

Abstract
BACKGROUND AND PURPOSE
The aim of this study was to characterize the functional impact of KCNQ1-encoded voltage-dependent potassium channels (Kv7.1) in the vasculature.
EXPERIMENTAL APPROACH
Mesenteric arteries, intrapulmonary arteries and thoracic aortae were isolated from adult rats. Kv7.1 channel expression was established by fluorescence immunocytochemistry. Wire myography determined functionality of these channels in response to selective blockers and activators. Xenopus oocytes expressing Kv7.1 channels were used to assess the effectiveness of selective Kv7.1 channel blockers.
KEY RESULTS
Kv7.1 channels were identified in arterial myocytes by immunocytochemistry. Kv7.1 blockers HMR1556, L-768,673 (10 µM) and JNJ39490282 (JNJ282; 1 µM) had no contractile effects in arteries, whereas the pan-Kv7 channel blocker linopirdine (10 µM) evoked robust contractions. Application of two compounds purported to activate Kv7.1 channels, L-364 373 (R-L3) and mefenamic acid, relaxed mesenteric arteries preconstricted by methoxamine. These responses were reversed by HMR1556 or L-768,673 but not JNJ282. Similar effects were observed in the thoracic aorta and intrapulmonary arteries.
CONCLUSIONS AND IMPLICATIONS
In contrast to previous assumptions, Kv7.1 channels expressed in arterial myocytes are functional ion channels. Although these channels do not appear to contribute to resting vascular tone, Kv7.1 activators were effective vasorelaxants.
Keywords: voltage-dependent K+ channel, KCNQ, Kv7, vascular smooth muscle, arterial tone
Introduction
KCNQ1 is a member of the KCNQ gene family (1–5) that encode for the pore-forming subunits of voltage-dependent potassium (K+) channels (termed Kv7.1–7.5). KCNQ1 is expressed abundantly in the heart and epithelia where it has well-established roles. In the heart, Kv7.1 channels co-assemble with the protein product of KCNE1 (minK) (Barhanin et al., 1996; Kang et al., 2008) to constitute the slow component of the delayed rectifier K+ current (IKs; reviewed, Jespersen et al., 2005). Mutation of KCNQ1 and KCNE1 underlie a large number of hereditary arrhythmias leading to long QT syndrome (Herbert et al., 2002; Jespersen et al., 2005). In colonic epithelia, Kv7.1 associates with the protein encoded by KCNE3 to form a constitutively active channel that regulates fluid flux (Schroeder et al., 2000). More recently, KCNQ1 expression has also been shown to be abundant in rodent and human vascular smooth muscle cells along with KCNQ4 and KCNQ5 (Ohya et al., 2003; Yeung et al., 2007; 2008; Greenwood and Ohya, 2009; Zhong et al., 2010; Jepps et al., 2011; Ng et al., 2011), but in contrast to its well-defined role highlighted earlier in non-vascular tissues, the functional impact of Kv7.1 in blood vessels remains enigmatic.
XE991 and linopirdine, which block all Kv7 channel isoforms, cause smooth muscle depolarization and contraction of rodent and human arteries (Yeung et al., 2007; 2008; Mackie et al., 2008; Joshi et al., 2009; Zhong et al., 2010; Jepps et al., 2011; Ng et al., 2011). However, in no smooth muscle study has chromanol 293B, a preferential Kv7.1 blocker (Bett et al., 2006; Lerche et al., 2007), been shown to mimic the effects of linopirdine or XE991. In addition, activation of Kv7 channels by compounds such as retigabine and the acrylamide S-1 have been shown to hyperpolarize smooth muscle cells and cause relaxation of blood vessels (Yeung et al., 2007; 2008; Mackie et al., 2008; Zhong et al., 2010; Jepps et al., 2011; Ng et al., 2011). These effects are mediated through Kv7.2–7.5 channels but not Kv7.1, which lacks the tryptophan residue in the fifth transmembrane domain that is required for the actions of these drugs (Schenzer et al., 2005; Wuttke et al., 2005; Bentzen et al., 2006). The lack of efficacy of chromanol 293B and the Kv7.1-independent effects of Kv7 channel activators has led to the assumption that Kv7.1 channels in blood vessels are most likely redundant and do not influence vascular tone. However, this conclusion relies heavily on the selectivity and responses of one agent, chromanol 293B, which may affect other ion channels. Indeed, the obvious expression of Kv7.1 channels in vascular tissue (Yeung et al., 2007; 2008; Mackie et al., 2008; Zhong et al., 2010; Jepps et al., 2011) as well as their critical function in cardiac myocytes leads to persisting questions regarding their role in smooth muscle. To address this issue, in the present study we utilized three structurally different IKs blockers [HMR1556, L-768,673 and JNJ39490282 (abbreviated to JNJ282) ] that have greater potency than chromanol 293B (IC50: 74, 6 and 1 nM for HMR1556, L-768,673 and JNJ282, respectively, compared with 6.9 µM for chromanol 293B) (Lerche et al., 2000; 2007; Towart et al., 2009), to confirm the impact of Kv7.1 channels on vascular tone. In addition, the effectiveness of activating Kv7.1 channels as an anti-contractile mechanism was assessed using two identified Kv7.1 activators, mefenamic acid (Busch et al., 1994; 1997) and L-364373 (abbreviated to R-L3) (Salata et al., 1998; Seebohm et al., 2003).
Methods
All genes are shown as KCNQ and all proteins as KV7 in agreement with the nomenclature in Alexander et al. (2011).
Tissue isolation
In accordance with the UK Animal (Scientific Procedures) Act 1986, male Wistar rats (200–225 g) (Charles River UK Ltd., Kent, U.K.), were killed by cervical dislocation. Thoracic aorta, third-order mesenteric arteries and intrapulmonary arteries were isolated and cleaned of all fat and connective tissue.
Isolation of arterial myocytes
Mesenteric artery and thoracic aorta smooth muscle cells were isolated as previously described (Yeung et al., 2008; Jepps et al., 2011). Briefly, third-order branches of mesenteric artery were cleaned of adherent connective tissue and transferred to Ca2+-free physiological saline solution (PSS, in mM: 6.0 KCl, 120 NaCl, 1.2 MgCl2, 10.0 d-glucose and 10.0 hepes, pH was adjusted to 7.3 with NaOH), supplemented with protease type X (0.5 mg·mL−1) and collagenase type IA (1.5 mg·mL−1), and incubated at 37 °C for 15 min. The thoracic aorta was cleaned of adherent connective tissue, cut into strips and transferred to Ca2+-free PSS supplemented with BSA (10 mg·mL−1), DTT (10 mg·mL−1) and papain (5 mg·mL−1). The tissue was incubated for 20 min at 37°C before being transferred to Ca2+-free PSS supplemented with BSA (10 mg·mL−1), and collagenase type IA (10 mg·mL−1) and incubated for a further 20 min at 37°C. Single cells were obtained by gentle agitation with a wide-bore pipette in PSS containing 0.1 mM Ca2+. The cells were gently spun down, the supernatant removed and replaced with 0.75 mM Ca2+ PSS, transferred to glass coverslips and kept for 20 min at room temperature to allow adherence of the cells.
Immunocytochemistry
Similar to previous experiments (Davis et al., 2010), enzymatically isolated smooth muscle cells were fixed in 4% paraformaldehyde solution for 1 min at room temperature and then permeablized for 5 min with antibody diluent (PBS containing 0.1% Triton X-100). Cells were subsequently co-incubated with anti Kv7.1 (1:50; AB5932 Millipore, Watford, UK) and anti sodium potassium ATPase (1:100; AB7671 Abcam Ltd., Cambridge, UK) antibodies in antibody diluent overnight in a humidified chamber at 4°C. For control experiments, primary antibodies were omitted. The following day, cells were incubated with TRITC donkey anti-rabbit IgG (1:100; 711-025-152 Jackson ImmunoResearch Europe Ltd., Suffolk, UK) and FITC sheep anti-mouse IgG (1:100; 515-095-062 Jackson ImmunoResearch Europe Ltd.) for 60 min at room temperature. Unbound secondary antibody was removed, and cells were stained with 4,6-diamidino-2-phenylindole (DAPI) for 5 min at room temperature. Single cells were imaged using a laser scanning confocal microscope (model LSM 510, Zeiss, Welwyn Garden City, UK). Images for each cell type were gain-matched to ensure accuracy between samples. The KV7.1 antibody had been validated by Western blot using KCNQ1 knockout mice (Knollmann et al., 2007).
Isometric tension myography
Segments (∼2 mm) of artery were mounted in a Mulvany-Halpern myograph (Danish Myo Technology, Aarhus, Denmark) containing Krebs solution (in mM: 125 NaCl, 4.6 KCl, 2.5 CaCl2, 15.4 NaHCO3, 1 Na2HPO4, 0.6 MgSO4, 10 d-glucose) constantly aerated with 95% O2/5% CO2 at 37°C. Following a 30-min equilibration period, arteries were placed under a tension equivalent to 90% of the diameter of the vessel at a distending pressure of 100 mmHg (thoracic aorta and mesenteric artery) or 20 mmHg (intrapulmonary artery). After a further equilibration period, vessels were exposed to an external solution containing 60 mM KCl to assess vessel viability. Measurement of contractions and relaxations to Kv7 blockers and activators were made following a plateau in response. Changes in arterial tension were recorded continuously on Chart (version 5, ADInstruments) in conjunction with PowerLab (ADInstruments, Oxford, UK).
KCNQ overexpression in Xenopus laevis oocytes
Human KCNQ1, KCNQ4 and KCNQ5 constructs were subcloned into the pTLN expression vector; capped RNA was transcribed using SP6 RNA polymerase in mMessage mMachine kit (Ambion, Austin, TX, USA) after linearization of the cDNA with HpaI. Individual stage V–VI oocytes were injected with KCNQ1, KCNQ4 and KCNQ5 encoding cRNA. Water-injected oocytes acted as controls. Oocytes were then kept at 17°C in ND96 solution (in mM: 96 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, 5 hepes, pH was adjusted to 7.4). The oocytes were purchased from EcoCyte Bioscience (Castrop-Rauxel, Germany).
Two-electrode voltage clamp electrophysiology
Two to three days after oocyte injection, two-electrode voltage clamp measurements were performed using an npi Turbotec amplifier (npi electronics, Tamm, Germany) and pClamp9 software (Axon Instruments, Sunnyvale, CA, USA). Oocytes were held at −80 mV and stepped to potentials between −80 mV and +40 mV for 2 s followed by repolarization to −40 mV. The effects of L-768,673, HMR1556 and JNJ282 (at 0.1, 1, 10 µM or DMSO equivalent in ND96) and R-L3 (at 1, 5 and 10 µM) were examined. All experiments were conducted at room temperature (approximately 22°C).
Drugs and solutions
All chemicals and drugs were purchased from Sigma-Aldrich (Dorset, UK) unless otherwise stated. 9,11-Dideoxy-11α,9α-epoxymethanoprostaglandin F2α (U46619) and L-364 373 (R-L3) were obtained from Tocris (Bristol, UK). L-768,673, HMR1556, and JNJ39490282 were supplied by Janssen Pharmaceuticals. All drugs were prepared in either DMSO or distilled water.
Statistics
All responses are expressed as mean ± SEM, where n equals the number of rats from which arteries were taken. Where appropriate, multiple comparisons were performed by one-way anova followed by Dunnett's post test. Changes in current of KCNQ overexpressed oocytes were determined by Student's paired t-test. A value of P < 0.05 was considered to indicate statistical significance.
Results
Expression of Kv7.1 channels
While KCNQ1 transcripts have been identified in rat aorta, mesenteric artery and pulmonary artery (Mackie et al., 2008; Joshi et al., 2009; Jepps et al., 2011) the only protein studies on Kv7.1 have been performed on rat cerebral artery (Zhong et al., 2010) or murine aorta and portal vein (Yeung et al., 2007; 2008). Single cell immunocytochemistry on smooth muscle cells isolated from rat mesenteric artery and thoracic aorta showed significant expression of Kv7.1 protein. In addition, co-staining with the membrane marker sodium potassium ATPase suggested that considerable Kv7.1 expression was localized to the plasma membrane (Figure 1).
Figure 1.
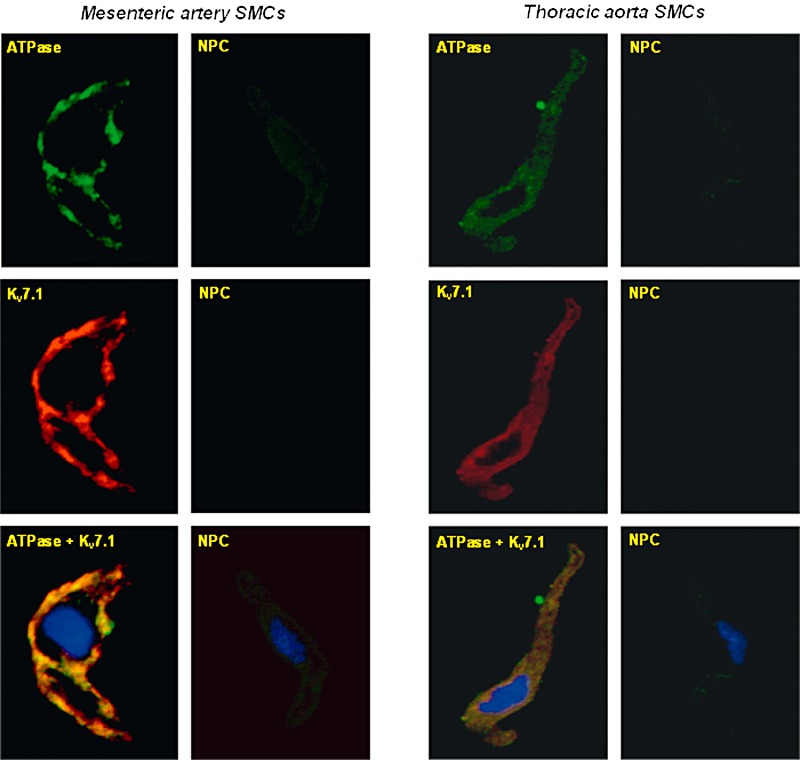
Identification of Kv7.1 protein in arterial myocytes. Representative fluorescence images from enzymatically isolated rat mesenteric artery and thoracic aorta SMCs co-incubated with the membrane marker anti sodium potassium ATPase (1:100; Abcam AB7671) and anti Kv7.1 (1:50 Millipore AB5932) or secondary antibodies only. Overlayed fluorescence image of sodium potassium ATPase, Kv7.1 and nucleus (DAPI) staining (bottom panels) indicates co-expression of Kv7.1 protein and sodium potassium ATPase. Fluorescence immunocytochemistry reveals expression of Kv7.1 protein in the plasma membrane of all tissues tested compared with minimal fluorescence in control cells. All images were time and gain matched. ATPase refers to sodium potassium ATPase. SMC, smooth muscle cell; NPC, no primary control.
Effects of Kv7 channel blockers on vascular tone
Previous studies have demonstrated that linopirdine and XE991, which block all types of Kv7 channel, evoke contractile responses in various rodent and human arterial preparations (Joshi et al., 2006; Yeung et al., 2007; Mackie et al., 2008; Zhong et al., 2010; Jepps et al., 2011; Ng et al., 2011). In the present study, using tension myography, 10 µM linopirdine did not significantly contract mesenteric artery under basal conditions but in the presence of minimal methoxamine-induced pre-tone (<10% KCl response), linopirdine caused a contraction similar to KCl (60 mM) responses (mean contraction was 107.1 ± 24.0%, n = 3; P < 0.01). In contrast, none of the selective Kv7.1 blockers, at concentrations that produce considerable block of K+ currents produced by the overexpression of KCNQ1 (10 µM L-768,673, 10 µM HMR1556, 1 µM JNJ282) (Towart et al., 2009 and present study), produced any contraction of mesenteric arteries significantly different from the vehicle control (Figure 2A and B; Table 1). In addition, 10 µM L-768,673 failed to contract mesenteric vessels following pretreatment with 20 mM KCl (0.5 ± 0.8% relative to 60 mM KCl contraction; n = 3). It is worth highlighting that L-768,673, HMR1556 and JNJ282 at the concentrations used did not alter contractions evoked by 60 mM KCl (data not shown) indicating that the lack of effect of these compounds was not due to a direct effect on voltage-dependent calcium channels.
Figure 2.
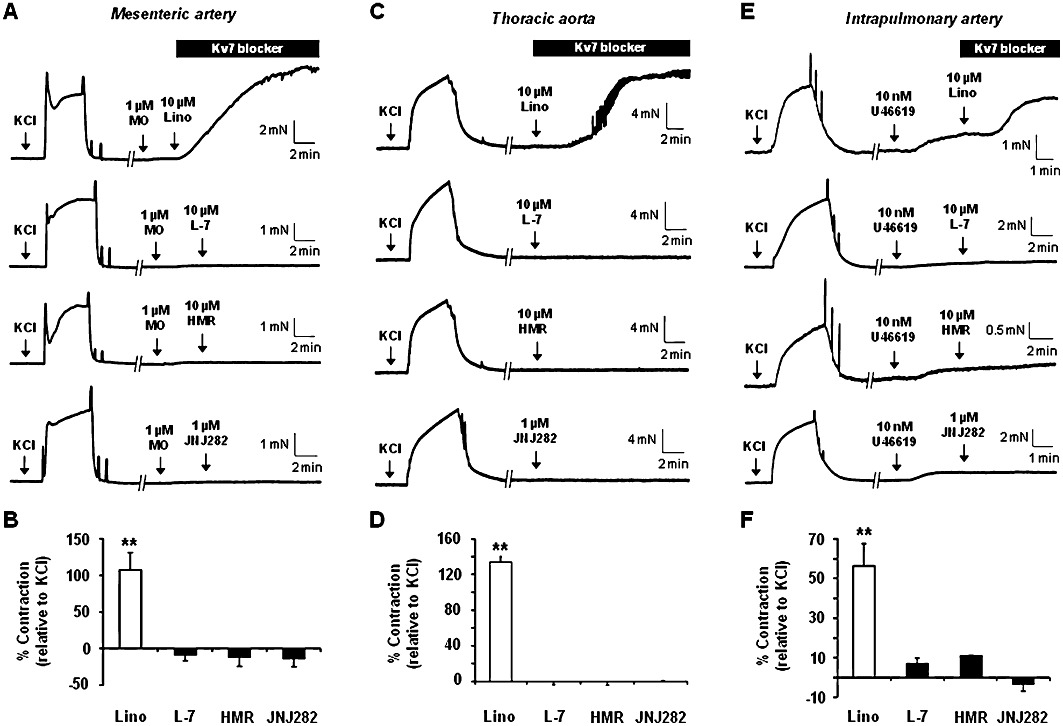
Effect of Kv7 blockers on arterial tone. Representative and mean smooth muscle contractions in response to 10 µM linopirdine or Kv7.1 blockers, 10 µM L-768,673 (L-7), 10 µM HMR1556 (HMR) and 1 µM JNJ282, in mesenteric arteries (A and B), thoracic aorta (C and D) and intrapulmonary arteries (E and F). It should be noted that mesenteric and intrapulmonary arteries were minimally preconstricted using methoxamine (MO) or U46619, respectively, before exposure to Kv7 blockers. **P < 0.01 compared with tone in the absence of any Kv7 blockers using Dunnet's post hoc test following anova (P = 0.001).
Table 1.
Effect of Kv7 modulators in rat arterial preparations
| Mesenteric artery | Thoracic aorta | Intrapulmonary artery | ||||
|---|---|---|---|---|---|---|
| Contraction (% of 60 mM KCl response) | % Reversal of RL-3 relaxation | Contraction (% of 60 mM KCl response) | % Reversal of RL-3 relaxation | Contraction (% of 60 mM KCl response) | % Reversal of RL-3 relaxation | |
| Linopirdine | 107.1 ± 24.0 | – | 133.6 ± 6.1 | – | 56.3 ± 11.3 | – |
| L-768,673 | −8.8 ± 7.6 | 106.8 ± 2.6 | 0.0 ± 0.0 | 71.2 ± 28.8 | 7.1 ± 2.8 | 52.6 ± 11.2 |
| HMR1556 | −12.1 ± 12.1 | 104.2 ± 5.9 | 0.0 ± 0.002 | 47.3 ± 20.6 | 11.1 ± 0.0 | 66.7 ± 33.3 |
| JNJ282 | −13.7 ± 10.3 | −7.6 ± 5.5 | 0.2 ± 0.5 | – | −3.3 ± 3.3 | – |
| pEC50 | Emax (%) | pEC50 | Emax (%) | pEC50 | Emax (%) | |
| RL-3 | 6.3 ± 0.4 | 83.4 ± 3.3 | 5.4 ± 0.2 | 53.9 ± 7.5 | 5.5 ± 0.2 | 41.5 ± 3.5 |
Application of linopirdine (10 µM) to segments of thoracic aorta caused robust contractions (approximately 130% of the KCl contraction; P < 0.01) but none of the Kv7.1 blockers had any effect on basal tone (Figure 2C and D). Intrapulmonary arteries have been described previously as preferential targets for Kv7 blockade compared with systemic arteries (Joshi et al., 2009). However in this study, 10 µM linopirdine had minimal contractile effects in the absence of any pre-tone but, like the mesenteric arteries, linopirdine evoked a substantial contraction (∼45% of KCl response; P < 0.01) when a low concentration of U46619 was applied. Similar to the aorta and mesenteric artery the Kv7.1 blockers did not contract intrapulmonary vessels under the same conditions where linopirdine produced a robust response (Figure 2E and F; Table 1).
Vasorelaxant effects of R-L3 and mefenamic acid
Having established that compounds that block either Kv7.1 or Kv7.1/KCNE1-encoded protein complexes (IKs) do not alter basal tension in mesenteric arteries, we investigated whether R-L3 and mefenamic acid, which augment Kv7.1 activity (Busch et al., 1997; Seebohm et al., 2003), had any functional impact. Application of the benzodiazepine R-L3 (0.1–3 µM) to segments of preconstricted mesenteric arteries produced concentration-dependent relaxations (pEC50: 6.3 ± 0.4; Emax: 83.4 ± 3.3%, n = 7) (Figure 3A and B). This relaxant effect was fully reversed by the Kv7.1 selective blockers L-768,673 and HMR1556 but not JNJ282 (Figure 3C) and was not affected by the removal of the endothelium (confirmed by lack of response to carbachol) with a mean relaxation of 88 ± 1% (n = 3) to 3 µM RL-3 under these conditions. It is worth highlighting that the responses to S-1, an agent that activates Kv7.2–7.5 but not Kv7.1 (Schenzer et al., 2005; Wuttke et al., 2005) and which has been shown previously to relax rat aorta and mesenteric artery (Jepps et al., 2011) were not effected by the Kv7.1 inhibitors. L-768,673 did not reverse relaxations (3.5 ± 4.5%; n = 3) evoked by 10 µM S-1 (Figure 3D), while tone generated in the presence of the Kv7.1 blockers was fully relaxed by 10 µM S-1 (n = 4, data not shown). Similar to the mesenteric artery, R-L3 (0.1–3 µM) caused concentration-dependent relaxation in the aorta (pEC50: 5.5 ± 0.2; Emax: 53.9 ± 7.5, n = 8; Figure 4A and B) and intrapulmonary arteries (pEC50: 5.5 ± 0.2; Emax: 41.5 ± 3.5%, n = 10; Figure 4C and D; Table 1), yet these responses were not as potent as those observed in the mesenteric arteries.
Figure 3.
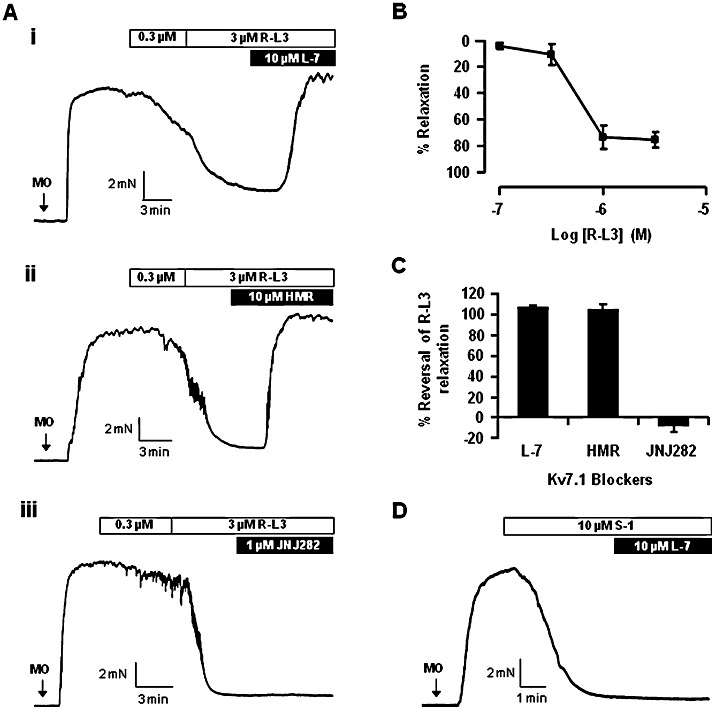
R-L3 evoked relaxation in mesenteric artery. Representative responses to R-L3 and subsequent exposure to Kv7.1 blockers, 10 µM L-768,673 (L-7; i), 10 µM HMR1556 (HMR; ii) or 1 µM JNJ282 (iii), in mesenteric arteries preconstricted with 10 µM methoxamine (A). Mean R-L3 concentration-dependent relaxation (B) and percentage reversal by Kv7.1 blockers (C). Representative relaxation to 10 µM S-1 in mesenteric arteries preconstricted with 10 µM methoxamine (MO) followed by subsequent exposure to 10 µM L-768,673 (L-7) is shown in (D).
Figure 4.
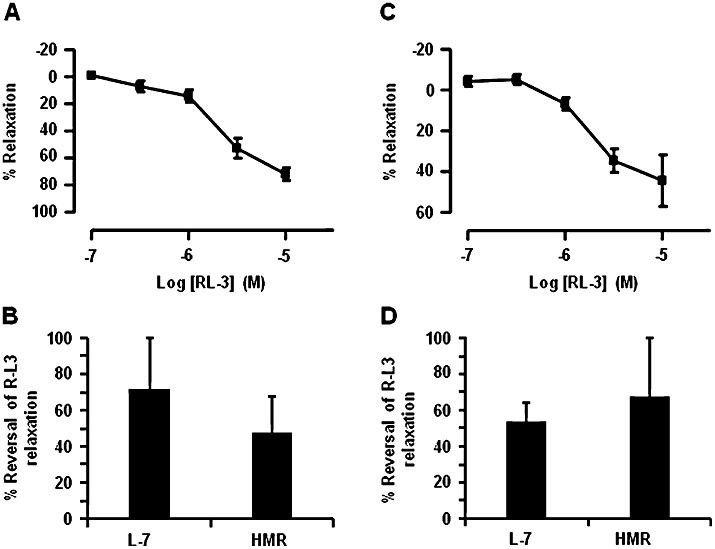
R-L3 evoked relaxation in thoracic aorta and intrapulmonary arteries. Mean R-L3 concentration-dependent relaxation and percentage reversal by Kv7.1 blockers, 10 µM L-768,673 and 10 µM HMR1556, in thoracic aorta (A and B) and intrapulmonary arteries (C and D) preconstricted with 3 µM methoxamine and 0.3 µM U46619, respectively.
In order to confirm the functional effect of activating Kv7.1 channels, an alternative Kv7.1 activator mefenamic acid was applied to preconstricted mesenteric arteries. Similar to RL-3, mefenamic acid (1–30 µM) relaxed these arteries in a concentration-dependent manner (Emax: 96.1 ± 0.8%, n = 3), which was reversed by 10 µM L-768,673 (Figure 5). These data are the first to show vascular effects of Kv7.1 stimulants and illustrates that both L-768,673 and HMR1556 were functionally active. Moreover, it provides strong evidence that Kv7.1-containing channel complexes are functionally relevant in the vasculature.
Figure 5.
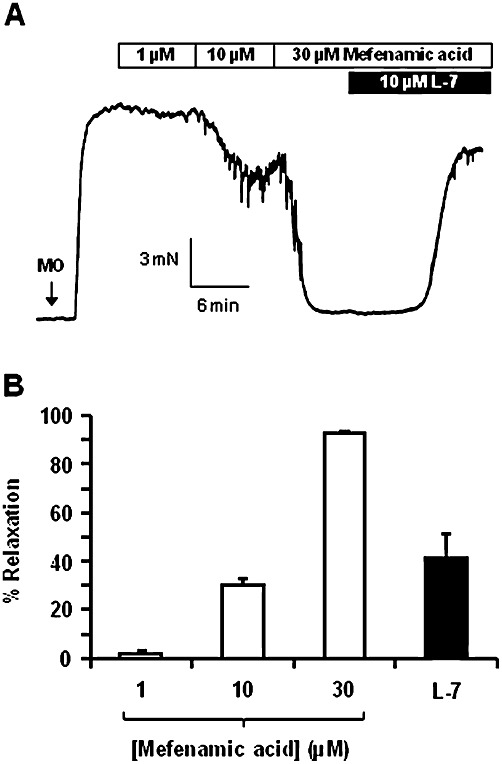
Mefenamic acid evoked relaxation in mesenteric artery. Representative trace (A) and mean data (B) of mefenamic acid concentration-responses in mesenteric arteries preconstricted with 10 µM methoxamine (MO) and subsequent blockade of Kv7.1 channels with 10 µM L-768,673 (L-7; n = 3).
Effect of Kv7.1 channel blockers on heterologously expressed KCNQ channels
Previous studies have shown L-768,673, HMR1556 and JNJ282 to be effective blockers of the current formed by the association of Kv7.1 with KCNE1-encoded proteins (termed IKs); the prominent repolarizing current in the heart (Seebohm et al., 2003; Towart et al., 2009). However, there are few data on the impact of these agents on Kv7.1 channels alone or on the other Kv7 channels expressed in the vasculature. In addition, the present functional data indicate that while L-768,673 and HMR1556 can reverse the effects of R-L3, JNJ282 cannot, therefore the differential effects of these compounds on Kv7.1 channel activity was investigated. Overexpression of KCNQ1 in Xenopus oocytes resulted in robust K+ currents in response to membrane depolarization (Figures 6 and 7). Application of 0.1 µM L-768,673 produced considerable inhibition of the KCNQ1-generated current at all test potentials (n = 8 oocytes). Application of 1 µM L-768,673 had no further effect on the evoked current. HMR1556 had a similar effect on KCNQ1-evoked K+ currents (data not shown). In contrast, JNJ282 only produced minimal inhibition at 1 and 10 µM, concentrations which are far higher than the IC50 for blockade of IKs by this compound (Towart et al., 2009). These experiments show that L-768,673 and HMR1556 were viable as Kv7.1 probes whereas JNJ282 was only effective against Kv7.1/KCNE1-encoded protein complexes. To provide more information about these agents the effect of L-768,673, HMR1556 and JNJ282 on currents produced by the overexpression of KCNQ4 and KCNQ5 was investigated. Figure 6 shows that none of the agents had an effect on the currents produced by the overexpression of KCNQ5 at concentrations that were functional in the isometric tension studies. However, L-768,673 but not HMR1556 nor JNJ282 had a pronounced inhibitory effect on KCNQ4 generated currents at potentials positive to 0 mV (Figure 6B, D and F). These studies provide a thorough assay of the selectivity of these agents for the different KV7 channels expressed in the vasculature.
Figure 6.
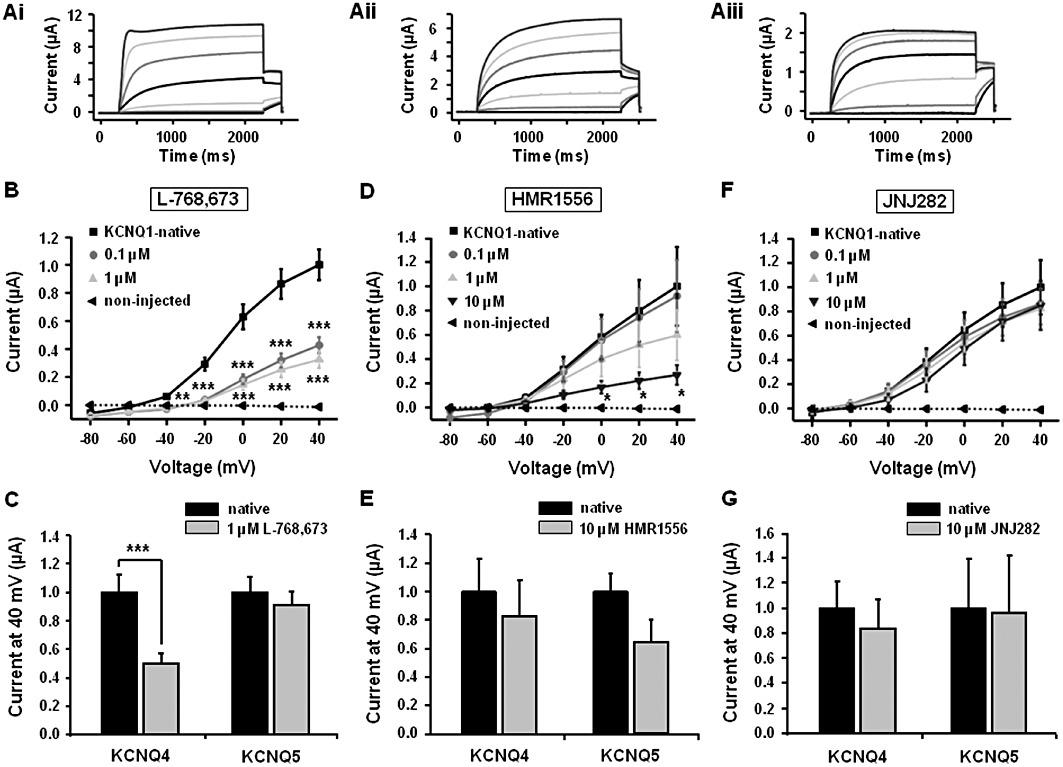
Effect of Kv7.1 blockers on currents produced by the overexpression of KCNQ1, 4 and 5 in Xenopus oocytes. Representative traces show currents produced by the overexpression of KCNQ1 (Ai), KCNQ4 (Aii) and KCNQ5 (Aiii) recorded at voltages between −80 mV and +40 mV. I/V curves of KCNQ1 as a summary of the current recordings obtained from oocytes in the presence of different concentrations of Kv7.1 channel blockers L-768,673 (n = 10; B), HMR 1556 (n = 9 to 10, D) and JNJ282 (n = 8; F). (C), (E) and (G) show the mean effects of L-768,673 (n = 8), HMR 1556 (n = 8 to 10) and JNJ282 (n = 2 to 5), respectively, on currents produced by the overexpression of KCNQ4 and KCNQ5 recorded at +40 mV. *P < 0.05, **P < 0.01 and ***P < 0.001 compared by Student's paired t-test.
Figure 7.
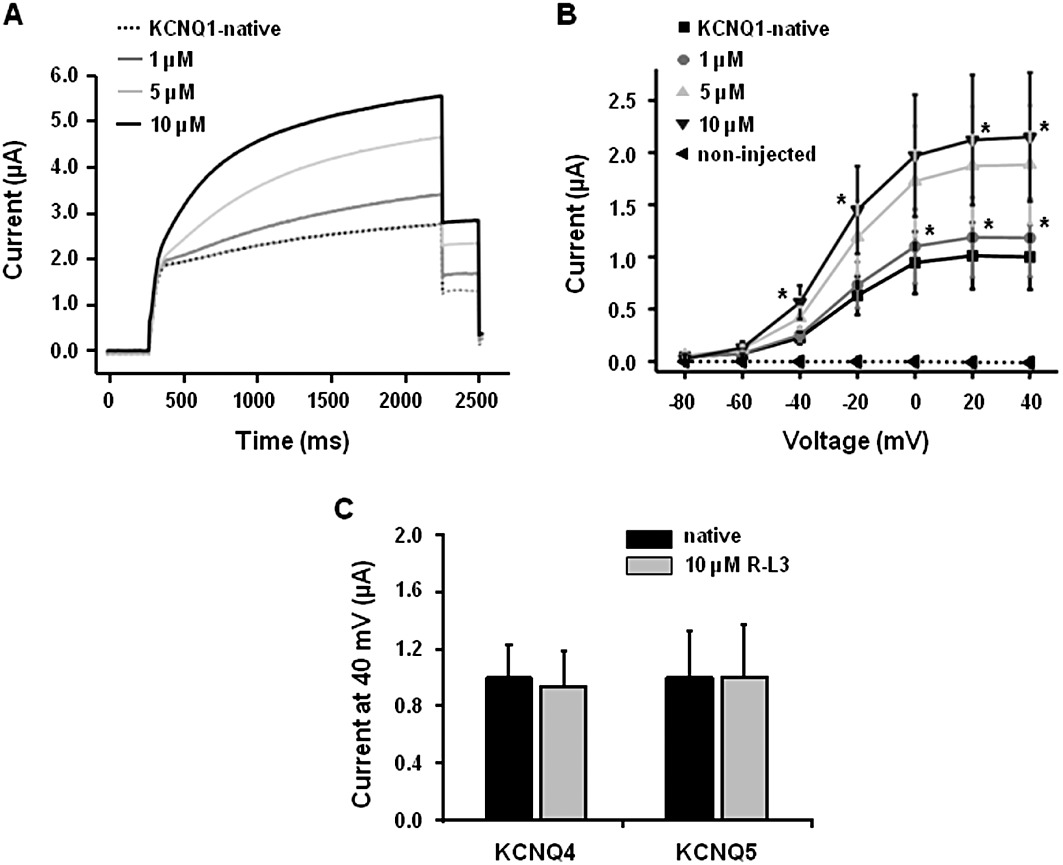
Effect of RL-3 on KCNQ1 expressed in Xenopus oocytes. (A) Representative effects of RL-3 (1–10 µM) on currents produced by the overexpression of KCNQ1. Oocytes were held at −80 mV stepped to +40 mV for 2 s and repolarized to −40 mV. Mean I/V data are shown in (B) (n = 7–9). (C) The lack of effect of 10 µM RL-3 on KCNQ4 and KCNQ5-generated currents (n = 7–9). *P < 0.05 compared by Student's paired t-test.
Effect of R-L3 on heterologously expressed KCNQ channels
The effectiveness of RL-3 as a vasorelaxant in all blood vessels tested provides considerable evidence that Kv7.1-containing channels are functionally relevant. However, the effects of this agent could be mediated by stimulation of the Kv7.4 and Kv7.5 channels that are present in the vasculature. Figure 7 demonstrates that at concentrations that had a pronounced effect on currents produced by the overexpression of KCNQ1 (Figure 7A and B) RL-3 had no effect on KCNQ4 or KCNQ5-generated currents (Figure 7C). These data support the functional studies with L-768,673 and HMR1556 that suggest the relaxant effect of RL-3 is due to enhanced Kv7.1 activity.
Discussion
The current study shows that Kv7.1 channels are expressed in the plasmalemmal membrane of smooth muscle cells from rat mesenteric arteries and thoracic aortae but three structurally different Kv7.1 channel or IKs blockers (L-768,673, HMR1556 and JNJ282) at concentrations in excess of their IC50 for these channels had no effect on resting vascular tone, in contrast to the effect of the pan-Kv7 blocker linopirdine. However, this study provides unique evidence that direct activation of Kv7.1 channels is a highly effective dilator mechanism in both systemic and pulmonary arteries.
KCNQ1-encoded (Kv7.1) channels in co-assembly with the translated protein of KCNE1 (kcne1 or MinK) comprise the slow component of the delayed rectifier K+ current (IKs) in cardiac myocytes (Barhanin et al., 1996; Sanguinetti et al., 1996). Mutations to KCNQ1 or KCNE1 underlie a number of hereditary arrhythmias leading to long QT syndrome (Herbert et al., 2002; Jespersen et al., 2005). Consequently, several IKs blockers including chromanol 293B and its more potent analogue HMR1556 (Gogelein et al., 2000), as well as the structurally different compound L-768,673, have been tested for use as potential anti-arrhythmic therapies. These drugs, as well as the highly potent compound JNJ282, reduce the IKs current and thus prolong repolarization and refractoriness leading to long QT (Antzelevitch, 2007). The effects of L-768,673, HMR1556 and JNJ282 on other cardiac ion channels (INa, ICa, IKr, Ito, IK1) reveal very high selectivity for IKs (Towart et al., 2009). The present study confirms that L-768,673 and HMR 1556 were good inhibitors of homomeric KCNQ1 channels, whereas JNJ282 was totally ineffective supporting the findings of Towart et al. (2009) that this compound is an inhibitor of the channel formed by the association of KCNQ1 + KCNE1 expression products that underlie the cardiac IKs. The results from the present study also show that neither HMR1556 nor JNJ282 affects currents generated by the expression of KCNQ4 or KCNQ5. L-768,673 also had no effect on KCNQ5-generated currents but reduced currents produced by KCNQ4 expression significantly. These studies provide an extensive addition to the pharmacological palette for studying KCNQ-encoded channels.
In previous studies it was observed that the selective Kv7.1 channel inhibitor chromanol 293B does not affect vascular tone, whereas under the same conditions the pan-Kv7 blockers linopiridne and XE991 contract arteries (Yeung and Greenwood, 2005; Yeung et al., 2007; Ng et al., 2011). These findings are now corroborated by the present study where linopirdine was shown to robustly contract systemic and pulmonary arteries, with the caveat that mesenteric and intrapulmonary vessels require a small degree of pre-tone, induced with a low concentration of methoxamine or U46619, respectively. This phenomenon has been observed previously in murine mesenteric arteries (Yeung et al., 2007), but appears to be contradictory to previous results in intrapulmonary vessels (Joshi et al., 2006) where contractions to XE991 and linopirdine did not require pre-tone and were of a similar magnitude to those elicited by 50 mM KCl. Although we cannot fully explain this discrepancy, it is worth noting that experimental conditions were not identical including differences in the normalized basal tension, which was ∼1 mN (equivalent of 20 mmHg), almost 80% lower than in previous studies (Joshi et al., 2006; 2009). Irrespective of these aspects, the simple message is that under the same conditions where linopiridne contracts blood vessels, compounds shown to selectively inhibit Kv7.1 channels have no effect. It is worth stressing that in the present study, none of the three different agents inhibited contractions elicited by 60 mM KCl proving that they did not block voltage-dependent calcium channels, which would preclude any contractile effect.
These data, in addition to previous observations with chromanol 293B, confirm that Kv7.1 channels are unlikely to contribute to resting vascular tone. However, due to the consistent expression of these channels in the vasculature, the persisting question of their function remains. A working scenario is that Kv7.1 channels under resting conditions are in a closed state and that direct activation would elicit a change in arterial tone. To test this hypothesis we used two IKs activators namely R-L3 and mefenamic acid (Busch et al., 1994; 1997). The benzodiazepine R-L3 at concentrations <10 µM potentiates IKs (Salata et al., 1998) by interacting with the fifth and sixth transmembrane domains of Kv7.1 subunits, binding sites that are distinct from where the benzodiazepine antagonist L-768,673 interacts (Seebohm et al., 2003). In the present study, RL-3 enhanced currents produced by the overexpression of KCNQ1 but had no effect on KCNQ4 or KCNQ5-generated currents and relaxed preconstricted arteries. Interestingly, the potency of R-L3 in mesenteric vessels was greater than the dilator effects of the Kv7.2–7.5 channel activators retigabine, S-1, BMS-205352 and flupirtine (Mackie et al., 2008; Jepps et al., 2011). The finding that R-L3 relaxations were completely reversed by HMR1556 or L-768,673 confirmed that the dilator responses observed were due to modulation of Kv7.1 channels. Similarly, mesenteric arteries were relaxed by mefenamic acid, although with less potency than R-L3; however, these responses were only partially reversed by L-768,673, suggesting some non-selective effects of mefenamic acid such as blockade of Ca2+-activated Cl− channels (Greenwood and Large, 1995) at the higher concentrations used. Preconstricted thoracic aortae and intrapulmonary arteries were relaxed by ∼70% and 45%, respectively, in response to 10 µM R-L3. These relaxations were reversed by 50–60% in the presence of HMR1556 or L-768,673 indicating that some effects of R-L3 at this higher concentration was not due to stimulation of Kv7.1 channels. Interestingly, R-L3 relaxations were not reversed by JNJ282 at concentrations that did not inhibit calcium channels. The lack of effect of JNJ282 cannot be fully explained, due to the lack of knowledge of the molecular mode of inhibition by the compound. However, in contrast to HMR1556 and L-768,673, which inhibit IKs as well as KCNQ1 alone (Seebohm et al., 2003; present study), the poor interaction of JNJ282 with monomeric Kv7.1 isoforms (Figure 6) while being an effective IKs blocker (Towart et al., 2009) may provide a possible explanation for the discrepency between the actions of these drugs. At present it is not possible to determine why Kv7.1-containing channels do not seem to contribute to the setting of the membrane potential in these blood vessels. One possibility is that in the vascular smooth muscle, Kv7.1 associates with the product of KCNE4 genes expressed in blood vessels (Yeung et al., 2007; Zhong et al., 2010), which are known to effectively silence Kv7.1 activity (Grunnet et al., 2003). Alternatively, a situation analogous to that seen in cardiac myocytes may exist where other potassium channels provide sufficient hyperpolarization to cover the lost control provided by KV7.1 blockade (termed ‘repolarization reserve’). Future work will concentrate on both of these aspects.
In conclusion, the results in this study dispel previous assumptions that Kv7.1 channels are functionally redundant in vascular smooth muscle. We have shown using a variety of potent Kv7.1 modulators that although these channels do not contribute to resting vascular tone they can evoke profound relaxation upon stimulation, particularly in resistance-like systemic arteries. Interestingly, it is unlikely to be the cardiac Kv7.1/MinK complex that exists in blood vessels. We speculate that vascular Kv7.1 channels may in part underlie vasodilatation pathways involved in rectifying vascular tone. It remains a challenge to determine the molecular architecture of vascular Kv7 channels and the respective contributions to physiological control of vascular tone.
Acknowledgments
We thank the staff at the Image Resource Facility for their technical assistance with the fluorescent microscopy, and the assistance of all the staff from the Biological Research Facility at St. George's, University of London. PSC and AJD were funded by British Heart Foundation grants to IAG. (PG/09/104 and PG\07\127\24235). TAJ was funded by a BBSRC-CASE studentship (BB/G016321/1) in association with NeuroSearch A/S. MS received funding from the Deutsche Forschungsgemeinschaft (SCHW866/4-1 and SFB877).
Glossary
- HMR1556
N-(6-cyano-3-hydroxy-2,2-dimethyl-3,4-dihydrochromen-4-yl)-N-methylethanesulfonamide
- JNJ39490282
2-(2-bromo-4-chlorophenoxy)-2-methyl-N-{5 [(methylsulfonyl)amino]tricyclo[3.3.1.13,7]dec-2-yl}propanamide
- L-768
6732-[2,4-bis(trifluoromethyl)phenyl]-N-[(3R)-2-oxo-5-phenyl-1-(2,2,2-trifluoroethyl)-3H-1,4-benzodiazepin-3-yl]acetamide
- RL-3
[(3-R)-1, 3-dihydro-5-(2-fluorophenyl)-3-(1H-indol-3-ylmethyl)-1-methyl-2H- 1,4-benzodiazepin-2-one]
Conflicts of interest
None.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th Edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antzelevitch C. Ionic, molecular, and cellular bases of QT-interval prolongation and torsade de pointes. Europace. 2007;9(Suppl. 4):iv4–i15. doi: 10.1093/europace/eum166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature. 1996;384:78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- Bentzen BH, Schmitt N, Calloe K, Brown DW, Grunnet M, Olesen SP. The acrylamide (S)-1 differentially affects Kv7 (KCNQ) potassium channels. Neuropharmacology. 2006;51:1068–1077. doi: 10.1016/j.neuropharm.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Bett GC, Morales MJ, Beahm DL, Duffey ME, Rasmusson RL. Ancillary subunits and stimulation frequency determine the potency of chromanol 293B block of the KCNQ1 potassium channel. J Physiol. 2006;576:755–767. doi: 10.1113/jphysiol.2006.116012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch AE, Herzer T, Wagner CA, Schmidt F, Raber G, Waldegger S, et al. Positive regulation by chloride channel blockers of IsK channels expressed in Xenopus oocytes. Mol Pharmacol. 1994;46:750–753. [PubMed] [Google Scholar]
- Busch AE, Busch GL, Ford E, Suessbrich H, Lang HJ, Greger R, et al. The role of the IsK protein in the specific pharmacological properties of the IKs channel complex. Br J Pharmacol. 1997;122:187–189. doi: 10.1038/sj.bjp.0701434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis AJ, Forrest AS, Jepps TA, Valencik ML, Wiwchar M, Singer CA, et al. Expression profile and protein translation of TMEM16A in murine smooth muscle. Am J Physiol Cell Physiol. 2010;299:C948–C959. doi: 10.1152/ajpcell.00018.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogelein H, Bruggemann A, Gerlach U, Brendel J, Busch AE. Inhibition of IKs channels by HMR 1556. Naunyn Schmiedebergs Arch Pharmacol. 2000;362:480–488. doi: 10.1007/s002100000284. [DOI] [PubMed] [Google Scholar]
- Greenwood IA, Large WA. Comparison of the effects of fenamates on Ca-activated chloride and potassium currents in rabbit portal vein smooth muscle cells. Br J Pharmacol. 1995;116:2939–2948. doi: 10.1111/j.1476-5381.1995.tb15948.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood IA, Ohya S. New tricks for old dogs: KCNQ expression and role in smooth muscle. Br J Pharmacol. 2009;156:1196–1203. doi: 10.1111/j.1476-5381.2009.00131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunnet M, Rasmussen HB, Hay-Schmidt A, Rosenstierne M, Klaerke DA, Olesen SP, et al. KCNE4 is an inhibitory subunit to Kv1.1 and Kv1.3 potassium channels. Biophys J. 2003;85:1525–1537. doi: 10.1016/S0006-3495(03)74585-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert E, Trusz-Gluza M, Moric E, Smilowska-Dzielicka E, Mazurek U, Wilczok T. KCNQ1 gene mutations and the respective genotype-phenotype correlations in the long QT syndrome. Med Sci Monit. 2002;8:RA240–RA248. [PubMed] [Google Scholar]
- Jepps TA, Chadha PS, Davis AJ, Harhun MI, Cockerill GW, Olesen SP, et al. Downregulation of Kv7.4 channel activity in primary and secondary hypertension. Circulation. 2011;124:602–611. doi: 10.1161/CIRCULATIONAHA.111.032136. [DOI] [PubMed] [Google Scholar]
- Jespersen T, Grunnet M, Olesen SP. The KCNQ1 potassium channel: from gene to physiological function. Physiology (Bethesda) 2005;20:408–416. doi: 10.1152/physiol.00031.2005. [DOI] [PubMed] [Google Scholar]
- Joshi S, Balan P, Gurney AM. Pulmonary vasoconstrictor action of KCNQ potassium channel blockers. Respir Res. 2006;7:31. doi: 10.1186/1465-9921-7-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi S, Sedivy V, Hodyc D, Herget J, Gurney AM. KCNQ modulators reveal a key role for KCNQ potassium channels in regulating the tone of rat pulmonary artery smooth muscle. J Pharmacol Exp Ther. 2009;329:368–376. doi: 10.1124/jpet.108.147785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang C, Tian C, Sonnichsen FD, Smith JA, Meiler J, George AL, et al. Structure of KCNE1 and implications for how it modulates the KCNQ1 potassium channel. Biochemistry. 2008;47:7999–8006. doi: 10.1021/bi800875q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knollmann BC, Sirenko S, Rong Q, Katchman AN, Casimiro M, Pfeifer K, et al. Kcnq1 contributes to an adrenergic-sensitive steady-state K+ current in mouse heart. Biochem Biophys Res Commun. 2007;360:212–218. doi: 10.1016/j.bbrc.2007.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerche C, Seebohm G, Wagner CI, Scherer CR, Dehmelt L, Abitbol I, et al. Molecular impact of MinK on the enantiospecific block of I(Ks) by chromanols. Br J Pharmacol. 2000;131:1503–1506. doi: 10.1038/sj.bjp.0703734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerche C, Bruhova I, Lerche H, Steinmeyer K, Wei AD, Strutz-Seebohm N, et al. Chromanol 293B binding in KCNQ1 (Kv7.1) channels involves electrostatic interactions with a potassium ion in the selectivity filter. Mol Pharmacol. 2007;71:1503–1511. doi: 10.1124/mol.106.031682. [DOI] [PubMed] [Google Scholar]
- Mackie AR, Brueggemann LI, Henderson KK, Shiels AJ, Cribbs LL, Scrogin KE, et al. Vascular KCNQ potassium channels as novel targets for the control of mesenteric artery constriction by vasopressin, based on studies in single cells, pressurized arteries, and in vivo measurements of mesenteric vascular resistance. J Pharmacol Exp Ther. 2008;325:475–483. doi: 10.1124/jpet.107.135764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng FL, Davis AJ, Jepps TA, Harhun MI, Yeung SY, Wan A, et al. Expression and function of the K+ channel KCNQ genes in human arteries. Br J Pharmacol. 2011;162:42–53. doi: 10.1111/j.1476-5381.2010.01027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohya S, Sergeant GP, Greenwood IA, Horowitz B. Molecular variants of KCNQ channels expressed in murine portal vein myocytes: a role in delayed rectifier current. Circ Res. 2003;92:1016–1023. doi: 10.1161/01.RES.0000070880.20955.F4. [DOI] [PubMed] [Google Scholar]
- Salata JJ, Jurkiewicz NK, Wang J, Evans BE, Orme HT, Sanguinetti MC. A novel benzodiazepine that activates cardiac slow delayed rectifier K+ currents. Mol Pharmacol. 1998;54:220–230. doi: 10.1124/mol.54.1.220. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, et al. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature. 1996;384:80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- Schenzer A, Friedrich T, Pusch M, Saftig P, Jentsch TJ, Grotzinger J, et al. Molecular determinants of KCNQ (Kv7) K+ channel sensitivity to the anticonvulsant retigabine. J Neurosci. 2005;25:5051–5060. doi: 10.1523/JNEUROSCI.0128-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder BC, Waldegger S, Fehr S, Bleich M, Warth R, Greger R, et al. A constitutively open potassium channel formed by KCNQ1 and KCNE3. Nature. 2000;403:196–199. doi: 10.1038/35003200. [DOI] [PubMed] [Google Scholar]
- Seebohm G, Pusch M, Chen J, Sanguinetti MC. Pharmacological activation of normal and arrhythmia-associated mutant KCNQ1 potassium channels. Circ Res. 2003;93:941–947. doi: 10.1161/01.RES.0000102866.67863.2B. [DOI] [PubMed] [Google Scholar]
- Towart R, Linders JT, Hermans AN, Rohrbacher J, van der Linde HJ, Ercken M, et al. Blockade of the I(Ks) potassium channel: an overlooked cardiovascular liability in drug safety screening? J Pharmacol Toxicol Methods. 2009;60:1–10. doi: 10.1016/j.vascn.2009.04.197. [DOI] [PubMed] [Google Scholar]
- Wuttke TV, Seebohm G, Bail S, Maljevic S, Lerche H. The new anticonvulsant retigabine favors voltage-dependent opening of the Kv7.2 (KCNQ2) channel by binding to its activation gate. Mol Pharmacol. 2005;67:1009–1017. doi: 10.1124/mol.104.010793. [DOI] [PubMed] [Google Scholar]
- Yeung SY, Greenwood IA. Electrophysiological and functional effects of the KCNQ channel blocker XE991 on murine portal vein smooth muscle cells. Br J Pharmacol. 2005;146:585–595. doi: 10.1038/sj.bjp.0706342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung SY, Pucovsky V, Moffatt JD, Saldanha L, Schwake M, Ohya S, et al. Molecular expression and pharmacological identification of a role for K(v)7 channels in murine vascular reactivity. Br J Pharmacol. 2007;151:758–770. doi: 10.1038/sj.bjp.0707284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung SY, Lange W, Schwake M, Greenwood IA. Expression profile and characterisation of a truncated KCNQ5 splice variant. Biochem Biophys Res Commun. 2008;371:741–746. doi: 10.1016/j.bbrc.2008.04.129. [DOI] [PubMed] [Google Scholar]
- Zhong XZ, Harhun MI, Olesen SP, Ohya S, Moffatt JD, Cole WC, et al. Participation of KCNQ (Kv7) potassium channels in myogenic control of cerebral arterial diameter. J Physiol. 2010;588:3277–3293. doi: 10.1113/jphysiol.2010.192823. [DOI] [PMC free article] [PubMed] [Google Scholar]