

Abstract

We describe here light-regulated swelling and degradation features of polymeric nanoparticles that are produced using an inverse microemulsion polymerization method. We demonstrate the phototriggered release characteristics of the nanoparticles by sequestering protein molecules and releasing them using light as a trigger. Furthermore, the intracellular translocation of the nanoparticles, along with its fluorescent protein payload, was achieved using a cell-penetrating peptide-based surface modification. We expect that the noncovalent encapsulation of proteins using nanoparticles and their photo triggered release using an external light would provide opportunities for achieving intracellular release of molecular therapeutics for on-demand requirements.
Introduction
Nanosized materials have emerged as effective delivery systems for therapeutic applications in recent years.1 In part, this development has been fuelled by their unique properties, including (i) greater cell penetration capability and (ii) the ability to passively accumulate near tumor cells through the enhanced permeation and retention (EPR) effect.2−4 Nanomaterials that are currently explored for delivery purposes are mainly lipids, surfactants, copolymers, and dendrimers.5−8 While the technology of utilizing polymeric assemblies (particularly self-assembled structures of copolymers) is rapidly growing, these nanovehicles are solely based on noncovalent interactions, which have considerably restricted stability, especially when transferred to large volumes of biological fluids.9−11 Furthermore, sequestering large drug molecules like proteins in these noncovalently assembled structures is a challenge. To address these issues, cross-linked polymer nanoparticles have emerged as potent nanosized delivery vehicles that are highly stable and capable of maintaining their structure even at diluted conditions.12−19 However, in initial studies, release of cargos from these polymeric materials was entirely diffusion controlled. To provide greater diversity, there is a growing interest in introducing functionalities that are responsive to specific triggers, as cross-linkers in these polymeric materials;20−22 not only to control the release kinetics, but also to trigger release after reaching targeted locations. This strategy of triggered de-cross-linking has positioned cross-linked polymeric materials as promising systems for controlled delivery of therapeutics. Herein, we present such de-cross-linking features using a light sensitive cross-linked polymeric nanoparticle system.
While there are a significant number of triggers such as pH, temperature, redox, proteins, light ,and magnetic field being explored,20−25 light has drawn much attention, because it provides the opportunity for the user to not only control release properties, but also to do so remotely in a spatiotemporal defined manner. Exploiting this advantage, light has been widely utilized to (i) pattern surfaces, (ii) activate caged biological entities and, (iii) spatially control the properties of gel-like scaffolds.26−31 Although there are a significant number of photocontrolled release systems, they are either exclusively employed in noncovalently assembled polymeric systems or based on cyto-incompatible shortwave UV light (∼250 nm).32−42 In this context, photolabile protecting groups are also utilized to de-cross-link polymer networks and control release of noncovalently encapsulated guest molecules.42,43 More recently, light-sensitive polymeric microgels have been reported using emulsion polymerization methods, which produce particles that are quite hydrophobic.44 A potential complementary approach to this is developing particles that are hydrophilic, since this renders them highly water-soluble and provides opportunities to sequester hydrophilic molecular entities such as proteins and DNA/RNAs. Such photocontrolled systems would allow us to carry out fundamental studies that offer unprecedented control over cellular responses to cargo release in in vitro experiments, and consequently provide basic understanding on cell properties. In this manuscript, we report water-soluble, cross-linked polymeric nanogels that exhibit photoinduced swelling and degradation properties in aqueous conditions with the potential ability to encapsulate and release proteins in response to light (Figure 1a).
Figure 1.
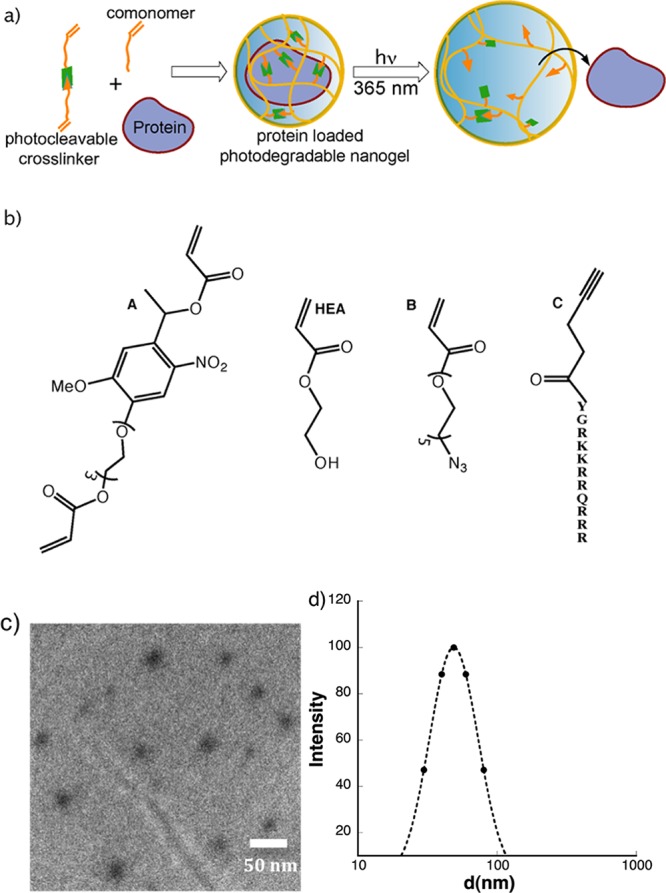
(a) Schematic illustration of protein encapsulation and its light-induced release from cross-linked polymeric nanogels. (b) Chemical structures of monomers used in the synthesis of photocontrolled nanogels. (c) TEM image of nanogels. (d) Hydrodynamic size of nanogel (0.2 mg/mL).
Experimental Section
Synthesis of Photodegradable Nanogels
The photodegradable nanogels used in this study were prepared through inverse microemulsion polymerization following the method developed by Mcallister et al.5 with modifications. The molar ratio of the hydroxyethylacrylate (HEA) to the cross-linker A in the emulsion was maintained at 95:5. Accordingly for the reference, 125 mg (1.08 mmol) of HEA and 22.6 mg of cross-linker A (0.05 mmol) were taken in a 20 mL vial and was diluted using 100 μL of distilled water. This vial was then protected from light to prevent degradation of the light sensitive cross-linker. Separately, the inverse microemulsion was prepared by mixing 5.0 g of n-heptane with 0.6 g of the surfactant, Laureth-4 (Brij 30, from Sigma Aldrich). The microemulsion solution was then added to the monomer mixture solution and then vortexed until a clear solution was obtained. Then, the contents of the vial were stirred and purged with argon for 4 min to remove oxygen from the reaction vial. The initiator, ammonium persulfate (APS) (10 mg) in water (60 μL) was added to the microemulsion-monomer mixture, followed by 10 μL of the activator, tetramethylethylenediamine (TEMED) under argon conditions. The reaction, while shielded from light, was conducted at room temperature under argon conditions for 12 h. To isolate the nanogels, 8 mL water was added to the emulsion and allowed to stir for 5 min. The contents were transferred to two 15 mL falcon tubes and were centrifuged for 15 min at 4400 rpm. After disposing the organic layer, the aqueous layer in each tube was then washed with 2 mL of n-butanol and centrifuged for 15 min at 4400 rpm. This wash was repeated two times to remove all the surfactants. The remaining aqueous layers of each tube were combined and poured into a 7K-dialysis membrane. The membrane was then dialyzed in 2.0 L of water for 24 h at 5 °C, replacing the water every 6 h. The nanogel solution was removed from the dialysis tube and then passed through 0.22 μm filter. The resulting solution was lyophilized to obtain the nanogels as a dry solid (yield: 40%).
Protein Encapsulation in Photodegradable Nanogels
For protein encapsulation, the same procedure described above for nanogel synthesis was followed with minor variations. Here the molar ratio of the HEA to the cross-linker A in the emulsion was kept at 97.5:2.5. Similarly, 131.0 mg (1.13 mmol) of HEA and 12.6 mg of cross-linker A (0.03 mmol) were taken in a 20 mL vial and diluted using 100 μL of distilled water. To this vial was added a solution of 2.5 mg of alkaline phosphatase (ALP) (or fluorescein-BSA) in 100 μL of water. Separately, the inverse microemulsion was prepared and added to the monomer mixture solution and then vortexed mildly. While stirring, the contents of the vial were purged with argon for 4 min to remove oxygen from the reaction vial. The initiator, APS (5 mg) in water (30 μL) and 5 μL of TEMED were added to the microemulsion–monomer mixture, and then the reaction was conducted at room temperature under argon conditions for 2 h. The nanogels were isolated according to the above procedure, but dialysis was carried out using phopshate buffered saline (PBS) buffer (pH 7.6) for 24 h at 5 °C and replacing the water for every 6 h. The protein loaded nanogel solution was removed from the dialysis tube and then passed through a 0.22 μm filter. The filtered resulting solution was directly used for ALP activity studies. The amount of nanogels in these solutions was obtained by lyophilization, and then the approximate concentrations were calculated. The concentration of nanogels before lyophilization was found to be 4 mg/mL, and for ALP activity studies the nanogel solution was directly used from dialysis.
ALP Activity/Release Studies of Photodegradable Nanogels
Approximately 4.0 mg/mL of nanogel solution loaded with ALP (NG-ALP) was used for the study and was passed through a 0.22 μm filter before experiments. Fifty microliters of these NG-ALP solutions was exposed to 365 nm (10 mW/cm2) light for different times. Then 8.0 μL of NG-ALP solutions (0, 2, and 4 min of light exposures) was added to 150 μL of 0.2 mM solution of ALP substrate (p-nitrophenyl phosphate (p-NPP)) in a 96 well plate. The final concentration of NG in each well was 0.2 mg/mL. The change in the absorbance at 400 nm was tracked over a period of 15 h, and the temperature was kept at 25 °C throughout the experiment.
Intracellular Uptake of Bovine Serum Albumin (BSA)–Fluorescein Loaded Nanogels
3T3 cells were plated at 50 000 cells per well in a six-well plate and then allowed to attach on a cover glass that was placed on the bottom of each well for 6 h. Then BSA-loaded nanogel solutions (each well contained 0.25 mg of nanogel) were added to the wells and continued the culturing for 12 h. After 12 h, the media was removed, and the cells were washed with media 2–3 times. The cover glass was removed from the well and imaged by confocal microscopy. For all cell experiments, the basic culture composition was low glucose Dulbecco’s modified Eagle's medium supplemented with 10% fetal bovine serum, 50 U/mL each of penicillin and streptomycin, and 1 μg/mL fungizone (all from invitrogen). The cells were kept at 37 °C and 5% CO2 for the duration of the experiments.
Results and Discussion
To obtain the photodegradable nanogel, we designed and synthesized a short photodegradable cross-linker A (Figure 1b). Here, a methoxy-nitrobenzyl ether derivative was selected as the photodegradable moiety because of its well-established chemistry and greater utilization in numerous biological applications due its absorbance at longer wavelengths of light.8 Also, a triethylene glycol spacer was introduced to the base nitro-benzyl unit in order to improve its solubility in water. The key photodegradable cross-linker A was obtained in six steps as shown in Scheme 1a. First the phenolic hydroxyl group of starting acetovanillone 1 was benzyl-protected for nitration and then deprotected using TFA. Then, the nitro-acetovanillone 2 was treated with monotosylated tri(ethylene glycol) (TEG-OTs) followed by keto-reduction and acrylation of terminal hydroxyl groups to yield the photodegradable cross-linker A. Having synthesized the main cross-linker A, we decided to assemble nanogels using an inverse microemulsion polymerization method,5 because it enables us to achieve particles that are: (i) water-soluble for the encapsulation of biologically relevant cargos such as proteins/enzymes and (ii) within the nanosized regime for efficient intracellular delivery. Photodegradable nanogels were obtained by copolymerizing the cross-linker A with HEA in an inverse microemulsion milieu using a free-radical-initiated polymerization. To initiate polymerization, APS was used as the initiator and TEMED as the activator (Supporting Information (SI)).
Scheme 1. (a) Synthesis of Key Photocleavable Crosslinker A. (b) Light Induced De-cross-linking of Photodegradable Nanogel.

To determine whether the photodegradable cross-linker was incorporated into the polymer network, we first used 1H NMR spectroscopy (SI). The presence of signals for the nitro-benzyl unit at the aromatic region clearly indicated incorporation of the photodegradable monomer in the polymeric backbone. Additionally, there were no signals corresponding to monomeric vinylic protons in the spectrum, suggesting very high levels of conversion. We then characterized the size and shape of the resulting nanogels using both transmission electron microscope (TEM) and dynamic light scattering studies (DLS). The TEM image in Figure 1c indicated a spherical morphology of the nanogels with sizes ranging from 25 to 40 nm. Since TEM images were obtained from dry surfaces, DLS was carried out to obtain the hydrodynamic sizes of the nanogels in solution. DLS results indicate that the nanogels have a relatively narrow size distribution, with an average diameter of 50 nm (Figure 1d).
To test the photoresponsive properties of the nanogels, we investigated changes in the hydrodynamic diameter as a function of exposure time using DLS. When aqueous solutions of the nanogel were exposed to 365 nm of light at 10 mW/cm2, we observed two prominent changes (Figure 2a): (i) a systematic increase in the size of the nanogels occurred with increasing exposure time, consistent with light-induced de-cross-linking and subsequent swelling of the nanogels (Scheme 1b); (ii) an increase in the hydrodynamic size of the nanogels was observed up to 5 min of light exposure. Interestingly, further light exposure did not lead to significant increases in the nanogel size, possibly due to either the existence of noncovalent larger aggregates or partial degradation/de-cross-linking of the cross-linked polymer. To verify this further, the degradation profiles of the nanogels were examined using gel permeation chromatography (GPC). Before exposure to light, the nanogels showed a single peak with a slight shoulder in the chromatogram (Figure 2b), but this peak gradually decreased in area with increasing light exposure, implying the disappearance of the starting nanogel due to its light-induced degradation. More importantly, this steady decrease in the original peak was concomitant with the emergence of a new peak at longer elution times (i.e., the lower molecular weight region), which corroborated the degradation of the starting nanogel into lower molecular weight polymers. As a control experiment, when nonphotodegradable nanogels, synthesized using diethylene glycol diacrylate as a cross-linker, were exposed to light, we did not observe any significant change in the GPC profile (SI). This further supports the idea that changes observed in the GPC for photodegradable nanogels were due to light-induced de-cross-linking of the polymer networks.
Figure 2.

Studies of photoresponsive properties and protein release: (a) Change in hydrodynamic size of nanogel (0.2 mg/mL) at different time of light exposures (2.5, 5 and 10 min) using DLS; (b) GPC traces outlining light induced degradation of nanogel (1.0 mg/mL); (c) Control over protein activity: Evolution of p-NP (λmax: 400 nm) upon exposing p-NPP substrate (0.2 mM) to ALP loaded nanogel (0.2 mg/mL) and its light exposed (2 min of exposure) counterpart in UV–vis; (d) SDS-PAGE electrophoresis depicting protein release: L - ladder; 1- native ALP; 2- ALP loaded nanogel; 3- ALP loaded nanogel after exposure to light (2 min); 4 - blank nanogel (not loaded with ALP).
Having explored the swelling and degradation profiles of the nanogels, we then entrapped protein molecules in the nanogels and tested the possibility of controlling protein release in response to light. For this demonstration, we chose ALP as a model protein, since ALP is a biologically important enzyme that is readily available and known to specifically cleave only phosphate esters. Hence, the release of ALP from the nanogel could be easily monitored by the change in the enzyme activity, but the ALP would have minimal ability to access its substrate when entrapped in the nanogel compartments. Once released, however, it would have easy access toward its substrate and its concentration can be readily inferred from activity measurements. Prior to test this hypothesis, we investigated size, shape, and loading efficiency of the ALP sequestered nanogels using TEM analysis. While being spherical, TEM image of the protein-loaded nanogels showed an increase in size, i.e., ranged from 100 to 200 nm compared to unloaded nanogels (Figure S1). The ALP loading efficiency of the nanogels was determined using micro BCA assay and found to be almost 15 μg of protein per mg of nanogel (∼50% loading efficiency) (SI). We then moved on to study the effect of light exposure on the release and enzymatic activity of ALP-loaded nanogels. Typically, the activity of ALP is monitored using p-NPP as the substrate, which releases p-nitrophenol (p-NP) upon the enzymatic cleavage of the phosphate ester. Here, the evolution in the absorbance of p-NP in Figure 2c shows that light-degraded ALP-loaded nanogels exhibit significantly increased enzyme activity as compared to samples that were not exposed to light. This result clearly suggests that the bioactivity of the ALP is (i) intact; and (ii) can be controlled using an external light. However, it is important to further determine whether this increase in enzymatic activity is due to (i) the light induced swelling of nanogels that results in increased diffusion of small molecule substrate into the nanogel or (ii) the release of protein from the nanogel compartments. For this, we have carried out protein gel electrophoresis, and the results are shown in Figure 2d. The ALP loaded nanogels before exposing to light did not show any well-defined protein band (lane 2) as compared to the distinctive band observed for native ALP (lane 1). After exposure to light, the appearance of a well-defined band (lane 3) that is comparable to the native ALP indicated that the protein molecules are indeed released from nanogel compartments that effect in increased enzymatic activity.
Finally, we investigated the intracellular uptake of nanogels loaded with fluorescein-labeled BSA using NIH 3T3 cells. Confocal imaging (Figure 3a) of the cells clearly indicated that there is minimal uptake of the nanogels by the 3T3 cells. However, if a cell penetrating TAT peptide is included on the surface of the nanogel, internalization is facile (Figure 3b). TAT is known to facilitate cellular uptake of nanoparticles, and functionalization was achieved using an azide-terminated acrylate B (Figure 1b), which was readily incorporated during the nanogel synthesis. The azide-containing particles were then treated with alkyne-tagged TAT peptide C (Figure 1b) under click chemistry conditions. When these TAT-incorporated nanogels were incubated with 3T3 cells, they were easily internalized and almost exclusively in the cell cytoplasm as shown in Figure 3b. This further demonstrates that these protein-loaded nanogels have the potential to be internalized by cells, and subsequent control over the intracellular release of cargos using an external light can be easily envisioned. Since nitrobenzyl molecular systems are known to cleave under two-photon light (740 nm),30 for such controlled intracellular applications, one can also utilize two-photon light. Moreover, both 740 and 365 nm light are known to be cyto-compatible over a range of intensities and exposure times.45,46
Figure 3.

Confocal images of 3T3 cells after being exposed to flourescein-BSA loaded nanogel: (a) without TAT peptide; (b) with TAT peptide.
Conclusion
In summary, we have designed and synthesized light controlled polymeric nanoparticles and explored their photoresponsive properties in aqueous conditions. The synthesis of photodegradable nanogels was achieved using a short photocleavable diacrylate as a cross-linker in an inverse microemulsion milieu. We demonstrate that light induces degradation and concomitant swelling of the nanogels, as observed by significant changes in the hydrodynamic size and GPC response/retention-time with exposure time. More importantly, we demonstrate that these nanomaterials are capable of encapsulating proteins, maintaining their activity, and releasing proteins in response to light. Since light is a remote-trigger and can be used externally, we envision that these photoresponsive nanosized materials might provide new opportunities for controlled/targeted delivery applications. Utilization of these nanomaterials for encapsulation of specific disease oriented proteins/RNA molecules and controlling cellular properties using light are current investigations in our laboratory.
Acknowledgments
The authors would like to thank the National Science Foundation (DM 1006711D) and the Howard Hughes Medical Institute for funding for this work. M.W.T. would like to thank the National Institute of Health (T32 GM-065103) and the Teets Family Endowed Doctoral Fellowship for funding assistance. S.J.R. would like to thank the UROP (UC-Boulder) for funding assistance. We would like to thank Jennifer Leight for helping with the gel electrophoresis.
Supporting Information Available
Syntheses and characterization of photocleavable cross-linker, TAT peptide, and its incorporation to the nanogel are presented. Also, methods for TEM, DLS, and GPC can be found here. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Namiki Y.; Fuchigami T.; Tada N.; Kawamura R.; Matsunuma S.; Kitamoto Y.; Nakagawa M. Acc. Chem. Res. 2011, 44, 1080–1093. [DOI] [PubMed] [Google Scholar]
- Chou L. Y. T.; Ming K.; Chan W. C. W. Chem. Soc. Rev. 2011, 40, 233–245. [DOI] [PubMed] [Google Scholar]
- Maeda H.; Matsumura Y.; Sasamoto K. Proc. Am. Assoc. Cancer Res. 1986, 27, 401–401. [Google Scholar]
- Maeda H.; Wu J.; Sawa T.; Matsumura Y.; Hori K. J. Controlled Release 2000, 65, 271–284. [DOI] [PubMed] [Google Scholar]
- Lawrence M. J.; Rees G. D. Adv. Drug Delivery Rev. 2000, 45, 89–121. [DOI] [PubMed] [Google Scholar]
- Haag R.; Kratz F. Angew. Chem., Int. Ed. 2006, 45, 1198–1215. [DOI] [PubMed] [Google Scholar]
- Kataoka K.; Harada A.; Nagasaki Y. Adv. Drug Delivery Rev. 2001, 47, 113–131. [DOI] [PubMed] [Google Scholar]
- Ihre H.; De Jesus O. L. P.; Frechet J. M. J. J. Am. Chem. Soc. 2001, 123, 5908–5917. [DOI] [PubMed] [Google Scholar]
- Bae Y. H.; Yin H. Q. J. Controlled Release 2008, 131, 2–4. [DOI] [PubMed] [Google Scholar]
- Kim S.; Shi Y. Z.; Kim J. Y.; Park K.; Cheng J. X. Expert Opin. Drug Delivery 2010, 7, 49–62. [DOI] [PubMed] [Google Scholar]
- Jiwpanich S.; Ryu J. H.; Bickerton S.; Thayumanavan S. J. Am. Chem. Soc. 2010, 132, 10683–10685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabanov A. V.; Vinogradov S. V. Angew. Chem., Int. Ed. 2009, 48, 5418–5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister K.; Sazani P.; Adam M.; Cho M. J.; Rubinstein M.; Samulski R. J.; DeSimone J. M. J. Am. Chem. Soc. 2002, 124, 15198–15207. [DOI] [PubMed] [Google Scholar]
- Joralemon M. J.; O’Reilly R.; Hawker C. J.; Wooley K. L. J. Am. Chem. Soc. 2005, 127, 16892–16899. [DOI] [PubMed] [Google Scholar]
- Oh J. K.; Drumright R.; Siegwart D. J.; Matyjaszewski K. Prog. Polym. Sci. 2008, 33, 448–477. [Google Scholar]
- Raemdonck K.; Demeester J.; De Smedt S. Soft Matter 2009, 5, 707–715. [Google Scholar]
- Ryu J. H.; Chacko R. T.; Jiwpanich S.; Bickerton S.; Babu R. P.; Thayumanavan S. J. Am. Chem. Soc. 2010, 132, 17227–17235. [DOI] [PubMed] [Google Scholar]
- Sasaki Y.; Akiyoshi K. Chem. Rec. 2010, 10, 366–376. [DOI] [PubMed] [Google Scholar]
- Oh J. K.; Siegwart D. J.; Lee H. I.; Sherwood G.; Peteanu L.; Hollinger J. O.; Kataoka K.; Matyjaszewski K. J. Am. Chem. Soc. 2007, 129, 5939–5945. [DOI] [PubMed] [Google Scholar]
- Murthy N.; Xu M. C.; Schuck S.; Kunisawa J.; Shastri N.; Frechet J. M. J. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 4995–5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrott M. C.; Luft J. C.; Byrne J. D.; Fain J. H.; Napier M. E.; DeSimone J. J. Am. Chem. Soc. 2010, 132, 17928–17932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen J.; Anderson S. M.; Du J. J.; Yan M.; Wang J.; Shen M. Q.; Lu Y. F.; Segura T. Adv. Mater. 2011, 23, 4549–4553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y.; Park S. Y.; Kim C.; Park T. G. J. Controlled Release 2009, 135, 89–95. [DOI] [PubMed] [Google Scholar]
- Hu Y. X.; He L.; Yin Y. D. Angew. Chem., Int. Ed. 2011, 50, 3747–3750. [DOI] [PubMed] [Google Scholar]
- Ulijn R. V. J. Mater. Chem. 2006, 16, 2217–2225. [Google Scholar]
- Holmes C. P. J. Org. Chem. 1997, 62, 2370–2380. [DOI] [PubMed] [Google Scholar]
- Park E. J.; Carroll G. T.; Turro N. J.; Koberstein J. T. Soft Matter 2009, 5, 36–50. [Google Scholar]
- Lemke E. A.; Summerer D.; Geierstanger B. H.; Brittain S. M.; Schultz P. G. Nat. Chem. Biol. 2007, 3, 769–772. [DOI] [PubMed] [Google Scholar]
- Luo Y.; Shoichet M. S. Nat. Mater. 2004, 3, 249–253. [DOI] [PubMed] [Google Scholar]
- Kloxin A. M.; Kasko A. M.; Salinas C. N.; Anseth K. S. Science 2009, 324, 59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deforest C. A.; Anseth K. S. Nat. Chem. 2011, 3, 925–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomatsu I.; Peng K.; Kros A. Adv. Drug Delivery Rev. 2011, 63, 1257–1266. [DOI] [PubMed] [Google Scholar]
- Pasparakis G.; Manouras T.; Argitis P.; Vamvakaki M. Macromol. Rapid Commun. 2012, 33, 183–198. [DOI] [PubMed] [Google Scholar]
- Yan B.; Boyer J.-C.; Branda N. R.; Zhao Y. J. Am. Chem. Soc. 2011, 133, 19714–19717. [DOI] [PubMed] [Google Scholar]
- Xiao W.; Chen W. H.; Xu X. D.; Li C.; Zhang L.; Zhuo R. X.; Zhang X. Z. Adv. Mater. 2011, 23, 3526–3530. [DOI] [PubMed] [Google Scholar]
- Han D. H.; Tong X.; Zhao Y. Macromolecules 2011, 44, 437–439. [Google Scholar]
- Lee H. I.; Wu W.; Oh J. K.; Mueller L.; Sherwood G.; Peteanu L.; Kowalewski T.; Matyjaszewski K. Angew. Chem., Int. Ed. 2007, 46, 2453–2457. [DOI] [PubMed] [Google Scholar]
- Schumers J. M.; Fustin C. A.; Gohy J. F. Macromol. Rapid Commun. 2010, 31, 1588–1607. [DOI] [PubMed] [Google Scholar]
- Babin J.; Pelletier M.; Lepage M.; Allard J. F.; Morris D.; Zhao Y. Angew. Chem., Int. Ed. 2009, 48, 3329–3332. [DOI] [PubMed] [Google Scholar]
- Yang H.; Jia L.; Wang Z. F.; Di-Cicco A.; Levy D.; Keller P. Macromolecules 2011, 44, 159–165. [Google Scholar]
- Jochum F. D.; Theato P. Chem. Commun. 2010, 46, 6717–6719. [DOI] [PubMed] [Google Scholar]
- Fomina N.; Sankaranarayanan J.; Almutairi A. Adv. Drug Delivery Rev. 2012, 10.1016/j.addr.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fomina N.; McFearin C.; Sermsakdi M.; Edigin O.; Almutairi A. J. Am. Chem. Soc. 2010, 132, 9540–9542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinger D.; Landfester K. Soft Matter 2011, 7, 1426–1440. [Google Scholar]
- Bryant S. J.; Nuttelman C. R.; Anseth K. S. J. Biomater. Sci. Polym. Ed. 2000, 11, 439–457. [DOI] [PubMed] [Google Scholar]
- DeForest C. A.; Polizzotti B. D.; Anseth K. S. Nat. Mater. 2009, 8, 659–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.