

Abstract
BACKGROUND AND PURPOSE
ATP, UTP and UDP act at smooth muscle P2X and P2Y receptors to constrict rat intrapulmonary arteries, but the underlying signalling pathways are poorly understood. Here, we determined the roles of the Ca2+-dependent chloride ion current (ICl,Ca), Cav1.2 ion channels and Ca2+ influx.
EXPERIMENTAL APPROACH
Isometric tension was recorded from endothelium-denuded rat intrapulmonary artery rings (i.d. 200–500 µm) mounted on a wire myograph.
KEY RESULTS
The ICl,Ca blockers, niflumic acid and 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid and the Cav1.2 channel blocker, nifedipine, reduced peak amplitude of contractions evoked by UTP and UDP by ∼45–50% and in a non-additive manner. Ca2+-free buffer inhibited responses by ∼70%. Niflumic acid and nifedipine similarly depressed contractions to ATP, but Ca2+-free buffer almost abolished the response. After peaking, contractions to UTP and UDP decayed slowly by 50–70% to a sustained plateau, which was rapidly inhibited by niflumic acid and nifedipine. Contractions to ATP, however, reversed rapidly and fully. Tannic acid contracted tissues per se and potentiated nucleotide-evoked contractions.
CONCLUSIONS AND IMPLICATIONS
ICl,Ca and Ca2+ influx via Cav1.2 ion channels contribute substantially and equally to contractions of rat intrapulmonary arteries evoked by UTP and UDP, via P2Y receptors. ATP also activates these mechanisms via P2Y receptors, but the greater dependence on extracellular Ca2+ most likely reflects additional influx through the P2X1 receptor pore. The lack of a sustained response to ATP is probably due to it acting at P2 receptor subtypes that desensitize rapidly. Thus multiple signalling mechanisms contribute to pulmonary artery vasoconstriction mediated by P2 receptors.
Keywords: pulmonary artery, P2Y receptor, P2X receptor, Ca2+-dependent chloride channels, Cav1.2 ion channels
Introduction
The cardiovascular actions of the endogenous nucleotides ATP, UTP and UDP are mediated by P2X and P2Y receptors (Burnstock and Kennedy, 1985; 1986; 2011; Erlinge and Burnstock, 2008), both of which are expressed in human pulmonary arteries (Liu et al., 1989b). P2X receptors are ligand-gated cation channels (Khakh et al., 2001) and P2X1 is the predominant subtype expressed in pulmonary vascular smooth muscle (Nori et al., 1998; Hansen et al., 1999; Lewis and Evans, 2001; Syed et al., 2010), where it mediates vasoconstriction (Liu et al., 1989a; Hasséssian and Burnstock, 1995; Rubino and Burnstock, 1996; Rubino et al., 1999; Chootip et al., 2002; Syed et al., 2010). In contrast, P2Y receptors are G protein-coupled (Abbracchio et al., 2006; Alexander et al., 2011), and in rat pulmonary vessels, P2Y agonists elicit vasodilatation via endothelial receptors and vasoconstriction via smooth muscle receptors (McCormack et al., 1989; Liu et al., 1989a; Hasséssian and Burnstock, 1995; Rubino and Burnstock, 1996; Hartley et al., 1998; Rubino et al., 1999; Chootip et al., 2002; Jernigan et al., 2006).
In healthy individuals, the delivery of de-oxygenated blood to the alveoli is maximized by a complex balance of factors that maintains pulmonary arteries in a dilated, low-resistance state (Barnes and Liu, 1995), and nucleotides appear to contribute to this state. For example, mM ATP is present in red blood cells (Erlinge and Burnstock, 2008) and is released during passage through the lungs, where it acts via endothelial P2Y receptors to induce a nitric oxide-dependent decrease in pulmonary vascular resistance (Sprague et al., 1996; 2003). Vasoconstriction mediated by the smooth muscle P2X and P2Y receptors is likely to become more prominent when endothelium-dependent relaxation is impaired, such as in hypoxia- or monocrotaline-induced pulmonary hypertension (Adnot et al., 1991; Mam et al., 2010) and chronic obstructive pulmonary disease (Dinh-Xuan et al., 1991). Indeed, extracellular ATP is elevated in the latter disease (Lommatzsch et al., 2010), which would increase the contribution of P2 receptors to artery regulation. The smooth muscle P2X and P2Y receptors also appear to play a role in hypoxic pulmonary vasoconstriction as the P2 receptor antagonist suramin inhibited this response in rabbit perfused lungs (Baek et al., 2008). Given these potential pathophysiological roles of nucleotides, it is important to understand the mechanisms that couple P2 receptors to the constrictor response.
We reported previously that UTP and UDP each constrict rat intrapulmonary arteries (IPA) via two P2Y subtypes, the P2Y6 and either P2Y2 or P2Y4 receptors, while ATP acts at P2X1 receptors and a P2Y receptor, most probably the P2Y2 subtype (Chootip et al., 2002; 2005; Kennedy et al., 2010). The signalling pathways through which these receptors produce their effects are, however, poorly characterized. P2Y receptor stimulation evokes release of intracellular Ca2+ stores in pulmonary artery smooth muscle cells (Bakhramov et al., 1996; Drummond and Tuft, 1999; Guibert et al., 1996; Jernigan et al., 2006; Baek et al., 2008), but the subsequent, downstream events that lead to vasoconstriction are unknown. An inward Ca2+-dependent, chloride ion current (ICl,Ca) that is also activated by P2Y agonists (Bakhramov et al., 1996; Guibert et al., 1997; Hartley et al., 1998; Chootip et al., 2005) might be predicted to contribute, but this remains to be confirmed, particularly as ICl,Ca is also activated by endothelin in rat IPA, but plays no role in endothelin's contractile activity (Kato et al., 1999). Activation of P2X1 receptors results in the opening of the ligand-gated cation channel and depolarization (Khakh et al., 2001), but again, how this leads to pulmonary vasoconstriction is unknown. The depolarization may promote opening of Cav1.2 ion channels, while the P2X1 pore is Ca2+ permeable (Egan and Khakh, 2004) and so Ca2+ could also potentially enter the cell directly through the pore to evoke contraction. Once more, however, the contributions of these pathways to vasoconstriction remain to be determined.
Clearly, multiple potential signalling mechanisms exist by which nucleotides may evoke pulmonary vasoconstriction, but the involvement and relative contributions of each are unknown. The aim of the present study was, therefore, to determine the extent to which the development and maintenance of nucleotide-induced constriction of the rat isolated IPA depend upon the influx of extracellular Ca2+ via Cav1.2 ion channels and the P2X1 pore, and the role of ICl,Ca in promoting Cav1.2 activity. We present evidence that ICl,Ca and Ca2+ influx via Cav1.2 ion channels contribute substantially and equally to both the peak and plateau of contractions evoked by UTP and UDP acting at P2Y receptors. ATP appears to act not only via P2Y receptors and these mechanisms, but also via P2X1 receptors.
Methods
Male Sprague–Dawley rats (200–250 g) were killed by cervical dislocation and exsanguination. The heart and lungs were removed en bloc and placed in a solution composed of (mM); NaCl 122, KCl 5, HEPES 10, KH2PO4 0.5, NaH2PO4 0.5, MgCl2 1, glucose 11, CaCl2 1.8, titrated to pH 7.3 with NaOH and bubbled with air (21% O2, 5% CO2, 74% N2). IPAs of internal diameter 200–500 µm were dissected, cleaned of connective tissue and their endothelium removed gently by passing a needle and thread through the lumen. They were then cut into 5-mm rings, mounted horizontally on a pair of intraluminal wires in 1 mL organ baths and equilibrated under a resting tension of 0.5 g for 60 min at 37°C. Tension was recorded with Grass FT03 isometric force transducers, connected to a PowerLab/4e system, using Chart 4.2 software (ADInstruments, Oxford, UK).
Experimental protocols
Drugs were added directly to the tissue bath and washed out by replacement with drug-free solution. Removal of the endothelium was confirmed by loss of the relaxation to ACh (10 µM) following precontraction with UDP, UTP or ATP. The nucleotide concentration–contraction curves in rat IPA do not reach a maximum (Chootip et al., 2002; 2005); therefore, they were applied at the equi-effective concentration of 300 µM to generate vasoconstriction and investigate the underlying signalling mechanisms. When studying peak contraction amplitude, each nucleotide was added for 5 min at 30 min intervals, as preliminary experiments showed that this protocol elicited highly reproducible contractions when agonist application was repeated six times.
To determine the effects of niflumic acid, 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS) and nifedipine, control responses to an agonist were obtained. Arteries were then incubated with niflumic acid or DIDS for 10 min, nifedipine for 15 min or nifedipine plus niflumic acid or DIDS for 15 min before the agonist was re-administered. The effects of tannic acid were measured in the same way after 15 min incubation and also against contractions evoked by KCl and phenylephrine. When investigating the role of extracellular Ca2+, control agonist responses were obtained in normal buffer, which was then replaced with Ca2+-free solution for 10 min, before the agonists were re-administered. When studying the plateau phase of the nucleotide-evoked contractions, UDP or UTP were added for 45 min and niflumic acid or nifedipine was applied 20 min after the agonist and their effects measured a further 10 min later. Similarly, tannic acid was added 20 min after UDP or UTP and the peak amplitude of the evoked contraction measured.
Drugs and solutions
Drugs were prepared as 10 or 100 mM stock solutions and diluted in the HEPES-based buffer before applying to the tissues. ATP (Na2 salt), UTP (Na3 salt), UDP (Na salt), phenylephrine hydrochloride and ACh chloride (Sigma, Dorset, UK) were dissolved in distilled water. Nifedipine, niflumic acid (Sigma) and DIDS (Na2 salt) (Invitrogen, Paisley, UK) were dissolved in dimethyl sulfoxide and tannic acid (Sigma) in ethanol. Isotonic 40 mM K+ solution was prepared by replacing NaCl in the HEPES-buffered solution with an equimolar amount of KCl to maintain osmolarity. Ca2+-free buffer was prepared by replacing the CaCl2 with 3.12 mM MgCl2 and 1 mM EGTA to give equimolar free [Mg2+]. Free and bound [Mg2+] were calculated using Maxchel (Chris Patten, Stanford University, Stanford, CA, USA).
Data analysis
Contractions are expressed as mg tension, a percentage of the peak amplitude or a percentage of the control response produced by a given agonist, as appropriate. Data are shown as mean ± SEM of experiments on vessels from n animals and were compared using Student's paired t-test or one-way anova with Tukey's comparison of the mg tension values, as appropriate. Values of P < 0.05 were considered to be statistically significant.
Results
Contribution of ICl,Ca, Cav1.2 ion channels and extracellular Ca2+ to peak contraction amplitude
The channel that mediates ICl,Ca, TMEM16A, has only recently been cloned (Caputo et al., 2008; Schroeder et al., 2008; Yang et al., 2008) and is functionally expressed in rat pulmonary artery smooth muscle cells (Manoury et al., 2010). Little is known of P2 receptor function in mouse pulmonary artery and mice lacking a functional TMEM16A gene die within days of birth (Rock et al., 2008). A pharmacological approach was, therefore, taken using the most potent and most commonly used ICl,Ca blocker, niflumic acid (Hogg et al., 1994b), and the structurally unrelated inhibitor, DIDS (Hogg et al., 1994a) to determine the role of TMEM16A in nucleotide-evoked pulmonary vasoconstriction in rat.
Contractions evoked by UDP (300 µM) and UTP (300 µM) reached a peak within 2–3 min (Figure 1), while responses to ATP (300 µM) peaked within 1–2 min (Figure 3A). Niflumic acid (1 µM) and DIDS (100 µM) had no effect on basal tone of rat IPA or on contractions to KCl (40 mM) (103 ± 11 % of control, n = 5 and 96.5 ± 3.9 % of control, n = 4, respectively), indicating that at these concentration they do not interact directly with Cav1.2 ion channels or the myofilaments to depress smooth muscle contractility. Both, however, reduced significantly the peak responses to UDP (P < 0.01) (Figures 1A, 2A) and UTP (P < 0.05) (Figures 1B, 2B) by approximately 40–55% of their control values. Niflumic acid (1 µM) depressed responses to ATP by a similar amount (P < 0.05) (Figure 3B) and the degree of inhibition did not differ significantly between the nucleotides. Higher concentrations of niflumic acid (10 and 100 µM) depressed contractions to KCl (40 mM), indicating an action of niflumic acid at other sites and so were not studied further.
Figure 1.
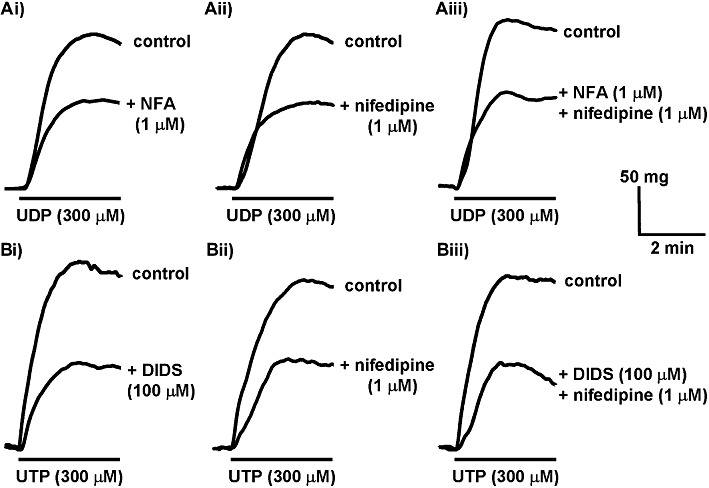
Inhibition of the peak amplitude of contractions evoked by UDP and UTP. (A) The superimposed traces shows typical contractions of endothelium-denuded rat isolated IPA evoked by (A) UDP (300 µM) in the absence of (upper traces) and after incubation with (Ai) niflumic acid (1 µM), (Aii) nifedipine (1 µM) and (Aiii) niflumic acid (1 µM) plus nifedipine (1 µM) for 10 min (lower traces). (B) The superimposed traces show responses evoked by UTP (300 µM) in the absence of (upper traces) and after incubation with (Bi) DIDS (100 µM), (Bii) nifedipine (1 µM) and (Biii) DIDS (100 µM) plus nifedipine (1 µM) for 10 min (lower trace). UDP and UTP were applied as indicated by the solid bar. Each pair of traces was obtained in a separate tissue.
Figure 3.
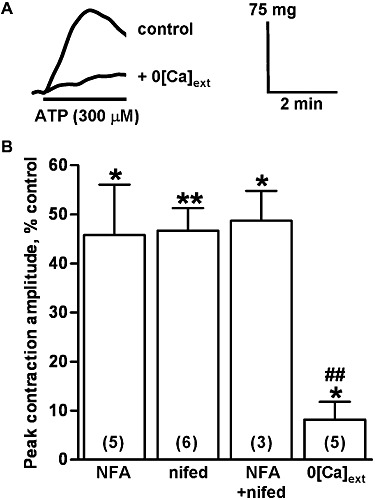
Inhibition of the peak amplitude of contractions evoked by ATP. (A) The superimposed traces show typical contractions of endothelium-denuded rat isolated IPA evoked by ATP (300 µM) in normal buffer (upper trace) and when bathed in Ca2+-free buffer for 10 min (lower trace). ATP was applied as indicated by the solid bar. (B) The mean peak amplitude of contractions evoked by ATP (300 µM) in the presence of niflumic acid (1 µM) (NFA), nifedipine (1 µM) (nifed), niflumic acid (1 µM) plus nifedipine (1 µM) (NFA + nifed) and in Ca2+-free buffer (0[Ca]ext), expressed as a percentage of control responses, is shown. The numbers in parentheses show n for each. Vertical lines show SEM. *P < 0.05, **P < 0.01 for responses after treatment compared with control. ##P < 0.01 for the response to ATP in Ca2+-free buffer compared with the other treatments.
Figure 2.
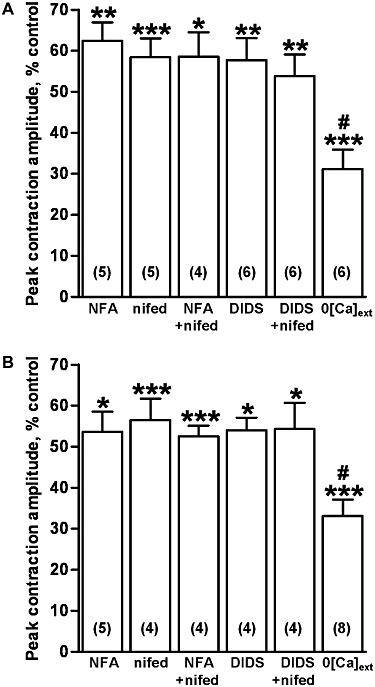
Inhibition of the peak amplitude of contractions evoked by UDP and UTP. The mean peak amplitude of contractions of endothelium-denuded rat isolated IPA evoked by (A) UDP (300 µM) and (B) UTP (300 µM) in the presence of niflumic acid (1 µM) (NFA), nifedipine (1 µM) (nifed), niflumic acid (1 µM) plus nifedipine (1 µM) (NFA + nifed), DIDS (100 µM), DIDS (100 µM) plus nifedipine (1 µM) (DIDS + nifed) and in Ca2+-free buffer (0[Ca]ext), expressed as a percentage of control responses, is shown. The numbers in parentheses show n for each. Vertical lines show SEM. *P < 0.05, **P < 0.01, ***P < 0.001 for responses after treatment compared with control. #P < 0.05 for responses in Ca2+-free buffer compared with the other treatments.
Nifedipine (1 µM), a concentration that maximally inhibits Cav1.2 ion channels in pulmonary arteries (Clapp and Gurney, 1991), had no effect on basal tone, but virtually abolished contractions to KCl (40 mM) (99 ± 1 % inhibition, n = 4) and significantly inhibited the peak response to UDP (P < 0.001) (Figures 1A, 2A), UTP (P < 0.001) (Figures 1B, 2B) and ATP (P < 0.01) (Figure 3B) by a similar amount as niflumic acid. There was no significant difference in the degree of inhibition of each nucleotide response. Co-administration of niflumic acid (1 µM) and nifedipine (1 µM) inhibited contractions to the same extent as either agent alone (Figures 2A,B, 3B). Likewise, adding DIDS (100 µM) and nifedipine (1 µM) together did not elicit greater inhibition of the responses to UDP (Figure 2A) or UTP (Figures 1B, 2B).
Bathing tissues in Ca2+-free buffer for 10 min had no effect on basal tone, but abolished contractions to KCl (40 mM) (n = 4, not shown) and significantly decreased the peak amplitude of the contractions to UDP (P < 0.001) (Figure 2A) and UTP (P < 0.001) (Figure 2B) by nearly 70%. Responses to ATP were depressed by more than 90% (P < 0.05) (Figure 3) and now appeared to take longer, 2–3 min, to reach peak. The inhibition of the ATP response was significantly greater than that of UTP and UDP (P < 0.05). For each nucleotide, removing extracellular Ca2+ caused a significantly larger reduction in the peak response than did the ion channel blockers (UDP and UTP, P < 0.05, ATP, P < 0.01). Thus, ICl,Ca, Cav1.2 ion channels and Ca2+ influx each make a similar contribution to the peak amplitude of contractions of rat IPA evoked by UDP and UTP, whereas influx of extracellular Ca2+ plays a relatively greater role in the response to ATP.
Contribution of ICl,Ca and Cav1.2 ion channels to contraction plateau
Once the contractions to UTP and UDP had reached a peak, they decayed slowly and significantly (P < 0.001), and with a similar time course over the next 10–15 min to a plateau of around 30–50% of the peak (Figure 4A,C). Thereafter, the responses were generally stable and while they tended to decrease slightly between 20, 30 and 40 min after agonist administration, the tensions recorded at these times were not significantly different. The contractions to ATP also decayed significantly from peak, but compared with UTP and UDP, the decay was faster and the tone returned to the basal level within 40 min (Figure 4B,D).
Figure 4.
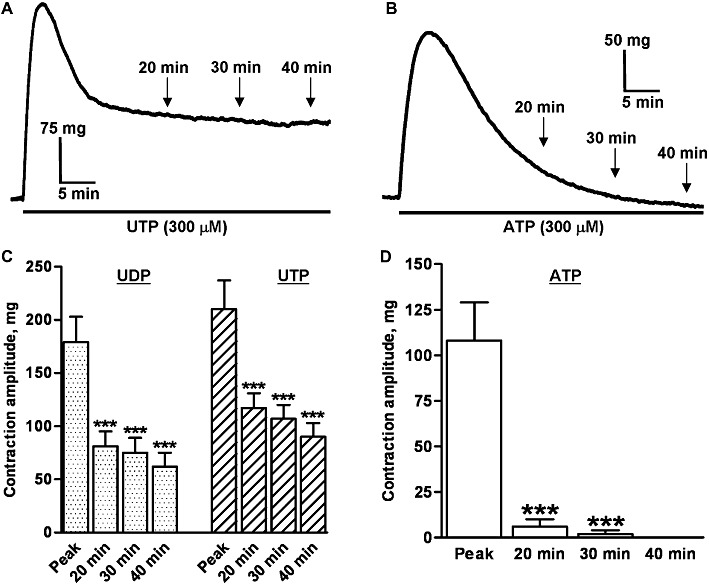
Time-course of contractions to UTP, UDP and ATP. The traces shows typical contractions of endothelium-denuded rat isolated IPA evoked by (A) UTP (300 µM) and (B) ATP (300 µM), applied as indicated by the solid bars. The mean amplitude of contractions evoked by (C) UDP (300 µM, n = 5) and UTP (300 µM, n = 6) and (D) ATP (300 µM, n = 4) at peak and 20, 30 and 40 min after agonist administration is shown. Vertical lines show SEM. ***P < 0.001 for contraction amplitude at 20, 30 or 40 min compared with peak.
The signalling mechanisms that underlie the initiation and development of contraction may differ from those that mediate the maintained response, therefore, the roles of ICl,Ca and Cav1.2 ion channels in maintaining the UTP and UDP plateaux was determined by adding the channel inhibitors 20 min after the agonists and measuring the tension a further 10 min later. Under these conditions, niflumic acid (1 µM) rapidly and significantly reduced the plateau response to both nucleotides by about half (P < 0.001) (Figure 5). Nifedipine (1 µM) significantly relaxed the tissue by a similar amount (UDP, P < 0.001; UTP, P < 0.01) and the effects of co-administration of the two inhibitors were not additive (Figure 5). Thus ICl,Ca and Cav1.2 ion channels contribute substantially and equally to the plateau of UTP- and UDP-evoked contractions of rat IPA.
Figure 5.
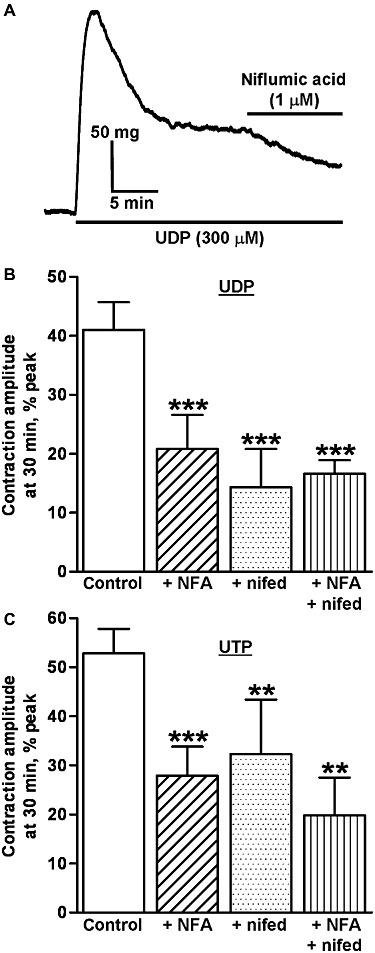
Inhibition of the plateau of UTP- and UDP-evoked contractions. (A) The trace show a typical contraction of endothelium-denuded rat isolated IPA evoked by UDP (300 µM) and the effect of adding niflumic acid (1 µM) for 10 min, 20 min after UDP. The drugs were applied as indicated by the solid bars. The mean amplitude of contractions evoked by (B) UDP (300 µM) and (C) UTP (300 µM) 30 min after their addition and expressed as a percentage of the peak amplitude, is shown as follows: control responses in the absence of inhibitor (UDP n = 5, UTP n = 6), responses in the presence of niflumic acid (1 µM) (NFA, UDP n = 6, UTP n = 7), in the presence of nifedipine (1 µM) (nif, UDP n = 6, UTP n = 6) and in the presence of niflumic acid (1 µM) plus nifedipine (1 µM) (NFA + nif, UDP n = 7, UTP n = 5). Vertical bars indicate SEM. **P < 0.01 and ***P < 0.001 for response in the presence of inhibitors compared with the control response in their absence.
Effects of tannic acid
Tannic acid was shown recently to inhibit ICl,Ca, with a maximal effect at 100 µM (Namkung et al., 2010; 2011). At this concentration it induced small, rapid, transient contractions in 18/24 tissues (mean = 26 ± 5 mg, range = 10 − 84 mg) and significantly potentiated the peak amplitude of the contractions evoked by UTP, UDP and ATP in all tissues tested, by approximately 30–40% (P < 0.05) (Figure 6A). Contractions to phenylephrine (1 µM) and KCl (40 mM) were, in contrast, significantly depressed by about 30% (P < 0.05) (Figure 6A). When applied during the plateau phase of the UTP- and UDP-induced contractions (two tissues each), tannic acid (100 µM) caused a significantly larger, rapid, transient contraction (mean = 78 ± 12 mg, range = 52 − 110 mg, n = 4, P < 0.001), but no relaxation (Figure 6B).
Figure 6.
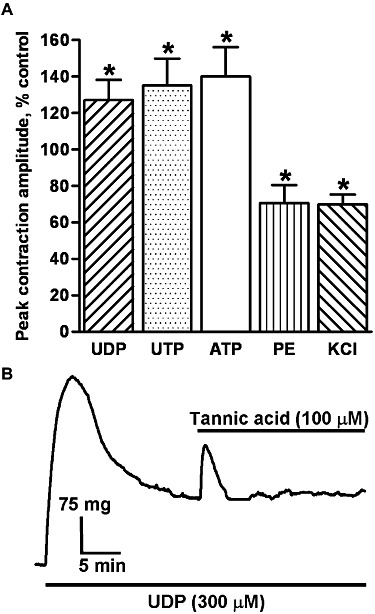
The effects of tannic acid. (A) The mean peak amplitude of contractions evoked by UDP (n = 5), UTP (n = 4), ATP (n = 5) (300 µM each), phenylephrine (PE) (1 µM) (n = 4) and KCl (40 mM) (n = 6) in the presence of tannic acid (100 µM), expressed as a percentage of the control response, is shown. Vertical lines show SEM. *P < 0.05 for responses after treatment compared with control. (B) The trace shows a typical contraction of endothelium-denuded rat isolated IPA evoked by UDP (300 µM) and the effect of adding tannic acid (100 µM) 20 min after UDP. Drugs were applied as indicated by the solid bars.
Discussion
In this study, the vasoconstriction of the rat isolated IPA induced by UTP and UDP was inhibited by almost half by the ICl,Ca blockers, niflumic acid and DIDS, suggesting that ICl,Ca plays a major role in the vasoconstriction elicited via P2Y receptors. The Cav1.2 Ca2+ channel blocker, nifedipine, depressed the contractions by a similar amount and co-application with either ICl,Ca blocker had no additional effect. This is consistent with a model whereby the depolarization induced by ICl,Ca causes voltage-dependent Cav1.2 Ca2+ channels to open and Ca2+ to enter the cells. The contractions evoked by ATP were likewise inhibited by niflumic acid and nifedipine, whereas bathing tissues in Ca2+-free buffer almost abolished the response. Moreover, contractions to ATP reached a peak more quickly and showed faster and greater decay following the peak, which suggests that ATP acts at both P2X and P2Y receptors. Thus, these data indicate that stimulation of P2X and P2Y receptors activates multiple signalling mechanisms to induce pulmonary artery vasoconstriction.
Signalling mechanisms underlying UDP- and UTP-evoked vasoconstriction
We reported that UDP and UTP activate ICl,Ca in rat IPA myocytes (Chootip et al., 2005) and here we show that both the peak and plateau of the contractions to UTP and UDP were depressed by around half by the ICl,Ca inhibitors niflumic acid and DIDS. This is the first demonstration that ICl,Ca makes an essential contribution to the development and maintenance of P2Y receptor-mediated contractions. It is notable because activation of ICl,Ca does not necessarily lead to vasoconstriction. For example, ICl,Ca is activated by endothelin in, but plays no role in the concomitant contraction of rat IPA (Kato et al., 1999). Thus ICl,Ca is an important, but not ubiquitous, mediator of pulmonary vasoconstriction induced by GPCRs. Higher concentrations of niflumic acid (≥10 µM) can affect sites other than ICl,Ca, such as BKCa ion channels, non-selective cation channels, voltage-gated K+ channels and COX (Kato et al., 1999; Cruickshank et al., 2003; Greenwood and Leblanc, 2007) and consistent with this, at 10 and 100 µM, it depressed contractions of rat IPA to KCl. In contrast, 1 µM niflumic acid and 100 µM DIDS had no effect on KCl-induced contractions, indicating selectivity of action at these concentrations.
The contractions evoked by UTP and UDP were also depressed by the Cav1.2 ion channel inhibitor, nifedipine and in a non-additive manner with niflumic acid and DIDS. This is consistent with a model in which ICl,Ca evokes contraction by depolarizing smooth muscle cells, causing Cav1.2 ion channels to open and Ca2+ to flow into the cell (Large and Wang, 1996; Kitamura and Yamazaki, 2001; Leblanc et al., 2005). The non-additivity of inhibition also indicates that ICl,Ca was the only stimulus causing Cav1.2 ion channels to open, consistent with the inability of UTP and UDP to elicit other types of depolarizing current in rat IPA myocytes (Hartley et al., 1998; Chootip et al., 2005). The concentrations of ICl,Ca blocker used were close to the IC50 values reported for niflumic acid (2.3 µM, Hogg et al., 1994b) and DIDS (210 µM, Hogg et al., 1994a) against ICl,Ca in rabbit portal vein smooth muscle cells, so it is perhaps surprising that they depressed contractions by a similar amount to a maximally effective concentration of nifedipine. The reason for this is not clear, but it may be that relatively small changes in depolarization produce much larger changes in the opening of voltage-dependent Ca2+ channels. Alternatively, it may reflect the tissue variability of the potency of the ICl,Ca blockers (see Large and Wang, 1996; Greenwood and Leblanc, 2007). For example, DIDS is nearly 13 times more potent against ICl,Ca in rat portal vein myocytes (IC50 = 16.5 µM; Baron et al., 1991) than in rabbit portal vein (IC50 = 210 µM, Hogg et al., 1994a). Similarly, while 10 µM niflumic acid depressed KCl-evoked contractions in the present study, it had no effect on KCl-induced vasoconstriction of rat aorta and mesenteric vascular bed (Criddle et al., 1996; 1997).
Signalling mechanisms underlying ATP-evoked vasoconstriction
The vasoconstriction evoked by ATP was also inhibited by niflumic acid in the present study, consistent with our earlier report that ATP activates ICl,Ca in rat IPA myocytes (Chootip et al., 2005). As seen with UTP and UDP, nifedipine inhibited ATP-induced contractions to a similar extent and in a non-additive manner. Thus, P2Y receptor-mediated activation of ICl,Ca and subsequent opening of Cav1.2 Ca2+ channels also plays a major role in the contractile response to ATP. In contrast to UTP and UDP, however, Ca2+-free buffer had a significantly greater inhibitory effect, almost abolishing the ATP response. Clearly, Ca2+ influx plays a greater role in the ATP vasoconstriction and appears to be via Cav1.2 ion channels, plus another route. This is most likely the P2X1 receptor, a Ca2+ permeable, ligand-gated cation channel (Egan and Khakh, 2004), which is the main P2X subtype expressed in rat IPA smooth muscle (Syed et al., 2010) and which our pharmacological studies showed was involved in the pulmonary vasoconstrictor action of ATP (Chootip et al., 2002; 2005). Consistent with an action at both P2X1 and P2Y receptors, ATP induced not only release of Ca2+ stores in rat pulmonary artery myocytes (responsible for activating ICl,Ca), but also Ca2+ influx (Guibert et al., 1996). A similar difference between the relative roles of Ca2+ influx in contractions evoked by ATP and UTP was reported in the rat isolated tail artery (McLaren et al., 1998) and that study also concluded that ATP acted via P2X and P2Y receptors to elicit vasoconstriction. A dual action on P2X and P2Y receptors may, therefore, be a general rule for ATP's contractile effects on vascular smooth muscle.
Signalling mechanisms underlying the decay of nucleotide-evoked vasoconstriction
The vasoconstriction elicited by UTP and UDP was biphasic, reaching a peak within 2–3 min and then decaying over 10–15 min to a plateau at about half of the peak response. The reason for the decay is unclear, but is unlikely to be due to the rapid decline seen in the amplitude of intracellular (Ca2+) oscillations (Guibert et al., 1996) and ICl,Ca (Hartley et al., 1998; Chootip et al., 2005) evoked by UTP and UDP, as both reached a steady state within 2–3 min, which is similar to the time taken for the contractions to reach their peak. Furthermore, in the present study, niflumic acid substantially relaxed precontracted tissues, indicating that ICl,Ca was active for at least 20 min after contractions peaked. Ca2+ oscillations and ICl,Ca have yet, however, to be recorded over this longer timescale, and we cannot rule out the possibility that a slower decline in the frequency and/or amplitude of one or both of these signals accounts for the contraction decay. Similarly, we cannot exclude that the transient and sustained phases of constriction reflect temporal changes in the relative contributions of Ca2+ release and Ca2+ influx to the contractions (Lee et al., 2002). Further experiments using Ca2+-sensitive dyes will shed light on these possibilities.
Alternatively, the decay of the contraction could be due to desensitization of the P2Y receptors that mediate the actions of UTP and UDP, rather than changes in Ca2+ signalling per se. We showed that both agonists act via two P2Y subtypes to evoke contraction in this tissue, most likely P2Y6, plus P2Y2 or P2Y4 receptors (Chootip et al., 2002; 2005; Kennedy et al., 2010). P2Y2 (Sromek and Harden, 1998; Flores et al., 2005; Lemon et al., 2005) and P2Y4 (Brinson and Harden, 2001) receptors both desensitize rapidly and with a time-course compatible with the decay seen in the present study, whereas the P2Y6 receptor desensitizes much more slowly, reaching a steady-state over 3–6 h (Robaye et al., 1997; Brinson and Harden, 2001). Thus, the activation of P2Y6 and P2Y2/P2Y4 receptors could together lead to the peak contraction, with the subsequent decay reflecting desensitization of P2Y2/P2Y4 receptors, to leave P2Y6 receptors dominating the plateau. This model will be investigated once potent, stable, receptor subtype-selective agonists become commercially-available.
The contraction of rat pulmonary artery evoked by ATP was transient, showing full reversal over 40 min in the continuous presence of ATP, even though ATP induces Ca2+ influx in addition to Ca2+ release in this tissue. This may be explained by ATP acting at both P2X1 and P2Y receptors (Chootip et al., 2005). The P2X1 receptor desensitizes rapidly and completely during agonist exposure (Evans and Kennedy, 1994; Khakh et al., 2001). The P2Y2 receptor is probably the second site of action of ATP and, as discussed above, it also desensitizes rapidly. ATP is not an agonist at the slowly desensitizing P2Y6 receptor (Abbracchio et al., 2006), consistent with the lack of a contraction plateau. Thus the rapid desensitization of the two sites of action of ATP can explain why its contractions reverse fully during agonist exposure.
Tannic acid
As tannic acid was shown recently to be a potent inhibitor of ICl,Ca carried by recombinant and native TMEM16A channels (Namkung et al., 2010; 2011), we used it as an additional probe for investigating ICl,Ca involvement in nucleotide-induced constriction of rat IPA. Unfortunately, it produced effects that are inconsistent with a selective inhibitory action on ICl,Ca, such as evoking transient contractions in 75% of unstimulated tissues and significantly potentiating, rather than inhibiting the nucleotide-evoked contractions. Moreover, although tannic acid inhibited the vasoconstriction induced by phenylephrine, which may also involve the activation of ICl,Ca (Yuan, 1997), it had the same effect on the vasoconstriction evoked by KCl. Thus, at the concentration used tannic acid does not appear to be a selective inhibitor of ICl,Ca in pulmonary artery.
The mechanism(s) underlying the excitatory actions of tannic acid are unclear, but it is notable that the drug inhibits a variety of ATPases, including gastric H+,K+-ATPase (Murakami et al., 1992), mitochondrial proton F0F1-ATPase (Zheng and Ramirez, 2000) and verapamil-stimulated ATPase activity in membrane vesicles (Kitagawa et al., 2007) and so appears to be a general ATPase inhibitor. Ecto-nucleoside triphosphate diphosphohydrolases (formerly known as ecto-ATPase), are a family of ecto-enzymes that inactivate ATP, UTP and UDP by dephosphorylation (Robson et al., 2006). In vascular smooth muscle these enzymes decrease nucleotide potency by up to two to three orders of magnitude (Evans and Kennedy, 1994; Kauffenstein et al., 2010) and inhibition of the ecto-enzymes by the specific inhibitor ARL67156 potentiated contractions of rat tail artery to UTP to a similar extent as tannic acid (McLaren et al., 1998). So inhibition of nucleotide breakdown by tannic acid might explain its ability to potentiate the nucleotide-evoked contractions, thereby masking any inhibitory effect that it might have on ICl,Ca. Further experiments are required to address this possibility.
Summary
In conclusion, these data indicate that UTP and UDP act at P2Y receptors to elicit release of Ca2+ from intracellular stores, which activates ICl,Ca, a depolarizing current. Our working hypothesis is that the depolarization in turn causes voltage-dependent Cav1.2 Ca2+ channels to open, allowing Ca2+ to flow into the cell and induce vasoconstriction. The constrictions caused by UDP and UTP probably involve rapidly desensitizing P2Y2/P2Y4 receptors and slowly desensitizing P2Y6 receptors, giving rise to an initial peak response that decays partially to a plateau. ATP acts at P2Y receptors to activate the same pathway, but also acts at the P2X1 receptor. Together these activated receptors elicit depolarization, Cav1.2 ion channel opening and Ca2+influx, as well as Ca2+ entry via the P2X1 receptor pore. The complete decay of the constrictor response to ATP following its peak is consistent with ATP acting at P2X1 and P2Y2 receptors, both of which desensitize rapidly. Thus purine and pyrimidine nucleotides employ different receptors and produce kinetically distinct constrictor responses in rat IPA, but in each case, both ICl,Ca and Ca2+ influx via Cav1.2 ion channels mediate a substantial proportion of the response. It was notable that Ca2+-free buffer inhibited the UDP- and UTP-evoked contractions significantly more than did blocking ICl,Ca and Cav1.2 ion channels, indicating that refilling of the intracellular Ca2+ stores by voltage-independent Ca2+ channels may also play a role. This possibility and the contribution of other signalling mechanisms, such as Ca2+-sensitization, in nucleotide-induced pulmonary vasoconstriction are currently being studied.
Acknowledgments
This study was supported by the British Heart Foundation (grant no. FS/04/070 to CK/AMG).
Glossary
- DIDS
4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid
- ICl,Ca
Ca2+-dependent chloride ion current
- IPA
intrapulmonary artery
Conflicts of interest
None.
References
- Abbracchio MP, Burnstock G, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, et al. International Union of Pharmacology LVIII: update on the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy. Pharmacol Rev. 2006;58:281–341. doi: 10.1124/pr.58.3.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adnot S, Raffestin B, Eddahibi S, Braquet P, Chabrier PE. Loss of endothelium-dependent relaxant activity in the pulmonary circulation of rats exposed to chronic hypoxia. J Clin Invest. 1991;87:155–162. doi: 10.1172/JCI114965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th Edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek EB, Yoo HY, Park SJ, Kim HS, Kim SD, Earm YE, et al. Luminal ATP-induced contraction of rabbit pulmonary arteries and role of purinoceptors in the regulation of pulmonary arterial pressure. Pflugers Arch. 2008;457:281–291. doi: 10.1007/s00424-008-0536-z. [DOI] [PubMed] [Google Scholar]
- Bakhramov A, Hartley SA, Salter KJ, Kozlowski RZ. Contractile agonists preferentially activate Cl− over K+ currents in arterial myocytes. Biochem Biophys Res Commun. 1996;1227:168–175. doi: 10.1006/bbrc.1996.1484. [DOI] [PubMed] [Google Scholar]
- Barnes PJ, Liu SF. Regulation of pulmonary vascular tone. Pharmacol Rev. 1995;47:87–131. [PubMed] [Google Scholar]
- Baron A, Pacaud P, Loirand G, Mironneau C, Mironneau J. Pharmacological block of Ca2+-activated Cl− current in rat vascular smooth muscle cells in short-term primary culture. Pflugers Arch. 1991;419:553–558. doi: 10.1007/BF00370294. [DOI] [PubMed] [Google Scholar]
- Brinson AE, Harden TK. Differential regulation of the uridine nucleotide-activated P2Y4 and P2Y6 receptors. J Biol Chem. 2001;276:11939–11948. doi: 10.1074/jbc.M009909200. [DOI] [PubMed] [Google Scholar]
- Burnstock G, Kennedy C. Is there a basis for distinguishing two types of P2-purinoceptor? Gen Pharmacol. 1985;16:433–440. doi: 10.1016/0306-3623(85)90001-1. [DOI] [PubMed] [Google Scholar]
- Burnstock G, Kennedy C. A dual function for adenosine triphosphate in the regulation of vascular tone: excitatory cotransmitter with noradrenaline from perivascular nerves and locally released inhibitory intravascular agent. Circ Res. 1986;58:319–330. doi: 10.1161/01.res.58.3.319. [DOI] [PubMed] [Google Scholar]
- Burnstock G, Kennedy C. P2X receptors in health and disease. Adv Pharmacol. 2011;61:333–372. doi: 10.1016/B978-0-12-385526-8.00011-4. [DOI] [PubMed] [Google Scholar]
- Caputo A, Caci E, Ferrera L, Pedemonte N, Barsanti C, Sondo E, et al. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science. 2008;322:590–594. doi: 10.1126/science.1163518. [DOI] [PubMed] [Google Scholar]
- Chootip K, Ness K, Wang J, Gurney AM, Kennedy C. Regional variation in P2 receptor expression in the rat pulmonary arterial circulation. Br J Pharmacol. 2002;137:637–646. doi: 10.1038/sj.bjp.0704915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chootip K, Gurney AM, Kennedy C. Evidence for multiple P2Y receptors coupled to calcium-dependent, chloride channels in smooth muscle cells of the rat pulmonary artery. Respir Res. 2005;26:124. doi: 10.1186/1465-9921-6-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapp LH, Gurney AM. Modulation of calcium movements by nitroprusside in isolated vascular smooth muscle cells. Pflugers Arch. 1991;418:462–470. doi: 10.1007/BF00497774. [DOI] [PubMed] [Google Scholar]
- Criddle DN, de Moura RS, Greenwood IA, Large WA. Effect of niflumic acid on noradrenaline-induced contractions of the rat aorta. Br J Pharmacol. 1996;118:1065–1071. doi: 10.1111/j.1476-5381.1996.tb15507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criddle DN, de Moura RS, Greenwood IA, Large WA. Inhibitory action of niflumic acid on noradrenaline- and 5-hydroxytryptamine-induced pressor responses in the isolated mesenteric vascular bed of the rat. Br J Pharmacol. 1997;12:813–818. doi: 10.1038/sj.bjp.0700981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruickshank SF, Baxter LM, Drummond RM. The Cl− channel blocker niflumic acid releases Ca2+ from an intracellular store in rat pulmonary artery smooth muscle cells. Br J Pharmacol. 2003;140:1442–1450. doi: 10.1038/sj.bjp.0705571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinh-Xuan AT, Higenbottam TW, Clelland CA, Pepke-Zaba J, Cremona G, Butt AY, et al. Impairment of endothelium-dependent pulmonary-artery relaxation in chronic obstructive lung disease. N Engl J Med. 1991;324:1539–1547. doi: 10.1056/NEJM199105303242203. [DOI] [PubMed] [Google Scholar]
- Drummond RM, Tuft RA. Release of Ca2+ from the sarcoplasmic reticulum increases mitochondrial [Ca2+] in rat pulmonary artery smooth muscle cells. J Physiol. 1999;516:139–147. doi: 10.1111/j.1469-7793.1999.139aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan TM, Khakh BS. Contribution of calcium ions to P2X channel responses. J Neurosci. 2004;24:3413–3420. doi: 10.1523/JNEUROSCI.5429-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlinge D, Burnstock G. P2 receptors in cardiovascular regulation and disease. Purinergic Signal. 2008;4:1–20. doi: 10.1007/s11302-007-9078-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans RJ, Kennedy C. Characterization of P2-purinoceptors in the smooth muscle of the rat tail artery: a comparison between contractile and electrophysiological responses. Br J Pharmacol. 1994;113:853–860. doi: 10.1111/j.1476-5381.1994.tb17071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores RV, Hernández-Pérez MG, Aquino E, Garrad RC, Weisman GA, Gonzalez FA. Agonist-induced phosphorylation and desensitization of the P2Y2 nucleotide receptor. Mol Cell Biochem. 2005;280:35–45. doi: 10.1007/s11010-005-8050-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood IA, Leblanc N. Overlapping pharmacology of Ca2+-activated Cl− and K+ channels. Trends Pharmacol Sci. 2007;28:1–5. doi: 10.1016/j.tips.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Guibert C, Pacaud P, Loirand G, Marthan R, Savineau JP. Effect of extracellular ATP on cytosolic Ca2+ concentration in rat pulmonary artery myocytes. Am J Physiol. 1996;271:450–458. doi: 10.1152/ajplung.1996.271.3.L450. [DOI] [PubMed] [Google Scholar]
- Guibert C, Marthan R, Savineau JP. Oscillatory Cl− current induced by angiotensin II in rat pulmonary arterial myocytes: Ca2+ dependence and physiological implication. Cell Calcium. 1997;21:421–429. doi: 10.1016/s0143-4160(97)90053-1. [DOI] [PubMed] [Google Scholar]
- Hansen MA, Dutton JL, Balcar VJ, Barden JA, Bennett MR. P2X (purinergic) receptor distributions in rat blood vessels. J Auton Nerv Syst. 1999;75:147–155. doi: 10.1016/s0165-1838(98)00189-1. [DOI] [PubMed] [Google Scholar]
- Hartley SA, Kato K, Salter KJ, Kozlowski RZ. Functional evidence for a novel suramin-insensitive pyrimidine receptor in rat small pulmonary arteries. Circ Res. 1998;83:940–946. doi: 10.1161/01.res.83.9.940. [DOI] [PubMed] [Google Scholar]
- Hasséssian H, Burnstock G. Interacting roles of nitric oxide and ATP in the pulmonary circulation of the rat. Br J Pharmacol. 1995;114:846–850. doi: 10.1111/j.1476-5381.1995.tb13281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg RC, Wang Q, Large WA. Effects of Cl channel blockers on Ca-activated chloride and potassium currents in smooth muscle cells from rabbit portal vein. Br J Pharmacol. 1994a;111:977–984. doi: 10.1111/j.1476-5381.1994.tb14891.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg RC, Wang Q, Large WA. Action of niflumic acid on evoked and spontaneous calcium-activated chloride and potassium currents in smooth muscle cells from rabbit portal vein. Br J Pharmacol. 1994b;112:977–984. doi: 10.1111/j.1476-5381.1994.tb13177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jernigan NL, Broughton BR, Walker BR, Resta TC. Impaired NO-dependent inhibition of store- and receptor-operated calcium entry in pulmonary vascular smooth muscle after chronic hypoxia. Am J Physiol. 2006;290:517–525. doi: 10.1152/ajplung.00308.2004. [DOI] [PubMed] [Google Scholar]
- Kato K, Evans AM, Kozlowski RZ. Relaxation of endothelin-1-induced pulmonary arterial constriction by niflumic acid and NPPB: mechanism(s) independent of chloride channel block. J Pharmacol Exp Ther. 1999;288:1242–1250. [PubMed] [Google Scholar]
- Kauffenstein G, Drouin A, Thorin-Trescases N, Bachelard H, Robaye B, D'Orléans-Juste P, et al. NTPDase1 (CD39) controls nucleotide-dependent vasoconstriction in mouse. Cardiovasc Res. 2010;85:204–213. doi: 10.1093/cvr/cvp265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy C, Mitchell C, Gurney AM. Pharmacological characterisation of P2Y receptor subtypes that mediate vasoconstriction of rat pulmonary artery. Purinergic Signal. 2010;6(Suppl. 1):S117–S118. doi: 10.1007/s11302-022-09895-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakh BS, Burnstock G, Kennedy C, King BF, North RA, Seguela P, et al. International union of pharmacology. XXIV. Current status of the nomenclature and properties of P2X receptors and their subunits. Pharmacol Rev. 2001;53:107–118. [PubMed] [Google Scholar]
- Kitagawa S, Nabekura T, Nakamura Y, Takahashi T, Kashiwada Y. Inhibition of P-glycoprotein function by tannic acid and pentagalloylglucose. J Pharm Pharmacol. 2007;59:965–969. doi: 10.1211/jpp.59.7.0008. [DOI] [PubMed] [Google Scholar]
- Kitamura K, Yamazaki J. Chloride channels and their functional roles in smooth muscle tone in the vasculature. Jpn J Pharmacol. 2001;85:351–357. doi: 10.1254/jjp.85.351. [DOI] [PubMed] [Google Scholar]
- Large WA, Wang Q. Characteristics and physiological role of the Ca2+-activated Cl− conductance in smooth muscle. Am J Physiol. 1996;271:435–454. doi: 10.1152/ajpcell.1996.271.2.C435. [DOI] [PubMed] [Google Scholar]
- Leblanc N, Ledoux J, Saleh S, Sanguinetti A, Angermann J, O'Driscoll K, et al. Regulation of calcium-activated chloride channels in smooth muscle cells: a complex picture is emerging. Can J Physiol Pharmacol. 2005;83:541–556. doi: 10.1139/y05-040. [DOI] [PubMed] [Google Scholar]
- Lee CH, Poburko D, Kuo KH, Seow CY, van Breemen C. Ca2+ oscillations, gradients, and homeostasis in vascular smooth muscle. Am J Physiol. 2002;282:H1571–H1583. doi: 10.1152/ajpheart.01035.2001. [DOI] [PubMed] [Google Scholar]
- Lemon G, Brockhausen J, Li GH, Gibson WG, Bennett MR. Calcium mobilization and spontaneous transient outward current characteristics upon agonist activation of P2Y2 receptors in smooth muscle cells. Biophys J. 2005;88:1507–1523. doi: 10.1529/biophysj.104.043976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis CJ, Evans R. P2X receptor immunoreactivity in different arteries from the femoral, pulmonary, cerebral, coronary and renal circulations. J Vasc Res. 2001;38:332–340. doi: 10.1159/000051064. [DOI] [PubMed] [Google Scholar]
- Liu SF, McCormack DG, Evans TW, Barnes PJ. Characterization and distribution of P2-purinoceptor subtypes in rat pulmonary vessels. J Pharmacol Exp Ther. 1989a;251:1204–1210. [PubMed] [Google Scholar]
- Liu SF, McCormack DG, Evans TW, Barnes PJ. Evidence for two P2-purinoceptor subtypes in human small pulmonary arteries. Br J Pharmacol. 1989b;98:1014–1020. doi: 10.1111/j.1476-5381.1989.tb14633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lommatzsch M, Cicko S, Müller T, Lucattelli M, Bratke K, Stoll P, et al. Extracellular adenosine triphosphate and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;181:928–934. doi: 10.1164/rccm.200910-1506OC. [DOI] [PubMed] [Google Scholar]
- Mam V, Tanbe AF, Vitali SH, Arons E, Christou HA, Khalil RA. Impaired vasoconstriction and nitric oxide-mediated relaxation in pulmonary arteries of hypoxia- and monocrotaline-induced pulmonary hypertensive rats. J Pharmacol Exp Ther. 2010;332:455–462. doi: 10.1124/jpet.109.160119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manoury B, Tamuleviciute A, Tammaro P. TMEM16A/anoctamin 1 protein mediates calcium-activated chloride currents in pulmonary arterial smooth muscle cells. J Physiol. 2010;588:2305–2314. doi: 10.1113/jphysiol.2010.189506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack DG, Barnes PJ, Evans TW. Purinoceptors in the pulmonary circulation of the rat and their role in hypoxic vasoconstriction. Br J Pharmacol. 1989;98:367–372. doi: 10.1111/j.1476-5381.1989.tb12606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren GJ, Burke KS, Buchanan KJ, Sneddon P, Kennedy C. Evidence that ATP acts at two sites to evoke contraction in the rat isolated tail artery. Br J Pharmacol. 1998;124:5–12. doi: 10.1038/sj.bjp.0701772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami S, Muramatsu M, Otomo S. Inhibitory effect of tannic acid on gastric H+,K+-ATPase. J Nat Prod. 1992;55:513–516. doi: 10.1021/np50082a022. [DOI] [PubMed] [Google Scholar]
- Namkung W, Thiagarajah JR, Phuan PW, Verkman AS. Inhibition of Ca2+-activated Cl− channels by gallotannins as a possible molecular basis for health benefits of red wine and green tea. FASEB J. 2010;24:4178–4186. doi: 10.1096/fj.10-160648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namkung W, Phuan PW, Verkman AS. TMEM16A inhibitors reveal TMEM16A as a minor component of calcium-activated chloride channel conductance in airway and intestinal epithelial cells. J Biol Chem. 2011;286:2365–2374. doi: 10.1074/jbc.M110.175109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nori S, Fumagalli L, Bo X, Bogdanov Y, Burnstock G. Coexpression of mRNAs for P2X1, P2X2 and P2X4 receptors in rat vascular smooth muscle: an in situ hybridization and RT-PCR study. J Vasc Res. 1998;35:179–185. doi: 10.1159/000025582. [DOI] [PubMed] [Google Scholar]
- Robaye B, Boeynaems JM, Communi D. Slow desensitization of the human P2Y6 receptor. Eur J Pharmacol. 1997;329:231–236. [PubMed] [Google Scholar]
- Robson SC, Sévigny J, Zimmermann H. The E-NTPDase family of ectonucleotidases: structure function relationships and pathophysiological significance. Purinergic Signal. 2006;2:409–430. doi: 10.1007/s11302-006-9003-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock JR, Futtner CR, Harfe BD. The transmembrane protein TMEM16A is required for normal development of the murine trachea. Dev Biol. 2008;321:141–149. doi: 10.1016/j.ydbio.2008.06.009. [DOI] [PubMed] [Google Scholar]
- Rubino A, Burnstock G. Evidence for a P2-purinoceptor mediating vasoconstriction by UTP, ATP and related nucleotides in the isolated pulmonary vascular bed of the rat. Br J Pharmacol. 1996;118:1415–1420. doi: 10.1111/j.1476-5381.1996.tb15554.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubino A, Ziabary L, Burnstock G. Regulation of vascular tone by UTP and UDP in isolated intrapulmonary arteries. Eur J Pharmacol. 1999;370:139–143. doi: 10.1016/s0014-2999(99)00150-8. [DOI] [PubMed] [Google Scholar]
- Schroeder BC, Cheng T, Jan YN, Jan LY. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell. 2008;134:1019–1029. doi: 10.1016/j.cell.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprague RS, Ellsworth ML, Stephenson AH, Lonigro AJ. ATP: the red blood cell link to NO and local control of the pulmonary circulation. Am J Physiol. 1996;271:H2717–H2722. doi: 10.1152/ajpheart.1996.271.6.H2717. [DOI] [PubMed] [Google Scholar]
- Sprague RS, Olearczyk JJ, Spence DM, Stephenson AH, Sprung RW, Lonigro AJ. Extracellular ATP signaling in the rabbit lung: erythrocytes as determinants of vascular resistance. Am J Physiol. 2003;285:H693–H700. doi: 10.1152/ajpheart.01026.2002. [DOI] [PubMed] [Google Scholar]
- Sromek SM, Harden TK. Agonist-induced internalization of the P2Y2 receptor. Mol Pharmacol. 1998;54:485–494. doi: 10.1124/mol.54.3.485. [DOI] [PubMed] [Google Scholar]
- Syed NH, Tengah A, Paul A, Kennedy C. Characterisation of P2X receptors expressed in rat pulmonary arteries. Eur J Pharmacol. 2010;649:342–348. doi: 10.1016/j.ejphar.2010.09.041. [DOI] [PubMed] [Google Scholar]
- Yang YD, Cho H, Koo JY, Tak MH, Cho Y, Shim WS, et al. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature. 2008;455:1210–1215. doi: 10.1038/nature07313. [DOI] [PubMed] [Google Scholar]
- Yuan XJ. Role of calcium-activated chloride current in regulating pulmonary vasomotor tone. Am J Physiol. 1997;272:L959–L968. doi: 10.1152/ajplung.1997.272.5.L959. [DOI] [PubMed] [Google Scholar]
- Zheng J, Ramirez VD. Inhibition of mitochondrial proton F0F1-ATPase/ATP synthase by polyphenolic phytochemicals. Br J Pharmacol. 2000;130:1115–1123. doi: 10.1038/sj.bjp.0703397. [DOI] [PMC free article] [PubMed] [Google Scholar]