucose with or without 10 µM NS8593. Continuous gap-free cell-attached recordings at pipette potentials of +60 mV (corresponding to −60 mV transmembrane potential) were performed for 1.5 min after gigaseal formation using ∼8 MΩ borosilicale pipettes filled with a divalent cation free solution containing 145 mM CsCl, 5 mM NMDG-HEPES (pH 7.4) and 200 µM Cs-HEDTA. A recording was considered positive when at least 1 burst of TRPM7 activity could be observed.
Functional characterization of TRPM3 was carried out in HBS (140 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM glucose, 10 mM HEPES, pH 7.4). The pipette solution contained 120 mM CsCl, 20 mM NaCl, 1 mM MgCl2, 2 mM EGTA and 10 mM HEPES, pH 7.4. Cells were activated by 10 µM PS. The extent of inhibition caused by NS8593 was quantified by interpolation of currents at +100 mV before the addition of the inhibitor and after complete wash-out.
Statistical analysis
Data are presented as means ± SEM. Unless stated otherwise, data were compared by Student's unpaired t-test. Significance was accepted at P≤ 0.05.
Results
Modulators of KCa2.1–2.3 (SK) channels inhibit TRPM7 activity
In light of the functional and structural similarity between pore-forming segments of TRPM7 and tetrameric K+ channels (Chubanov et al., 2007; Mederos y Schnitzler et al., 2008), we wondered whether known modulators of K+ channels would also affect TRPM7. To this end, we employed a ‘chemical genomics’ approach by investigating a subset of small compounds that have been shown to target distinct types of K+ channels, that is, KIR, KV, KCa2.1–2.3, KCa1.1 (BK) and intermediate conductance K+ (IK or KCa3.1) channels (Figure 1A). In these experiments, we took advantage of a recently developed bioluminescent assay that allows monitoring of Ca2+ influx mediated by recombinant TRPM7 stably expressed in HEK 293 cells under the control of a ponasteron A-inducible promoter sequence (Chubanov et al., 2007). As shown in Figure 1B, in the absence of Mg2+ in the external bath solution, a one-step elevation of the external Ca2+ level ([Ca2+]e) from 1 to 5 mM elicited a fast and sustained rise of intracellular Ca2+ in stimulated HEK 293 cells. Intracellular Ca2+ levels ([Ca2+]i) were not increased in unstimulated cells or if external Ca2+ was constantly maintained at 1 mM (data not shown). TRPM7-mediated Ca2+ entry was abolished when 5 mM Ca2+ was co-applied with 10 mM Mg2+, due to Mg2+-dependent channel block (Nadler et al., 2001). Furthermore, we observed that a select array of compounds targeting KCa2.1–2.3 channels (CyPPA, Dequalinium, NS8593, Quinine, and UCL 1684) inhibited TRPM7 to various degrees when applied externally at 30 µM (Figure 1B, C). In contrast, numerous modulators of KIR, KV, KCa1.1 and KCa3.1 channels (listed in Figure 1A) had no effect on TRPM7-mediated Ca2+ influx (data not shown). Since the sensitivity of TRPM7 to only one group of modulators was unexpected, we extended our screen to compounds affecting more distantly related channels, such as epithelial Na+ (ENaC) channels (amiloride, triamterene), voltage-gated Na+ (NaV) channels (BIA 2–093, disopyramide, lidocaine, mexiletine, phenytoin, prilocaine, procainamide, procaine, R(-)-Me5, tetracaine, tocainide) and voltage-gated Ca2+ (CaV) channels (amiodarone, bepridil, cilnidipine, cinnarizine, diltiazem, ethosuximide, felodipine, gabapentin, FPL 64176, lercanidipine, nemadipine-A, nifedipine, nimodipine, nitrendipine, methoxyverapamil, plunarizine, SKF 96365, verapamil). We found that TRPM7 is insensitive to these compounds (30 µM) (data not shown). Thus, beyond inhibitors of KCa2.1–2.3 channels, TRPM7 displays a rather low cross-sensitivity to modulators targeting different channel groups.
NS8593 directly blocks the TRPM7 channel
In order to investigate if KCa2.1–2.3 inhibitors had direct effects on TRPM7 currents, we employed the patch-clamp technique. Figure 2 illustrates whole-cell currents measured in HEK 293 cells transiently transfected with TRPM7 cDNA. Removal of internal Mg2+ using a divalent-free pipette solution induced large currents with a current–voltage relationship, reflecting the typical signature of TRPM7: steep outward rectification, a reversal potential of about 0 mV, minute inward currents with divalent cations in the bath solution (Figure 2A, B). After currents had activated to saturation, we perfused cells with either 1 or 30 µM NS8593. We found that 1 µM NS8593 effectively reduced TRPM7 currents (Figure 2A). Of note, the suppression of TRPM7 currents was fully reversible and reproducible. The current–voltage (I–V) relationship of TRPM7 currents in the presence of 1 µM NS8593 was indistinguishable from I–V characteristics of TRPM7 in untreated cells (Figure 2A), indicating that under the experimental conditions chosen, the inhibitory effect of NS8593 does not appear to be voltage-dependent. This notion was further supported by recordings of voltage ramps up to 200 mV, thus ruling out substantial voltage-dependence of the NS8593 block (Supporting Information Figure S1, Appendix S1 and Appendix S2). In line with the Ca2+ imaging experiments, we observed that 30 µM NS8593 attenuated TRPM7 whole-cell currents in transfected HEK 293 cells (Figure 2B).
Figure 2.
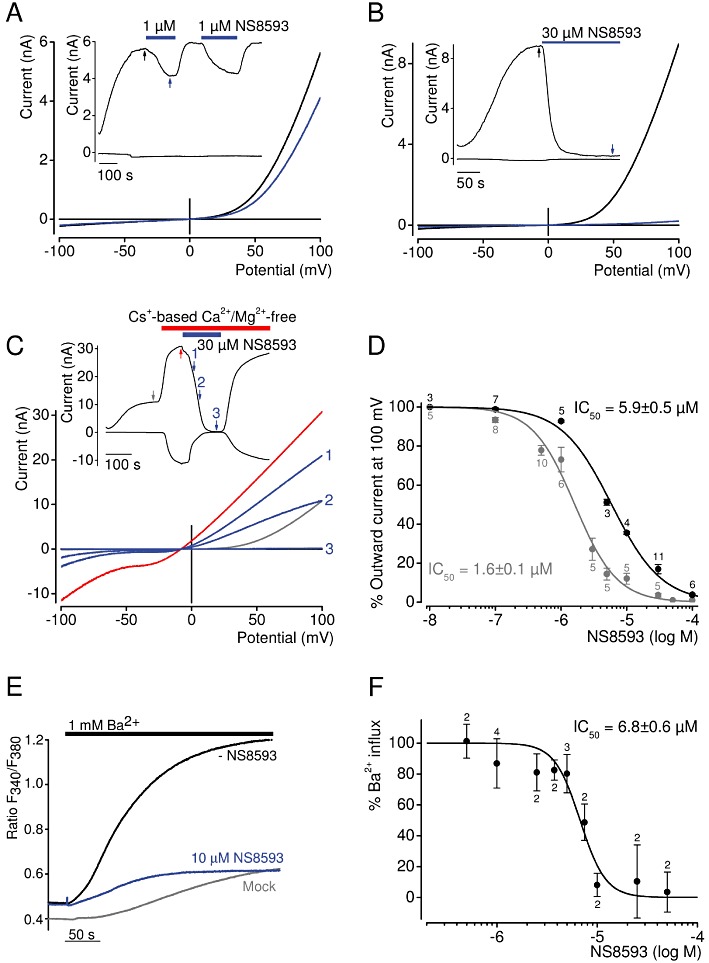
Inhibition of TRPM7 channel by NS8593. (A–B) Whole-cell TRPM7 currents measured in HEK 293 cells transiently expressing TRPM7. Representative current–voltage (I–V) relationships of TRPM7 currents were acquired before and after external application of 1 µM (A) or 30 µM (B) NS8593 as indicated by arrows in the corresponding currents over time recordings (shown in inserts) at −100 and 100 mV. (C) Representative traces of whole-cell currents measured as in (A–B) except that 30 µM NS8593 were applied when cells were perfused with divalent cation-free bath solution. (D) Concentration-dependent suppression of TRPM7 currents measured with internal solutions containing 0 or 300 µM Mg2+. Numbers above dots indicate the number of cells measured. (E–F) The effect of NS8593 on Ba2+ influx in TRPM7-transfected HEK 293 cells. (E) Representative measurements of fura-2 fluorescence (ratio F340/380) in cells incubated in divalent cation-free external bath solution followed by addition of 1 mM Ba2+ without or with 10 µM NS8593. Traces obtained with mock-transfected cells are shown. (F) Concentration-dependent effect of NS8593 on TRPM7-mediated Ba2+ entry. Measurements were performed as in (E), and Δ ratio F340/380 values (calculated from datasets acquired at 25 and 250 s) were used to evaluate the inhibitory effect of NS8593. Numbers above dots indicate independent measurements.
External divalent cations (Mg2+ and Ca2+) are permeant blockers of TRPM7 resulting in very small inward currents at physiological membrane potentials carried by divalent cations. However, in the presence of divalent cation-free external solutions, TRPM7 is well permeable to monovalent cations such as Na+ and Cs+, giving rise to large inward currents. Consequently, we asked whether NS8593 would be equally effective in blocking such inward currents. However, our measurements performed in Na+-based external saline revealed that initial monovalent cation currents exhibited a fast run-down, suggesting that under these experimental conditions Na+ behaves as a blocker of TRPM7 similar to its effect on the genetically related TRPM3 channel (Oberwinkler et al., 2005) (Supporting Information Figure S2). By contrast, whole-cell recordings in the presence of Cs+-based external bath solutions yielded sustained inward and outward TRPM7 currents, allowing for a reliable analysis of NS8593 effects (Figure 2C). We found that 30 µM NS8593 completely suppressed both the inward and outward component of monovalent TRPM7 currents. Analogous to recordings with divalents in the bath solution (Figure 2A), NS8593 had no influence on the shape of the I–V relationship of Cs+ currents (Figure 2C), supporting the notion that NS8593 acts by targeting the gating process of the channel rather than the permeation properties of the channel pore.
Next, we determined the concentration-dependence of the NS8593 effect under standard experimental conditions (i.e. Mg2+-free internal solutions) (Figure 2D). Fitting of experimental data with the Hill equation resulted in an IC50 value of 1.6 ± 0.1 µM. Previously, NS8593 was described as a ‘negative gating modulator’ of KCa2.1–2.3 channels, because channel inhibition was prominent at low internal Ca2+ concentrations and undetectable at 30 µM [Ca2+]i (Jenkins et al., 2011). Therefore, we tested whether internal Mg2+ would effect the sensitivity of TRPM7 to NS8593. Thus, we investigated the concentration-dependent suppression of TRPM7 activity by NS8593 in the presence of 300 µM internal Mg2+, a subphysiological concentration that causes approximately half-maximal inhibition of TRPM7 currents (Mederos y Schnitzler et al., 2008). Under these conditions, we observed a 3.7-fold rightward shift of the IC50 value (5.9 ± 0.5 µM), suggesting that NS8593 has a lower apparent affinity to the inactive (closed) channel; that is, the compound interferes with Mg2+-dependent gating of the channel.
To investigate the inhibitory effect of NS8592 under more physiological conditions, we chose an imaging approach such that [Mg2+]i was not modified by patch pipette solutions. It has previously been shown that TRPM7 is well permeable to Ba2+ (Monteilh-Zoller et al., 2003; Mederos y Schnitzler et al., 2008). Therefore, we measured Ba2+ influx into TRPM7-expressing HEK 293 labelled with the fluorescent dye fura-2 (Figure 2E, F). Addition of 1 mM Ba2+ to the bath resulted in fast and sustained Ba2+ influx (Figure 2E). NS8593 10 µM suppressed Ba2+ entry to levels observed in untransfected HEK 293 cells. Consequently, we systematically monitored concentration-dependent inhibition of TRPM7-mediated Ba2+ entry and determined an IC50 value of 6.5 ± 0.6 µM (Figure 2F), similar to the IC50 value obtained in patch-clamp experiments with 300 µM [Mg2+]i (Figure 2D).
Next, we aimed to evaluate the inhibitory effect of NS8593 on TRPM7 at the single channel level using experimental conditions employed to study properties of endogenous TRPM7-like currents in Jurkat T-lymphocytes and RBL cells (referred to as Mg2+ inhibited currents or MIC) (Kerschbaum and Cahalan, 1999; Braun et al., 2001; Kozak et al., 2002). Accordingly, we performed continuous cell-attached recordings of monovalent cation currents at transmembrane potentials of −60 mV for 1.5 min after gigaseal formation (Figure 3). In TRPM7-transfected HEK 293 cells, we observed characteristic bursts of channel activity strikingly resembling MIC in RBL cells (Braun et al., 2001; Prakriya and Lewis, 2002; Kerschbaum et al., 2003). These bursts were not detectable in mock-transfected HEK 293 cells. Since under these experimental conditions the opening of TRPM7 was infrequent and short, we have compared the burst frequency in patches recorded from control and treated cells (Figure 3). We noted that characteristic single channel activities were not detectable when cells had been pre-exposed to 10 µM NS8593.
Figure 3.
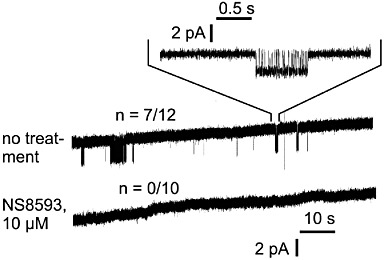
Cell-attached recordings of TRPM7 currents in HEK 293 cells. Before as well as during recording, cells were superfused with normal bath solution or bath solution supplemented with 10 µM NS8593. Inward monovalent currents through recombinant TRPM7 are visible as intermittent deflections from baseline level in 7 out of 12 untreated patches, but in 0 out of 10 patches from NS8593-pretreated cells.
Next, we performed a set of experiments to uncover the binding site of NS8593. It was shown that the kinase domain regulates the sensitivity of TRPM7 channel to [Mg2+]i. Therefore, we investigated whether the kinase domain is required for the NS8593 block of TRPM7 currents. In these experiments, we used two mutant cDNA variants: TRPM7 carrying the point mutation K1646R resulting in complete loss of kinase activity (TRPM7-K1646R) (Schmitz et al., 2003; Matsushita et al., 2005) and TRPM7 containing yellow fluorescent protein (YFP) instead of the kinase domain (residues 1464–1863 of mouse TRPM7, TRPM7-Δkinase-YFP). We found that TRPM7-K1646R and TRPM7-Δkinase-YFP gave rise to characteristic TRPM7 currents that could be suppressed by 10 µM NS8593 (data not shown), indicating that the kinase domain is not required for the action of NS8593 on TRPM7.
It was demonstrated that in the case of KCa2.1–2.3 channels, NS8593 directly interacts with serine and alanine residues located in the pore-forming loop and the S6 helix, implying that NS8593 occupies the inner pore vestibule near the selectivity filter, thus blocking the gate of KCa2.1–2.3 channels (Jenkins et al., 2011). Therefore, we determined whether NS8593 would interact with TRPM7 in a similar fashion. We have recently shown (Mederos y Schnitzler et al., 2008) that the putative pore-forming loop of mouse TRPM7 harbours conserved residues (E1047 and Y1049) essential for the permeability of divalent cations. While the E1047Q mutant was as sensitive to NS8593 as wild-type TRPM7 (data not shown), TRPM7-Y1049P displayed a substantially increased apparent affinity of the channel to NS8593 (Table 2). The Y1049P mutation resulted in an approximately fourfold reduction of IC50 values obtained either with 0 or 300 µM Mg2+ in the patch pipette when compared with wild-type TRPM7 (Table 2). These results suggest that the TRPM7 pore loop is involved in the interaction of TRPM7 and NS8593.
Table 2.
Apparent affinity of wild-type and mutant TRPM7 channels to NS8593
| IC50 (µM) | ||
|---|---|---|
| TRPM7 variants | [Mg2+]I free | 300 µM [Mg2+]i |
| Wild type | 1.6 ± 0.1 | 5.9 ± 0.6 |
| Y1049P | 0.47 ± 0.03 | 1.9 ± 0.2 |
Taken together, we conclude that NS8593 directly and reversibly interacts with the pore-forming segment of activated TRPM7 channels resulting in a complete block.
Effect of modulators of KCa2.1–2.3 channels on TRPM7 currents
Our primary screening experiments suggested that chemically unrelated modulators of KCa2.1–2.3 channels are able to suppress TRPM7-mediated Ca2+ influx, in contrast to numerous modulators of other K+ channel types (Figure 1B). In order to further substantiate these findings, we performed patch-clamp experiments to monitor the effects of commonly used inhibitors of KCa2.1–2.3, KCa3.1 and KCa1.1 channels on TRPM7 currents (Figure 4A, Supporting Information Figure S3). Analogous to experiments with NS8593, we assessed the modulators in the presence (Figure 4A) and absence (Figure 4B) of divalent cations in the external bath solution. These experiments confirmed that external (30 µM) quinine, dequalinium, CyPPA and UCL 1684 are able to suppress TRPM7 currents with different efficacies (Figure 4). We also observed that quinidine (stereoisomer of quinine) and clotrimazole exerted only modest inhibitory effects (5.6% and 7.5%, respectively) on TRPM7 currents. Interestingly, a peptide toxin of the Indian red scorpion, tamapin, that specifically blocks the pore of KCa2.1–2.3 channels by binding to an external site of the channel subunits (Pedarzani et al., 2002), elicited a modest, but reproducible suppression of TRPM7 currents. In contrast, apamin, the bee venom peptide toxin, failed to inhibit TRPM7 activity, neither did paxilline and TRAM 34 (Figure 4A), known modulators of KCa1.1 and KCa3.1 channels, respectively.
Figure 4.
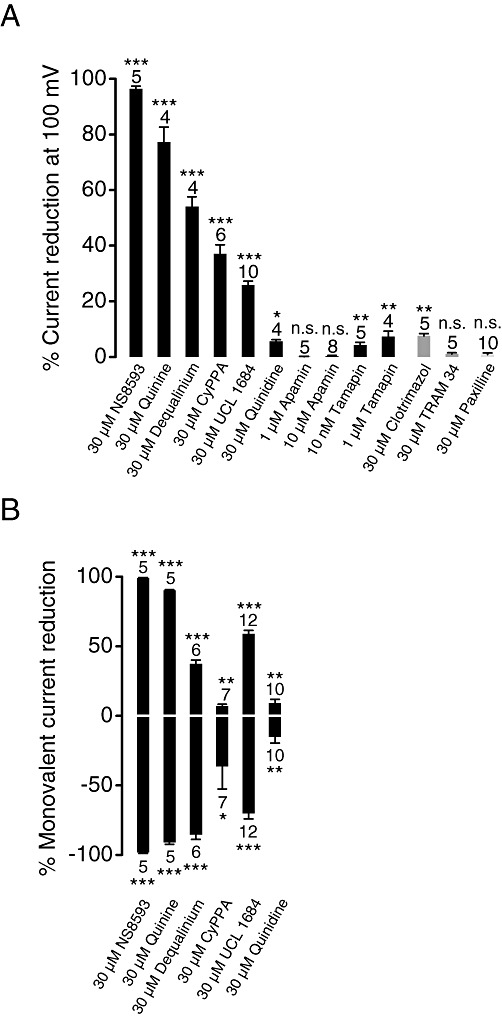
Assessment of effects of the KCa2.1–2.3 (SK) inhibitors on TRPM7 currents. (A) Whole-cell TRPM7 currents were measured in the presence of divalent cations in the same bath solutions as in Figure 2A. Inhibitory effects (%) were quantified by means of current amplitudes acquired at +100 mV before and after application of the indicated compounds. Effects of the compounds acting on KCa3.1 (IK) and KCa1.1 (BK) channels are illustrated. Numbers above columns indicate the number of cells measured. (B) Outward (upper panel) and inward (lower panel) monovalent cation currents were measured at +100 and −100 mV, respectively, in divalent cation-free bath solutions as in Figure 2C. ***P≤ 0.001; **P≤ 0.01; *P≤ 0.05; n.s., not significantly different (t-test).
Assessment of NS8593-related compounds
Structure–activity relationships (SAR) provide critical information for drug improvement. NS8593 contains benzimidazole and naphthylamine groups (Figure 5A). In order to get initial insights into SAR of NS8593, we tested several small compounds representing distinct chemical moieties of NS8593, such as 1,2,3,4-tetrahydro-1-naphthylamine (THN) and ABI. Patch-clamp experiments revealed that 30 µM ABI significantly (21%) suppressed TRPM7 currents, while THN had no influence on channel activity (Figure 5B), indicating that the aminobenzimidazole moiety is critical for the activity of NS8593. This conclusion was further supported by the observation that 30 and 100 µM BMB, containing two covalently linked aminobenzimidazole groups, inhibited TRPM7 (Figure 5B). Furthermore, we found that CBI is as effective as ABI, suggesting that an amino group at position 2 of benzimidazole is not essential for the sensitivity of TRPM7 to benzimidazoles.
Figure 5.
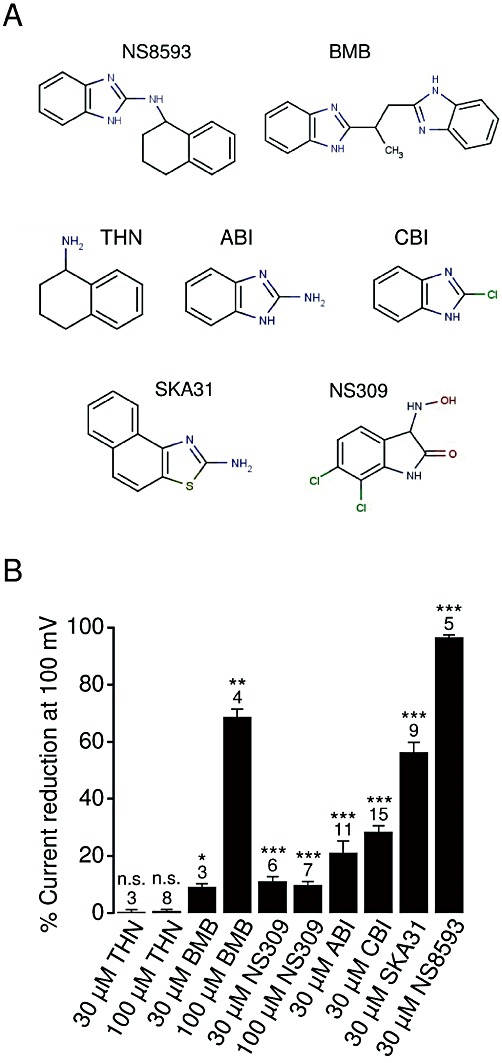
Characterization of NS8593-related compounds. (A) Chemical structures of the compounds examined. (B) Whole-cell TRPM7 currents were measured in the presence of divalent cations in bath solutions as in Figure 2A. Inhibitory effects (%) were quantified by means of current amplitudes acquired at +100 mV before and after the application of the indicated compounds. Numbers above columns indicate the number of cells measured. ***P≤ 0.001; **P≤ 0.01; *P≤ 0.05; n.s., not significantly different (t-test).
The benzimidazole group is present in several compounds with known biological activity such as ABT 724 (dopamine D4 receptor antagonist), clemizole (histamine H1 receptor antagonist), DMAT (casein kinase 2 inhibitor 2), d-ribofuranosylbenzimidazole (inhibitor of RNA synthesis), droperidol (dopamine D2 receptor antagonist), IRAK-1/4 Inhibitor I (inhibitor of interleukin-1 receptor-associated kinases 1/4), rabenzazole (fungicide), Ro 90-7501 (inhibitor of amyloid β42 fibril formation) and TBBz (inhibitor of casein kinase 2). To test the potential cross-reactivity of these molecules, we employed the bioluminescent assay of TRPM7 channel activity. We found no detectable effects of the latter compounds at 30 µM on TRPM7 (data not shown) and, therefore, excluded them from further biophysical analysis. We conclude that the benzimidazole group of NS8593 is primarily required for targeting of TRPM7 channels. Nevertheless, the overall structure of benzimidazole-derived compounds also appears to play a role in the inhibitory effect on TRPM7.
We noted that the benzimidazole group of NS8593 resembles known positive modulators of KCa2.1–2.3 channels such as NS309 and SKA31 (Figure 5A). Therefore, we tested whether these compounds (30 µM) were able to positively modulate TRPM7. However, we found that SKA31 suppressed TRPM7 currents (56%), whereas NS309 caused only a minor reduction (11%) of channel activity (Figure 5B).
Effect of NS8593 on other TRP channels
We determined whether NS8593 can discriminate between TRPM7 and other genetically related TRP channels. TRPM7 and TRPM6 are paralogues sharing ∼64% amino acid sequence similarity within their channel segments (Chubanov et al., 2004). However, we were unable to monitor the effect of NS8593 on TRPM6, since our previous efforts (Chubanov et al., 2004; 2007) to express recombinant functional TRPM6 channels either in mammalian cells or Xenopus oocytes failed and attempts to measure endogenous TRPM6 currents were not successful so far (Li et al., 2006; Ryazanova et al., 2010).
TRPM3, the closest non-kinase-bearing relative of TRPM7 within the TRPM family, is a constitutively active cation channel highly permeable to Ca2+ and can further be potentiated by sulfated steroids like PS (Wagner et al., 2008). To this end, we performed whole-cell measurements of PS-induced currents in HEK 293 cells transiently transfected with TRPM3 cDNA (Figure 6). In line with previous publications (Wagner et al., 2008), external application of 10 µM PS induced large cation currents. At ramp potentials of +100 mV, coapplication of NS8593 in the range of 10–100 µM resulted in suppression of PS-induced TRPM3 currents with an IC50 value of 26.7 ± 3.9 µM. During voltage ramps from −100 to +200 mV, we observed an increase in current suppression at highly positive potentials (Supporting Information Figure S1B). This behaviour was only rudimentarily seen in analogous experiments with TRPM7. To assess whether this phenomenon would reduce the ability of the compound to discriminate between the two related ion channels at more physiological membrane potentials, we performed fluorescence-based Ca2+ entry experiments with fura-2 loaded cells (Supporting Information Figure S4A). NS8593 at 10 µM has no measurable influence on PS-induced Ca2+ influx in TRPM3-expressing cells. We conclude that moderate concentrations of NS8593 are selective for TRPM7.
Figure 6.

Effect of NS8593 on PS-induced TRPM3 currents. (A, B) Representative traces of whole-cell TRPM3 currents measured at −100 and +100 mV over time induced by extracellular application of 10 µM PS. NS8593 10 µM (A) or 100 µM (B) were coapplied with PS as indicated. (C) Current–voltage relationships of PS-induced TRPM3 currents acquired from measurements before and after application of 100 µM NS8593 as indicated by arrows in (B). (D) Concentration-dependent suppression of PS-induced TRPM3 currents by NS8593. Numbers above symbols indicate the number of cells measured.
Next, we investigated the effect of NS8593 on TRPM channels more distantly related to TRPM7 in terms of primary sequence similarity and function. TRPM8 is Ca2+ -permeable cation channel activated by cold temperatures and several agonists such as menthol and icilin (Peier et al., 2002). TRPM2 is activated by free intracellular ADP-ribose in synergy with free intracellular Ca2+ (Perraud et al., 2001; Starkus et al., 2007). TRPM2 also acts as a sensor for reactive oxygen species including H2O2 (Hara et al., 2002). Using a Ca2+ imaging approach, we tested whether 10 µM NS8593 would negatively modulate H2O2-activated TRPM2 and icilin-activated TRPM8 channels (Supporting Information Figure S4B, C). These experiments demonstrated that NS8593 does not suppress TRPM8 and TRPM2 channel activity. TRPM5 is a monovalent-selective cation channel, directly gated by intracellular calcium that rises upon stimulation of GPCRs linked to PLC (Hofmann et al., 2003). We observed that 10 µM NS8593 has no effect on TRPM5 currents (Supporting Information Figure S5A). TRPC6 is a Ca2+-permeable cation channel directly gated by diacylglycerol produced by PLC after activation of GPCR such as histamine H1 receptors (Hofmann et al., 1999). We found that TRPC6 currents induced by stimulation of H1 receptors are insensitive to externally applied 10 µM NS8593 (Supporting Information Figure S5B). Finally, we studied the effects of NS8593 on TRPV1 and TRPA1, which are receptive to a very broad range of natural and synthetic compounds (Vriens et al., 2008) (Supporting Information Figure S6). We observed that Ca2+ entry in HEK 293 cells mediated by capsaicin-induced TRPV1 as well as allyl isothiocyanate (AITC)-stimulated TRPA1 is not affected by 10–30 µM NS8593. Thus, NS8593 specifically targets the TRPM7 channel among TRP channels representing distinct genetic and functional branches of TRP channels.
Assessment of long-term effects of NS8593
So far, we had studied short-term pharmacological properties of NS8593. In order to clarify if NS8593 was a useful suppressor of TRPM7 function in a cell biological context, we performed additional experiments addressing long-term effects of NS8593. Overexpression of human TRPM7 in a tetracycline-inducible HEK 293 cell line results in detachment and consequently cell death (Nadler et al., 2001). Therefore, we wondered whether NS8593 would be able to forestall this cellular phenotype (Figure 7). We induced TRPM7 expression in a HEK 293 cell line with tetracycline and confirmed channel expression using a TRPM7-specific antibody (Figure 7A). In line with previous reports, we found that after 18 h of induction with 1 µM tetracycline, TRPM7-expressing cells developed a rounded morphology (Figure 7A), while patch-clamp measurements revealed at the same time that stimulated cells exhibited very large TRPM7 currents (Figure 7B). Most of the cells stimulated for 36 h lost adherence to the substratum and formed floating cell clusters (Figure 7C, D). Next, we added different concentrations of NS8593 to the culture medium and noted that 5–10 µM NS8593 preserved cell adherence and viability, thus completely reversing the cellular phenotype caused by TRPM7 overexpression (Figure 7C, D). Thus, application of NS8593 for a longer time period allows for the suppression of TRPM7 function without any obvious cell toxicity. Finally, we tested whether other TRPM7 inhibitors, like quinine and UCL 1684 (Figure 4, Supporting Information Figure S3), were able to rescue the phenotype of stimulated HEK 293 cells. We found that the application of both compounds (10 µM) to the cell culture medium prevented detachment of tetracycline-induced cells (Supporting Information Figure S7). Thus, structurally unrelated inhibitors of TRPM7 currents were able to protect HEK 293 cells overexpressing recombinant TRPM7 from resultant morphological changes.
Figure 7.
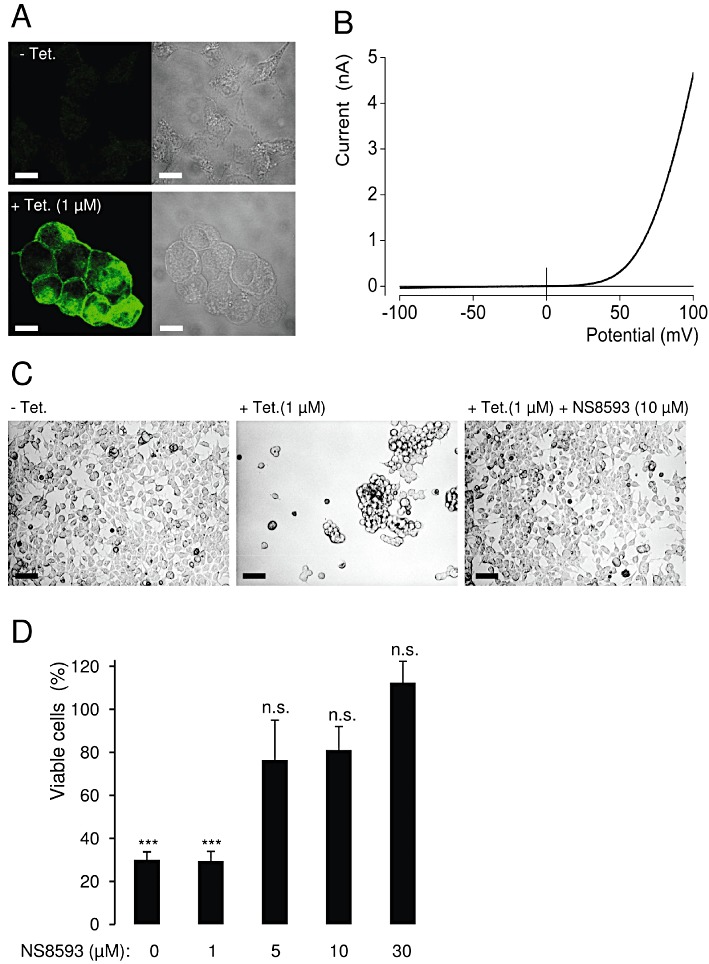
Effect of NS8593 on the viability of HEK 293 cells overexpressing recombinant TRPM7. (A) Tetracyclin(Tet)-induced (1 µM, 18 h) expression of the human TRPM7 protein as assessed by immunofluorescence staining using anti-TRPM7 antibody. Scale bars – 10 µm. (B) Representative I–V relationship of fully developed whole-cell TRPM7 currents measured in stimulated HEK 293 cells (1 µM tetracycline, 18–24 h). (C) Effect of NS8593 (10 µM) on the morphology of tetracycline-induced (1 µM, 36 h) HEK 293 cells. Scale bars – 50 µm. (D) Viability of HEK 293 cells expressing TRPM7 in the presence of different concentrations of NS8593. Experiments were performed as in (C), and the total number of viable cells was compared with that of unstimulated cells (– tetracycline). Data from three independent experiments are shown. ***P≤ 0.001; n.s., not significantly different (t-test).
Targeting of endogenous TRPM7 currents by NS8593
We determined whether NS8593 is able to inhibit endogenous TRPM7 channels. TRPM7 is a ubiquitously expressed protein and, consistently, patch-clamp experiments showed that native TRPM7-like currents are present in all cells examined so far, including HEK 293 cells (Nadler et al., 2001; Chubanov et al., 2007). Figure 8 illustrates the effect of NS8593 on native cation currents with an I–V relationship very similar to that of recombinant TRPM7. Currents were induced by removal of cytosolic Mg2+ as shown in Figure 2. We found that 10 or 30 µM NS8593 effectively suppressed whole-cell currents to background levels measured immediately after establishment of the whole-cell configuration (Figure 8A). We next studied the affect of 10 µM NS8593 on endogenous TRPM7 currents in mouse smooth muscle cells freshly isolated from brain arteries, primary mouse podocytes and primary human ventricular myocytes (Figure 9). TRPM7-like currents were persistently detectable in all cell types examined. We observed that – like in HEK 293 cells – 10 µM NS8593 caused a full and reversible suppression of native TRPM7-like currents (Figure 9). Thus, NS8593 appears to be equally efficient in targeting recombinant and native TRPM7 channels.
Figure 8.
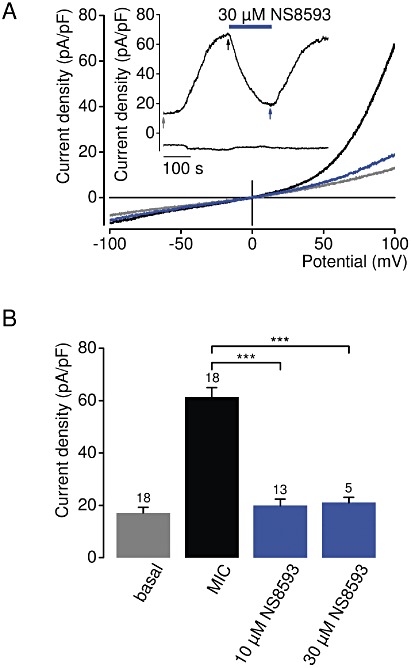
Inhibition of native TRPM7-like currents in HEK 293 cells by NS8593. (A) Representative current density–voltage relationships of native whole-cell currents (MIC) induced by depletion of internal Mg2+ in HEK 293 cells. Measurements were performed as in Figure 2A. The insert shows the corresponding currents over time measured at −100 and +100 mV. Current density–voltage relationships were acquired at time points as indicated by arrows. (B) Analysis of current density amplitudes at +100 mV acquired before and after current induction or after application of NS8593 as indicated by arrows in (A). Numbers above columns indicate the number of cells measured; ***P≤ 0.001 (t-test).
Figure 9.
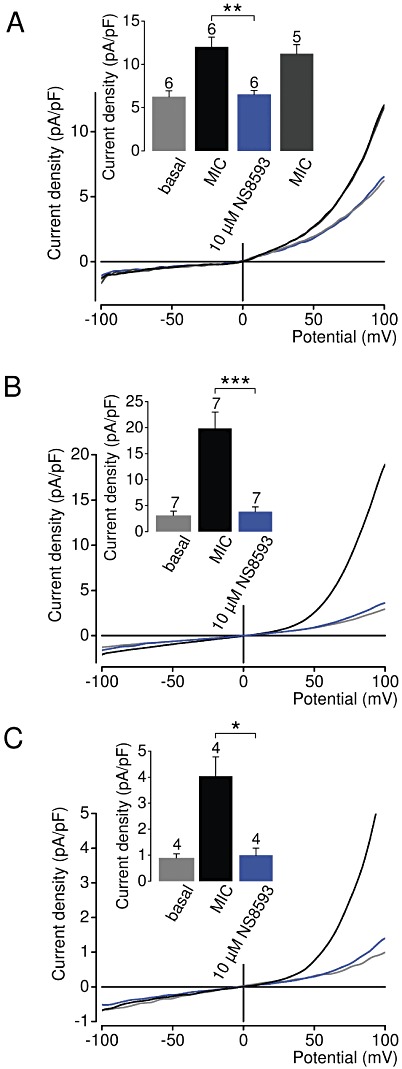
Inhibition of native TRPM7-like currents in vascular smooth muscle cells, podocytes and ventricular myocytes by NS8593. Representative current density–voltage relationships of native whole-cell currents (MIC) induced by depletion of internal Mg2+ in mouse smooth muscle cells freshly isolated from brain arteries (A), primary mouse podocytes (B) and primary human ventricular myocytes (C). Measurements were performed as in Figure 2A. The insert shows corresponding current densities at +100 mV acquired before and during current induction or in the presence of NS8593. The insert in (A) shows recovered currents, if pretreated cells were reperfused with NS8593-free solution. Numbers above columns indicate the number of cells measured; ***P≤ 0.001; **P≤ 0.01; *P≤ 0.05 (t-test).
Subsequently, we determined whether pharmacological inhibition of the native TRPM7 protein by NS8593 would affect TRPM7-dependent cellular processes such as cell motility (Clark et al., 2006; Su et al., 2006; 2011) and proliferation (Nadler et al., 2001; Schmitz et al., 2003; Sahni and Scharenberg, 2008), which are affected by a depletion of TRPM7 or by RNAi approaches. Specifically, we investigated if NS8593 would influence growth and motility of HEK 293 cells displaying the highest level of native TRPM7 currents (compare Figure 8 and Figure 9). Figure 10A depicts HEK 293 cells cultured for 24 h without or with 10–30 µM NS8593. Under control conditions, we observed groups of cells with characteristic protrusions as well as numerous single spreading cells. We noted that 10 µM NS8593 suppressed motility of HEK 293 cells, because most treated cells remained in colonies and displayed either shorter protrusions or a round shape. Furthermore, cells cultured in the presence of 30 µM NS8593 often formed very tight round colonies (Figure 10A, insert), which were never observed in control cells. These morphological changes were not found, when 1 µM apamin was added to the culture medium, strongly arguing against an involvement of KCa2.1–2.3 channels (Supporting Information Figure S8). Next we investigated whether benzimidazol derivatives, like BMB (Figure 5B), might suppress the motility of HEK 293 cells as well. Previously, we found that 30 µM BMB elicited only a minor (∼10%) inhibitory effect on TRPM7 currents (Figure 5B). Consistently, application of 10–30 µM BMB to the cell culture medium had no influence on cell morphology (Supporting Information Figure S8), indicating that the effect of NS8593 on HEK 293 cells was due to inactivation of TRPM7 currents rather than KCa2.1–2.3 channels.
Figure 10.

Effects of NS8593 on motility and proliferation of HEK 293 cells. (A) Images of living HEK 293 cells cultured without or with indicated concentrations of NS8593 (24 h). In the presence of 30 µM NS8593, cell colonies frequently displayed a tight and round shape. Insert illustrates at a higher magnification the colony indicated by the red arrow. Scale bars – 30 µm. (B, C) Effect of 30 µM NS8593 on cell motility as assessed by a wound healing assay. (B) Representative images of HEK 293 cells acquired either immediately or 24 h after wounding. Outline of the wound and percentage of the closure area (%) were determined using ImageJ. Scale bars – 200 µm. (C) Data from six independent assays are shown; **P≤ 0.01 (t-test). (D) Viability of HEK 293 cells cultured (48 h) in the presence of indicated concentrations of NS8593. The total number of viable cells was compared with that of untreated cells (100%). Data from three independent experiments are shown, *P≤ 0.05; n.s., not significantly different (t-test).
In order to further evaluate the influence of NS8593 on the motility of HEK 293 cells, we performed a wound healing assay (Figure 10B, C). In line with our previous experiments, wound closure was substantially more efficient in control cells as compared with cells cultured for 24 h in the presence of 30 µM of NS8593 (P≤ 0.01, Figure 10C). Finally, we examined whether NS8593 would influence the proliferation of HEK 293 cells, because numerous studies reported that TRPM7 deficiency profoundly interferes with cell viability (Nadler et al., 2001; Schmitz et al., 2003; Sahni and Scharenberg, 2008; Ryazanova et al., 2010). NS8539 10–30 µM did not significantly reduce the total number of HEK 293 cells cultured for 24 h (data not shown), ruling out the possibility that the reduced motility of NS8593-treated cells (Figure 10A–C) is secondary to cellular toxicity of the drug. However, after 48 h, cell proliferation was reduced (17%, P≤ 0.05) in the presence of 30 µM NS8593 (Figure 10D). Thus, NS8593 has a rather modest influence on the proliferation of HEK 293 cells, which is fully compatible with the aforementioned concept that NS8593 preferentially targets active TRPM7 channels. However, we cannot exclude the possibility that inhibition of both the channel and kinase moieties of TRPM7 would impart a more pronounced effect on the proliferation of HEK 293 cells.
Overall, our cell biological experiments demonstrate that moderate concentrations of NS8593, at which the drug discriminates well amongst TRPM channels and is non-toxic to cells in culture, selectively inhibit native TRPM7 channels and elicit characteristic cellular phenotypes. Consequently, NS8593 may be a promising compound to pharmacologically target TRPM7 in vivo.
Discussion and conclusions
We present the results of a hypothesis-driven ‘chemical genomics’ approach to identify non-toxic inhibitors of the ubiquitously expressed Mg2+-sensitive TRPM7 cation channel. Our data indicate that structurally unrelated modulators of KCa2.1–2.3 channels are able to suppress TRPM7 currents, in stark contrast to numerous compounds targeting other channel types. Thus, the broad array of known KCa2.1–2.3 modulators opens up the possibility for a pharmacological assessment of still poorly understood aspects of TRPM7 function. As a proof-of-concept, we further investigated NS8593. This drug turned out to be specific for TRPM7 when compared with other TRP channels, interfered with Mg2+ dependent activation of TRPM7 most probably due to interaction with the channel pore, and is capable of modulating cell motility.
Identification of new TRPM7 modulators
There is mounting evidence that TRPM channels share several structural and functional characteristics with tetrameric K+ channels. For instance, intrinsic voltage-dependence of temperature-sensitive TRPM8 and TRPM5 was found to be mediated by positively charged residues located in the fourth transmembrane helix, analogous to a voltage sensor of voltage-gated K+ channels (Nilius et al., 2007). Due to a homology of the pore-forming regions of TRPM and K+ channels, ion permeation mechanisms of several TRPM channels have recently been uncovered (Mederos y Schnitzler et al., 2008). Furthermore, the control of TRPM7 channel activity by Mg2+ and, ATP and PIP2 (Nadler et al., 2001; Runnels et al., 2002; Kerschbaum et al., 2003) is also a functional hallmark of several types of K+ channels (Figure 1) (Stanfield et al., 1994; Wible et al., 1994; Taglialatela et al., 1995).
Against this background, we investigated whether known inhibitors and activators of Mg2+-sensitive K+ channels would target TRPM7. We tested a subset of compounds targeting different types of cation channels and identified several organic compounds exhibiting inhibitory effects on TRPM7 activity when applied at 30 µM. The identified inhibitors can be grouped into three different classes according to their chemical structure. Quinine, dequalinium and UCL 1684 contain quinolinium groups; NS8593, SKA31, THN and ABI are benzimidazole derivates; finally, CyPPA represents a third scaffold – this molecule comprises cyclohexyl, dimethylpyrazol and methylpyrimidin groups. Previous SAR studies concentrating on these three scaffolds (Sorensen et al., 2008) yielded dozens of related compounds, which can be used in future to improve the affinity and specificity of TRPM7 inhibitors (reviewed in Weatherall et al., 2010). Interestingly, some of these compounds are known therapeutic agents. For instance, quinine is a plant alkaloid known as the first effective antimalaria drug, which is still employed to treat the disease in certain critical situations
In addition to the identification of new organic modulators of TRPM7, our screening experiments highlight an unexpected relationship between TRPM7 and KCa2.1–2.3 channels. Thus, we observed that TRPM7 is not sensitive to numerous modulators targeting KIR, KV, KCa1.1, KCa3.1, ENaC, NaV, CaV channels, arguing that TRPM7 has a rather low cross-sensitivity to these types of cation channel modulators. By contrast, chemically unrelated inhibitors of KCa2.1–2.3 channels were able to inactivate TRPM7. The most straightforward mechanistic explanation for this finding is that despite the low degree of primary amino acid sequence homology, the channel segments of TRPM7 and KCa2.1–2.3 harbour a similar drug binding site interfering with the gating of these channels. We cannot exclude the possibility that common endogenous metabolites regulate KCa2.1–2.3 and TRPM7 channels, thereby causing cross-sensitivity of KCa2.1–2.3 and TRPM7 to the same synthetic ligands.
NS8593 as a negative gating modulator of TRPM7
The mechanism underlying the Mg2+-dependent regulation of TRPM7 channel activity is still incompletely understood, unlike the Ca2+-dependent gating of KCa2.1–2.3 channels, which occurs via a ‘chemical-coupling’ mechanism. The C-terminus of KCa2.1–2.3 channels is constitutively attached to calmodulins. Interaction of Ca2+ with calmodulin induces a conformational rearrangement of the C-terminus and, subsequently, of the channel gate (Berkefeld et al., 2010). In addition, intracellular Mg2+ negatively regulates KCa2.1–2.3 channels acting via a conserved serine pore loop residue (S359 in rat KCa2.2, Figure 1) (Soh and Park, 2002). It has been shown recently that this serine residue directly interacts with NS8593 (Jenkins et al., 2011). It was postulated that NS8593 acts as negative gating modulator of KCa2.1–2.3 channels, since the inhibitory potential of NS8593 was highly dependent on intracellular Ca2+: inhibition was prominent at low Ca2+ concentrations and abolished at 30 µM Ca2+ (Strobaek et al., 2006).
A similar scenario may also underlie the inhibitory effects of NS8593 on TRPM7. Our experiments revealed that the effect of NS8593 on TRPM7 is modulated by internal Mg2+: the IC50 value of NS8593 was increased by 3.7-fold when the pipette solution was supplemented with 300 µM Mg2+. The inhibition of TRPM7 currents by NS8593 was voltage-independent and equally potent for monovalent and divalent cation currents carried by TRPM7. Furthermore, we were able to detect the inhibitory effect of NS8593 on TRPM7 channels at the single channel level. We also determined a possible NS8593 interaction site within TRPM7. Apparently, the kinase domain of TRPM7 is not required for targeting of TRPM7 by NS8593, since two kinase deficient mutants, TRPM7-K1646R and TRPM7-Δkinase-YFP, were fully suppressed by NS8592. However, we observed that a point mutation in the pore-forming loop (Y1049P) of TRPM7 increased sensitivity of the channel to NS8593 since TRPM7-Y1049P elicited a fourfold smaller IC50 value. Finally, we performed an initial SAR analysis of NS8593, suggesting that the benzimidazole group is required for the pharmacological action of NS8593 on TRPM7. However, TRPM7 was found to be insensitive to a subset of biologically active compounds containing the benzimidazole group, arguing that interaction of TRPM7 with NS8593 is relatively specific.
Taken together, we hypothesize that similar to KCa2.1–2.3 channels NS8593 occupies the inner pore vestibule distally to the predicted selectivity filter of TRPM7. The molecular interaction is most probably mediated by the pore-forming loop of TRPM7 and the imidazole ring of NS8593 and is critically dependent on intracellular Mg2+. A plausible explanation for the observed Mg2+ dependence is that the inactive (closed) TRPM7 channel (high concentration of intracellular Mg2+) is less accessible to NS8593 than the active (open) channel (low intracellular Mg2+).
Recently, Zierler et al. (2011) showed that a natural compound, waixenicin A, inhibits TRPM7-mediated entry of divalent cations (IC50= 12 µM) as assessed by Fura 2. Interestingly, patch-clamp experiments revealed that waixenicin A irreversibly blocks TRPM7 currents (IC50= 7 µM and 16 nM at 0 and 700 µM [Mg2+]i). Thus, unlike other TRPM7 inhibitors (Table 1), NS8593 and waixenicin A act on TRPM7 in a [Mg2+]i-dependent manner, which may be instrumental in differentiating gating mechanisms.
Pharmacological potential of NS8593
TRPM7 plays a crucial role in several pathological conditions. For instance, TRPM7 regulates proliferation and survival of several types of tumour cells (Hanano et al., 2004; Jiang et al., 2007; Kim et al., 2008; Guilbert et al., 2009; Mishra et al., 2009). Furthermore, a role of TRPM7 was postulated in hypertension (Touyz, 2008), neuronal cell death following ischaemic injury (Aarts et al., 2003), multiple sclerosis and Alzheimer's disease (Tseveleki et al., 2010). More recently, up-regulation of TRPM7 function was found to be associated with fibrogenesis leading to atrial fibrillation in the failing heart (Du et al., 2010). These findings highlight TRPM7 as a promising drug target. However, since ubiquitously expressed TRPM7 plays an indispensible role in the cell cycle, an indiscriminate pharmacological inactivation of TRPM7 will most likely be toxic implying that use-dependent TRPM7 blockers would have a better therapeutic perspective. In the present study, we present data to support the notion that the development of this type of organic compounds is feasible. Specifically, we found that NS8593 reversibly inhibits recombinant TRPM7 channels in a Mg2+-dependent manner, allowing for preferential suppression of active TRPM7 channels. Our results indicate that NS8593 at concentrations (10 µM) sufficient to fully suppress TRPM7 activity does not affect other TRP channels, like TRPM2, TRPM3, TRPM5, TRPM8, TRPC6, TRPV1 and TRPA1. We have also shown that NS8593 is able to elicit a sustained effect on TRPM7, since addition of NS8593 to the cell culture medium completely reversed the cell toxic effect of TRPM7 overexpression in HEK 293 cells. In addition, we showed that 10 µM NS8593 fully inhibit native TRPM7 currents in HEK 293 cells, resulting in a substantial inhibition of cell motility, indicating that inactivation of the channel activity of the bifunctional TRPM7 protein is sufficient to induce this phenotype. Finally, we demonstrated that 10 µM NS8593 block endogenous TRPM7 currents in mouse smooth muscle cells freshly isolated from brain arteries, primary mouse podocytes and primary human ventricular myocytes. Together, our findings suggest that NS8593 is well-suited to study recombinant and native TRPM7 channels in vitro and may be a first step towards the development of non-toxic drugs targeting TRPM7 in vivo.
NS8593 was originally described as a reversible inhibitor of apamin-sensitive Ca2+-activated potassium channels with IC50 values 0.42, 0.60 and 0.73 µM at 0.5 µM Ca2+ for KCa2.1–2.3 subtypes (Strobaek et al., 2006). Interestingly, the inhibitory effects of NS8593 on KCa2.1–2.3 were undetectable at 30 µM [Ca2+]i (Jenkins et al., 2011). In excitable cells, KCa2.1–2.3 channels mediate hyperpolarizing K+ currents to modulate repetitive electrical activity and firing patterns (Berkefeld et al., 2010). In nonexcitable cells, such as endothelial cells, hyperpolarizing KCa2.1–2.3 currents maintain the driving force for Ca2+ influx (Taylor et al., 2003; Berkefeld et al., 2010). The pharmacological potential of NS8593 in vivo has already been demonstrated for the modulation of the firing rate of dopaminergic neurons (Herrik et al., 2010) and the termination of atrial fibrillation (Diness et al., 2010). Of note, the cross-reactivity of NS8593 might be beneficial for these pharmacological actions: The suppression of ubiquitously present TRPM7 by NS8593 may reduce intracellular concentrations of divalent cations including Ca2+, which in turn may further enhance the NS8593-induced block of KCa2.1–2.3 channels. Future studies will have to specifically address this issue.
Acknowledgments
This study was supported by the Deutsche Forschungsgemeinschaft, Wissenschaftliches Herausgeberkollegium der Münchener Medizinischen Wochenschrift e.V and Friedrich-Baur-Stiftung Muenchen. TH was supported by the Emmy-Noether-Programm of the DFG (DFG-Ho-3869). We thank Anna-Lena Forst for preparation of podocytes, Renate Heilmair and Joanna Zaißerer for excellent technical assistance and Ursula Storch for critical discussion. We thank Carsten Schmitz, National Jewish Health and University of Colorado Denver, for providing a HEK 293 cell line stably expressing human TRPM7.
Glossary
- BK
channels, high-conductance Ca2+-activated K+ channels
- IK
channels, intermediate conductance Ca2+-activated K+ channels
- KCa2.1–2.3
(SK) channels, small conductance Ca2+-activated K+ channels
- TRPM7
melastatin-related TRP cation channel 7
Conflicts of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Voltage dependence of the NS8593 block of TRPM7 and TRPM3 channels. Representative current–voltage (I–V) relationships of whole-cell currents measured in HEK 293 cells transiently transfected with TRPM7 or TRPM3 cDNAs. (A) TRPM7 currents acquired before and a few seconds after external application of 5 μM NS8593. Measurements were performed as in Figure 2B applying voltage ramps from −100 to 200 mV. (B) PS (10 μM) induced TRPM3 currents acquired before and a few seconds after external application of 30 μM NS8593. Measurements were performed as in Figure 6B applying voltage ramps from −100 to +200 mV.
Figure S2 Run-down of TRPM7 currents in Na+-based external saline. Whole-cell TRPM7 currents measured in HEK 293 cells transiently transfected with TRPM7 cDNA. Representative currents over time acquired at −100 and 100 mV (left) and corresponding I–V relationships (right) are shown. (A) Effect of a Na+-based external solution on fully induced TRPM7 currents. Note that perfusion of cells with a Na+-based saline results in transient increases of inward and outward currents followed by their fast run-down, which was reversible if cells were perfused again by physiological saline (containing 1 mMMg2+ and 2 mM Ca2+). (B) Measurements were performed similar to (A) using a Cs+-based saline. Monovalent TRPM7 cation currents were fully sustained during recordings in Cs+-based saline.
Figure S3 Effects of SK inhibitors on TRPM7 currents in HEK 293 cells. Representative traces of TRPM7 currents from experiments shown in Figure 4A. Inhibitory effects of 30 μM quinine (A), dequalinium (B), CyPPA (C), UCL 1684 (D) and 10 nM and 1 μM tamapin (E, F) on TRPM7 whole-cell currents are illustrated.
Figure S4 Evaluation of NS8593 on Ca2+ influx mediated by TRPM3, TRPM8 and TRPM2 channels using fura-2. (A) (Left) Representative traces obtained with mock- and TRPM3 cDNA-transfected cells stimulated with 10 μM PS; (right) mean values of Ca2+ transients (Δ ratio F340/F380 of fura-2) obtained from three independent transfections. (B) (Left) Representative traces obtained with mock- and TRPM8 cDNA-transfected cells stimulated with 10 μM icillin; (right) mean values of Ca2+ transients (Δ ratio F340/F380 of fura-2) obtained from three independent transfections. (C) (Left) Representative traces (aequorin-based assay) obtained with mock-and TRPM2 cDNA-transfected cells stimulated with 1 mMH2O2; (right) mean values of Ca2+ transients obtained from three independent transfections.
Figure S5 Evaluation of NS8593 on TRPM5 and TRPC6 currents. HEK 293 cells were transfected with mouse TRPM5 (Hofmann et al., 2003) or a YFP-labelled human TRPC6 (Hofmann et al., 1999; 2002). Representative recordings are shown. (A) TRPM5 cells were perfused with a pipette solution containing calculated 3 μM Ca2+. In the right panel, cells were superfused with 10 μM NS8593 as indicated by the application bar (n = 12), in the left panel without the inhibitor (n = 4). (B) TRPC6-expressing cells were stimulated with a moderate concentration of histamine (1 μM) acting at a coexpressed H1 histamine receptor, and superfused with 10 μM NS8593, as indicated by application bars (n = 3).
Figure S6 Evaluation of NS8593 on Ca2+ influx mediated by TRPA1 and TRPV1 channels in HEK293 cells using an aequorin-based approach. (A) (Left) Representative traces obtained with TRPA1 cDNA-transfected cells stimulated with 10 μM AITC; (right) mean values of Ca2+ rises obtained from three independent transfections. (B) (Left) Representative traces obtained with TRPV1 cDNA-transfected cells stimulated with 1 μM capsaicin; (right) mean values of Ca2+ rises obtained from three independent transfections.
Figure S7 Impact of NS8593, Quinine and UCL 1684 on the viability of HEK 293 cells overexpressing recombinant TRPM7. Experiments with tetracycline-induced (1 μM, 40 h) expression of human TRPM7 were performed as described in Methods. Scale bars – 60 μm
Figure S8 Effects of NS8593, BMB and apamin on the morphology of HEK 293 cells. The images show living HEK 293 cells cultured with or without the compounds indicated for 48 h. Note that in the presence of 30 μM NS8593, HEK 293 cells form tight colonies. Scale bars represent 30 μm each.
Appendix S1 Supplementary methods.
Appendix S2 Supplementary References.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Aarts M, Iihara K, Wei WL, Xiong ZG, Arundine M, Cerwinski W, et al. A key role for TRPM7 channels in anoxic neuronal death. Cell. 2003;115:863–877. doi: 10.1016/s0092-8674(03)01017-1. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th Edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baubet V, Le Mouellic H, Campbell AK, Lucas-Meunier E, Fossier P, Brulet P. Chimeric green fluorescent protein-aequorin as bioluminescent Ca2+ reporters at the single-cell level. Proc Natl Acad Sci U S A. 2000;97:7260–7265. doi: 10.1073/pnas.97.13.7260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkefeld H, Fakler B, Schulte U. Ca2+-activated K+ channels: from protein complexes to function. Physiol Rev. 2010;90:1437–1459. doi: 10.1152/physrev.00049.2009. [DOI] [PubMed] [Google Scholar]
- Brauchi S, Krapivinsky G, Krapivinsky L, Clapham DE. TRPM7 facilitates cholinergic vesicle fusion with the plasma membrane. Proc Natl Acad Sci U S A. 2008;105:8304–8308. doi: 10.1073/pnas.0800881105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun FJ, Broad LM, Armstrong DL, Putney JW., Jr Stable activation of single Ca2+ release-activated Ca2+ channels in divalent cation-free solutions. J Biol Chem. 2001;276:1063–1070. doi: 10.1074/jbc.M008348200. [DOI] [PubMed] [Google Scholar]
- Chen HC, Xie J, Zhang Z, Su LT, Yue L, Runnels LW. Blockade of TRPM7 channel activity and cell death by inhibitors of 5-lipoxygenase. PLoS ONE. 2010b;5:e11161. doi: 10.1371/journal.pone.0011161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Numata T, Li M, Mori Y, Orser BA, Jackson MF, et al. The modulation of TRPM7 currents by nafamostat mesilate depends directly upon extracellular concentrations of divalent cations. Mol Brain. 2010a;3:38. doi: 10.1186/1756-6606-3-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chubanov V, Waldegger S, Mederos y Schnitzler M, Vitzthum H, Sassen MC, Seyberth HW, et al. Disruption of TRPM6/TRPM7 complex formation by a mutation in the TRPM6 gene causes hypomagnesemia with secondary hypocalcemia. Proc Natl Acad Sci U S A. 2004;101:2894–2899. doi: 10.1073/pnas.0305252101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chubanov V, Mederos y Schnitzler M, Waring J, Plank A, Gudermann T. Emerging roles of TRPM6/TRPM7 channel kinase signal transduction complexes. Naunyn Schmiedebergs Arch Pharmacol. 2005;371:334–341. doi: 10.1007/s00210-005-1056-4. [DOI] [PubMed] [Google Scholar]
- Chubanov V, Schlingmann KP, Waring J, Heinzinger J, Kaske S, Waldegger S, et al. Hypomagnesemia with secondary hypocalcemia due to a missense mutation in the putative pore-forming region of TRPM6. J Biol Chem. 2007;282:7656–7667. doi: 10.1074/jbc.M611117200. [DOI] [PubMed] [Google Scholar]
- Clark K, Langeslag M, van Leeuwen B, Ran L, Ryazanov AG, Figdor CG, et al. TRPM7, a novel regulator of actomyosin contractility and cell adhesion. EMBO J. 2006;25:290–301. doi: 10.1038/sj.emboj.7600931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demeuse P, Penner R, Fleig A. TRPM7 channel is regulated by magnesium nucleotides via its kinase domain. J Gen Physiol. 2006;127:421–434. doi: 10.1085/jgp.200509410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich A, Mederos y Schnitzler M, Gollasch M, Gross V, Storch U, Dubrovska G, et al. Increased vascular smooth muscle contractility in TRPC6-/- mice. Mol Cell Biol. 2005;25:6980–6989. doi: 10.1128/MCB.25.16.6980-6989.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diness JG, Sorensen US, Nissen JD, Al-Shahib B, Jespersen T, Grunnet M, et al. Inhibition of small-conductance Ca2+-activated K+ channels terminates and protects against atrial fibrillation. Circ Arrhythm Electrophysiol. 2010;3:380–390. doi: 10.1161/CIRCEP.110.957407. [DOI] [PubMed] [Google Scholar]
- Du J, Xie J, Zhang Z, Tsujikawa H, Fusco D, Silverman D, et al. TRPM7-mediated Ca2+ signals confer fibrogenesis in human atrial fibrillation. Circ Res. 2010;106:992–1003. doi: 10.1161/CIRCRESAHA.109.206771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilbert A, Gautier M, Dhennin-Duthille I, Haren N, Sevestre H, Ouadid-Ahidouch H. Evidence that TRPM7 is required for breast cancer cell proliferation. Am J Physiol Cell Physiol. 2009;297:C493–C502. doi: 10.1152/ajpcell.00624.2008. [DOI] [PubMed] [Google Scholar]
- Gwanyanya A, Amuzescu B, Zakharov SI, Macianskiene R, Sipido KR, Bolotina VM, et al. Magnesium-inhibited, TRPM6/7-like channel in cardiac myocytes: permeation of divalent cations and pH-mediated regulation. J Physiol. 2004;559:761–776. doi: 10.1113/jphysiol.2004.067637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanano T, Hara Y, Shi J, Morita H, Umebayashi C, Mori E, et al. Involvement of TRPM7 in cell growth as a spontaneously activated Ca2+ entry pathway in human retinoblastoma cells. J Pharmacol Sci. 2004;95:403–419. doi: 10.1254/jphs.fp0040273. [DOI] [PubMed] [Google Scholar]
- Hara Y, Wakamori M, Ishii M, Maeno E, Nishida M, Yoshida T, et al. LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol Cell. 2002;9:163–173. doi: 10.1016/s1097-2765(01)00438-5. [DOI] [PubMed] [Google Scholar]
- Hermosura MC, Nayakanti H, Dorovkov MV, Calderon FR, Ryazanov AG, Haymer DS, et al. A TRPM7 variant shows altered sensitivity to magnesium that may contribute to the pathogenesis of two Guamanian neurodegenerative disorders. Proc Natl Acad Sci U S A. 2005;102:11510–11515. doi: 10.1073/pnas.0505149102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrik KF, Christophersen P, Shepard PD. Pharmacological modulation of the gating properties of small conductance Ca2+-activated K+ channels alters the firing pattern of dopamine neurons in vivo. J Neurophysiol. 2010;104:1726–1735. doi: 10.1152/jn.01126.2009. [DOI] [PubMed] [Google Scholar]
- Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature. 1999;397:259–263. doi: 10.1038/16711. [DOI] [PubMed] [Google Scholar]
- Hofmann T, Schaefer M, Schultz G, Gudermann T. Subunit composition of mammalian transient receptor potential channels in living cells. Proc Natl Acad Sci U S A. 2002;99:7461–7466. doi: 10.1073/pnas.102596199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann T, Chubanov V, Gudermann T, Montell C. TRPM5 is a voltage-modulated and Ca2+-activated monovalent selective cation channel. Curr Biol. 2003;13:1153–1158. doi: 10.1016/s0960-9822(03)00431-7. [DOI] [PubMed] [Google Scholar]
- Hofmann T, Chubanov V, Chen X, Dietz AS, Gudermann T, Montell C. Drosophila TRPM channel is essential for the control of extracellular magnesium levels. PLoS ONE. 2010;5:e10519. doi: 10.1371/journal.pone.0010519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins DP, Strobaek D, Hougaard C, Jensen ML, Hummel R, Sorensen US, et al. Negative gating modulation by (R)-N-(Benzimidazol-2-yl)-tetrahydro-1-naphtylamine (NS8593) depends on residues in the inner pore vestibule: pharmacological evidence of deep-pore gating of KCa2 channels. Mol Pharmacol. 2011;79:899–909. doi: 10.1124/mol.110.069807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Li M, Yue L. Potentiation of TRPM7 inward currents by protons. J Gen Physiol. 2005;126:137–150. doi: 10.1085/jgp.200409185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Li MH, Inoue K, Chu XP, Seeds J, Xiong ZG. Transient receptor potential melastatin 7-like current in human head and neck carcinoma cells: role in cell proliferation. Cancer Res. 2007;67:10929–10938. doi: 10.1158/0008-5472.CAN-07-1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Desai BN, Navarro B, Donovan A, Andrews NC, Clapham DE. Deletion of Trpm7 disrupts embryonic development and thymopoiesis without altering Mg2+ homeostasis. Science. 2008;322:756–760. doi: 10.1126/science.1163493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerschbaum HH, Cahalan MD. Single-channel recording of a store-operated Ca2+ channel in Jurkat T lymphocytes. Science. 1999;283:836–839. doi: 10.1126/science.283.5403.836. [DOI] [PubMed] [Google Scholar]
- Kerschbaum HH, Kozak JA, Cahalan MD. Polyvalent cations as permeant probes of MIC and TRPM7 pores. Biophys J. 2003;84:2293–2305. doi: 10.1016/S0006-3495(03)75035-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BJ, Jeon JH, Kim SJ, So I, Kim KW. Regulation of transient receptor potential melastatin 7 (TRPM7) currents by mitochondria. Mol Cells. 2007;23:363–369. [PubMed] [Google Scholar]
- Kim BJ, Park EJ, Lee JH, Jeon JH, Kim SJ, So I. Suppression of transient receptor potential melastatin 7 channel induces cell death in gastric cancer. Cancer Sci. 2008;99:2502–2509. doi: 10.1111/j.1349-7006.2008.00982.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak JA, Cahalan MD. MIC channels are inhibited by internal divalent cations but not ATP. Biophys J. 2003;84:922–927. doi: 10.1016/S0006-3495(03)74909-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak JA, Kerschbaum HH, Cahalan MD. Distinct properties of CRAC and MIC channels in RBL cells. J Gen Physiol. 2002;120:221–235. doi: 10.1085/jgp.20028601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak JA, Matsushita M, Nairn AC, Cahalan MD. Charge screening by internal pH and polyvalent cations as a mechanism for activation, inhibition, and rundown of TRPM7/MIC channels. J Gen Physiol. 2005;126:499–514. doi: 10.1085/jgp.200509324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Jiang J, Yue L. Functional characterization of homo- and heteromeric channel kinases TRPM6 and TRPM7. J Gen Physiol. 2006;127:525–537. doi: 10.1085/jgp.200609502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita M, Kozak JA, Shimizu Y, McLachlin DT, Yamaguchi H, Wei FY, et al. Channel function is dissociated from the intrinsic kinase activity and autophosphorylation of TRPM7/ChaK1. J Biol Chem. 2005;280:20793–20803. doi: 10.1074/jbc.M413671200. [DOI] [PubMed] [Google Scholar]
- Mederos y Schnitzler M, Waring J, Gudermann T, Chubanov V. Evolutionary determinants of divergent calcium selectivity of TRPM channels. FASEB J. 2008;22:1540–1551. doi: 10.1096/fj.07-9694com. [DOI] [PubMed] [Google Scholar]
- Mishra R, Rao V, Ta R, Shobeiri N, Hill CE. Mg2+- and MgATP-inhibited and Ca2+/calmodulin-sensitive TRPM7-like current in hepatoma and hepatocytes. Am J Physiol Gastrointest Liver Physiol. 2009;297:G687–G694. doi: 10.1152/ajpgi.90683.2008. [DOI] [PubMed] [Google Scholar]
- Monteilh-Zoller MK, Hermosura MC, Nadler MJ, Scharenberg AM, Penner R, Fleig A. TRPM7 provides an ion channel mechanism for cellular entry of trace metal ions. J Gen Physiol. 2003;121:49–60. doi: 10.1085/jgp.20028740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadler MJ, Hermosura MC, Inabe K, Perraud AL, Zhu Q, Stokes AJ, et al. LTRPC7 is a Mg.ATP-regulated divalent cation channel required for cell viability. Nature. 2001;411:590–595. doi: 10.1038/35079092. [DOI] [PubMed] [Google Scholar]
- Nilius B, Mahieu F, Karashima Y, Voets T. Regulation of TRP channels: a voltage-lipid connection. Biochem Soc Trans. 2007;35:105–108. doi: 10.1042/BST0350105. [DOI] [PubMed] [Google Scholar]
- Numata T, Shimizu T, Okada Y. Direct mechano-stress sensitivity of TRPM7 channel. Cell Physiol Biochem. 2007a;19:1–8. doi: 10.1159/000099187. [DOI] [PubMed] [Google Scholar]
- Numata T, Shimizu T, Okada Y. TRPM7 is a stretch- and swelling-activated cation channel involved in volume regulation in human epithelial cells. Am J Physiol Cell Physiol. 2007b;292:C460–C467. doi: 10.1152/ajpcell.00367.2006. [DOI] [PubMed] [Google Scholar]
- Oancea E, Wolfe JT, Clapham DE. Functional TRPM7 channels accumulate at the plasma membrane in response to fluid flow. Circ Res. 2006;98:245–253. doi: 10.1161/01.RES.0000200179.29375.cc. [DOI] [PubMed] [Google Scholar]
- Oberwinkler J, Lis A, Giehl KM, Flockerzi V, Philipp SE. Alternative splicing switches the divalent cation selectivity of TRPM3 channels. J Biol Chem. 2005;280:22540–22548. doi: 10.1074/jbc.M503092200. [DOI] [PubMed] [Google Scholar]
- Parnas M, Peters M, Dadon D, Lev S, Vertkin I, Slutsky I, et al. Carvacrol is a novel inhibitor of Drosophila TRPL and mammalian TRPM7 channels. Cell Calcium. 2009;45:300–309. doi: 10.1016/j.ceca.2008.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedarzani P, D'Hoedt D, Doorty KB, Wadsworth JD, Joseph JS, Jeyaseelan K, et al. Tamapin, a venom peptide from the Indian red scorpion (Mesobuthus tamulus) that targets small conductance Ca2+-activated K+ channels and afterhyperpolarization currents in central neurons. J Biol Chem. 2002;277:46101–46109. doi: 10.1074/jbc.M206465200. [DOI] [PubMed] [Google Scholar]
- Peier AM, Moqrich A, Hergarden AC, Reeve AJ, Andersson DA, Story GM, et al. A TRP channel that senses cold stimuli and menthol. Cell. 2002;108:705–715. doi: 10.1016/s0092-8674(02)00652-9. [DOI] [PubMed] [Google Scholar]
- Perraud AL, Fleig A, Dunn CA, Bagley LA, Launay P, Schmitz C, et al. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature. 2001;411:595–599. doi: 10.1038/35079100. [DOI] [PubMed] [Google Scholar]
- Prakriya M, Lewis RS. Separation and characterization of currents through store-operated CRAC channels and Mg2+-inhibited cation (MIC) channels. J Gen Physiol. 2002;119:487–507. doi: 10.1085/jgp.20028551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runnels LW, Yue L, Clapham DE. TRP-PLIK, a bifunctional protein with kinase and ion channel activities. Science. 2001;291:1043–1047. doi: 10.1126/science.1058519. [DOI] [PubMed] [Google Scholar]
- Runnels LW, Yue L, Clapham DE. The TRPM7 channel is inactivated by PIP2 hydrolysis. Nat Cell Biol. 2002;4:329–336. doi: 10.1038/ncb781. [DOI] [PubMed] [Google Scholar]
- Ryazanov AG, Pavur KS, Dorovkov MV. Alpha-kinases: a new class of protein kinases with a novel catalytic domain. Curr Biol. 1999;9:R43–R45. doi: 10.1016/s0960-9822(99)80006-2. [DOI] [PubMed] [Google Scholar]
- Ryazanova LV, Rondon LJ, Zierler S, Hu Z, Galli J, Yamaguchi TP, et al. TRPM7 is essential for Mg2+ homeostasis in mammals. Nat Commun. 2010;1:109. doi: 10.1038/ncomms1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahni J, Scharenberg AM. TRPM7 ion channels are required for sustained phosphoinositide 3-kinase signaling in lymphocytes. Cell Metab. 2008;8:84–93. doi: 10.1016/j.cmet.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlondorff D. Preparation and study of isolated glomeruli. Methods Enzymol. 1990;191:130–140. doi: 10.1016/0076-6879(90)91011-t. [DOI] [PubMed] [Google Scholar]
- Schmitz C, Perraud AL, Johnson CO, Inabe K, Smith MK, Penner R, et al. Regulation of vertebrate cellular Mg2+ homeostasis by TRPM7. Cell. 2003;114:191–200. doi: 10.1016/s0092-8674(03)00556-7. [DOI] [PubMed] [Google Scholar]
- Shi J, Cui J. Intracellular Mg2+ enhances the function of BK-type Ca2+-activated K+ channels. J Gen Physiol. 2001;118:589–606. doi: 10.1085/jgp.118.5.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soh H, Park CS. Localization of divalent cation-binding site in the pore of a small conductance Ca2+-activated K+ channel and its role in determining current-voltage relationship. Biophys J. 2002;83:2528–2538. doi: 10.1016/S0006-3495(02)75264-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen US, Strobaek D, Christophersen P, Hougaard C, Jensen ML, Nielsen EO, et al. Synthesis and structure-activity relationship studies of 2-(N-substituted)-aminobenzimidazoles as potent negative gating modulators ofsmall conductance Ca2+-activated K+ channels. J Med Chem. 2008;51:7625–7634. doi: 10.1021/jm800809f. [DOI] [PubMed] [Google Scholar]
- Stanfield PR, Davies NW, Shelton PA, Sutcliffe MJ, Khan IA, Brammar WJ, et al. A single aspartate residue is involved in both intrinsic gating and blockage by Mg2+ of the inward rectifier, IRK1. J Physiol. 1994;478((Pt 1)):1–6. doi: 10.1113/jphysiol.1994.sp020225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starkus J, Beck A, Fleig A, Penner R. Regulation of TRPM2 by extra- and intracellular calcium. J Gen Physiol. 2007;130:427–440. doi: 10.1085/jgp.200709836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strobaek D, Hougaard C, Johansen TH, Sorensen US, Nielsen EO, Nielsen KS, et al. Inhibitory gating modulation of small conductance Ca2+-activated K+ channels by the synthetic compound (R)-N-(benzimidazol-2-yl)-1,2,3,4-tetrahydro-1-naphtylamine (NS8593) reduces afterhyperpolarizing current in hippocampal CA1 neurons. Mol Pharmacol. 2006;70:1771–1782. doi: 10.1124/mol.106.027110. [DOI] [PubMed] [Google Scholar]
- Su LT, Agapito MA, Li M, Simonson WT, Huttenlocher A, Habas R, et al. TRPM7 regulates cell adhesion by controlling the calcium-dependent protease calpain. J Biol Chem. 2006;281:11260–11270. doi: 10.1074/jbc.M512885200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su LT, Liu W, Chen HC, Gonzalez-Pagan O, Habas R, Runnels LW. TRPM7 regulates polarized cell movements. Biochem J. 2011;434:513–521. doi: 10.1042/BJ20101678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taglialatela M, Ficker E, Wible BA, Brown AM. C-terminus determinants for Mg2+ and polyamine block of the inward rectifier K+ channel IRK1. EMBO J. 1995;14:5532–5541. doi: 10.1002/j.1460-2075.1995.tb00240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MS, Bonev AD, Gross TP, Eckman DM, Brayden JE, Bond CT, et al. Altered expression of small-conductance Ca2+-activated K+ (SK3) channels modulates arterial tone and blood pressure. Circ Res. 2003;93:124–131. doi: 10.1161/01.RES.0000081980.63146.69. [DOI] [PubMed] [Google Scholar]
- Touyz RM. Transient receptor potential melastatin 6 and 7 channels, magnesium transport, and vascular biology: implications in hypertension. Am J Physiol Heart Circ Physiol. 2008;294:H1103–H1118. doi: 10.1152/ajpheart.00903.2007. [DOI] [PubMed] [Google Scholar]
- Tseveleki V, Rubio R, Vamvakas SS, White J, Taoufik E, Petit E, et al. Comparative gene expression analysis in mouse models for multiple sclerosis, Alzheimer's disease and stroke for identifying commonly regulated and disease-specific gene changes. Genomics. 2010;96:82–91. doi: 10.1016/j.ygeno.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vriens J, Nilius B, Vennekens R. Herbal compounds and toxins modulating TRP channels. Curr Neuropharmacol. 2008;6:79–96. doi: 10.2174/157015908783769644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner TF, Loch S, Lambert S, Straub I, Mannebach S, Mathar I, et al. Transient receptor potential M3 channels are ionotropic steroid receptors in pancreatic beta cells. Nat Cell Biol. 2008;10:1421–1430. doi: 10.1038/ncb1801. [DOI] [PubMed] [Google Scholar]
- Weatherall KL, Goodchild SJ, Jane DE, Marrion NV. Small conductance calcium-activated potassium channels: from structure to function. Prog Neurobiol. 2010;91:242–255. doi: 10.1016/j.pneurobio.2010.03.002. [DOI] [PubMed] [Google Scholar]
- Wei C, Wang X, Chen M, Ouyang K, Song LS, Cheng H. Calcium flickers steer cell migration. Nature. 2009;457:901–905. doi: 10.1038/nature07577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wible BA, Taglialatela M, Ficker E, Brown AM. Gating of inwardly rectifying K+ channels localized to a single negatively charged residue. Nature. 1994;371:246–249. doi: 10.1038/371246a0. [DOI] [PubMed] [Google Scholar]
- Wu Y, Yang Y, Ye S, Jiang Y. Structure of the gating ring from the human large-conductance Ca2+-gated K+ channel. Nature. 2010;466:393–397. doi: 10.1038/nature09252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H, Matsushita M, Nairn AC, Kuriyan J. Crystal structure of the atypical protein kinase domain of a TRP channel with phosphotransferase activity. Mol Cell. 2001;7:1047–1057. doi: 10.1016/s1097-2765(01)00256-8. [DOI] [PubMed] [Google Scholar]
- Yang H, Shi J, Zhang G, Yang J, Delaloye K, Cui J. Activation of Slo1 BK channels by Mg2+ coordinated between the voltage sensor and RCK1 domains. Nat Struct Mol Biol. 2008;15:1152–1159. doi: 10.1038/nsmb.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zierler S, Yao G, Zhang Z, Kuo WC, Poerzgen P, Penner R, et al. Waixenicin A inhibits cell proliferation through magnesium-dependent block of TRPM7. J Biol Chem. 2011;286:39328–39335. doi: 10.1074/jbc.M111.264341. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.