

Abstract
BACKGROUND AND PURPOSE
Terfenadine has been reported to cause cardiac death. Hence, we investigated its pro-arrhythmic potential in various in vitro models.
EXPERIMENTAL APPROACH
Pro-arrhythmic effects of terfenadine were investigated in rabbit isolated hearts and left ventricular wedge preparations. Also, using whole-cell patch-clamp recording, we examined its effect on the human ether-à-go-go-related gene (hERG) current in HEK293 cells transfected with hERG and on the INa current in rabbit ventricular cells and human atrial myocytes.
KEY RESULTS
Terfenadine concentration- and use-dependently inhibited INa in rabbit myocytes and in human atrial myocytes and also inhibited the hERG. In both the rabbit left ventricular wedge and heart preparations, terfenadine at 1 µM only slightly prolonged the QT- and JT-intervals but at 10 µM, it caused a marked widening of the QRS complex, cardiac wavelength shortening, incidences of in-excitability and non-TdP-like ventricular tachycardia/fibrillation (VT/VF) without prolongation of the QT/JT-interval. At 10 µM terfenadine elicited a lower incidence of early afterdepolarizations versus non- Torsades de Pointes (TdP)-like VT/VF (100% incidence), and did not induce TdPs. Although the concentration of terfenadine in the tissue-bath was low, it accumulated within the heart tissue.
CONCLUSION AND IMPLICATIONS
Our data suggest that: (i) the induction of non-TdP-like VT/VF, which is caused by slowing of conduction via blockade of INa (like Class Ic flecainide), may constitute a more important risk for terfenadine-induced cardiac death; (ii) although terfenadine is a potent hERG blocker, the risk for non-TdP-like VT/VF exceeds the risk for TdPs; and (iii) cardiac wavelength (λ) could serve as a biomarker to predict terfenadine-induced VT/VF.
Keywords: conduction time, INa, QRS, QT, terfenadine, VT/VF: ventricular tachycardia/fibrillation, TdPs: Torsades de Pointes
Introduction
Terfenadine has been reported to cause cardiac death, in at least 125 and 14 cases in the United States and UK, respectively (Rangno, 1997), and the Food and Drug Administration (FDA) recommended to remove it from the market in 1997 due to its pro-arrhythmic risk for long QT-related Torsades de Pointes (TdPs) (US Food and Drug Administration, 1997). However, most of the terfenadine-related fatality cases were reported without ECG measurements and the authors speculated that these mortalities were the result of TdPs, presumably due to its known effects on the human ether-à-go-go-related gene (hERG). In some case reports, so called ‘TdPs’ occurred in the absence of significant QT prolongation (Woosley et al., 1993; June and Nasr, 1997). Recent detailed reviewing of the FDA records suggested that terfenadine-induced ventricular tachycardia/ventricular fibrillation (non-TdP-like: VT/VF) may largely exceed its incidence of TdPs (Hondeghem et al., 2011). Moreover, it has been shown that it is difficult to induce TdPs in preclinical models with terfenadine, but very easy to induce non-TdP-like cardiac arrhythmias such as VT/VF (Lu et al., 2000; Batey and Coker, 2002; Fish and Antzelevitch, 2003; Hondeghem et al., 2011).
Therefore, the question arose: were these speculated terfenadine-induced TdPs in man really TdPs, or were they, at least in part, the result of other cardiac events such as non-TdP-like VT/VF? Indeed, clinical symptoms of non-TdP-like VT/VF and TdPs are similar. In fact, in a clinical trial, administration of terfenadine was associated with a very small QTc prolongation in healthy volunteers and even in patients with cardiovascular diseases (McTavish et al., 1990), and it has been suggested that terfenadine-induced cardiac death was due to direct cardiac arrest (non-TdPs) (Pratt et al., 1994; 1996). In experimental in vivo models, terfenadine alone causes cardiotoxic effects/cardiac death, but this is not associated with QT prolongation and the occurrence of TdPs, but with marked widening of the QRS complex and other cardiac arrhythmias (Lu et al., 2000; Batey and Coker, 2002). Moreover, terfenadine reduced the maximal upstroke velocity (Vmax) and induced in-excitability in dog isolated Purkinje fibres and guinea-pig ventricular muscles without prolonging the action potential duration (APD) (Lang et al., 1993; Tanaka et al., 1996). These findings indicate that terfenadine-induced cardiac events/death could be caused by its blocking effect on the sodium current (INa) (responsible for slowing conduction, widening the QRS complex and reduction of Vmax), rather than its inhibition of the rapidly activating delayed rectifier potassium current (IK(R)) (responsible for prolonging the QT/ incidence of TdPs).
In a recent study, we showed that a drug with ‘bad’INa-blocking activities like flecainide can indeed slow cardiac conduction (widening QRS) and therefore induce non-TdP-like VT/VF (Lu et al., 2010). However, a suitable biomarker for detecting the ability of these INa-blockers to induce VT/VF is at present unavailable, and an appropriate biomarker to predict this potential is also lacking. Hence, we speculated that cardiac wavelength (λ) (Girouard et al., 1996; Girouard and Rossenbaum, 2001; Hondeghem et al., 2011) may serve as a parameter to predict a compound's potential to induce non-TdP-like VT/VF.
Cardiac wavelength (λ) is defined as the distance travelled by the depolarization wave during the functional refractory period (Mines, 1913; Robert et al., 1999). A direct local estimate of λ is determined by the multiplication of conduction velocity (CV) by the effective refractory period (ERP): λ= CV × ERP or ERP/QRS duration (Karagounis et al., 1995; Girouard and Rossenbaum, 2001). Drug-induced changes in λ are used to determine their arrhythmogenic or anti-arrhythmic properties. Drugs that increase λ tend to be anti-arrhythmic, and drugs that decrease λ tend to be arrhythmogenic, at least in atria (Jacquemet et al., 2005). The wavelength has been used as a parameter to study ventricular re-entrant tachycardia and atrial fibrillation (Robert et al., 1996; Aidonidis et al., 2009), but it is less well known whether it could be used as an index of drug-induced VT/VF.
In the present study, we investigated the effects of terfenadine on cardiac INa not only in rabbit isolated ventricular myocytes, but also in human atrial myocytes, in order to investigate how the effects of terfenadine-induced INa blockade in animals would translate to humans. Furthermore, we investigated the pro-arrhythmic potential of terfenadine in rabbit isolated, Langendorff-perfused hearts and isolated, arterially perfused left ventricular wedge preparations. Our present results suggest that terfernadine is indeed a ‘bad’INa blocker like Class Ic flecainide, causing non-TdP-like VT/VF. Furthermore, our data suggest that cardiac wavelength (λ) can be used as a biomarker to predict drug-induced VT/VF.
Methods
All animal care and experimental procedures were performed in accordance with ‘The provision of the European Convention’ on the protection of vertebrate animals that are used for experimental and other scientific purposes, and with ‘the Appendices A and B’, made at Strasbourg on March 18, 1986 (Belgian Act of October 18, 1991).
Effects of terfenadine on cardiac INa in rabbit isolated left ventricular myocytes and in human atrial myocytes
As described previously (Guo et al., 2008), rabbit ventricular myocytes were isolated enzymatically from rabbit hearts (New Zealand, female, 2.3–2.8 kg). The total number of rabbits used was 33. Human atrial myocytes were isolated enzymatically from human atrial appendages, which were obtained from 2 female patients during open-heart surgery. Informed consent was obtained from these patients for use of their cells and the experiments were approved by IRB.
Aliquots of cell-containing solution (about 0.1 mL) were added to a 1.5 mL bath chamber on a stage of an inverted microscope and INa was recorded at room temperature (22 ± 0.5°C) using a whole-cell patch-clamp technique. Cells were superfused at 2 mL·min−1 with a bath solution containing (in mM): CsCl 130, NaCl 10, MgCl2 1.0, CaCl2 1.0, HEPES 5, glucose 10, CdCl2 0.3 (pH was adjusted to 7.4 with CsOH). Command pulses were generated by a Digidata 1320A (Axon Instruments, Foster City, CA, USA) controlled by pClamp 8 (Axon Instruments). Pipettes (made using Model P80 puller, Sutter Instrument, Novato, CA, USA) with a resistance of 3 to 4 MΩ when filled with a pipette solution were selected. The composition of the pipette solution was (in mM): NaCl 10, CsF 110, CsCl 20, EGTA 5, HEPES 5, ATP-Mg 5 (pH adjusted to 7.4 with CsOH). Liquid junction potentials were zeroed before the formation of the membrane-pipette seal, and passing to the whole-cell mode was obtained by applying a light negative pressure suction. The series resistance was compensated electronically 70–80%. INa, defined as tetrodotoxin (TTX)-sensitive current, was recorded during an 80 ms pulse duration (or 10 ms for the use-dependent protocol) depolarizing voltage step from a holding potential (Vh) of −100 mV to test potentials (Vt) between −80 mV and +40 mV in a 20 mV increment.
During each experiment, the holding potential was maintained at −100 mV. Current-voltage (I-V) relationships were obtained 4 min after cell membrane rupture as INa(0), and then as INa at 4 min after initiation of the terfenadine perfusion at each concentration. The INa/INa(0) values obtained at a voltage of −40 mV were plotted against concentration values. IC50 was determined by fitting the individual data point values by using the equation: I/Io= 1/[1 + ([C]/[IC50])nH], where nH is Hill coefficient, [C] is the corresponding treatment concentration; and [IC50] is the concentration at which INa(0) is reduced by 50%.
The use-dependent effect of terfenadine on INa was assessed by eliciting a train (20 pulses) of 10 ms depolarizing pulses from the holding potential of −100 to −40 mV at different frequencies (0.5 and 2 Hz) at concentrations of 0.1, 1 and 10 µM of terfenadine. The two frequencies were tested in the same cell with a 2 min rest between stimulations. The first pulse represented the tonic block, whereas the last pulse (20th pulse) was used for calculating the IC50 at that frequency. Relative current, INa/INa(0) {normalized to the current before test article perfusion [INa(0)] as control} was plotted as a function of frequencies.
Effects of terfenadine on hERG current
The experimental approach we used is similar to that employed in our previous study (Lu et al., 2010). A HEK cell line (HEK293) with a stable transfection of hERG was used (purchased from Dr Z. Zhou, University of Wisconsin, Madison, USA).
After disruption of the membrane, the cell capacitance and the series resistance were compensated (90%) using the circuit of the EPC-9 patch clamp amplifier. The holding potential was −80 mV. The hERG-mediated current (K+-selective outward current) was determined as the maximal tail current at −40 mV after a 2 s depolarization to +60 mV. Pulse cycling rate was 15 s. Before each test pulse a short pulse (0.5 s) to −60 mV was given to determine leak current. The protocol consisted of a 5 min equilibration period (no pulses), 5 min in control solution and then 5 min for each concentration of the drug. Six concentrations of terfenadine (0.003, 0.01, 0.03, 0.1, 0.3 and 1 µM) were tested to obtain the IC50 value (n= 5 per concentration; up to three concentrations per cell). The effect of the drug was measured after 5 min of drug application by dividing the measured current by the extrapolated current.
Effects of terfenadine in rabbit isolated, Langendorff-perfused hearts
The method for determining various cardiac electrophysiological properties was similar to, and has been described in detail as, the rabbit isolated, Langendorff-perfused heart with total AV block (Lu et al., 2010). Briefly, the heart was paced at 1 Hz and perfused at a constant pressure of 80 cmH2O with a bicarbonate buffer (at 37°C). Recording electrodes were placed in the left ventricular endocardium, left and right epicardium for monophasic action potentials (MAPs), and for ECG recording, respectively.
APD90 or APD60, triangulation (APD90–APD30), short-term beat-to-beat instability of the APD90, intraventricular conduction time (CT) and coronary flow were measured. An ECG from Lead II was recorded. ECG parameters such as QRS duration, QT interval, JT interval, Tp–Te = QTend–QTpeak, rTp–Te (Tp–Te/QT–interval*100) (Yan and Antzelevitch, 1999; Liu et al., 2006) were also measured. CT was defined as the interval between the electrical stimulation and the start of the depolarization of the action potential (upstroke of the action potential). The hearts were perfused with terfenadine (0.1, 1 and 10 µM) or its solvent for 30 min per concentration (n= 6 per group). A random stimulation protocol was applied, for a short period, to the heart at baseline, and at 17 min after each concentration. All data were taken at a stimulation rate of 1000 ms.
Three additional experiments with terfenadine were performed using the same protocol, in order to measure the concentration of terfenadine in the tissue bath and in the heart tissue. Samples were taken at the end of each perfusion for quick analysis of the concentration of terfenadine by means of LC-MS.
Effects of terfenadine in rabbit left ventricular arterially perfused wedge preparation
The methods used for isolation, perfusion, and recording of transmembrane activity from the arterially perfused ventricular wedge preparation from female rabbits (weighing 2.5 to 3 kg), as well as the viability and electrical stability of the preparation, have been described in previous studies (Yan and Antzelevitch, 1999; Fish and Antzelevitch, 2003; Liu et al., 2006). The preparation was paced at basic cycle lengths (BCLs) of 1000 and 2000 ms. A brief period (30 to 60 s) of faster pacing at a BCL of 500 ms or less was introduced between BCLs of 1000 and 2000 ms using bipolar silver electrodes.
From the ECG, we measured transmural dispersion of repolarization (TDR), which is approximately equal to the interval from the peak to the end of the T-wave (QTend–QTpeak) and rTp–Te (=Tp–Te/QT*100) (Yan and Antzelevitch, 1999; Liu et al., 2006). The QT interval was defined as the time from the onset of the QRS to the point at which the final downslope of the T wave crosses the isoelectric line. In order to estimate CV, the size of each preparation was measured.
The CV was calculated by the equation: CV (cm·s−1) = the longest distance from the edge of the preparation to the pacing electrode divided by the QRS duration(s). The programmed stimulation at twofold the threshold from the endocardium was used to determine the ERP: eight regular stimulations (S1) were followed by an extrastimulus (S2). The interval of S1 to S2 was changed decrementally by 5 ms until the stimulation failed to initiate excitation of the preparation. This S1–S2 interval was then considered as the ERP. The impulse wavelength (λ) was calculated by ERP (ms) × CV (cm·s−1) (Girouard and Rossenbaum, 2001) or ERP/QRS ratio (Karagounis et al., 1995). All parameters were recorded at a BCL of 500 and 2000 ms.
Terfenadine (0.1, 1 and 10 µM) or solvent (maximal 0.1% DMSO) was perfused for 30 min per concentration (n= 6 per group).
VT was defined as four or more consecutive ventricular premature beats, VF was defined as a ventricular tachyarrhythmia without identifiable ECG, and in-excitability was defined as a preparation that did not respond to electrical stimulation.
Compounds
Terfenadine was obtained from Sigma and dissolved in either pyrogen-free water (acidified with tartaric acid to obtain a pH of approximately 4) for the study in isolated hearts, or in dimethyl sulfoxide (DMSO; final maximal bath concentration up to 0.1%) for the ion channel tests and the study in the wedge preparations. Similar solutions without compound were used as solvent controls. The concentrations of terfenadine up to 10 µM, selected for these studies, were based on previously published IC50 values for Na+- or hERG-current blockade and on its reported effects in a pro-arrhythmic in vitro model (Crumb et al., 1995; Roy et al., 1996; Fish and Antzelevitch, 2003).
Data analysis
All values are expressed as mean and SEM. Statistically significant differences between solvent and compound were calculated based on their changes from baseline with the Wilcoxon–Mann–Whitney test. Two-tailed probabilities of less than 0.05 were considered to indicate statistically significant differences.
Results
Effects of terfenadine on cardiac INa in rabbit isolated, left ventricular myocytes and human atrial myocytes
Effect of terfenadine on INa current in rabbit ventricular myocytes
Terfenadine concentration-dependently inhibited INa in rabbit ventricular myocytes. This effect was partially reversible upon washout. At the test potential of −40 mV, terfenadine inhibited INa by 2.8 ± 0.9%, 16.8 ± 1.2% and 59.6 ± 2.8% (mean ± SEM, n= 5) at concentrations of 0.1, 1 and 10 µM, respectively (Figure 1). The estimated IC50 value for INa blockade in ventricular myocytes at 0.1 Hz (tonic blockade) was 6.9 ± 0.7 µM (mean ± SEM, n= 5).
Figure 1.
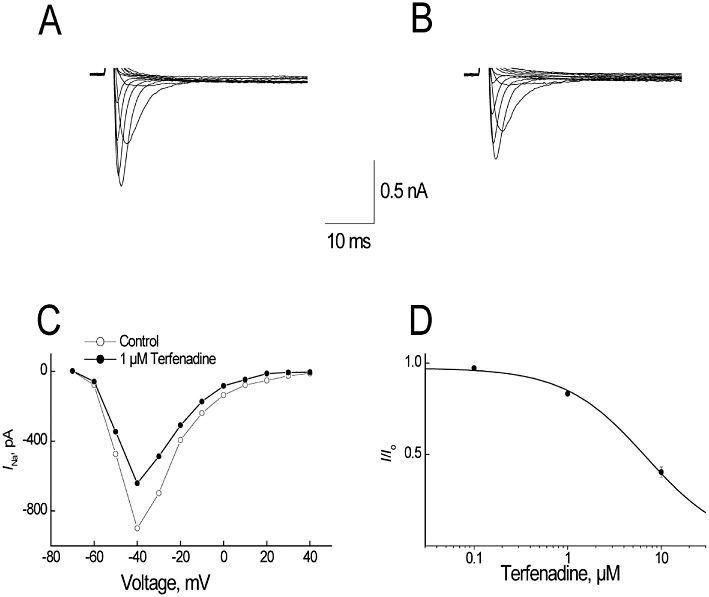
Effect of terfenadine on sodium currents (INa) in rabbit ventricular myocytes. (A,B) The responses of superimposed currents to step depolarizations ranging from −80 to +40 mV from a holding potential of −100 mV were obtained under control (A) conditions and after 2 min of superfusion with 1 µM terfenadine (B). (C) Effect of 1 µM terfenadine on the peak current-voltage relationships of INa. I–V curves before and after 2 min superfusion with 1 µM terfenadine. (D) Concentration–response relationship of terfenadine on INa. Data were fitted with an equation I/Io= 1/(1 +[C]/[IC50]). All data are expressed as mean ± SEM, n= 5.
Terfenadine inhibited INa in a use-dependent manner in rabbit ventricular myocytes. At a frequency of 0.5 Hz, there was no significant cumulative reduction in INa magnitude during consecutive pulses. However, at a frequency of 2 Hz, in addition to a reduction in INa during the first pulse, there was a significant cumulative reduction in INa magnitude during consecutive pulses. At steady state (i.e. the 20th pulse), the estimated IC50 values were 6.9 ± 0.7 µM, 6.8 ± 0.7 µM and 1.3 ± 0.1 µM at frequencies of 0.1, 0.5 and 2 Hz (mean ± SEM, n= 5), indicating terfenadine has a strong use-dependent effect on INa in rabbit isolated, ventricular myocytes.
Effect of terfenadine on INa current in human isolated, atrial myocytes
Similar to the effects of terfenadine in the rabbit ventricular myocytes, terfenadine exhibited concentration- and use-dependent inhibition of INa in human isolated atrial myocytes: inhibition of current by 3.9 ± 0.4%, 16.2 ± 1.1% and 56.5 ± 1.1% at 0.1 µM, 1 and 10 µM respectively (n= 6 preparations from two subjects per concentration vs. 1.1 ± 0.3% with solvent, n= 6; P < 0.05). The estimated IC50 value for INa blockade was 8.1 ± 0.4 µM at 0.1 Hz (tonic blockade). Terfenadine also exhibited a strong use-dependent inhibition of INa in the human atrial myocytes as the estimated IC50 value at 2 Hz was 1.7 ± 0.2 µM (vs. 8.1 ± 0.4 µM at 0.1 Hz; P < 0.05) (Figure 2).
Figure 2.

Terfenadine-induced use-dependent blockade of INa in human atrial myocytes. (A,B) Representative current traces using 20 consecutive pulses at the frequency of 0.5 Hz (A) and 2.0 Hz (B). (C) Relative INa was plotted as a function of pulse number. A train of 20 depolarizing pulses to −40 mV was applied at different frequencies (0.5. and 2.0 Hz) in the presence of 1 µM terfenadine. (D) Relative INa of frequency-dependent blockade at steady-state (the 20th pulse) was plotted against concentrations. (E) The IC50 values of terfenadine on INa at corresponding stimulation frequencies.
Effects of terfenadine on hERG
Terfenadine reduced the hERG-mediated current from low, nanomolar concentrations with an IC50 of 26 nM; it inhibited hERG current by 8.4 ± 3.4%, 27.0 ± 4.0%, 53.0 ± 3.8%, 79.6.0 ± 3.6%, 94.8.0 ± 1.7% and 98.4 ± 0.4% at 3, 10, 30 and 100 nM, 0.3 and 1 µM, respectively (vs. <6.0% with solvent control (n= 5 cells per concentration); P < 0.05 at 10 nM and higher concentrations) (Figure 3).
Figure 3.

Effect of terfenadine on the hERG current. Top: the pulse protocol, which was repeated every 15 s, consisted of a small test pulse to −60 mV to determine the (linear) leak current, a depolarizing pulse to +60 mV to activate the hERG current, a pulse to −40 mV to see the large tail current (which is used to determine the amplitude of the hERG current) and then the cells were clamped back to the holding potential of −80 mV eliciting a small tail current. Bottom: current traces obtained in different conditions superimposed (control solution, 3 and 30 nM and 1 µM of terfenadine).
Effects of terfenadine on isolated, Langendorff-perfused rabbit hearts
Compared with solvent (n= 6), terfenadine (n= 6) at 0.1 µM did not significantly change the duration of the QT interval, JT interval, QRS duration and rTp–Te of the T wave. At 1 µM, terfenadine significantly prolonged the QT interval by 21% (vs. +1% of baseline with solvent; P < 0.05) and JT interval by 30% from baseline (vs. +1% of baseline with solvent; P < 0.05) and increased rTp–Te by 48% (vs. +9% of baseline with solvent; P < 0.05), but terfenadine at this concentration did not significantly change QRS duration. At 10 µM, terfenadine reduced the prolongation of JT interval (+1% of baseline vs. +3% of baseline with solvent; P > 0.05) and markedly increased QRS duration by 89% from baseline (vs. +6% from baseline with solvent; P < 0.05) and Tp – Te – by 64% from baseline (vs. +3% from baseline with solvent; P < 0.05) (Figure 5). Terfenadine increased coronary flow (+17% and +17% from baseline at 0.1 and 1 µM vs. −1 and −10% with the time-matched solvent; P > 0.05). At 30 and 60 min perfusion with terfenadine at 10 µM, the coronary flow could not be accurately measured due to its strong effects on the heart, but it was found to be increased by 28% in one heart. Compared to the solvent terfenadine was not found to have any statistically significant effects on coronary flow due to the large variations in this parameter.
Figure 5.

Effects of terfenadine on QT interval, JT interval, QRS duration and dispersion of the ventricular repolarization (rTp–Te) in the isolated, Langendorff-perfused rabbit hearts. Terfenadine prolonged the JT interval only at 1 µM, markedly widened QRS duration only at 10 µM, and significantly increased dispersion both at 1 and 10 µM. *P < 0.05 versus solvent control group.
At 0.1 µM, terfenadine did not elicit early afterdepolarizations (EADs), VF and in-excitability. At 1 µM, terfenadine elicited EADs in two out of the six hearts (vs. 0 out of the 6 hearts with solvent) (Figure 4). The incidence of EADs was associated with a prolongation of the ventricular repolarization time. At 1 µM, terfenadine elicited in-excitability in one out of the six hearts (vs. 0 out of the 6 hearts with solvent; P > 0.05). At 10 µM, terfenadine elicited EADs in one (P > 0.05 vs. solvent), but induced VT and VF in 6 and 5 (P < 0.05 vs. 0 out of the 6 hearts with solvent) and in-excitability in three out of the six hearts (P > 0.05 vs. 0 out of the 6 hearts with solvent) (see an example in Figure 4).
Figure 4.

An example: effects of terfenadine in an isolated, Langendorff-perfused rabbit heart. After a 10 min perfusion with terfenadine at 1 µM, early afterdepolarization (EAD) occurred. Ventricular fibrillation (VF: non-TdP -like) developed after 18 min of perfusion with 10 µM terfenadine without QT prolongation. Terfenadine at 1 µM prolonged MAP duration at 90% repolarization (APD90) from 200 to 242 ms with EAD. However, at 10 µM, it shortened APD90 to 175 ms and largely increased QRS duration before induction of VF. ECG, electrocardiogram recording; ES, electrical stimulation at 1 Hz; MAP, epicardial monophasic action potential (MAP) recording.
The concentrations of terfenadine in three additional hearts were much higher than the target concentrations: being 2650 ng·g−1 (≍5.6 µM; assuming 1 g of wet tissue corresponds to 1 mL), 14 200 ng·g−1 (≍30 µM) and 376 000 ng·g−1 (≍800 µM) of the target concentrations of 0.1, 1 and 10 µM, respectively, at the end of 30 min perfusion (n= 3). These data indicate that terfenadine accumulated in the heart tissue. The concentration of terfenadine in one remaining heart without VF was 76 300 ng·g−1 (≍162 µM) of the target concentration of 10 µM (at end of 60 min perfusion). The concentrations in the tissue bath were low (18, 21 and 47% of the target concentrations at 0.1, 1 and 10 µM, respectively).
Effects of terfenadine on rabbit arterially perfused left ventricular wedge preparations
Relative to solvent (n= 7), terfenadine (n= 7) at 0.1 µM caused a slight but significant increase in the duration of the QT interval (+7 ms from baseline vs. +2 ms of baseline with solvent; P < 0.05), JT interval (+7 ms from baseline vs. +2 ms of baseline with solvent; P < 0.05), Tp–Te (+4 ms from baseline vs. 0 ms of baseline with solvent; P < 0.05) and rTp–Te (+1 vs. 0 with solvent; P < 0.05). At 1 µM, terfenadine significantly prolonged the QT interval and JT interval (+20 ms and +17 ms from baseline vs. +4 ms and +4 ms of baseline with solvent; P < 0.05), slightly increased QRS duration (+2 ms of baseline vs. 0 ms with solvent; P < 0.05), Tp–Te (+9 ms of baseline vs. +1 ms with solvent; P < 0.05) and rTp–Te (+2 ms vs. 0 ms; P < 0.05). At 30 min after onset of perfusion with the highest concentration (10 µM), terfenadine shortened the QT interval (−14 ms from baseline vs. +2 ms of baseline with solvent; P < 0.05) and JT interval (−28 ms from baseline vs. +2 ms of baseline with solvent; P < 0.05), largely widened the QRS complex (+18 ms vs. 0 ms with solvent; P < 0.05), and decreased Tp–Te (−12 ms of baseline vs. o ms with solvent; P < 0.05) and rTp–Te (−3 of baseline vs. 0 with solvent; P < 0.05) (Figure 6A). It should be mentioned that terfenadine not only markedly widened QRS duration, but also flattened the T wave and therefore Tp–Te could not be accurately measured (Figure 8). Therefore, the transmural dispersion of repolarization (Tp–Te and rTp–Te) might not be an accurate measurement in this study. The large widening of the QRS complex by terfenadine at 10 µM was rate-dependent (+26% of baseline at 0.5 Hz vs. +55% of baseline at 2 Hz; P < 0.05) (Figure 6B).
Figure 6.

(A) Effects of terfenadine (n= 7) and solvent (n= 7) on QT interval, JT interval, QRS duration, Tp–Te and rTp–Te in the rabbit isolated, arterially perfused left ventricular wedge preparation. Values are expressed as changes from baseline (in ms, except for rTp–Te) and taken at a stimulation cycle length of 2000 ms, *P < 0.05 versus solvent. (B) Rate-dependent effects of terfenadine (n= 7) on QRS duration in the left ventricular wedge preparation.*P < 0.05 versus solvent (≤1% changes from baseline).**P < 0.05 at a stimulation rate of 500 ms versus solvent at 500 ms and also P < 0.05 versus the respective value at 2000 ms. Values are mean ± SEM.
Figure 8.
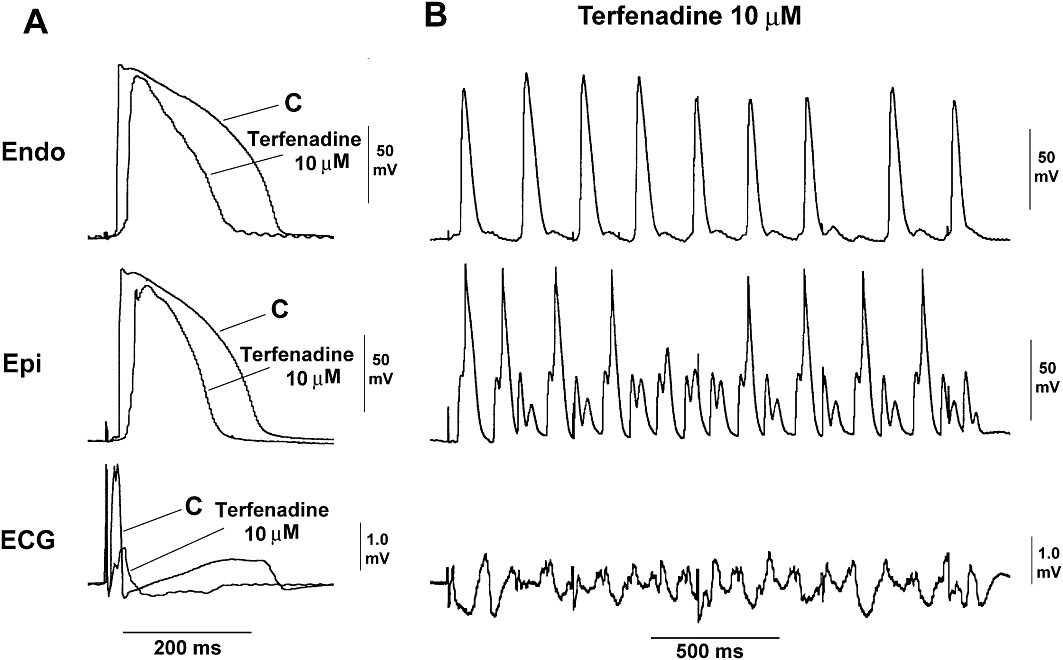
Original endocardial (Endo), epicardial (Epi) action potential and ECG tracings recorded from a rabbit left ventricular wedge preparation (A) during a control period (C) and in the presence of terfenadine at 10 µM. (B) After terfenadine (10 µM), and an incidence of VT/VF (non-TdP-like) occurred on pacing 500 ms. (A) The ECG showed that terfenadine at 10 µM shortened APD and QT, largely reduced upstroke of the action potential and increased QRS, before it induced VT/VF.
After the second 30 min perfusion period with terfenadine at 10 µM, the parameters were unmeasurable in most preparations due to the strong effects of the compound.
Terfenadine at 0.1 µM did not significantly change ERP, ERP/QRS ratio and the impulse wavelength (λ) [λ= ERP (ms) × CV (cm·s−1)] at a stimulation rate of 2000 ms or 500 ms. At 1 µM and at a stimulation rate of 500 ms, terfenadine slightly but significantly reduced ERP/QRS ratio and the wavelength, but not at a stimulation rate of 2000 ms (Figure 7). Terfenadine at 1 µM significantly increased ERP by 18 ms and 9 ms at a stimulation rate of 2000 and of 500 ms, respectively. At 10 µM (at end of 30 min), terfenadine did not significantly change ERP, but significantly decreased the ERP/QRS ratio from 5.3 ± 0.4 at baseline to 3.9 ± 0.2 (vs. from 5.5 ± 0.5 of baseline to 5.5 ± 0.5 with solvent, P < 0.05), and λ from 8.4 cm at baseline to 6.1 cm (vs. 8.5 cm to 8.5 cm with the time-matched solvent; P < 0.05). Importantly, the reductions in ERP/QRS ratio and λ by terfenadine were rate-dependent (Figure 7). At 500 ms, the reduction of the ERP/QRS ratio by the compound was similar (−1, −6 and −40% from baseline at 0.1, 1 and 10, respectively vs. ≤1% of baseline with solvent). The reduction of λ caused by the compound was mainly due to a slowing of conduction (CV: −1, −5 and −21% from baseline at a stimulation rate of 2000 ms and 0, −10 and −34% from baseline at 500 ms at 0.1, 1 and 10 µM, respectively, vs. <1% of baseline with solvent). Terfenadine slightly but significantly reduced ERP only at 10 µM at 60 min perfusion (−10% from baseline both at 2000 ms and 500 ms vs. <1% of baseline with solvent), probably due to its shortening of the APD/QT-interval.
Figure 7.
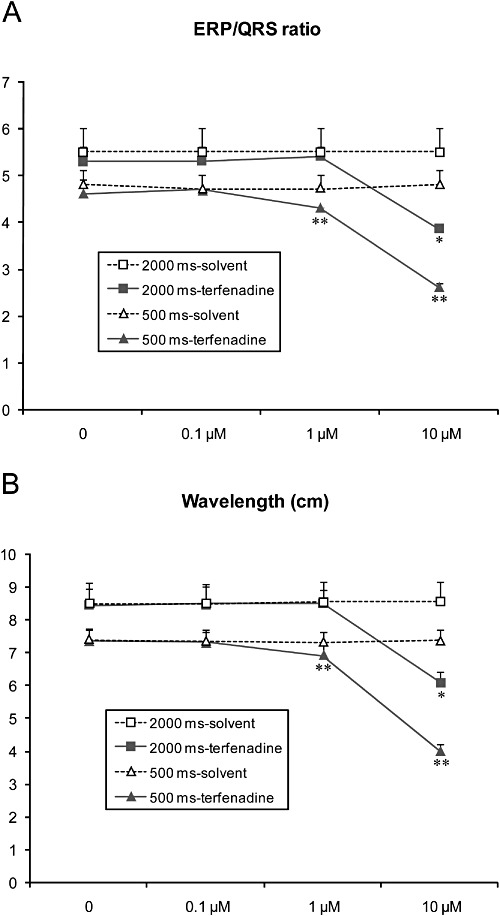
Effects of terfenadine (n= 7) on (A) ERP/QRS ratio and (B) wavelength (λ) in the rabbit isolated, arterially perfused left ventricular wedge preparation. ERP: effective refractory period. Values are mean ± SEM. *P < 0.05 versus solvent. **P < 0.05 at a stimulation rate of 500 ms versus solvent at 500 ms and also P < 0.05 versus the respective value at 2000 ms.
At 0.1 and 1 µM, terfenadine did not elicit EADs, VF and in-excitability. At 10 µM, terfenadine did not elicit EADs, but induced VT/VF in five out of the seven preparations (P < 0.05 vs. solvent) (see an example in Figure 8) and in-excitability in three out of the seven preparations after 30 min perfusion (P > 0.05 vs. 0 out of the 6 hearts with solvent).
Discussion and conclusions
Our data in isolated rabbit ventricular wedge and heart preparations showed that terfenadine at 10 µM elicited a high incidence of non-TdP-like VT/VF, associated with a marked slowing of conduction or widening of the QRS complex, without notable prolongation of QT and JT intervals. The slowing of conduction or QRS widening was clearly due to its blocking activities on cardiac INa. Indeed, terfenadine reduced this Na+-current in a strong use-dependent manner with IC50s of 6.9 µM (0.1 Hz) to 1.3 µM (2 Hz) in rabbit isolated ventricular cells. The INa-blocking property of terfenadine was also confirmed in human atrial cells (IC50 of 8.1 µM at 0.1 Hz and 1.7 µM at 2 Hz). The effect of terfenadine on INa was use-dependent in both rabbit and human cardiac cells. Terfenadine largely slowed down ventricular conduction without significantly changing ERP in the rabbit isolated wedge preparations (at 10 µM). As such terfenadine presents itself with a phenotype that can be classified as a Class Ic-like anti-arrhythmic similar to flecainide, according to Vaughan Williams classification (1970). The induction of non-TdP-like VT/VF was associated with a decrease in cardiac wavelength (λ). This parameter could potentially serve as an important biomarker for terfenadine-induced non-TdP-like VT/VF.
Cardiac wavelength plays an important role in terfenadine-induced non-TdP like VT/VF
In the present study in the rabbit isolated ventricular wedge, the induction of non-TdP-like VT/VF was associated with a significant decrease in cardiac wavelength λ at a concentration of 10 µM terfenadine (λ= ERP × CV or λ= ERP/QRS-duration) (Girouard and Rossenbaum, 2001; Karagounis et al., 1995) (Figure 7). The decrease in cardiac wavelength by shortening ERP and/or by slowing of conduction with terfenadine may therefore be a prerequisite for the initiation of non-TdP- like VT/VF.
Indeed, changes in conduction, ERP or both, promote re-entrant arrhythmias (Robert et al., 1996; 1999; Aidonidis et al., 2009). Drugs that increase wavelength tend to increase the risk for TdPs (Hondeghem, 2008), whereas agents that decrease the wavelength tend to increase the risk for non-TdP VT/VF (Robert et al., 1996; 1999; Aidonidis et al., 2009; Hondeghem et al., 2011). In 1913, Mines first suggested the role of λ in the mechanism of re-entry: re-entrant excitation was only possible if the λ of the propagating impulse was shorter than the re-entrant path length, i.e. that there is an excitable gap within the re-entrant circuit (Mines, 1913). Later on, other studies also suggested that decreases in λ are associated with the initiation and maintenance of re-entry, which lead to re-entrant VT or fibrillation (Robert et al., 1996; 1999; Aidonidis et al., 2009). Robert et al. reported that levcromakalim-induced re-entry VT in rabbit isolated hearts was associated with a shortening in wavelength due to a direct decrease in ERP without changes in CV (Robert et al., 1999). Aidonidis et al., (2009) reported that bimakalim (another KATP channel opener) shortened ERP and induced re-entrant non-TdP-like VT/VF in chronic infarct anaesthetized pigs. Therefore, the decrease in the cardiac wavelength (λ) may play an important role in terfenadine-induced non-TdP- like VT/VF.
In the present study, we showed that terfenadine-induced non-TdP- like VT/VF was associated with a widening of the QRS complex or a slowing of conduction. Slowing of conduction is associated with sudden cardiac arrhythmic death in both man and in experimental models (Fish and Antzelevitch, 2003; Antzelevitch, 2006; Lu et al., 2010). Drugs that slow conduction, thereby decreasing λ and associated VT/VF, (i.e. terfenadine) could be interpreted as ‘bad’INa blockers like flecainide (Class Ic) (Lu et al., 2010). Two limitations of the measurement of λ were identified in the present study: (i) we only measured λ at one site in the isolated left ventricular wedge preparation, not at different sites throughout the heart; and (ii) we did not use mapping due to technical limitations. However, changes in ECG parameters such as a marked slowing of conduction and a mild shortening of the QT interval by terfenadine at 10 µM, indirectly confirm the reduction in λ in both the left ventricular wedge preparation and the rabbit isolated heart. Our data confirm previous findings, in the rabbit isolated heart with terfenadine, with no direct measurements of ERP (Hondeghem et al., 2011).
Terfenadine-induced non-TdP-like VT/VF exceeds EADs/TdPs, and is therefore unlikely to be linked to its ability to prolong QT
Although terfenadine is a potent hERG blocker, some studies have already questioned the link between the pro-arrhythmic effects of terfenadine and a long QT/TdP (Woosley et al., 1993; Lu et al., 2000; Batey and Coker, 2002; Hove-Madsen et al., 2006; Hondeghem et al., 2011). FDA post marketing data also indicate that terfenadine caused non-TdP-like cardiac arrhythmias that exceed the TdP incidence (Hondeghem et al., 2011). Our present results and other studies (Fish and Antzelevitch, 2003; Hondeghem et al., 2011), which showed that terfenadine induced non-TdP-like VT/VF at concentrations that did not significantly prolong the QT interval confirm the earlier so-called ‘TdP’ cases that occurred with terfenadine without significant QT-prolongation (Woosley et al., 1993; June and Nasr, 1997). It has been demonstrated that drug-induced long QT and TdPs via a IK(R)/hERG blocking mechanism are more likely to occur in females than in males (Larsen and Kadish, 1998). This gender difference was not observed with terfenadine in vitro (Fish and Antzelevitch, 2003) and, in fact, VT was found to be more prevalent in males than in females in an epidemiological study with terfenadine (Hanrahan et al., 1995).
The IC50 value for inhibition of hERG current by terfenadine has been reported to be approximately 50 nM (Salata et al., 1995), which is similar to the results from our present study (26 nM), and in the range of the value obtained by Lacerda et al., 2001 (56 nM). However, the IC50 for hERG inhibition should not be considered as an absolute criterion for estimation of its TdP risk in other experimental models or in man. The extrapolation of hERG data into more integrated models in vitro and in vivo, where other ion channels are present, is extremely difficult. It is well known that not all hERG blockers are prone to prolong QT/APD in functional models. We have shown that only 55.4% of hERG-blocking compounds prolong APD/QT in functional models (Lu et al., 2010). Verapamil, an L-type Ca2+ channel blocker (IC50 of 246 nM; Freeze et al., 2006), is an example of a compound that has a large effect on hERG (IC50 of 143 nM, Zhang et al., 1999) but has not been reported clinically to prolong QT and related TdP. Therefore, it is not possible to estimate a drug's pro-arrhythmic potential based on only its IC50 value for a single ion channel such as hERG. Moreover, not all arrhythmias are linked to hERG blocking activities.
Terfenadine elicited EADs in two out of the six hearts at 1 µM. At 10 µM, the incidence of EADs (1/6) was much lower than the incidence of VT/VF (VT in 6/6 and VF in 5/6), with little QT prolongation, but with a marked slowing of conduction. These results show that with terfenadine there is only a small risk of a prolonged QT and TdPs, but a higher risk for non-TDP-like VT/VF, similar to the findings of others in rabbit isolated hearts (Hondeghem et al., 2011). Moreover, data from experimental in vivo models further show that terfenadine causes a marked widening of the QRS complex and cardiac arrest in the absence of QT prolongation and TdPs (Lu et al., 2000; Batey and Coker, 2002).
These in vitro and in vivo data indicate that terfenadine may represent a pro-arrhythmic substance for non-TdP-like VT/VF, which may be related its INa-blocking activities. Antzelevitch's group reported that terfenadine-induced non-TdP-like VT/VF, is similar to Brugada syndrome (which is caused by a genetic defect in the INa channel) (Fish and Antzelevitch, 2003). This hypothesis is in line with our present data, in which the induction of non-TdP- like VT/VF by terfenadine may be the result of its direct INa-blocking activities (slowing conduction without prolongation of JT-interval), and also with recent data by others in the isolated heart (Hondeghem et al., 2011). Terfenadine was shown to be an inhibitor of sodium channels in canine isolated atrial myocytes with an IC50 of 0.93 µM (at 0.1 Hz) and to act in a strong use-dependent manner (Lu and Wang, 1999). Our results in human atrial and rabbit ventricular myocytes were similar to these results. Also, the INa blocking activities of terfenadine are comparable with those of flecainide (Ming and Nordin, 1995; Lu and Wang, 1999). Flecainide was shown to have pro-arrhythmic potential in the clinic [Cardiac Arrhythmia Suppression Trial (CAST)] (The CAST Investigators, 1989), and in experimental models (Kou et al., 1987; Lu et al., 2010). In the CAST, INa blockers such as flecainide (Class Ic) were associated with an increased incidence of sudden cardiac death in post-infarct patients (The CAST Investigators, 1989). Our results indicate that terfenadine increases the risk for non-TdP-like VT/VT, relative to the risk for TdPs. The cases of terfenadine-induced TdPs reported were related to one or more factors that would be expected to cause excessively high concentrations of the drug such as co-medication with macrolide antibiotics (some of which have their own QT prolonging actions) or alcohol abuse (Woosley et al., 1993).
Terfenadine also blocks L-type calcium currents with an IC50 of 185 nM in human isolated atrial myocytes (Hove-Madsen et al., 2006), and blocks Ca2+ currents in guinea-pig isolated ventricular myocytes with an IC50 of 3 µM (Ming and Nordin, 1995). However, non-TdP-like-VT/VF and shortening of the QT interval by terfenadine are unlikely to be directly linked to its Ca2+-blocking activities. Indeed, in the rabbit isolated heart model, the Ca2+ antagonists nifedipine and verapamil exerted different effects from terfenadine: no induction of VT/VF, but a significant shortening of QT/APD without an increase in QRS, and a large increase in coronary flow started at 0.01 µM (unpublished data).
Are there ‘gaps’ between the Cmax in man and concentrations of terfenadine inducing cardiac effects in experimental models?
The concentrations of terfenadine used all in pre-clinical studies (McTavish et al., 1990; Lang et al., 1993; Woosley et al., 1993; Ming and Nordin, 1995; Tanaka et al., 1996; Fish and Antzelevitch, 2003; Hove-Madsen et al., 2006; Hondeghem et al., 2011) are always higher than its maximal therapeutic plasma level (Cmax) in man. Therefore, it seems that there is a large margin between the concentrations needed to block hERG and Na+ current or to change ECG or APD, and the free therapeutic plasma level in humans. To explain these apparent discrepancies, we propose the following.
Firstly, it is known that the free plasma levels in humans may reach values of 1 nM after dosing with terfenadine alone up to 59-fold (or more) when combined with ketoconazole or in conditions of drug-drug interaction and heart/liver disease (Von Moltke et al., 1994). Pro-arrhythmic effects induced by terfenadine are rare and mostly observed in conditions where the exposures are substantially increased (repeated doses in many days or overdoses), and in conditions where other drugs or heart/liver diseases are present.
Secondly, terfenadine has a low solubility and is known to be a compound that sticks to the tubings used in perfusion systems (Bridgland-Taylor et al., 2006). In our other in vitro studies with terfenadine (data not shown), we found that the recovery of terfenadine in the bath solution was ±10% of the target concentration. Therefore, the in vitro observations are an underestimate of the true effects of terfenadine. Furthermore, our data indicate that terfenadine accumulated in the heart-tissue (to about 80 times higher than the target concentration of 10 µM). Another earlier study (Cavero et al., 1999) showed that antihistamines including terfenadine accumulate in the heart of dogs and guinea-pigs as well (ratio for terfenadine was found to be approximately 260 higher in the heart-tissue after 2 h perfusion). Moreover, oral administration of terfenadine (30 mg·kg−1) to dogs and cats did not prolong the QT-interval and QRS-duration on the first day after its administration, but a significantly prolonged QT-interval and QRS-duration was observed after 7 days (Kii et al., 2003). Acute high i.v. doses of terfenadine (17 times higher than clinical plasma level) did not significantly changes ECG parameters in conscious dogs (Fossa et al., 2002). The cardiac side effects of terfenadine were only observed after repeat-doses for several days, after an overdose, in conditions where patients were taking other drugs (i.e. ketoconazole) or in cases of heart/liver disease (Von Moltke et al., 1994; Rangno, 1997). These results indirectly indicate that the time allowed for terfenadine to accumulate in heart tissue may play any important role in its effects on the heart.
Thirdly, terfenadine needs a long incubation-time to produce its effects in vitro (time-dependent effect). No changes in APD were reported in isolated cardiac tissues when terfenadine was perfused for 40 min at 10 or 20 µM (Masumiya et al., 2004). The cardiac effects of terfenadine were weaker when it was applied to the extracellular side of the cell membrane than when applied to the intracellular side (Nishio et al., 1998). Its pro-arrhythmic effects were more marked after a 450 min perfusion period, compared with a 10 or 30 min perfusion time in the isolated heart (Hondeghem et al., 2011). In addition, data from isolated perfused ventricular wedge preparations (terfenadine at 5 µM for 120 min) (Fish and Antzelevitch, 2003), and our unpublished data obtained in rabbit isolated Purkinje fibres show that the effects of terfenadine on APD are evident only after a long perfusion time of 90 to 120 min. In the present study, the effects of terfenadine on the electrophysiological parameters were much greater at 60 min than at 30 min, and were absent at 15 min (data not shown).
Therefore, these specific factors, including the much higher plasma levels found in man under pro-arrhythmic conditions, an underestimate of the actual concentrations present within the in vitro test systems (accumulation in the heart; ‘stickiness’ of the compound to the perfusion system), and the longer incubation time needed to show its effects, may all contribute to the apparent discord between the in vitro effects of terfenadine and its clinic effects, and may explain the so-called ‘high’ concentrations of terfenadine needed to observe cardiac effects in these in vitro models. In fact, there is a small ratio (less than 10) between the concentration that blocked the hERG current (in term of QT/JT-prolongation) and that needed to block INa (in terms of slowing conduction) in our functional models, rabbit isolated hearts and wedge preparations.
In conclusion, the present in vitro findings suggest that there is a high risk that terfenadine will elicit non-TdP-like VT/VF, relative to its risk to induce long QT and TdPs. This potential of terfenadine to induce non-TdP- like VT/VF is probably associated with its ability to slow conduction via its INa- blocking activities. Cardiac wavelength (λ) may constitute a potentially important biomarker to predict drug-induced re-entry VT/VF. Researchers should be prudent about selecting a new drug using ‘safety margins’, which are only based on the ratio of the hERG or INa IC50 and the free therapeutic plasma level, especially in cases where the compound has low solubility and has time-dependent effects (time-dependent accumulation within the heart tissue).
Acknowledgments
The authors wish to thank Dr R.Towart (Janssen Pharmaceutical Companies of Johnson & Johnson) and Dr B. Loenders (Janssen Pharmaceutical Companies of Johnson & Johnson) for their scientific comments, and Mr D. Borgers (K15, Belgium) for the appropriate figures. We thank Drs Guo D and Yan GX (Lankenau Institute for Medical Research) for the Heart Rhythm Solutions and for doing contract work.
Glossary
- APD
duration of the action potential
- BCL
basic cycle length
- CAST
Cardiac Arrhythmia Suppression Trial
- CT
conduction time
- DMSO
dimethyl sulfoxide
- EAD
early afterdeploarization
- ERP
effective refractory period
- hERG
human ether-à-go-go-related gene
- IK(R)
the rapidly activating delayed rectifier potassium current
- INa
sodium current
- MAP
monophasic action potential
- TdPs
Torsades de Pointes
- TDR
maximal transmural dispersion of repolarization
- Tp–Te
QTmax–Tmin (measurement of transmural dispersion)
- VF
ventricular fibrillation
- VT
ventricular tachycardia
Conflicts of interest
None. All authors are employees of Janssen Pharmaceutica NV.
References
- Aidonidis I, Poyatza A, Stamatiou G, Lymberi M, Stamatoyannis N, Molyvdas P-A. Does-related shortening of ventricular tachycardia cycle length after administration of the KATP channel opener bimakalim in a 4-day-old chronic anesethetized pig model. J Cardiovasc Pharmacol Ther. 2009;14:2222–2230. doi: 10.1177/1074248409338929. [DOI] [PubMed] [Google Scholar]
- Antzelevitch C. Brugada syndrome. Pacing Clin Electrophysiol. 2006;29:1130–1159. doi: 10.1111/j.1540-8159.2006.00507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batey AJ, Coker SJ. Proarrhythmic potential of halofantrine, terfenadine and cofilium in a modified in vivo model of torsade de pointes. Br J Pharmacol. 2002;135:1003–1012. doi: 10.1038/sj.bjp.0704550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridgland-Taylor MH, Hargreaves AC, Easter A, Orme A, Henthorn DC, Davis AM, et al. Optimisation and validation of a medium-throughput electrophysiology-based hHERG assay using IonWorks ™ HT. J Pharmacol Toxicol Methods. 2006;54:189–199. doi: 10.1016/j.vascn.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Cavero I, Mestre M, Guillon J-M, Heuillet E, Roach AG. Preclinical in vitro cardiac electrophysiology: a method of predicting arrhythmogenic potential of anti-histamines in humans. Drug Safety. 1999;21(Suppl. I):19–21. doi: 10.2165/00002018-199921001-00004. [DOI] [PubMed] [Google Scholar]
- Crumb WJ, Jr, Wible B, Arnold DJ, Payne JP, Brown AM. blockade of multiple human cardiac potassium currents by the antihistamine terfenadine: possible mechanism for terfenadine-associated cardiotoxicity. Mol Pharmacol. 1995;47:181–190. [PubMed] [Google Scholar]
- Fish JM, Antzelevitch C. Cellular and ionic basis for the sex-related difference in the manifestation of the Brugada syndrome and progressive conduction disease phenotypes. J Electrocardiol. 2003;36(Suppl. 1):173–179. doi: 10.1016/j.jelectrocard.2003.09.054. [DOI] [PubMed] [Google Scholar]
- Fossa AA, Depasqule MJ, Raunig DL, Avery MJ, Leishman DJ. The relastionship of clinical QT prolongation to outcome in the conscious dog using a beat-to-beat QT-RR interval assessment. J Pharmacol Exp Ther. 2002;202:828–833. doi: 10.1124/jpet.102.035220. [DOI] [PubMed] [Google Scholar]
- Freeze BS, McNulty MM, Hanck DA. State-dependent verapamil block of the cloned human Cav3.1 T-type Ca2+ channel. Mol Pharmacol. 2006;70:718–726. doi: 10.1124/mol.106.023473. [DOI] [PubMed] [Google Scholar]
- Girouard SD, Rossenbaum DS. Role of wavelength adaptation in the initiation, maintenance, and pharmacologic suppression of reentry. J Cardiovasc Electrophysiol. 2001;12:697–707. doi: 10.1046/j.1540-8167.2001.00697.x. [DOI] [PubMed] [Google Scholar]
- Girouard SD, Pastore JM, Laurita KR, Gregory KW, Rosenbaum DS. Optical mapping in new guinea-pig model of ventricular tachycardia reveals mechanisms for multiple wavelengths in a single reentrant circuit. Circulation. 1996;93:603–613. doi: 10.1161/01.cir.93.3.603. [DOI] [PubMed] [Google Scholar]
- Guo D, Zhou J, Zhao X, Gupta P, Kowey PR, Martin J, et al. L-type calcium current recovery versus ventricular repolarization: preserved membrane-stabilizing mechanism for different QT intervals across species. Heart Rhythm. 2008;5:271–279. doi: 10.1016/j.hrthm.2007.09.025. [DOI] [PubMed] [Google Scholar]
- Hanrahan JP, Choo PW, Carlson W, Greineder D, Faich GA, Platt R. Terfenadine-associated ventricular arrhythmias and QTc interval prolongation. A retrospective cohort comparison with other antihistamines among members of a health maintenance organization. Ann Epidemiol. 1995;5:201–209. doi: 10.1016/1047-2797(94)00039-v. [DOI] [PubMed] [Google Scholar]
- Hondeghem LM. QT prolongation is an unreliable predictor of ventricular arrhythmia. Heart Rhythm. 2008;5:1210–1212. doi: 10.1016/j.hrthm.2008.05.006. [DOI] [PubMed] [Google Scholar]
- Hondeghem LM, Dujardin K, Hoffmann P, Dumotier B, De clerk F. Drug-induced QTc prolongation dangerously underestimates proarrhythmic potential: lessons from terfenadine. J Cardiovasc Pharmacol. 2011;57:589–597. doi: 10.1097/FJC.0b013e3182135e91. [DOI] [PubMed] [Google Scholar]
- Hove-Madsen L, LIach A, Molina CE, Prat-Vidal CP, Farre J, Roura S, et al. The proarrhythmic antihistamine drug terfenadine increases spontaneous calcium release in human atrial myocytes. Eur J Pharmacol. 2006;553:215–221. doi: 10.1016/j.ejphar.2006.09.023. [DOI] [PubMed] [Google Scholar]
- Jacquemet V, Virag N, Kappenberger L. Wavelength and vulnerability to atrial fibrillation: insights from a computer model of human atria. Europace. 2005;7(s2):S83–S92. doi: 10.1016/j.eupc.2005.03.017. [DOI] [PubMed] [Google Scholar]
- June RA, Nasr I. Torsades de pointes with terfenadine ingestion. Am J Emerg Med. 1997;15:542–543. doi: 10.1016/s0735-6757(97)90206-0. [DOI] [PubMed] [Google Scholar]
- Karagounis LA, Anderson JL, Allen A, Osborn JS. Electrophysiological effects of antiarrhythmic drug therapy in the prediction of successful suppression of induced ventricular tachycardia. Am Heart J. 1995;129:343–349. doi: 10.1016/0002-8703(95)90017-9. [DOI] [PubMed] [Google Scholar]
- Kii Y, Nakatsji K, Nose I, Yabuuchi M, Matsuda M, Ito T. Effects of antihistamines, ebastine and terfenadine, on electrocardiogram in conscious dogs and cats. Drug Dev Res. 2003;58:209–217. [Google Scholar]
- Kou WH, Nelson SD, Lynch JJ, Montgomery DG, DiCarlo L, Lucchesi BR. Effect of flecainide acetate on prevention of electrical induction of ventricular tachycardia and occurrence of ischemic ventricular fibrillation during the early postmyocardial infarction period: evaluation in an conscious canine model of sudden death. J Am Coll Cardiol. 1987;9:359–365. doi: 10.1016/s0735-1097(87)80389-3. [DOI] [PubMed] [Google Scholar]
- Lacerda AE, Kramer J, Shen KZ, Thomas D, Brown AM. Comparison of block among cloned cardiac potassium channels by non-antiarrhythmic drugs. Eur Heart J Suppl. 2001;3(Suppl. K):K23–K30. [Google Scholar]
- Lang DG, Wang CM, Wenger TL. Terfenadine alters action potential in isolated canine Purkinjes more than acrivastine. J Cardiovasc Pharmacol. 1993;22:438–442. doi: 10.1097/00005344-199309000-00014. [DOI] [PubMed] [Google Scholar]
- Larsen JA, Kadish AH. Effects of gender on cardiac arrhythmias. J Cardiovasc Electrophysiol. 1998;9:655–664. doi: 10.1111/j.1540-8167.1998.tb00950.x. [DOI] [PubMed] [Google Scholar]
- Liu TG, Brown BS, Wu Y, Antzelevitch C, Kowey PR, Yan GX. Blinded validation of the isolated arterially perfused rabbit ventricular wedge in preclinical assessment of drug-induced proarrhytmias. Heart Rhythm. 2006;3:948–956. doi: 10.1016/j.hrthm.2006.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu HR, Remeysen P, De Clerck F. Nonselectrive IKr blockers do not induce torsades de pointes in the anesthetized rabbit during alpha1-adrenoceptor stimulation. J Cardiovasc Pharmacol. 2000;36:728–736. doi: 10.1097/00005344-200012000-00007. [DOI] [PubMed] [Google Scholar]
- Lu HR, Rohrbacher J, Vlaminckx E, Van Ammel K, Yan G-X, Gallacher DJ. Predicting drug-induced lowing of conduction and proarrhythmias: identifying the ‘bad’ sodium channel blockers. Br J Pharmacol. 2010;160:60–76. doi: 10.1111/j.1476-5381.2010.00646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Wang Z. Terfenadine block of sodium current in canine atrial mypcytes. J Cardiovasc Pharmacol. 1999;33:507–513. doi: 10.1097/00005344-199903000-00023. [DOI] [PubMed] [Google Scholar]
- Masumiya H, Saito M, Ito M, Matsuda T, Noguchi K, Lida-Tanaka N, et al. Lack of action potential-prolongation effect of terfenadine on rabbit myocardial tissue preparations. Biol Pharm Bull. 2004;27:131–135. doi: 10.1248/bpb.27.131. [DOI] [PubMed] [Google Scholar]
- McTavish D, Goa KL, Ferrill M. Terfenadine. An updated review of its pharmacological properties and therapeutic efficacy. Drugs. 1990;39:552–574. doi: 10.2165/00003495-199039040-00006. [DOI] [PubMed] [Google Scholar]
- Mines GR. On dynamic equilibrium in the heart. J Physiol. 1913;46:349–383. doi: 10.1113/jphysiol.1913.sp001596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming Z, Nordin C. Terfenadine blocks time-dependent Ca2+, Na+, and K+ channels in guinea-pig ventricular myocytes. J Cardiovasc Pharmacol. 1995;26:761–769. doi: 10.1097/00005344-199511000-00013. [DOI] [PubMed] [Google Scholar]
- Nishio M, Habuchi Y, Tanaka H, Morikawa J, Yamamoto T, Kashima K. Blockage by terfenadine of the adenosine triphosphate (ATP)-sensitive K+ current in rabbit ventricular myocytes. J Pharmacol Exp Ther. 1998;287:293–300. [PubMed] [Google Scholar]
- Pratt CM, Hertz RP, Ellis BE, Crowell SP, Louv W, Moye L. Risk of developing life-threatening ventricular arrhythmia associated with terfenadine in comparison with over-the-counter antihistamine, ibuprofen and clemastine. Am J Cardiol. 1994;73:346–352. doi: 10.1016/0002-9149(94)90006-x. [DOI] [PubMed] [Google Scholar]
- Pratt CM, Ruberg S, Morganroth J, McNutt B, Woodward J, Harris S, et al. Dose-response relation between terfenadine (Seldane) and the QTc interval on the scalar electrocardiogram: distinguishing a drug effect from spontaneous variability. Am Heart J. 1996;131:472–480. doi: 10.1016/s0002-8703(96)90525-6. [DOI] [PubMed] [Google Scholar]
- Rangno R. Terfenadine therapy: can we justify the risks? Can Med Assoc J. 1997;157:37–38. [PMC free article] [PubMed] [Google Scholar]
- Robert E, Bruelle P, de la Coussaye JE, Juan JM, Brugada J, Peray P, et al. Electrophysiologic and proarrhythmogenic effects of therapeutic and toxic doses of imipramine; a study with high resolution ventricular epicardial mapping in rabbit hearts. J Pharmacol Exp Ther. 1996;278:170–178. [PubMed] [Google Scholar]
- Robert E, Aya AG, de la Coussaye JE, Peray P, Juan JM, Brugada J, et al. Dispersion-based reentry: mechanism of initiation of ventricular tachycardia in isolated rabbit hearts. Am J Physiol. 1999;276:H413–H423. doi: 10.1152/ajpheart.1999.276.2.H413. [DOI] [PubMed] [Google Scholar]
- Roy ML, Dumaine R, Brown AM. HERG, a primary human ventricular target of the non-sendating antihistamine terfenadine. Circulation. 1996;94:817–823. doi: 10.1161/01.cir.94.4.817. [DOI] [PubMed] [Google Scholar]
- Salata JJ, Jurkiewicz NK, Wallace AA, Stupienski RF, Guinosso PJ, Jr, Lynch JJ., Jr Cardiac electrophysiological actions of the histamine H1-recetptor antagonists astermizole and terfenadine compared with chlorpheniramine and pyrilamine. Circ Res. 1995;76:110–119. doi: 10.1161/01.res.76.1.110. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Masumiya H, Kato Y, Shigenobu K. Inhibitory effects of terfenadine on the rising phase of action potentials and sinus rates in isolated guinea-pig myocardium. Gen Pharmacol. 1996;27:337–340. doi: 10.1016/0306-3623(95)02004-7. [DOI] [PubMed] [Google Scholar]
- The CAST Investigators. Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infacrtion. N Engl J Med. 1989;321:406–412. doi: 10.1056/NEJM198908103210629. [DOI] [PubMed] [Google Scholar]
- US Food and Drug Administration. 1997. FDA proposes to withdraw seldane approval [FDA Talk Paper T97-30] Jan 13.
- Vanghan Willaaims EM. Classification of anti-arrhythmic drugs. In: Sandfte E, Flensted-Jensen E, Olesen KH, editors. Symposium Cardiac Arrhythmias. Sodertalje, Sweden: AB Astra; 1970. pp. 449–472. [Google Scholar]
- Von Moltke LL, Greenblatt DJ, Duan SX, Harmatz JS, Shader RI. In vitro prediction of the terfenadine-ketoconazole pharmacokinetic interaction. J Clin Pharmacol. 1994;34:1222–1227. doi: 10.1002/j.1552-4604.1994.tb04735.x. [DOI] [PubMed] [Google Scholar]
- Woosley RL, Chen Y, Freiman JP, Gillis RA. Mechanism of the cardiotoxic actions of terfenadine. JAMA. 1993;269:1532–1536. [PubMed] [Google Scholar]
- Yan GX, Antzelevitch C. Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST-segment elevation. Circulation. 1999;100:1660–1666. doi: 10.1161/01.cir.100.15.1660. [DOI] [PubMed] [Google Scholar]
- Zhang S, Zhou Z, Gong Q, Makielski JC, January CT. Mechanism of block and identification of the verapamil binding domain to HERG potassium channels. Circ Res. 1999;84:989–998. doi: 10.1161/01.res.84.9.989. [DOI] [PubMed] [Google Scholar]