

Abstract
BACKGROUND AND PURPOSE
Recent studies detected the expression of proteins involved in cholinergic metabolism in airway epithelial cells, although the function of this non-neuronal cholinergic system is not known in detail. Thus, this study focused on the effect of luminal ACh as a regulator of transepithelial ion transport in epithelial cells.
EXPERIMENTAL APPROACH
RT-PCR experiments were performed using mouse tracheal epithelial cells for ChAT and organic cation transporter (OCT) transcripts. Components of tracheal airway lining fluid were analysed with desorption electrospray ionization (DESI) MS. Effects of nicotine on mouse tracheal epithelial ion transport were examined with Ussing-chamber experiments.
KEY RESULTS
Transcripts encoding ChAT and OCT1–3 were detected in mouse tracheal epithelial cells. The DESI experiments identified ACh in the airway lining fluid. Luminal ACh induced an immediate, dose-dependent increase in the transepithelial ion current (EC50: 23.3 µM), characterized by a transient peak and sustained plateau current. This response was not affected by the Na+-channel inhibitor amiloride. The Cl−-channel inhibitor niflumic acid or the K+-channel blocker Ba2+ attenuated the ACh effect. The calcium ionophore A23187 mimicked the ACh effect. Luminal nicotine or muscarine increased the ion current. Experiments with receptor gene-deficient animals revealed the participation of muscarinic receptor subtypes M1 and M3.
CONCLUSIONS AND IMPLICATIONS
The presence of luminal ACh and activation of transepithelial ion currents by luminal ACh receptors identifies a novel non-neuronal cholinergic pathway in the airway lining fluid. This pathway could represent a novel drug target in the airways.
Keywords: airway epithelium, acetylcholine, acetylcholine receptors, muscarinic receptors, nicotinic receptors, non-neuronal cholinergic system, transepithelial ion transport, auto-/paracrine
Introduction
The function of ACh as a neurotransmitter has long been known and is well established. Additionally, recent studies indicate that ACh might also play a role as an auto- and/or paracrine mediator in many non-nervous tissues and cells that modulate cell proliferation and differentiation (Wessler et al., 2003). In contrast to its role as a neurotransmitter, ACh that is synthesized in and secreted from non-nervous cells is termed non-neuronal ACh and the metabolism of ACh in these cells is referred to as the non-neuronal cholinergic system (nnCS) (Wessler et al., 2003). Components of the nnCS consisting of all enzymes, transporters and receptors needed for ACh synthesis, release and degradation have been identified in placenta (Wessler et al., 2001; 2003), urothelium (Zarghooni et al., 2007) and airway epithelia (Kummer et al., 2008). The basic components comprising the nnCS are similar to the ones found in the neuronal cholinergic system (Wessler et al., 2003; Kummer et al., 2008).
ACh receptors responsible for initiating the cellular responses of ACh are differentiated as muscarinic and nicotinic ACh receptors. Five different G-protein coupled muscarinic ACh receptor subtypes (M1–M5) are known (Racke and Matthiesen, 2004). In contrast to muscarinic ACh receptors, nicotinic ACh receptors form pentamers of different homologous subunits (α1–10, β2–4, γ, δ, ε) with a central membrane-spanning pore and thus constitute ion channels that allow cations, especially Na+ and Ca2+, to pass through the membrane upon activation (Dani, 2001; Racke and Matthiesen, 2004).
On the one hand, the cholinergic system of the lungs is primarily known as a mediator of the contraction of bronchial smooth muscle cells and its involvement in obstructive pulmonary diseases like asthma and chronic obstructive pulmonary disease (COPD) (Belmonte, 2005; Gosens et al., 2006). However, different muscarinic and nicotinic receptor subunits have also been identified in pulmonary epithelial cells (Mak et al., 1992; Maus et al., 1998; Kummer et al., 2008; Klein et al., 2009). In addition, organic cation transporters (OCTs), possible candidates for the release of ACh from the cells, have been found in these epithelia (Kummer et al., 2006; 2008), as well as the ACh synthesizing enzyme ChAT (Klapproth et al., 1997; Kummer et al., 2006; 2008) and ACh itself (Klapproth et al., 1997; Kummer et al., 2006). Nevertheless, knowledge about the particular function of the nnCS in airway epithelia remains unknown.
The activation of nicotinic and muscarinic receptors, as known from excitable cells, is connected with the activation of ion channels or the modulation of ion channel activity (Albuquerque et al., 1997; Dani, 2001; Racke and Matthiesen, 2004). Therefore it is reasonable to assume that ACh receptors expressed in non-excitable cells such as airway epithelial cells might also interfere with ion transport processes. So far there is no detailed knowledge of the action of non-neuronal ACh on the ion transport processes in tracheal epithelium. Accordingly, the possibility that ion transport mechanisms in the tracheal epithelium are influenced by non-neuronal ACh, still needs to be elucidated.
Investigation of the connections between the action of ACh and a possible regulation of ion transport mechanisms may be important for further developments of treatments for diseases in which ion transport in the lung is pathologically altered, like, for example, cystic fibrosis (CF). A connection between the nnCS and CF disease has been detected in the lung and blood cells (Wessler et al., 2007). CF patients had a reduced ACh content in bronchi, lung parenchyma and peripheral leucocytes (Wessler et al., 2007). Taking this into account, the aim of this study was to perform a basic characterization of the effect of non-neuronal (luminal) ACh on ion transport processes in mouse tracheal epithelium to gain a better physiological understanding of the functional role of the nnCS in epithelial physiology. Our results add evidence to the existence of the nnCS in mouse tracheal epithelium and demonstrate that luminal ACh modulates Ca2+-dependent luminal chloride and basolateral potassium secretions. These findings identify a novel non-neuronal cholinergic pathway in the airway lining fluid.
Methods
Animals
C57Bl/6 mice (10–17 weeks) were obtained from the local animal breeding facility (Justus-Liebig-University, Giessen, Germany) or Charles River (Sulzfeld, Germany). M1 and M3 muscarinic ACh receptor-deficient mice (M1R−/−, M3R−/−) were acquired from the local animal breeding facility. The generation of M1R−/− and M3R−/− mice was as described previously (Miyakawa et al., 2001; Yamada et al., 2001; Fisahn et al., 2002). All mice were kept under standard conditions and killed with an overdose of the narcotic isoflurane (Abbot, Wiesbaden, Germany) followed by aortic exsanguination. All animal care and experimental procedures were carried out in accordance with the law on the protection of animals in Germany.
RT-PCR experiments
Epithelial cells from tracheae of C57Bl/6 mice were abraded by rolling cotton swabs over the epithelial layer. Total RNA was extracted using the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's protocol. Contaminating DNA was degraded using 1 U DNase-I (Invitrogen, Karlsruhe, Germany) µg−1 total RNA. RNA was reverse-transcribed with superscript II reverse transcriptase (Invitrogen) for 50 min at 42°C. The cDNAs were amplified with gene-specific primer pairs (Table 1; MWG, Ebersberg, Germany). For subsequent RT-PCR, 2.5 µL buffer II, 1.5–2 µL MgCl2 (25 mM), 0.625 µL dNTP (10 mM), 0.625 µL primer (20 pM), and 0.125 µL AmpliTaq Gold polymerase (5 U·µL−1; Applied Biosystems, Branchburg, NJ, USA) were supplemented with H2O to a final volume of 25 µL. Cycling conditions were 12 min at 95°C, 45 s at the gene-specific annealing temperature between 57–61°C, and 45 s at 72°C, and a final extension at 72°C for 7 min. PCR products were separated by electrophoresis on a 1.25% TRIS-acetate-EDTA gel. Performed controls were: (1) samples processed without reverse transcription (–RT); (2) RT-PCR without template (H2O); (3) primers for glyceraldehyde-3-phosphate-dehydrogenase (GAPDH) and β-microglobulin (βMG); and (4) appropriate mouse tissues with confirmed expression (not depicted in the figures).
Table 1.
Oligonucleotide primers for OCT1, OCT2, OCT3, ChAT, βMG and GAPDH in RT-PCR analysis
| Primer | Sequence | Product length (bp) | Accession number | |
|---|---|---|---|---|
| OCT1 | Forward | GTAAGCTCTGCCTCCTGGTG | 186 | NM_009202 |
| Reverse | GCTGTCGTTCTCCTGTAGCC | |||
| OCT2 | Forward | TACCGGAGTCTCCAAGATGG | 169 | NM_013667 |
| Reverse | GACCAAGTCCAGGAACGAAG | |||
| OCT3 | Forward | CAGATATGGCAGGCTCATCA | 160 | NM_011395 |
| Reverse | TCACGATCACGAAGCAAGTC | |||
| ChAT | Forward | CCTGCCAGTCAACTCTAGCC | 183 | NM_009891 |
| Reverse | TCAGGGCAGCCTCTCTGTAT | |||
| ChAT | Forward | GCCCACCTATGAGAGTGCAT | 178 | NM_009891 |
| Reverse | GTACTGAGTTTGGGCCTGGA | |||
| βMG | Forward | ATGGGAAGCCGTAACATACTG | 176 | NM_205783 |
| Reverse | CAGTCTCAGTGGGGGTGAAT | |||
| GAPDH | Forward | CGTCTTCACCACCATGGAGA | 299 | AF106860 |
| Reverse | CGGCCATCACGCCACAGCTT |
Desorption electrospray ionization (DESI) measurements
For measurements of the ACh content, tracheae from two mice were removed immediately after they had been killed, cut open, placed on a glass slide and frozen on dried ice. DESI measurement experiments were conducted as established by Takats et al. (2004). The tracheal preparations were placed in front of the spray capillary and the aqueous solvent was sprayed on the tracheal samples at a defined flow rate under the influence of a voltage of 4 kV. The resulting electrostatic and pneumatic forces, led to desorption of ions from the sample. The ions were transferred to the mass spectrometer (Finnigan LTQ Orbitrap Discovery, Thermo Fisher Scientific, Waltham, MA, USA) via an atmospheric pressure ion transfer line. Mass spectra were recorded by using LTQ Tune and Xcalibur.
Ussing chamber
Transepithelial tracheal ion transport of C57Bl/6, M1R−/− and M3R−/− mice was investigated with the Ussing-chamber technique. Freshly isolated, longitudinally opened tracheae were mounted into the Ussing-chamber and both compartments were continuously perfused with buffer solution (in mM): 145 NaCl; 5 d-glucose; 5 HEPES; 1.6 K2HPO4; 1.3 Ca2+-gluconate; 1 MgCl2; 0.4 KH2PO4; pH 7.4. Experiments were performed at 37°C.
The Ussing-chamber was connected to a voltage-clamp amplifier via Ag/AgCl-electrodes linked to the bathing compartments with bridges of 2% agar in 3 M KCl. All experiments were carried out under short-circuit conditions. The short-circuit current (ISC) was monitored on a strip chart recorder (Phillips, Amsterdam, Netherlands) and recorded via a MacLab/2e interface and the Chart program (ADInstruments, Spechbach, Germany) on a Macintosh LCII computer.
Chemicals
NaCl, d-glucose, MgCl2, Ca2+-gluconate, BaCl2 and ouabain were obtained from Fluka (Taufkirchen, Germany), KH2PO4 and K2HPO4 from SERVA (Heidelberg, Germany) and HEPES, nicotine-d-di-tartrate-salt, muscarine chloride, amiloride, niflumic acid (NFA), tetrodotoxin (TTX), carbachol, ACh chloride, mannitol, apamin, clotrimazole and 2-aminoethyl-diphenyl-borinate (2-APB) from SIGMA (Taufkirchen, Germany). Iberiotoxin, NPPB and A23187 were purchased from TOCRIS (Eching, Germany) and charybdotoxin from Santa Cruz Biotechnology (Heidelberg, Germany).
Statistics
Values are presented as mean ± SEM, n represents the number of experiments. Statistical significance was evaluated by use of Student's paired or the unpaired t-test. Significantly different values (P < 0.05) are marked with asterisks (*).
Nomenclature
The nomenclature of the drugs and molecular targets represented by ion channels and receptors conforms to the Guide to Receptors and Channels of the British Journal of Pharmacology (Alexander et al., 2011).
Results
Expression of components belonging to the cholinergic system
To investigate if components of the nnCS are expressed in mouse tracheal epithelial cells, reverse transcription PCR (RT-PCR) experiments were performed using primers directed against transcripts of the ChAT and OCT1–3 all of which belong to the nnCS.
ChAT is the main synthesizing enzyme for ACh. In our RT-PCR experiments with two different ChAT primer pairs (ChAT1 and ChAT2; Figure 1A) products of appropriate sizes (approximately 200 bp) were visible in their respective lanes. Transcripts for ChAT were detected with both primers in five donors. There was no band in the H2O control.
Figure 1.
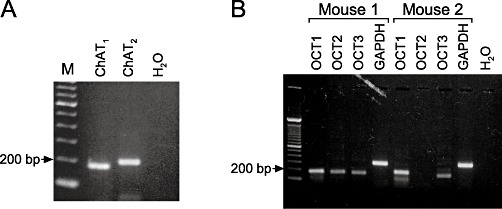
RT-PCR of components of the nnCS in mouse tracheal epithelial cells. (A) RT-PCR of ChAT in mouse tracheal epithelial cells. RT-PCR experiments showed a prominent ChAT band at about 200 bp with two different primers (ChAT1, ChAT2). H2O control is without template (M: size marker). (B) RT-PCR of OCT in mouse tracheal epithelium. Depicted are RT-PCR results of OCT1-3 from two of three mice. A RT-PCR product for OCT1 and OCT3 was visible at a size of about 200 bp in all tissue samples. For OCT2 the band at 200 bp only occurred in two out of three tissue samples. GAPDH and H2O denote the controls (M: size marker).
OCTs are known to represent one possible way of releasing ACh from epithelial cells (Kummer et al., 2006). Therefore, RT-PCR with primers against the subtypes OCT1, OCT2 and OCT3 was performed. The results obtained with samples from two different animals are depicted in Figure 1B. The results of mouse 3 were the same as for mouse 1 (data not shown). OCT1 and OCT3 transcripts were detected in samples of all mice, whereas OCT2 transcripts were only found in samples derived from two of three animals. GAPDH served as positive control for the efficiency of RNA isolation and cDNA synthesis and H2O was used in RT-PCR runs without template.
In summary transcripts of the ACh-synthesizing enzyme ChAT and OCT1-3 are expressed in mouse tracheal epithelium.
Detection of ACh with DESI
To investigate whether ACh is present on the luminal side of the mouse trachea, we analysed the surface of the tracheal epithelium with DESI measurements. These mass spectrometry experiments detected the presence of ACh as evidenced by the peak at the size of m/z 146.1170 (Figure 2). Thus, ACh is not only synthesized by ChAT in the tracheal epithelial cells but also luminally released and present in the airway lining fluid and thus is able to act on the epithelium from the luminal side.
Figure 2.
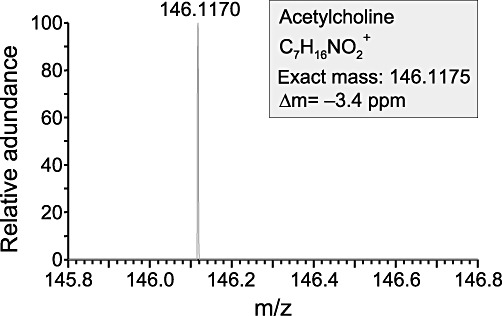
Positive DESI mass spectrum of the luminal surface of the mouse trachea. The y axis of the spectrum shows the relative abundance, after automatic scaling of the intensity of the highest signal in the depicted mass range to 100%. ACh was identified in the signal at m/z 146.1170. Since ACh was the only signal detected in the depicted mass range, it was automatically scaled to 100%. The label Δm =−3.4 expresses the exactness of the measured mass of ACh compared with the theoretically calculated mass of ACh.
Luminal ACh activates ISC in tracheal epithelia of mice
For functional characterization of the influence of ACh on the ion transport of mouse tracheal epithelium 100 µM ACh was applied to the luminal side of the epithelium for approximately 5 min. ACh application induced an increase in the transepithelial ISC (Figure 3A). This ACh-induced rise in ISC consisted of an immediate fast transient peak with an increase in IACh of 26.4 ± 1.5 µA cm−2, thereby doubling the baseline ISC (109% of control, n= 102, P < 0.001; Figure 3B) followed by a lower plateau phase (before wash out of ACh) with an IACh of 8.9 ± 0.7 µA cm−2 (38% of control, n= 102, P < 0.001; Figure 3B). The ISC dropped to baseline level upon wash out of ACh demonstrating that the effect of ACh on ISC was completely reversible (data not shown). The ACh effect was dose-dependent with an EC50 of 23.3 µM (Figure 3C).
Figure 3.
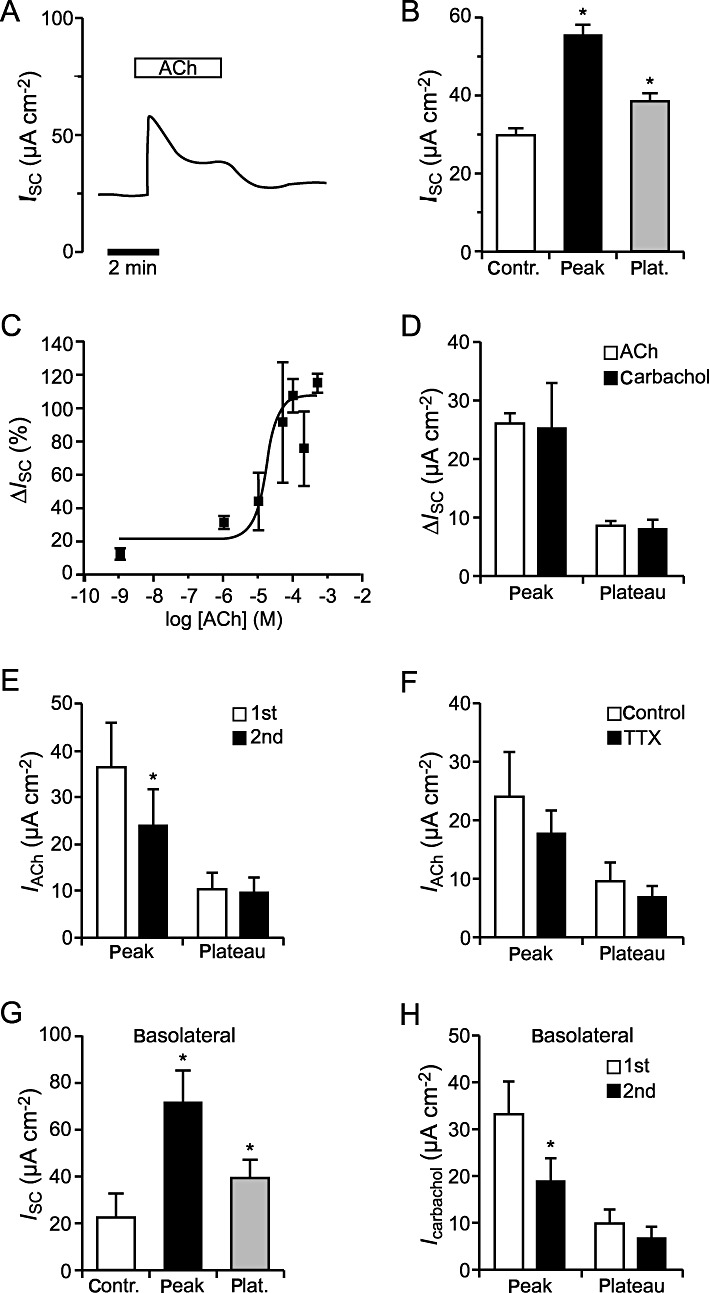
Effects of ACh and carbachol on the ISC of mouse tracheal epithelium. (A) Luminal application of 100 µM ACh led to an increase in the ISC that consisted of a transient peak and a lower plateau, as visible in the typical current trace. (B) Statistical analysis showed a significantly elevated level of the ISC in the presence of ACh compared with baseline current. This was the case for the peak (n= 102, P < 0.001) as well as the plateau component (n= 102, P < 0.001). Data are presented as mean ± SEM. (C) The effect of luminally applied ACh was dose-dependent with an EC50 of 23.3 µM. (D) Comparison of the applications of carbachol (n= 6) and ACh (n= 102) showed no significant differences (peak: P= 0.91, plateau: P= 0.65). (E) The ACh-induced peak current was significantly reduced upon the second application of ACh (n= 7, P < 0.05), whereas the plateau currents were similar (n= 7, P= 0.63). (F) The ACh-induced current increase (IACh) in the presence of 1 µM TTX basolateral was not different from control conditions (n= 7, peak: P= 0.47, plateau: P= 0.52). (G) Similar to luminal ACh application, basolateral application of 100 µM ACh led to a significant increase in the ISC compared with baseline current. This was the case for the peak (n= 5, P < 0.05) as well as the plateau component (n= 5, P < 0.05). (H) Comparison of the current increase induced by repeated application of the cholinoceptor agonist carbachol to the basolateral side of the epithelium, revealed that the carbachol-induced peak current was significantly reduced upon the second application of carbachol (n= 5, P < 0.01), whereas the plateau currents were similar (n= 5, P= 0.39).
Similar results were obtained with a different cholinergic agonist. Application of 100 µM carbachol (n= 6) to the luminal side of the epithelium led to a transient current increase that was followed by a lower plateau and was not significantly different from the ACh-evoked currents (n= 102), neither the peak (P= 0.91) nor the plateau (P= 0.65) component (Figure 3D).
In order to test whether the observed ACh effect desensitizes, ACh (100 µM, luminal) was applied twice to the same tracheal epithelium and washed out between both applications. Upon the second application of ACh, a similar current increase was observed, although the transient peak current of this second ACh response was significantly reduced (n= 7, P < 0.05; Figure 3E). In contrast to the peak currents, no significant changes in the plateau currents were detected (n= 7, P= 0.63; Figure 3E).
Additionally we conducted experiments with TTX (1 µM, basolateral), an inhibitor of ACh release from cholinergic nerve terminals. Comparison of the ACh (100 µM, luminal) induced changes of the ISC in the presence of TTX and under control conditions (without TTX) showed no statistical difference (peak: n= 7, P= 0.47; plateau: n= 7, P= 0.52; Figure 3F). Thus additional stimulation of cholinergic nerves in our experimental setup can be excluded from these experiments.
For additional characterization of the influence of ACh application on the mouse tracheal epithelium, we applied 100 µM ACh to the basolateral side of the epithelium (n= 5; Figure 3G). This resulted in a significant current increase with an ACh-induced peak (IACh: 49.35 ± 12.34 µA cm−2) and a plateau phase (IACh: 16.98 ± 4.49 µA cm−2) that was about twice as large as the luminal ACh effect, indicating involvement of slightly different ion channels and ACh receptors on the luminal and basolateral side of the epithelium. Similar to the luminal application of ACh, repeated basolateral administration of ACh receptor agonists (100 µM carbachol) resulted in a significant reduction of the carbachol-induced current upon the second application (n= 5, P < 0.01; Figure 3H), whereas the plateau component was not altered (n= 5, P= 0.39), indicating a desensitization of the basolateral cholinergic peak effect.
ACh activates K+ and Cl− conductances
Per convention an increase in the ISC, as observed in our experiments with ACh, may be due to an increased absorption of cations or to an enhanced secretion of anions. In the sodium absorbing mouse tracheal epithelium the epithelial sodium channel (ENaC) is well known to be highly involved in cation absorption (Schreiber and Kunzelmann, 2005). Thus, we investigated if the ENaC was involved in the observed ACh effect by adding 10 µM amiloride (a specific ENaC inhibitor) (Mall et al., 1998a) to the luminal side of the mouse tracheal epithelium in the Ussing-chamber. This led to an instant decrease in the ISC by 30% (Figure 4A). Application of 100 µM ACh to the luminal side in the presence of amiloride (n= 6) resulted in the typical response of the ISC that was not significantly altered compared with control conditions without amiloride (n= 7), with P= 0.20 for the peak and P= 0.11 for the plateau currents (Figure 4B).
Figure 4.
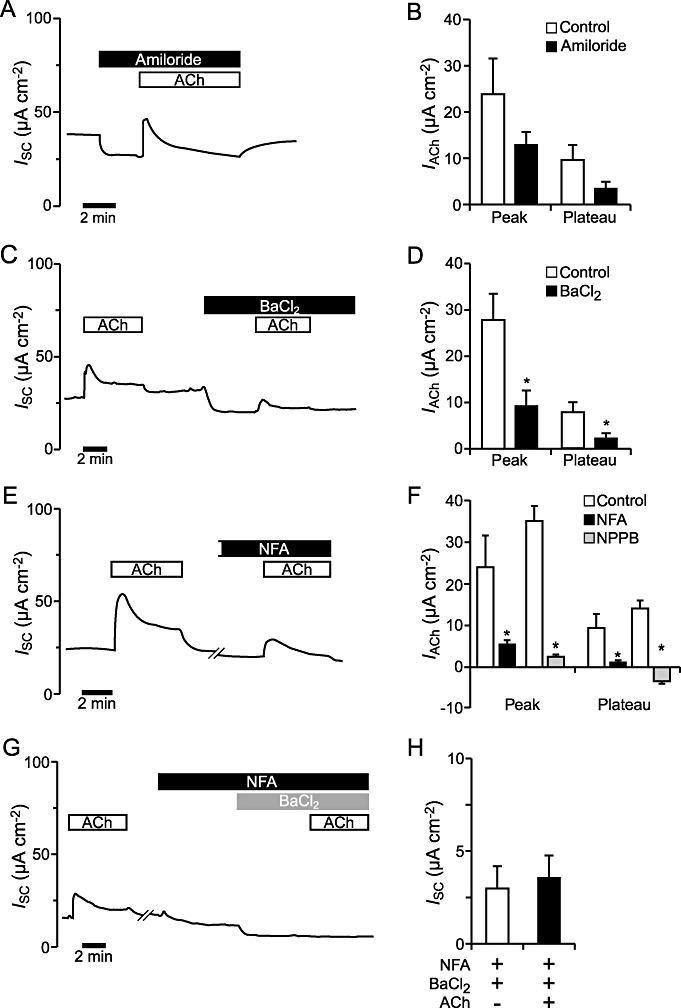
Effect of the ion channel modulators amiloride, BaCl2 NFA and NPPB on the ACh effect in mouse tracheal epithelium. (A) Upon application of 10 µM amiloride (luminal), the ISC decreased. When ACh (100 µM, luminal) was added as well, the typical ACh effect consistent of a transient peak followed by a lower plateau was visible. (B) The ACh-induced current (IACh) in presence of amiloride (n= 6) was not significantly different from the control effect (n= 7) (peak: P= 0.20, plateau: P= 0.11). (C) A typical trace for the effect of ACh (100 µM, luminal) before and after basolateral perfusion of 5 mM BaCl2. Application of ACh on the luminal side of the epithelium in the presence of BaCl2 on the basolateral side, led to a current increase. (D) The ACh effect was significantly decreased in the presence of basolateral BaCl2, the peak as well as the plateau component (n= 6, P < 0.05). (E) The current trace shows the effect of the application of 100 µM ACh on the luminal side of the epithelium, both under control conditions and in the presence of NFA (100 µM, luminal). Perfusion with NFA led to a current decrease. The tissue was pre-incubated for approximately 8 min with NFA before ACh application. (F) Both components of the ACh effect in the presence of NFA (n= 6) were significantly reduced (P < 0.05) compared with the control ACh effect (n= 7). Similarly, the ACh mediated peak current was significantly reduced in the presence of the chloride channel inhibitor NPPB (100 µM, luminal) compared with control conditions (n= 7, P < 0.05) and the plateau current was abolished (n= 7, P < 0.05). (G) Application of ACh (100 µM, luminal) led to the typical biphasic increase of the ISC. In the presence of NFA (100 µM, luminal) and BaCl2 (5 mM, basolateral), additional application of ACh had no effect. The tissue was pre-incubated with NFA for 12 min and with BaCl2 for 6 min before the ACh application, resulting in a time gap of 20 min between the first and the second ACh application, including the washing time. (H) The presence of luminal NFA and basolateral BaCl2 had no significant effect on the ACh-induced current (n= 4, P= 0.39).
Further, we tested if the ACh effect could be influenced by the presence of the non-specific potassium channel blocker Ba2+ (applied as BaCl2). Ba2+ inhibits cAMP-activated and Ca2+-activated K+-channels (Greger, 1996; Mall et al., 2000; Cowley and Linsdell, 2002). Administration of Ba2+ (5 mM, basolateral) led to a decrease in the ISC of 24% (Figure 4C). Additional luminal application of 100 µM ACh revealed that Ba2+ significantly reduced the peak and plateau current to ACh compared with the representative controls, which used mannitol for osmotic compensation (n= 6, P < 0.05; Figure 4D).
To investigate whether activation of anion transport through the luminal membrane of the epithelium was contributing to the ACh effect, 100 µM NFA was applied luminally; NFA is a non-specific chloride channel inhibitor that blocks the CF transmembrane conductance regulator (CFTR) (Scott-Ward et al., 2004) as well as Ca2+-activated chloride channels like the recently identified TMEM16 proteins (Moura et al., 2005). In the presence of NFA, the ISC declined to 25% below baseline current (Figure 4E). When 100 µM ACh was added to the luminal side of the tissue in the presence of NFA (n= 6), the ACh-induced changes in ISC were significantly reduced compared with control conditions (ACh effect without NFA, n= 7, P < 0.05), although a residual effect was still visible (Figure 4F). Similar results were obtained when ACh was administered to the epithelium in the presence of the chloride channel inhibitor NPPB (Malekova et al., 2007). Application of NPPB (100 µM, luminal) led to a decrease in the ISC of 41% below baseline current. Administration of ACh (100 µM, luminal) in the presence of NPPB (NPPB pre-incubation time of 5 min) significantly reduced the ACh-induced peak current (n= 7) compared with control conditions (ACh effect without NPPB, n= 7, P < 0.05) and to an abolishment of the ACh-induced plateau component (Figure 4F).
Next, we tested if co-application of Ba2+ and NFA affected the ACh effect. Therefore we applied 100 µM NFA to the luminal side and subsequently added 5 mM BaCl2 on the basolateral side of the mouse tracheal epithelium. With both components a partial decrease in the ISC was observed (Figure 4G). When ACh was applied to the tissue under these conditions, no significant changes in the ISC were detected in the presence of ACh (n= 4, P= 0.39; Figure 4H).
To further characterize the subtypes of potassium channels involved, we used several inhibitors of Ca2+-dependent K+ channels. Up to now several subtypes of Ca2+-dependent K+ channels have been identified in airway epithelium: KCa2.1, KCa3.1, KCa1.1 (Bardou et al., 2009). Apamin is known to inhibit KCa2.1, KCa2.2 and KCa2.3 channels (Grunnet et al., 2001). Application of apamin (2 µM, basolateral) showed no significant effect on the ACh response (n= 5) compared with control (n= 7) (peak: P= 0.31, plateau: P= 0.80, Figure 5A). Iberiotoxin has been described as a blocker of KCa1.1 channels (Giangiacomo et al., 1992; Jovanovic et al., 2003). The presence of iberiotoxin (250 nM, basolateral, n= 5) had no effect on the ACh-induced current increase compared with control experiments (n= 7) (peak: P= 0.65, plateau: P= 0.56; Figure 5B). Similarly application of ACh in the presence of charybdotoxin (200 nM, basolateral, n= 5), an antagonist of KCa1.1 and KCa3.1 channels (Vergara et al., 1998), did not alter the ACh effect compared with control measurements (n= 7) (peak: P= 0.94, plateau: P= 0.77; Figure 5C). Thus, none of the peptide inhibitors of Ca2+-dependent K+ channels was able to modulate the ACh effect. Since it was previously observed that a potassium channel with similar properties to the KCa3.1 channel was insensitive to charybdotoxin but sensitive to clotrimazole (Castillo et al., 2005), we performed additional experiments with the KCa3.1 channel inhibitor clotrimazole (Wilson et al., 2006). In contrast to the results observed with the peptide inhibitors, application of ACh in the presence of clotrimazole (n= 5) led to a significantly decreased ACh-induced current, with a reduced peak (P= 0.05) and plateu component (P < 0.05) compared with control conditions (n= 7). These data indicate that basolateral KCa3.1 channels are involved in the ACh-mediated activation of basolateral K+ channels.
Figure 5.
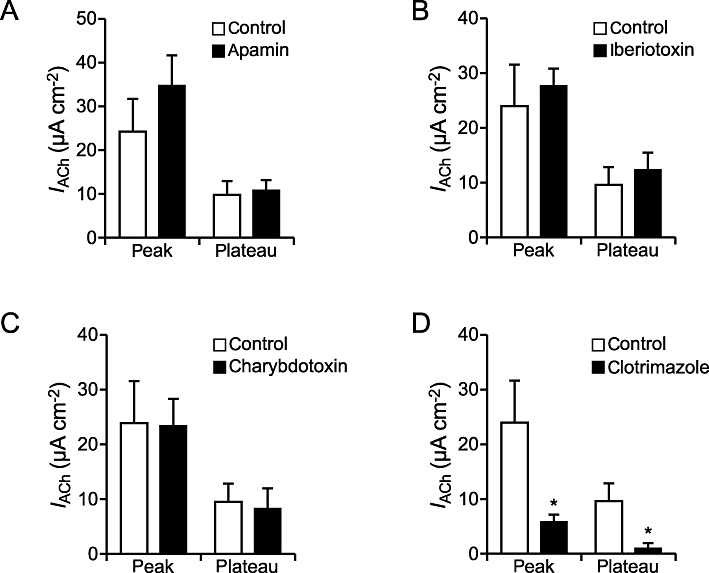
Influence of inhibitors of Ca2+ dependent K+ channels on the ACh effect. (A) The presence of the KCa2.1, KCa2.2 and KCa2.3 channel inhibitor apamin (2 µM, basolateral, n= 5) resulted in an unchanged ACh-induced current increase compared with control conditions (n= 7) (peak: P= 0.31, plateau: P= 0.80). (B) The ACh-induced current (IACh) in the presence of the KCa1.1 channel inhibitor iberiotoxin (250 nM, basolateral, n= 5) was not significantly different from the control effect (n= 7) (peak: P= 0.65, plateau: P= 0.56). (C) In the presence of the KCa1.1 and KCa3.1 channel inhibitor charybdotoxin (200 nM, basolateral, n= 5) the ACh-induced current was not significantly altered compared with the control effect (n= 7) (peak: P= 0.94, plateau: P= 0.77). (D) Both components of the ACh effect in the presence of the KCa3.1 channel inhibitor clotrimazol (n= 5) were significantly reduced (P≤ 0.05) compared with the control ACh effect (n= 7).
In summary, our investigations with the ion channel modulators showed that luminal ACh has no significant influence on Na+ transport and instead activates luminal Cl− and basolateral K+ channels, particularly basolateral KCa3.1 channels.
The ACh effect is modulated by the intracellular Ca2+ concentration
ACh is known for being able to act via an increase in intracellular Ca2+ levels ([Ca2+]i). Nicotinic receptors have been shown to form a Ca2+-permeable pore (Decker and Dani, 1990) and muscarinic ACh receptors act on intracellular calcium levels via activation of the PLC (Racke and Matthiesen, 2004). Therefore experiments were performed to investigate the ACh effect in the presence of the calcium ionophore A23187. Luminal application of 2 µM A23187 resulted in a transient current increase of 78% that declined slowly afterwards and did not have a plateau (Figure 6A). When adding 100 µM ACh to the epithelium in the presence of A23187, no significant change of the ISC occurred (n= 5, P= 0.12; Figure 6B). It is well known that muscarinic ACh receptors are able to activate the inositol-1,4,5-triphospate (IP3) signalling cascade after binding to ACh. To test, whether the IP3 signalling cascade was involved in the ACh effect, we performed experiments with the IP3 receptor inhibitor 2-APB (Dutta et al., 2009). Luminal application of 75 µM 2-APB led to a current decrease of 52% (Figure 6C). When 100 µM ACh was added luminally to the epithelium in the presence of 2-APB the peak component was significantly reduced compared with control conditions (n= 5, P < 0.05) and the plateau was completely abolished (Figure 6D). These results indicate that ACh increases [Ca2+]i and that this increase partly involves the IP3 signalling pathway.
Figure 6.
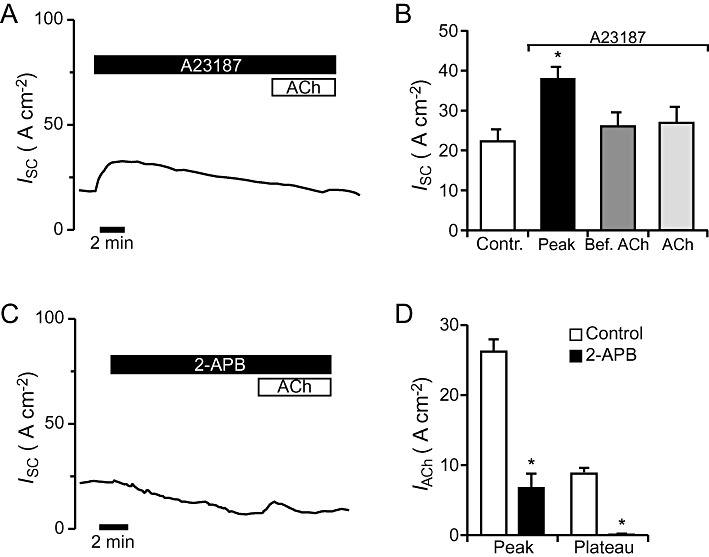
Effect of Ca2+-modulators on the response to ACh. (A) As visible from the representative current trace, application of the Ca2+-ionophore A23187 (2 µM, luminal) to the mouse tracheal epithelium led to a transient current increase. Addition of 100 µM ACh (luminal) in the presence of A23187 did not change the course of the current. (B) Application of A23187 led to a significant transient increase in the ISC (n= 5, P < 0.05). No significant response to ACh occurred in the presence of A23187 (n= 5, P= 0.12). (C) Luminal application of 75 µM of the IP3-receptor antagonist 2-APB led to a current decrease. Additional application of 100 µM ACh led to a current increase consisting of a transient peak. (D) Statistical analysis revealed that 2-APB significantly reduced the transient ACh-induced peak current (n= 5, P < 0.05) and abolished the plateau component.
Nicotinic and muscarinic ACh receptors mediate the ACh effect
For characterization of the ACh receptors involved in the ACh effect in mouse tracheal epithelium, the effect of nicotine on the ISC was tested. Luminal application of nicotine (100 µM) led to an increase in the ISC, similar to the observed ACh-induced current changes (Figure 7A). On comparing the peak currents induced by nicotine with those induced by ACh no significant changes were observed (n= 6, P= 0.85; Figure 7B). In contrast to this, the plateau current detected with nicotine was significantly reduced compared with the plateau values for ACh (n= 6, P < 0.05). This finding clearly demonstrates the involvement of nicotinic receptors in the ACh effect.
Figure 7.
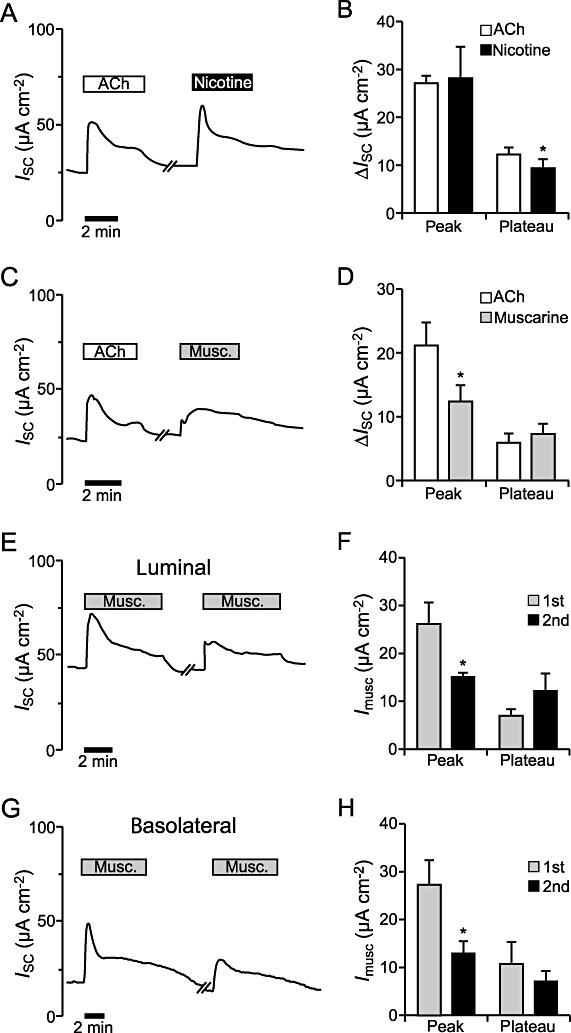
Influence of muscarine and nicotine on the ion transport in mouse tracheal epithelium. (A) The current trace shows a typical course of the ISC when 100 µM ACh was applied to the luminal side of the tracheal epithelium, washed out for 5 min, and then nicotine (100 µM, luminal) was added. (B) There was no significant difference between the ACh- and the nicotine-induced peak (n= 6, P= 0.85). The nicotine-induced plateau current was significantly lower than the ACh-induced plateau current (n= 6, P < 0.05). (C) Luminal application of 100 µM muscarine to the tissue led to an increase in the ISC, similar to the response observed to ACh (100 µM, luminal). The washing time between ACh and muscarine application was 5–10 min. (D) The muscarine-induced initial current increase was significantly reduced compared with the ACh peak (n= 6, P < 0.05). The plateau currents induced by ACh and muscarine were similar (n= 6, P= 0.55). (E) Repeated application of 100 µM muscarine on the luminal side of the epithelium led to a current increase that was divided into a transient peak and a plateau phase upon both applications. The washing time between the two applications of muscarine was 10 min. (F) The peak current of the second muscarine effect (n= 5) was significantly reduced compared with the first application (n= 7, P < 0.05), whereas the plateau currents of the first (n= 7) and the second effect (n= 5) were similar (P= 0.15). (G) Basolateral application of 100 µM muscarine led to a biphasic current increase that was divided into a transient peak and a plateau phase and repeatable upon a second application. A time period of about 10 min lay between the repeated muscarine administrations. (H) The peak current of the second basolateral muscarine effect was significantly reduced compared with the first application (n= 5, P < 0.05), whereas the plateau currents were similar (n= 5, P= 0.21).
To investigate if muscarinic receptors also contribute to the ACh effect, muscarine was used as a specific ligand of muscarinic ACh receptors. Luminal application of 100 µM muscarine led to a current increase, which in contrast to the ACh application showed no prominent peak phase (Figure 7C). Compared with the ACh-induced response, the initial muscarinic increase (peak) was reduced (n= 6, P < 0.05), whereas the muscarine-induced plateau was not different from the one occurring in the presence of ACh (n= 6, P= 0.55; Figure 7D). This shows that muscarinic ACh receptors are mainly responsible for the ACh-induced plateau component. To test if the reduced peak current of the muscarine-effect was due to desensitization, muscarine (100 µM, luminal) was applied twice to the same epithelium. This led to a split current increase with a peak and a plateau component, similar to the ACh effect (Figure 7E). Comparison of the first (n= 7) and the second muscarine response (n= 5) showed a significantly reduced peak component in the second response (P < 0.05), whereas the plateau currents where similar (P= 0.15; Figure 7F). Thus, the desensitizing component of the ACh effect (Figure 3E) is due to desensitization of muscarinic ACh receptors. To further characterize the influence of muscarine application on the tracheal epithelium, we applied 100 µM muscarine to the basolateral side of the epithelium (n= 5; Figure 7G). This resulted in a current increase with a significant muscarine-induced peak (n= 5, P < 0.01; Figure 7H) and a plateau phase (n= 5, P < 0.05; Figure 7H) that was similar to the luminal muscarine effect (Figure 7F). Analogous to the observations upon repeated luminal muscarine application (Figure 7F), repeated basolateral muscarine application (100 µM) resulted in a reduced peak component upon the second application (n= 5, P < 0.05; Figure 7H), whereas the plateau component was not altered (n= 5, P= 0.21). These data indicate a similar response to muscarine and the involvement of similar ion channels and muscarinic receptors on the luminal and basolateral side.
For closer investigation of the muscarinic ACh receptor subtypes involved we used tracheal epithelia of M3R−/− and M1R−/− mice. Application of 100 µM muscarine to the luminal side of the tracheal epithelium of M3R−/− mice resulted in a sustained current increase of approximately 22% (n= 4) but no peak current was observed. In three out of four animals, the application of muscarine resulted in a current trace as depicted in Figure 8A, whereas in one animal a clear muscarine-induced current was visible. Comparing the means of all four M3R−/− mice with the wild-type (WT) mice (n= 7), the muscarine-induced current in M3R−/− mice was significantly decreased (P < 0.05; Figure 8B). Application of 100 µM muscarine to the tracheal epithelium of M1R−/− mice led to an increase in the ISC that consisted of a transient peak and a lower plateau (n= 4; Figure 8C). However, both components were also significantly reduced compared with the tracheal epithelia of WT mice (n= 7, P < 0.05; Figure 8D). As the muscarine-induced effect in M1R−/− mice was more pronounced than in M3R−/− it seems that the M3R subtype is the major determinant of the muscarinic responses induced by ACh. In accordance with this observation the peak component of the ACh-induced current increase (100 µM, luminal) was significantly reduced in M3R−/− mice compared with WT mice (n= 7, P < 0.01; Figure 8E), whereas the peak component of the ACh effect was not altered in the M1R−/− mice (n= 7, P= 0.76). Compared with WT control mice the plateau component of the ACh-induced current change was not altered in either the M3R−/− mice (n= 7, P= 0.56) or in the M1R−/− mice (n= 7, P= 0.51). This indicates that the lack of the M3 muscarinic ACh receptors in the M3R−/− mice is not fully compensated for by nicotinic receptors, whereas the loss of the M1 muscarinic ACh receptor can be compensated for by the simultaneous activation of nicotinic receptors and M3 muscarinic ACh receptors, as ACh binds to nicotinic as well as to muscarinic receptors.
Figure 8.
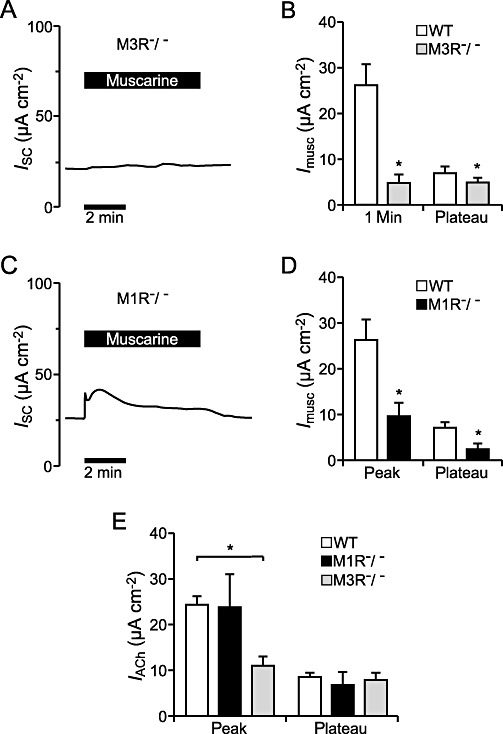
Effect of muscarine on the ion transport of the tracheal epithelium of M3R−/− and M1R−/− mice. (A) Typical current observed in a M3R−/− tracheal epithelium upon application of 100 µM muscarine luminal. Application of 100 µM muscarine led to a sustained current increase. (B) The muscarine-induced current increase was significantly reduced in the tracheal epithelium of M3R−/− mice (n= 4) compared with WT mice (n= 7, P < 0.05) and the initial peak occurring in WT mice could not be detected. (C) The representative current trace shows perfusion of the tracheal epithelium of M1R−/− mice with 100 µM muscarine luminal, which resulted in an increase in the ISC that consisted of a transient peak and a lower plateau. (D) The muscarine-induced current increase occurring in M1R−/− mice (n= 4) was significantly reduced compared with WT mice (n= 7, P < 0.05 for peak and plateau). (E) The peak current increase mediated by ACh (100 µM, luminal) was significantly reduced in M3R−/− mice compared with WT controls (n= 7, P < 0.01) and not altered in the M1R−/− mice (n= 7, P= 0.76). The ACh-induced plateau current increase was not changed in M3R−/− mice (n= 7, P= 0.56) or in M1R−/− mice (n= 7, P= 0.51) compared with control ACh effects in WT mice.
In summary our results show that luminal ACh binds to nicotinic and muscarinic M3 and M1 ACh receptors present in the tracheal epithelium.
Discussion and conclusions
Detection of ChAT, OCTs and luminal ACh in mouse trachea
The RT-PCR experiments using mouse tracheal epithelial cells confirmed the presence of components of the nnCS in mouse tracheal epithelium. By detecting ChAT transcripts we provide evidence that ACh can be synthesized in mouse tracheal epithelial cells. This observation agrees with other studies detecting ChAT transcripts or ChAT protein in airway epithelia of human, rat and mouse (Klapproth et al., 1997; Kummer et al., 2006; 2008; Lips et al., 2007). Additionally, a ChAT activity assay confirmed the ability of ChAT for ACh synthesis in airway epithelial cells (Klapproth et al., 1997). Since OCT1-3 have been demonstrated to transport ACh (Wessler et al., 2001; Lips et al., 2005; Koepsell et al., 2007), the consistent detection of OCT1 and OCT3 transcripts in our study indicates that mouse tracheal epithelial cells are able to release ACh. The detection of OCT transcripts in this study corresponds to the detection of OCT transcripts or protein in human and rat tracheal epithelial cells as well as in bronchial epithelium of BALB/cj and FVB mice (Lips et al., 2005; 2007; Kummer et al., 2006). As ChAT and OCT transcripts have so far not been characterized in the tracheal epithelium of C57Bl/6 mice, we additionally show the presence of these transcripts representing parts of the nnCS.
In addition to the detection of ChAT and OCT expression, the DESI experiments revealed the presence of luminal ACh in mouse trachea. There are no intra-epithelial nerve endings (Pack et al., 1980) and therefore no luminal cholinergic nerve fibres in this tissue. Thus, our results provide strong evidence for nnCS function in mouse tracheal epithelium, by identifying the luminal presence of ACh that is likely to be released from the epithelial cells.
Luminal ACh activates transepithelial ion transport
In addition to our findings the existence of the nnCS in airway epithelia has been described earlier (Klapproth et al., 1997; Wessler et al., 2003; Kummer et al., 2008). Since cholinergic nerve endings are only present on the basolateral side of the epithelium (see earlier discussion), the basolateral side can be targeted by neuronal and non-neuronal ACh. Therefore we applied ACh to the luminal side of the tracheal epithelium to mimic the effects of non-neuronal ACh. Indeed, the ion transport changes induced by luminal ACh or carbachol application confirmed a functional nnCS in this tissue. Experiments with TTX eliminated the possibility for additional stimulation of basolateral cholinergic nerve fibres.
The activation of transepithelial ion currents by luminal ACh provides new insights concerning the function of the airway epithelium. The ACh-induced biphasic current increase and the reduced second ACh effect indicate desensitization of the ACh response, indicating the involvement of different ACh receptors. Desensitization is characteristic of several ACh receptors, such as nicotinic α7 receptors (Albuquerque et al., 1997) as well as muscarinic M1 and M3 receptors (Stope et al., 2003; Gomez-Ramos et al., 2009). The EC50 value (23.3 µM ACh) is in good agreement with other observations from different tissues. For the muscarinic receptors, ACh-induced EC50 values were ∼61 nM (M1) and 3.5 nM (M3) (Gomez-Ramos et al., 2009) and for nicotinic receptors they ranged from 0.48 µM to 442.9 µM (Gerzanich et al., 1995; Chavez-Noriega et al., 1997). As the EC50 of ACh is species- and subtype-dependent, the EC50 in mouse tracheal epithelium represents a mixture of the affinity of ACh for all receptor subtypes present in this tissue.
Ca2+ dependent activation of chloride and potassium channels
The potent ENaC inhibitor amiloride (Mall et al., 1998a) did not influence the ACh effect, demonstrating that ENaCs are not involved. Similar results were reported from mouse tracheal epithelium, where basolateral application of carbachol did not influence the amiloride-sensitive Na+ current (Kunzelmann et al., 2002).
Instead, our results suggest a synergistic activation of basolateral potassium and luminal chloride channels by luminal ACh in mouse tracheal epithelium as visible from the reduced ACh effect upon application of BaCl2 or/and NFA or NPPB. Basolateral K+ channels are described as modulators of luminal chloride secretion (McCann and Welsh, 1990; Greger, 1996; Bernard et al., 2003), because their activation hyperpolarizes the membrane potential and thereby increases the driving force for Cl− secretion. Possible candidates are Ca2+-dependent K+ channels. Members of all three subtypes of these channels (KCa1.1, KCa3.1 and KCa2.1) that are grouped according to their conductance properties have been detected in airway epithelial cells (Greger, 1996; Ridge et al., 1997; Vergara et al., 1998; Bernard et al., 2003; Thompson-Vest et al., 2006; Bardou et al., 2009). Investigation of the different subtypes of Ca2+-dependent K+ channels with the inhibitors iberiotoxin, apamin, charybdotoxin and clotrimazole revealed the involvement of basolateral KCa3.1 channels in the ACh effect. This is in good agreement with existing literature, since the KCa3.1 has been identified as an abundant Ca2+-dependent K+ channel subtype in epithelia (Jensen et al., 2001). Similarly Mall et al. (2003) reported a contribution of basolateral KCa3.1 channels to a luminal Cl− secretion in nasal epithelia that was evoked by extracellular nucleotides.
Additionally, two channel types facilitate luminal Cl− secretion: Ca2+-activated Cl− channels formed by members of the TMEM protein family (Rock et al., 2009) and cAMP-dependent chloride channels represented by CFTR (Schreiber and Kunzelmann, 2005). Both Cl− channel types are inhibited by NFA and NPPB (Uyekubo et al., 1998; Scott-Ward et al., 2004; Moura et al., 2005; Zholos et al., 2005). So far activation of both by luminal ACh cannot be excluded, as in human colon epithelium activation of Ca2+- and cAMP-dependent Cl− channels has been observed in response to carbachol, indicating the involvement of cAMP- and Ca2+-dependent signalling pathways (Mall et al., 1998b). In contrast to this, Roomans et al. (2002) found reduced Na+ and Cl− contents in the airway surface liquid after administration of nicotine in the drinking water of rats. These contradicting results may be explained by the use of different cholinergic agonists (nicotine vs. ACh) and the indirect administration of nicotine via the drinking water compared with direct administration of ACh. However, our findings of a synergistic activation of K+- and Cl−-channels by luminal ACh are consistent with the synergistic activation of luminal Cl−- and basolateral K+-channels in response to basolateral carbachol application in human colon epithelium (Mall et al., 1998b) and upon basolateral ACh application in sheep trachea (Acevedo, 1994).
Concerning the mechanisms involved, the attenuation of the ACh effect in the presence of the Ca2+-ionophore A23187 indicates that the bulk of the effect is mediated via an increase in [Ca2+]i. Similarly Mall et al. (1998b) reported a partly Ca2+-dependent carbachol-induced chloride secretion in human colon. An increase in [Ca2+]i was identified in response to nicotine in human nasal epithelium (Blank et al., 1997) and to muscarine in rat colon epithelium (Schultheiss et al., 2005). The experiments with 2-APB further support the involvement of [Ca2+]i, which is at least partly mediated by the IP3 signalling pathway pointing to the participation of muscarinic ACh receptors, although a Ca2+ entry mediated by nicotinic receptors cannot be excluded.
ACh acts via nicotinic and muscarinic M1 and M3 receptors
Our finding that nicotinic receptors were involved in the ACh effect is supported by previous studies. Nicotinic α3α5β2 receptors promote wound repair in respiratory epithelium (Tournier et al., 2006). Transcripts for the α2-, α3-, α4-, α5-, α6-, α7-, α9-, α10-, β2- and β4-nicotinic receptor subunits have been detected in human and rodent bronchial epithelial cells (Maus et al., 1998; Kummer et al., 2008; Hollenhorst et al., 2012). Additionally functional nicotinic receptors have been characterized in mouse tracheal epithelial cells, in human isolated nasal epithelial cells as well as in human and monkey bronchial epithelial cells (Blank et al., 1997; Fu et al., 2009; Hollenhorst et al., 2012). These studies together with our results demonstrate that airway epithelial cells possess functional nicotinic receptors, although the exact composition and stoichiometry of these nicotinic receptor(s) remains unknown.
The role of muscarinic ACh receptors in the contraction of airway smooth muscles and bronchoconstriction is well established (Racke et al., 2006). Additionally, Klein et al. (2009) showed that M3, M2 and M1 muscarinic ACh receptors are able to modulate cilia-driven particle transport.
Further, our results suggest that functional muscarinic ACh receptors in the luminal membrane of tracheal epithelial cells are involved in transepithelial ion transport processes. The desensitization observed to a second ACh application seems to be due to the muscarinic ACh receptors, because similar observations were made with muscarine. This feature is characteristic for M1 and M3 muscarinic ACh receptors (Stope et al., 2003; Gomez-Ramos et al., 2009), which represent the most abundant muscarinic ACh receptor subtypes in mouse tracheal epithelium (Klein et al., 2009), whereas the M2 subtype is absent in this tissue (Klein et al., 2009). Our studies with M3R−/− and M1R−/− mice suggest that both receptors are involved in the ACh effect, as they displayed a reduced muscarine response compared with WT animals. The reduction of the muscarine effect was stronger in M3R−/− than in M1R−/− mice. Additionally the peak component of the ACh effect was reduced in the M3R−/− mice only and was not affected in the M1R−/− mice. Thus the effect is mainly mediated by M3 muscarinic ACh receptors and the M1 subtype contributes to a lesser extent. Our findings correspond to observations from mouse colon epithelium, which show involvement of M1R and M3R in Cl− secretion (Haberberger et al., 2006). Additionally it can be speculated that the results observed in the M3R−/− and M1R−/− mice could be due to heterodimers of the M1 and M3 muscarinic ACh receptors, since Goin and Nathanson (2006) demonstrated heterodimerization of M1 and M3 muscarinic ACh receptors in HEK293 cells.
However, under physiological conditions ACh serves as ligand for nicotinic and muscarinic receptors and the ACh effect represents an overlay involving the activation of different receptors – including nicotinic receptors as well as M1 and M3 muscarinic ACh receptors.
Perspectives
Conclusively, our findings provide new insights into the role of the airway cholinergic system and indicate that the nnCS is part of a novel signalling machinery relevant for epithelial airway functions. The ability to regulate airway epithelial ion transport indicates that nnCS function is related to the regulation of mucociliary clearance and thereby the innate immune defence. From this perspective, targeting the nnCS in the airways is of interest for therapy improvements of different pathological conditions: (i) Smoking might lead to a nicotine-induced uncontrolled nnCS activation in airway epithelium, which might contribute to the destructive effects in the lungs; (ii) activation of Ca2+-dependent Cl− secretion as observed by luminal ACh represents a possible strategy for CF treatment by activating alternative chloride secreting pathways to improve mucociliary clearance; and (iii) anticholinergic bronchodilators used in CF and COPD patients may impair mucociliary clearance (Restrepo, 2007), indicating disturbances of ion transport processes possibly by interference with the nnCS. Thus, the role of anticholinergics as therapeutics needs further evaluation in the context of a possible influence on ion transport and the nnCS in the airway epithelium.
Acknowledgments
The authors would like to thank Martin Bodenbenner for excellent technical support. The present study was supported by a scholarship from the Konrad-Adenauer-Stiftung to MI Hollenhorst and the Universities of Giessen and Marburg Lung Center.
Glossary
- 2-APB
2-aminoethyl-diphenyl-borinate
- [Ca2+]i
intracellular Ca2+ level
- CF
cystic fibrosis
- CFTR
cystic fibrosis transmembrane conductance regulator
- COPD
chronic obstructive pulmonary disease
- DESI
desorption electrospray ionization
- ENaC
epithelial sodium channel
- ISC
short-circuit current
- M1R−/−
muscarinic receptor subtype 1-deficient mouse
- M3R−/−
muscarinic receptor subtype 3-deficient mouse
- NFA
niflumic acid
- nnCS
non-neuronal cholinergic system
- NPPB
5-nitro-2-(3-phenylpropylamino)benzoic acid
- OCT
organic cation transporter
- TTX
tetrodotoxin
- WT
wild type
Conflict of interest
The authors report no conflict of interest.
References
- Acevedo M. Effect of acetyl choline on ion transport in sheep tracheal epithelium. Pflugers Arch. 1994;427:543–546. doi: 10.1007/BF00374272. [DOI] [PubMed] [Google Scholar]
- Albuquerque EX, Alkondon M, Pereira EF, Castro NG, Schrattenholz A, Barbosa CT, et al. Properties of neuronal nicotinic acetylcholine receptors: pharmacological characterization and modulation of synaptic function. J Pharmacol Exp Ther. 1997;280:1117–1136. [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th Edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardou O, Trinh NT, Brochiero E. Molecular diversity and function of K+ channels in airway and alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2009;296:L145–L155. doi: 10.1152/ajplung.90525.2008. [DOI] [PubMed] [Google Scholar]
- Belmonte KE. Cholinergic pathways in the lungs and anticholinergic therapy for chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2005;2:297–304. doi: 10.1513/pats.200504-043SR. [DOI] [PubMed] [Google Scholar]
- Bernard K, Bogliolo S, Soriani O, Ehrenfeld J. Modulation of calcium-dependent chloride secretion by basolateral SK4-like channels in a human bronchial cell line. J Membr Biol. 2003;196:15–31. doi: 10.1007/s00232-003-0621-3. [DOI] [PubMed] [Google Scholar]
- Blank U, Ruckes C, Clauss W, Weber WM. Effects of nicotine on human nasal epithelium: evidence for nicotinic receptors in non-excitable cells. Pflugers Arch. 1997;434:581–586. doi: 10.1007/s004240050439. [DOI] [PubMed] [Google Scholar]
- Castillo K, Bacigalupo J, Wolff D. Ca2+-dependent K+ channels from rat olfactory cilia characterized in planar lipid bilayers. FEBS Lett. 2005;579:1675–1682. doi: 10.1016/j.febslet.2005.01.079. [DOI] [PubMed] [Google Scholar]
- Chavez-Noriega LE, Crona JH, Washburn MS, Urrutia A, Elliott KJ, Johnson EC. Pharmacological characterization of recombinant human neuronal nicotinic acetylcholine receptors h alpha 2 beta 2, h alpha 2 beta 4, h alpha 3 beta 2, h alpha 3 beta 4, h alpha 4 beta 2, h alpha 4 beta 4 and h alpha 7 expressed in Xenopus oocytes. J Pharmacol Exp Ther. 1997;280:346–356. [PubMed] [Google Scholar]
- Cowley EA, Linsdell P. Characterization of basolateral K+ channels underlying anion secretion in the human airway cell line Calu-3. J Physiol. 2002;538:747–757. doi: 10.1113/jphysiol.2001.013300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani JA. Overview of nicotinic receptors and their roles in the central nervous system. Biol Psychiatry. 2001;49:166–174. doi: 10.1016/s0006-3223(00)01011-8. [DOI] [PubMed] [Google Scholar]
- Decker ER, Dani JA. Calcium permeability of the nicotinic acetylcholine receptor: the single-channel calcium influx is significant. J Neurosci. 1990;10:3413–3420. doi: 10.1523/JNEUROSCI.10-10-03413.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta AK, Khimji AK, Sathe M, Kresge C, Parameswara V, Esser V, et al. Identification and functional characterization of the intermediate-conductance Ca(2+)-activated K(+) channel (IK-1) in biliary epithelium. Am J Physiol Gastrointest Liver Physiol. 2009;297:G1009–G1018. doi: 10.1152/ajpgi.00223.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisahn A, Yamada M, Duttaroy A, Gan JW, Deng CX, McBain CJ, et al. Muscarinic induction of hippocampal gamma oscillations requires coupling of the M1 receptor to two mixed cation currents. Neuron. 2002;33:615–624. doi: 10.1016/s0896-6273(02)00587-1. [DOI] [PubMed] [Google Scholar]
- Fu XW, Lindstrom J, Spindel ER. Nicotine activates and up-regulates nicotinic acetylcholine receptors in bronchial epithelial cells. Am J Respir Cell Mol Biol. 2009;41:93–99. doi: 10.1165/rcmb.2008-0352OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerzanich V, Peng X, Wang F, Wells G, Anand R, Fletcher S, et al. Comparative pharmacology of epibatidine: a potent agonist for neuronal nicotinic acetylcholine receptors. Mol Pharmacol. 1995;48:774–782. [PubMed] [Google Scholar]
- Giangiacomo KM, Garcia ML, McManus OB. Mechanism of iberiotoxin block of the large-conductance calcium-activated potassium channel from bovine aortic smooth muscle. Biochemistry. 1992;31:6719–6727. doi: 10.1021/bi00144a011. [DOI] [PubMed] [Google Scholar]
- Goin JC, Nathanson NM. Quantitative analysis of muscarinic acetylcholine receptor homo- and heterodimerization in live cells: regulation of receptor down-regulation by heterodimerization. J Biol Chem. 2006;281:5416–5425. doi: 10.1074/jbc.M507476200. [DOI] [PubMed] [Google Scholar]
- Gomez-Ramos A, Diaz-Hernandez M, Rubio A, Diaz-Hernandez JI, Miras-Portugal MT, Avila J. Characteristics and consequences of muscarinic receptor activation by tau protein. Eur Neuropsychopharmacol. 2009;19:708–717. doi: 10.1016/j.euroneuro.2009.04.006. [DOI] [PubMed] [Google Scholar]
- Gosens R, Zaagsma J, Meurs H, Halayko AJ. Muscarinic receptor signaling in the pathophysiology of asthma and COPD. Respir Res. 2006;7:73–87. doi: 10.1186/1465-9921-7-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greger R. The membrane transporters regulating epithelial NaCl secretion. Pflugers Arch. 1996;432:579–588. doi: 10.1007/s004240050173. [DOI] [PubMed] [Google Scholar]
- Grunnet M, Jensen BS, Olesen SP, Klaerke DA. Apamin interacts with all subtypes of cloned small-conductance Ca2+-activated K+ channels. Pflugers Arch. 2001;441:544–550. doi: 10.1007/s004240000447. [DOI] [PubMed] [Google Scholar]
- Haberberger R, Schultheiss G, Diener M. Epithelial muscarinic M1 receptors contribute to carbachol-induced ion secretion in mouse colon. Eur J Pharmacol. 2006;530:229–233. doi: 10.1016/j.ejphar.2005.11.055. [DOI] [PubMed] [Google Scholar]
- Hollenhorst MI, Lips KS, Weitz A, Krasteva G, Kummer W, Fronius M. Evidence for functional atypical nicotinic receptors that activate K+ dependent Cl− secretion in mouse tracheal epithelium. Am J Respir Cell Mol Biol. 2012;46:106–114. doi: 10.1165/rcmb.2011-0171OC. [DOI] [PubMed] [Google Scholar]
- Jensen BS, Strobaek D, Olesen SP, Christophersen P. The Ca2+-activated K+ channel of intermediate conductance: a molecular target for novel treatments? Curr Drug Targets. 2001;2:401–422. doi: 10.2174/1389450013348173. [DOI] [PubMed] [Google Scholar]
- Jovanovic S, Crawford RM, Ranki HJ, Jovanovic A. Large conductance Ca2+-activated K+ channels sense acute changes in oxygen tension in alveolar epithelial cells. Am J Respir Cell Mol Biol. 2003;28:363–372. doi: 10.1165/rcmb.2002-0101OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klapproth H, Reinheimer T, Metzen J, Munch M, Bittinger F, Kirkpatrick CJ, et al. Non-neuronal acetylcholine, a signalling molecule synthesized by surface cells of rat and man. Naunyn Schmiedebergs Arch Pharmacol. 1997;355:515–523. doi: 10.1007/pl00004977. [DOI] [PubMed] [Google Scholar]
- Klein MK, Haberberger RV, Hartmann P, Faulhammer P, Lips KS, Krain B, et al. Muscarinic receptor subtypes in cilia-driven transport and airway epithelial development. Eur Respir J. 2009;33:1113–1121. doi: 10.1183/09031936.00015108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koepsell H, Lips K, Volk C. Polyspecific organic cation transporters: structure, function, physiological roles, and biopharmaceutical implications. Pharm Res. 2007;24:1227–1251. doi: 10.1007/s11095-007-9254-z. [DOI] [PubMed] [Google Scholar]
- Kummer W, Wiegand S, Akinci S, Wessler I, Schinkel AH, Wess J, et al. Role of acetylcholine and polyspecific cation transporters in serotonin-induced bronchoconstriction in the mouse. Respir Res. 2006;7:65–76. doi: 10.1186/1465-9921-7-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kummer W, Lips KS, Pfeil U. The epithelial cholinergic system of the airways. Histochem Cell Biol. 2008;130:219–234. doi: 10.1007/s00418-008-0455-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunzelmann K, Schreiber R, Cook D. Mechanisms for the inhibition of amiloride-sensitive Na+ absorption by extracellular nucleotides in mouse trachea. Pflugers Arch. 2002;444:220–226. doi: 10.1007/s00424-002-0796-y. [DOI] [PubMed] [Google Scholar]
- Lips KS, Volk C, Schmitt BM, Pfeil U, Arndt P, Miska D, et al. Polyspecific cation transporters mediate luminal release of acetylcholine from bronchial epithelium. Am J Respir Cell Mol Biol. 2005;33:79–88. doi: 10.1165/rcmb.2004-0363OC. [DOI] [PubMed] [Google Scholar]
- Lips KS, Luhrmann A, Tschernig T, Stoeger T, Alessandrini F, Grau V, et al. Down-regulation of the non-neuronal acetylcholine synthesis and release machinery in acute allergic airway inflammation of rat and mouse. Life Sci. 2007;80:2263–2269. doi: 10.1016/j.lfs.2007.01.026. [DOI] [PubMed] [Google Scholar]
- Mak JC, Baraniuk JN, Barnes PJ. Localization of muscarinic receptor subtype mRNAs in human lung. Am J Respir Cell Mol Biol. 1992;7:344–348. doi: 10.1165/ajrcmb/7.3.344. [DOI] [PubMed] [Google Scholar]
- Malekova L, Tomaskova J, Novakova M, Stefanik P, Kopacek J, Lakatos B, et al. Inhibitory effect of DIDS, NPPB, and phloretin on intracellular chloride channels. Pflugers Arch. 2007;455:349–357. doi: 10.1007/s00424-007-0300-9. [DOI] [PubMed] [Google Scholar]
- Mall M, Bleich M, Greger R, Schreiber R, Kunzelmann K. The amiloride-inhibitable Na+ conductance is reduced by the cystic fibrosis transmembrane conductance regulator in normal but not in cystic fibrosis airways. J Clin Invest. 1998a;102:15–21. doi: 10.1172/JCI2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mall M, Bleich M, Schurlein M, Kuhr J, Seydewitz HH, Brandis M, et al. Cholinergic ion secretion in human colon requires coactivation by cAMP. Am J Physiol. 1998b;275:G1274–G1281. doi: 10.1152/ajpgi.1998.275.6.G1274. [DOI] [PubMed] [Google Scholar]
- Mall M, Wissner A, Schreiber R, Kuehr J, Seydewitz HH, Brandis M, et al. Role of K(V)LQT1 in cyclic adenosine monophosphate-mediated Cl(-) secretion in human airway epithelia. Am J Respir Cell Mol Biol. 2000;23:283–289. doi: 10.1165/ajrcmb.23.3.4060. [DOI] [PubMed] [Google Scholar]
- Mall M, Gonska T, Thomas J, Schreiber R, Seydewitz HH, Kuehr J, et al. Modulation of Ca2+-activated Cl− secretion by basolateral K+ channels in human normal and cystic fibrosis airway epithelia. Pediatr Res. 2003;53:608–618. doi: 10.1203/01.PDR.0000057204.51420.DC. [DOI] [PubMed] [Google Scholar]
- Maus AD, Pereira EF, Karachunski PI, Horton RM, Navaneetham D, Macklin K, et al. Human and rodent bronchial epithelial cells express functional nicotinic acetylcholine receptors. Mol Pharmacol. 1998;54:779–788. doi: 10.1124/mol.54.5.779. [DOI] [PubMed] [Google Scholar]
- McCann JD, Welsh MJ. Regulation of Cl− and K+ channels in airway epithelium. Annu Rev Physiol. 1990;52:115–135. doi: 10.1146/annurev.ph.52.030190.000555. [DOI] [PubMed] [Google Scholar]
- Miyakawa T, Yamada M, Duttaroy A, Wess J. Hyperactivity and intact hippocampus-dependent learning in mice lacking the M1 muscarinic acetylcholine receptor. J Neurosci. 2001;21:5239–5250. doi: 10.1523/JNEUROSCI.21-14-05239.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moura CT, Bezerra FC, de Moraes IM, Magalhaes PJ, Capaz FR. Increased responsiveness to 5-hydroxytryptamine after antigenic challenge is inhibited by nifedipine and niflumic acid in rat trachea in vitro. Clin Exp Pharmacol Physiol. 2005;32:1119–1123. doi: 10.1111/j.1440-1681.2005.04308.x. [DOI] [PubMed] [Google Scholar]
- Pack RJ, Al Ugaily LH, Morris G, Widdicombe JG. The distribution and structure of cells in the tracheal epithelium of the mouse. Cell Tissue Res. 1980;208:65–84. doi: 10.1007/BF00234174. [DOI] [PubMed] [Google Scholar]
- Racke K, Matthiesen S. The airway cholinergic system: physiology and pharmacology. Pulm Pharmacol Ther. 2004;17:181–198. doi: 10.1016/j.pupt.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Racke K, Juergens UR, Matthiesen S. Control by cholinergic mechanisms. Eur J Pharmacol. 2006;533:57–68. doi: 10.1016/j.ejphar.2005.12.050. [DOI] [PubMed] [Google Scholar]
- Restrepo RD. Inhaled adrenergics and anticholinergics in obstructive lung disease: do they enhance mucociliary clearance? Respir Care. 2007;52:1159–1173. [PubMed] [Google Scholar]
- Ridge FP, Duszyk M, French AS. A large conductance, Ca2+-activated K+ channel in a human lung epithelial cell line (A549) Biochim Biophys Acta. 1997;1327:249–258. doi: 10.1016/s0005-2736(97)00073-4. [DOI] [PubMed] [Google Scholar]
- Rock JR, O'Neal WK, Gabriel SE, Randell SH, Harfe BD, Boucher RC, et al. Transmembrane protein 16A (TMEM16A) is a Ca2+-regulated Cl− secretory channel in mouse airways. J Biol Chem. 2009;284:14875–14880. doi: 10.1074/jbc.C109.000869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roomans GM, Vanthanouvong V, Dragomir A, Kozlova I, Wroblewski R. Effects of nicotine on intestinal and respiratory epithelium. J Submicrosc Cytol Pathol. 2002;34:381–388. [PubMed] [Google Scholar]
- Schreiber R, Kunzelmann K. Purinergic P2Y6 receptors induce Ca2+ and CFTR dependent Cl− secretion in mouse trachea. Cell Physiol Biochem. 2005;16:99–108. doi: 10.1159/000087736. [DOI] [PubMed] [Google Scholar]
- Schultheiss G, Siefjediers A, Diener M. Muscarinic receptor stimulation activates a Ca(2+)-dependent Cl(-) conductance in rat distal colon. J Membr Biol. 2005;204:117–127. doi: 10.1007/s00232-005-0757-4. [DOI] [PubMed] [Google Scholar]
- Scott-Ward TS, Li H, Schmidt A, Cai Z, Sheppard DN. Direct block of the cystic fibrosis transmembrane conductance regulator Cl(-) channel by niflumic acid. Mol Membr Biol. 2004;21:27–38. doi: 10.1080/09687680310001597758. [DOI] [PubMed] [Google Scholar]
- Stope MB, Kunkel C, Kories C, Schmidt M, Michel MC. Differential agonist-induced regulation of human M2 and M3 muscarinic receptors. Biochem Pharmacol. 2003;66:2099–2105. doi: 10.1016/s0006-2952(03)00580-x. [DOI] [PubMed] [Google Scholar]
- Takats Z, Wiseman JM, Gologan B, Cooks RG. Mass spectrometry sampling under ambient conditions with desorption electrospray ionization. Science. 2004;306:471–473. doi: 10.1126/science.1104404. [DOI] [PubMed] [Google Scholar]
- Thompson-Vest N, Shimizu Y, Hunne B, Furness JB. The distribution of intermediate-conductance, calcium-activated, potassium (IK) channels in epithelial cells. J Anat. 2006;208:219–229. doi: 10.1111/j.1469-7580.2006.00515.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tournier JM, Maouche K, Coraux C, Zahm JM, Cloez-Tayarani I, Nawrocki-Raby B, et al. alpha3alpha5beta2-Nicotinic acetylcholine receptor contributes to the wound repair of the respiratory epithelium by modulating intracellular calcium in migrating cells. Am J Pathol. 2006;168:55–68. doi: 10.2353/ajpath.2006.050333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uyekubo SN, Fischer H, Maminishkis A, Illek B, Miller SS, Widdicombe JH. cAMP-dependent absorption of chloride across airway epithelium. Am J Physiol. 1998;275:L1219–L1227. doi: 10.1152/ajplung.1998.275.6.L1219. [DOI] [PubMed] [Google Scholar]
- Vergara C, Latorre R, Marrion NV, Adelman JP. Calcium-activated potassium channels. Curr Opin Neurobiol. 1998;8:321–329. doi: 10.1016/s0959-4388(98)80056-1. [DOI] [PubMed] [Google Scholar]
- Wessler I, Roth E, Deutsch C, Brockerhoff P, Bittinger F, Kirkpatrick CJ, et al. Release of non-neuronal acetylcholine from the isolated human placenta is mediated by organic cation transporters. Br J Pharmacol. 2001;134:951–956. doi: 10.1038/sj.bjp.0704335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessler I, Kilbinger H, Bittinger F, Unger R, Kirkpatrick CJ. The non-neuronal cholinergic system in humans: expression, function and pathophysiology. Life Sci. 2003;72:2055–2061. doi: 10.1016/s0024-3205(03)00083-3. [DOI] [PubMed] [Google Scholar]
- Wessler I, Bittinger F, Kamin W, Zepp F, Meyer E, Schad A, et al. Dysfunction of the non-neuronal cholinergic system in the airways and blood cells of patients with cystic fibrosis. Life Sci. 2007;80:2253–2258. doi: 10.1016/j.lfs.2007.01.042. [DOI] [PubMed] [Google Scholar]
- Wilson SM, Brown SG, McTavish N, McNeill RP, Husband EM, Inglis SK, et al. Expression of intermediate-conductance, Ca2+-activated K+ channel (KCNN4) in H441 human distal airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2006;291:L957–L965. doi: 10.1152/ajplung.00065.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M, Miyakawa T, Duttaroy A, Yamanaka A, Moriguchi T, Makita R, et al. Mice lacking the M3 muscarinic acetylcholine receptor are hypophagic and lean. Nature. 2001;410:207–212. doi: 10.1038/35065604. [DOI] [PubMed] [Google Scholar]
- Zarghooni S, Wunsch J, Bodenbenner M, Bruggmann D, Grando SA, Schwantes U, et al. Expression of muscarinic and nicotinic acetylcholine receptors in the mouse urothelium. Life Sci. 2007;80:2308–2313. doi: 10.1016/j.lfs.2007.01.046. [DOI] [PubMed] [Google Scholar]
- Zholos A, Beck B, Sydorenko V, Lemonnier L, Bordat P, Prevarskaya N, et al. Ca(2+)- and volume-sensitive chloride currents are differentially regulated by agonists and store-operated Ca2+ entry. J Gen Physiol. 2005;125:197–211. doi: 10.1085/jgp.200409161. [DOI] [PMC free article] [PubMed] [Google Scholar]