

Abstract
Fibrosis, the result of excess deposition of extracellular matrix (ECM), in particular collagen, leads to scarring and loss of function in tissues that include the heart, lung, kidney and liver. The second messenger cAMP can inhibit the formation and extent of ECM during this late phase of inflammation, but the mechanisms for these actions of cAMP and of agents that elevate tissue cAMP levels are not well understood. In this article, we review the fibrotic process and focus on two recently recognized aspects of actions of cAMP and its effector Epac (Exchange protein activated by cAMP): (a) blunting of epithelial–mesenchymal transformation (EMT) and (b) down-regulation of Epac expression by profibrotic agents (e.g. TGF-β, angiotensin II), which may promote tissue fibrosis by decreasing Epac-mediated antifibrotic actions. Pharmacological approaches that raise cAMP or blunt the decrease in Epac expression by profibrotic agents may thus be strategies to block or perhaps reverse tissue fibrosis.
LINKED ARTICLES
This article is part of a themed section on Novel cAMP Signalling Paradigms. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2012.166.issue-2
Keywords: cyclic AMP, Epac, fibrosis, G protein-coupled receptors, protein kinase A, MDCK cell, myofibroblast, protein kinase A, TGF-β
Introduction to tissue fibrosis and cAMP signalling
Tissue fibrosis
Tissue fibrosis (scarring) results from the excess deposition of extracellular matrix (ECM) and occurs as part of normal physiology (e.g. aging) and following injury, in particular, with recurrent or persistent stimulation of the inflammatory process. Tissue fibrosis often alters tissue function. For example, cardiac fibrosis contributes to diastolic dysfunction and a decrease in cardiac output that accompany advanced age (Lakatta, 2003; Chen and Frangogiannis, 2010). Inflammation that follows tissue injury produces a series of acute and late-phase responses. In its acute phase, inflammation is characterized by several cardinal features [calor, rubor, dolor and tumour (heat, redness, pain and swelling)], which can be treated by pharmacological agents, in particular non-steroidal anti-inflammatory drugs (NSAIDs), whose major action is inhibition of COXs and thus the synthesis of prostaglandins.
Much less is known regarding the mechanisms that mediate the late phase of inflammation during which resolution of the tissue infiltration of acute inflammatory cells occurs, but tissue fibrosis can be initiated. Recent efforts have helped identify mechanisms of the resolution of inflammation (Serhan, 2010; Maskrey et al., 2011; Wynn, 2011; Yates et al., 2011). Glucocorticoids, the principal class of drugs used to treat this phase of the inflammatory process, block protein synthesis, thereby decreasing the accumulation of collagens and other ECM proteins. However, the use of glucocorticoids is associated with numerous side effects. Pirfenidone is a recently available antifibrotic agent whose mechanism of action is, at least in part, inhibition of the production and activity of TGF-β, a cytokine with strong profibrotic actions (Cho and Kopp, 2010). The wide prevalence of diseases associated with tissue fibrosis and the increasing age of the population (which is accompanied by such disorders) provide a strong impetus for the discovery of new antifibrotic agents.
A key goal in developing treatments for fibrosis is the avoidance of tissue damage and resolution of the remodelling that occur as a consequence of deposition of ECM with a resultant decrease in tissue parenchyma and function. Although certain types of tissue fibrosis can be reversed, it is generally irreversible, especially if its extent produces substantial loss of tissue parenchyma (Wallace et al., 2007; Marian, 2009; Ramachandran and Iredale, 2009; Chen and Frangogiannis, 2010; Dussaule et al., 2011). The main cell responsible for tissue remodelling during fibrosis is the myofibroblast. Myofibroblasts, α-smooth muscle actin–expressing cells that secrete ECM components and collagen type I and III, originate from a number of sources that depend on the physiological stimulus and the tissue in which the insult occurs. The mechanisms that contribute to the increase in the number of myofibroblasts in fibrosis are outlined in Figure 1 and include (Krenning et al., 2010; Pinzani and Macias-Barragan, 2010; Meran and Steadman, 2011) (1) mitosis of tissue-resident fibroblasts and their phenotypic ‘switch’ to profibrotic myofibroblasts (Hinz, 2010); (2) altered stem cell differentiation or de-differentiation of other cell types into fibroblasts (Brack et al., 2007); (3) activation and migration of stromal cells, pericytes, particularly in kidney fibrosis (Armulik et al., 2011; Schrimpf and Duffield, 2011); (4) conversion of epithelial or endothelial cells to fibroblasts, via epithelial–mesenchymal transformation (termed EMT) or endothelial–mesenchymal transformation, respectively; and (5) entry of bone marrow-derived, circulating fibrocytes into inflammatory sites (Grieb et al., 2011). The relative contribution of these various mechanisms to fibrosis is a topic of considerable current debate along with the question as to whether myofibroblasts derived in different ways and from different tissues exhibit different characteristics and functions; since myofibroblasts produce mediators that contribute to the fibrotic process, attenuation of their recruitment and activation are approaches to treat tissue fibrosis (Hinz, 2010; Humphreys et al., 2010; Krenning et al., 2010; Pinzani and Macias-Barragan, 2010; Kriz et al., 2011; Meran and Steadman, 2011).
Figure 1.
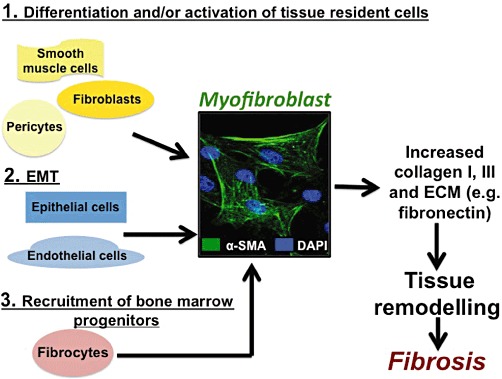
Heterogeneous origin of myofibroblasts. Myofibroblasts, which synthesize and secrete ECM, including collagens I and III, and contribute to fibrosis, originate from various sources, which include the following: (1) Differentiation of fibroblasts, activation of pericytes (e.g. hepatic stellate cells) and de-differentiation of muscle cells; (2) EMT or endothelial–mesenchymal transformation (transition); (3) Recruitment of circulating fibrocytes derived from the bone marrow.
cAMP signalling
The second messenger cAMP is as a regulator of fibroblast function. cAMP is generated by ACs in response to activation of GPCRs and degraded by cyclic nucleotide PDEs. cAMP acts via three mechanisms to modulate tissue responses: PKA, Epac and cyclic nucleotide-gated (CNG) ion channels. The cAMP effector, Epac (Exchange protein activated by cAMP) (Gloerich and Bos, 2010; Grandoch et al., 2010; Breckler et al., 2011), has recently been identified as an important mediator of the antifibrotic effect of cAMP and will be discussed in more detail below. As reviewed in the references cited above, the two forms of Epac, Epac1 and Epac2, respectively possess one and two cAMP binding sites and function as guanine nucleotide exchange factors for the low molecular weight G-protein Rap (e.g. Rap1 and Rap2). Epac proteins regulate numerous cellular responses through their ability to promote the exchange of GTP for GDP on Raps and perhaps other G-proteins (Gloerich and Bos, 2010; Grandoch et al., 2010; Breckler et al., 2011). cAMP analogues that selectively activate PKA or Epac (Bos, 2006; Holz et al., 2008) can aid in defining their role as effectors of cellular responses.
Increases in cAMP levels inhibit fibroblast function and produce antifibrotic effects that include (Table 1) decrease in fibroblast proliferation, stimulation of the death of fibroblasts and inhibition of ECM protein synthesis. Such effects can occur in response to increases in cAMP by agonists/antagonists of GPCRs, activation of ACs, inhibition of PDEs or use of cAMP analogues. The precise mechanisms for the antifibrotic effects of cAMP that are summarized in Table 1 are not clearly defined. We will discuss two aspects of cAMP/Epac-regulated actions that influence tissue fibrosis: (1) inhibition by cAMP of EMT and an associated decrease in synthesis of ECM components by fibroblasts and (2) inhibition by profibrotic agents of Epac expression, thereby blunting its antifibrotic actions. In addition, we discuss the roles of cAMP effectors in fibrosis and provide some therapeutic implications of these actions.
Table 1.
Antifibrotic actions of cAMP
| Action | References | Cell type |
|---|---|---|
| Inhibition of formation and action of profibrotic growth factors/hormones | Windmeier and Gressner, 1997 | Rat hepatic fibroblasts |
| Kothapalli et al., 1998 | Rat kidney interstitial fibroblasts (NRK-49F) | |
| Heusinger-Ribeiro et al., 2001 | Immortalized human renal fibroblasts | |
| Yu et al., 2002 | Rat-1 Fibroblasts | |
| Swaney et al., 2005 | Rat cardiac fibroblasts | |
| Liu et al., 2006 | Rat cardiac fibroblasts | |
| Black et al., 2007 | Human gingival fibroblasts | |
| Clancy et al., 2007 | Human Fetal Cardiac Fibroblasts | |
| Decrease in fibroblast proliferation and fibroblast –> myofibroblast conversion | Dunkern et al., 2007 | Human lung fibroblasts, fibroblast cell line IMR-90, fetal human fibroblast cell line GM06114 |
| Huang et al., 2008 | Human fetal lung fibroblast cell line IMR-90, primary human lung fibroblasts | |
| Xing and Bonanno, 2009 | Primary rabbit corneal keratocytes | |
| Sandbo et al., 2009 | Human pulmonary fibroblasts | |
| Selige et al., 2010 | Human lung fibroblasts | |
| Liu et al., 2004; 2005; 2010 | Rat pulmonary fibroblasts, murine pulmonary fibroblasts | |
| Stumm et al., 2011 | Murine lung fibroblasts | |
| Promotion of cell death of fibroblasts | Huang et al., 2009 | Human fetal lung fibroblast cell line IMR-90, primary human lung fibroblasts |
| Insel et al., 2012b | Various | |
| Zhang et al., 2011 | Rat cardiac fibroblasts | |
| Decrease in fibroblast motility | Sandulache et al., 2006; 2007 | Human fetal dermal fibroblast, human adult dermal fibroblast |
| Yokoyama et al., 2008 | Rat cardiac fibroblasts | |
| Togo et al., 2009 | Human fetal lung fibroblasts (HFL-1) | |
| Kohyama et al., 2009 | Human fetal lung fibroblasts (HFL-1) | |
| Decrease in synthesis/release/function of ECM components | Parekh et al., 2007 | Human fetal dermal fibroblast, human adult dermal fibroblast |
| Sachs et al., 2007 | Murine lung fibroblasts | |
| Liu et al., 2008 | Rat cardiac fibroblasts | |
| Parekh et al., 2009 | Human fetal dermal fibroblast, human adult dermal fibroblast | |
| Huang et al., 2008 | Human fetal lung fibroblast cell line IMR-90, primary human lung fibroblasts | |
| Liu et al., 2004; 2005; 2010 | Rat pulmonary fibroblasts, murine pulmonary fibroblasts | |
| Bauman et al., 2010 | Murine lung fibroblasts, human fetal lung fibroblast cell line IMR-90 | |
| Chan et al., 2010 | Murine cardiac fibroblasts | |
| D'Souza et al., 2011 | Human cardiac fibroblasts | |
| Okunishi et al., 2011 | Murine lung fibroblasts | |
| Miller et al., 2011 | Murine and rat fibroblasts |
Myofibroblast transformation is inhibited by cAMP via Epac and PKA
EMT
EMT has been proposed as a mechanism for tissue fibrosis in numerous settings, including in development, cancer and diseases of many organs (Hinz, 2010; Humphreys et al., 2010; Krenning et al., 2010; Chaffer and Weinberg, 2011; Kriz et al., 2011; Meran and Steadman, 2011). A commonly used model to study EMT is to ligate one of the ureters and observe the resultant changes in the kidney. Other studies of EMT have used renal epithelial cells, such as the distal tubule/collecting duct-derived MDCK cells. MDCK cells treated with TGF-β show changes that occur with EMT (Zhang et al., 2006a,b; Park et al., 2007; Cufíet al., 2010; Li et al., 2010; Figure 2): loss of expression of E-cadherin (and other proteins characteristic of epithelial cells) and increased expression of α-smooth muscle actin (α-SMA) [and also vimentin, fibronectin and collagens (proteins characteristic of fibroblasts; data not shown)]. We observe such changes in parental MDCK cells treated with TGF-β but not in MDCK-D1 cells, a clonal isolate that has enriched expression of α1b- and β2-adrenoceptors (Klijn et al., 1991; data not shown). Parental MDCK cells represent a mixture of renal cell types; an extensive literature documents that clonal isolates can have properties that differ from those present in the parental cells (e.g. Meier et al., 1983).
Figure 2.
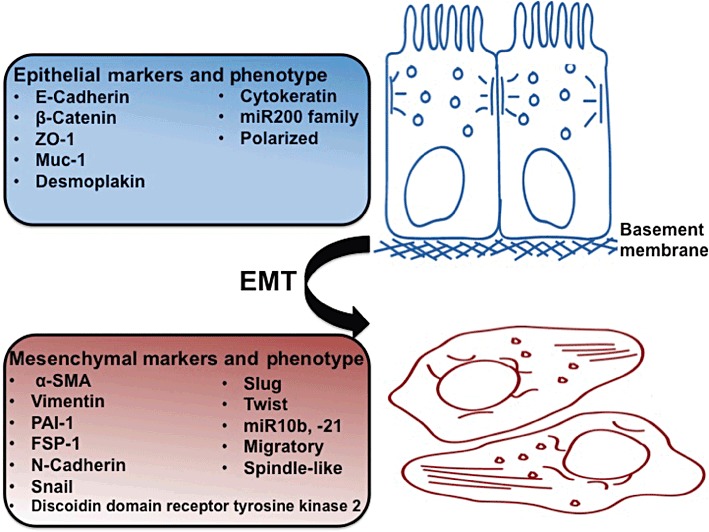
Markers of EMT. EMT involves a transition of polarized epithelial cells, which normally interact with the basement membrane, into migratory spindle-like, ECM-producing mesenchymal cells. The figure lists some commonly used markers that distinguish the two phenotypes.
Increases in cAMP by agents that activate AC, PDE inhibitors or cAMP analogues inhibits EMT in MDCK cells (Zhang et al., 2006a,b; Figures 3 and 4). Based on their epithelial phenotype, parental MDCK cells have high expression of E-cadherin but express little or no α-SMA: TGF-β induces transition to a mesenchymal phenotype by increasing α-SMA and decreasing E-cadherin expression (Figures 3 and 4). Treatment of these MDCK cells with a cAMP derivative that selectively activates Epac (8-Me-cAMP, 50 µM) but not with a PKA-selective cAMP agonist (N6-cAMP, 50 µM) blunts the increase in α-SMA (Figures 3 and 4). By contrast, both 8-Me-cAMP and N6-cAMP attenuate the TGF-β-promoted inhibition of E-cadherin expression. Thus, inhibition of aspects of EMT by cAMP in MDCK cells involves both Epac and PKA.
Figure 3.
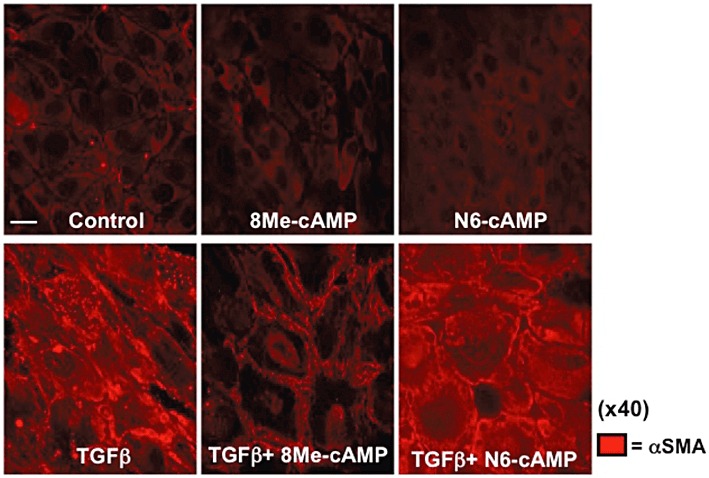
Activation of Epac, but not PKA, attenuates the TGF-β-induced increase in α-SMA in MDCK cells. Immunocytochemistry demonstrates that Epac activation (8Me-cAMP, 50 µM, 72 h), but not PKA activation (N6-cAMP, 50 µM, 72 h) inhibits the increase in α-SMA (stained red) induced by TGF-β1 (5ng·mL−1, 72 h), 40× objective. Data are representative of three separate experiments. Bar equals 10 µm.
Figure 4.
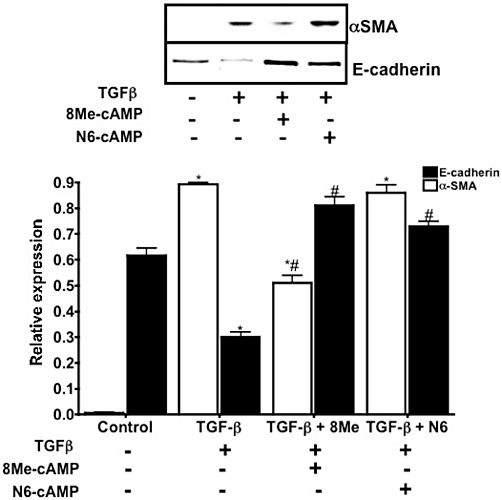
Activation of either Epac or PKA reverses characteristics of EMT in MDCK cells. Representative immunoblot reveals that MDCK cells treated with TGF-β1 (5ng·mL−1, 72 h) show changes associated with EMT (increased α-SMA and loss of E-cadherin). Epac activation (8Me-cAMP, 50 µM, 72 h) inhibits the increase in α-SMA and the decrease in E-cadherin, whereas PKA activation (N6-cAMP, 50 µM, 72 h) only prevents the decrease in E-cadherin. Lower panel, immunoblots were quantified and normalized to GAPDH expression. n= 3; *P < 0.05 versus control; #P < 0.05, versus TGF-β.
Fibroblast-myofibroblast transformation
Studies with adult rat cardiac fibroblasts (CF) reveal that increasing cAMP formation by the overexpression of AC6 or by activating AC with forskolin blunts collagen synthesis, TGF-β- or angiotensin II-stimulated increase in α-SMA expression, actin/focal adhesion assembly and, in parallel, the transformation of fibroblasts to myofibroblasts (Swaney et al., 2005). Experiments conducted with N6-cAMP and 8-Me-cAMP indicate that activation of either PKA or Epac inhibit expression of collagens I and III; by contrast, Epac and PKA have opposing effects on CF migration (Yokoyama et al., 2008). Such data are consistent with results observed with other fibroblasts, such as those from the lung (Huang et al., 2008).
Regulation of Epac expression by profibrotic agents
Our studies with MDCK cells (Figure 5) and primary cultures of rat cardiac fibroblasts (Yokoyama et al., 2008) reveal that treatment with profibrotic agents decreases the expression of Epac, results confirmed by others (Basoni et al., 2005). Prevention of this decrease in Epac expression inhibits profibrotic response (Yokoyama et al., 2008), thus implying the importance of the decrease in Epac for this response.
Figure 5.
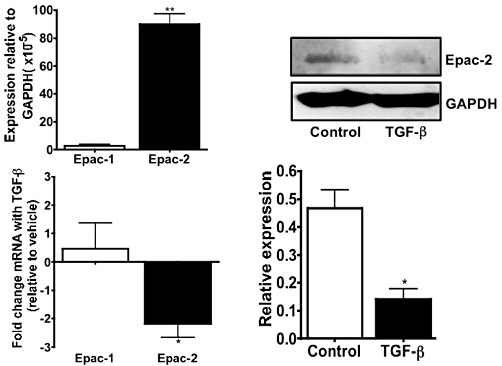
Epac2 expression is markedly decreased in MDCK cells by TGF-β. Upper left panel, Epac-1 and Epac2 mRNA expression in MDCK cells (by real-time PCR, results normalized to GAPDH) shows that Epac2 is expressed at higher levels than is Epac1 n= 3; **P < 0.001 compared with Epac1. Lower left panel, treatment with 5 ng·mL−1 TGFβ1 for 72 h decreases Epac2 mRNA expression (by real-time PCR) n= 3; *P < 0.05 versus vehicle. The data are expressed as fold change relative to vehicle-treated MDCKs. Upper right panel, a representative immunoblot shows that Epac2 protein expression is decreased in MDCK cells treated with 5 ng·mL−1 TGF-β1 for 72 h. Lower right panel, immunoblots were quantified and normalized to GAPDH expression. n= 3; *P < 0.05, versus vehicle.
Treatment of MDCK cells with profibrotic agents decreases expression of Epac2, which is expressed at much higher levels than Epac1 in these cells (Figure 5), but in fibroblasts from multiple tissues, profibrotic agents only decrease expression of Epac1 (Yokoyama et al., 2008). Thus, profibrotic agents can down-regulate expression of both isoforms of Epac and Epac-mediated signalling, such as activation of Rap-1, but alter expression of different isoforms in different tissues.
The roles of cAMP and Epac in tissue fibrosis and therapeutic implications
The information provided above implies that therapeutic approaches that increase cAMP or that activate Epac or increase its expression have the potential to decrease tissue fibrosis (Figure 6). Although PKA was initially thought to be the exclusive mediator of cAMP action (Insel et al., 1975), Epac mediates numerous effects of cAMP (Gloerich and Bos, 2010; Grandoch et al., 2010; Breckler et al., 2011). One such effect is the inhibition of EMT, although both PKA and Epac regulate features characteristic of EMT in MDCK cells (Figures 3 and 4). The contribution of EMT to tissue fibrosis in vivo is controversial (Hinz, 2010; Humphreys et al., 2010; Kriz et al., 2011; Quaggin and Kapus, 2011). Even so, the activation of Epac, potentially either Epac1 or Epac2 in different tissues, can regulate profibrotic responses, including collagen and DNA synthesis and other functional activities of fibroblasts. Data in rat cardiac, lung and skin fibroblasts show Epac1 is preferentially decreased in response to profibrogenic agonists; however, Epac2 is decreased by the same stimuli in MDCK cells (Figure 5; Yokoyama et al., 2008). Other data implicate increases in cAMP as a means to inhibit EMT (Zhang et al., 2006a,b; Kim et al., 2009; Kolosionek et al., 2009) and of PKA-mediated phosphorylation to influence proteins that regulate EMT (MacPherson et al., 2010).
Figure 6.
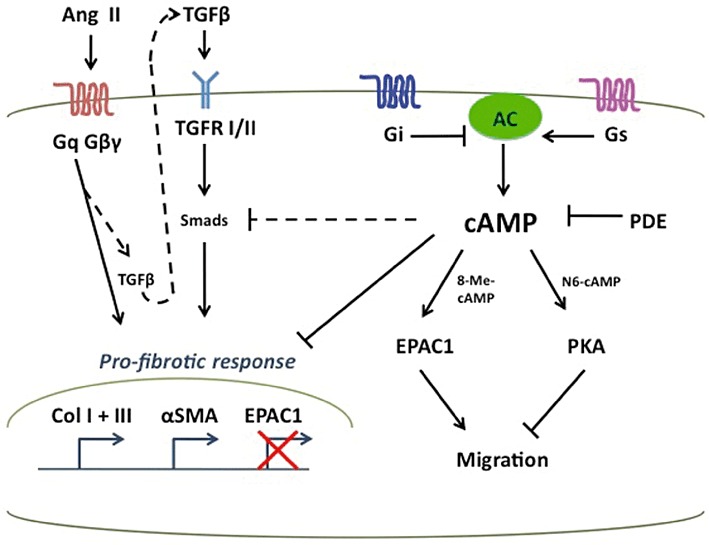
Model of the antifibrotic effects of cAMP. After injury, profibrotic stimuli (e.g. Ang II and TGF-β1) activate fibroblasts, increase ECM synthesis and deposition (e.g. collagens I and III), and regulate expression of fibrotic genes (e.g. α-SMA). Pharmacological agents that increase intracellular cAMP (Gs-linked GPCR agonists, Gi-linked GPCR antagonists, AC activators, or PDE inhibitors) can inhibit the profibrotic effects of Ang II and TGF-β1, potentially through SMAD inhibition. cAMP activation of Epac1 and PKA lowers expression of collagens I and III but has opposite effects on cell migration.
Recent information emphasizes the contribution of various transcription factors (e.g. Snail, Slug, Twist, etc.) to EMT and mechanisms that regulate their expression. EMT is thought to play an important role in pathophysiology in cancer and cancer cell metastases, with EMT-associated features representing signals expressed after the formation of primary tumours (Chaffer and Weinberg, 2011). The contribution of EMT to tissue fibrosis is controversial in a number of other settings, in part because not all features of the ‘EMT phenotype’ are consistently observed, especially between in vitro and in vivo models. A ‘two-hit’ model has been proposed that involves (1) disruption of intercellular contact between epithelial cells and (2) promotion of fibrotic features by TGF-β (Masszi et al., 2010). A consequence of the disruption of intercellular contact is the nuclear translocation of myocardin-related transcription factor (MRTF), which binds to a region in the α-SMA gene promoter. This promoter region contains TGF-β-responsive Smad3-binding elements. Smad3 has been proposed to be a critical timer/delayer of development of EMT and to differentially affect various components of EMT (Masszi et al., 2010; Masszi and Kapus, 2011). It will thus be of interest to determine the impact of cAMP, Epac and PKA on these recently recognized regulators of EMT. Of note, Smad3 and the regulatory subunits of PKA have been shown to coimmunoprecipitate and cAMP via PKA inhibits SMAD-mediated transcription (Schiller et al., 2003; Yang et al., 2008).
The ability of profibrotic stimuli to inhibit expression of Epac likely contributes to cardiac fibrosis because overexpression of Epac blocks profibrotic response and Epac expression decreases at sites that have fibrotic activity (Figure 5; Yokoyama et al., 2008). The mechanisms that regulate Epac expression and inhibition of its expression by profibrotic stimuli remain to be determined; little is known regarding the transcriptional and post-transcriptional regulation of Epac expression (Basoni et al., 2005; Brown et al., 2011). Epac also has other roles, including altering cell migration, that may relate to its actions in fibrosis (Yokoyama et al., 2008; Grandoch et al., 2010; Breckler et al., 2011; Mironov et al., 2011; Stokman et al., 2011). siRNA-mediated inhibition of Epac1 (but not Epac2) in rat cardiac fibroblasts and its downstream mediator Rap-1 decreases cAMP-induced migration of cardiac fibroblasts, implying a role for Epac1 signalling via Rap-1 in the antifibrotic actions of cAMP (Yokoyama et al., 2008). In contrast, Rap1-targeted siRNA did not affect Epac-induced inhibition of collagen and DNA synthesis, thus suggesting a role for other downstream mediators of Epac1 and requires further investigation.
The ability of cAMP acting via Epac to exert antifibrotic effects and of profibrotic agents to inhibit Epac expression suggests that strategies to increase Epac expression and activity may have therapeutic potential in preventing or treating tissue fibrosis. However, since Epac regulates numerous effects of cAMP (Gloerich and Bos, 2010; Grandoch et al., 2010; Breckler et al., 2011), it may be difficult to selectively alter Epac expression and actions in fibroblasts. This problem is heightened by the lack of pharmacological inhibitors of Epac and the difficulty in achieving fibroblast-specific targeting of genes (Liu et al., 2010; Österreicher et al., 2011).
Might other approaches be used to increase cAMP and activate Epac in fibroblasts? One possibility is to take advantage of fibroblast-selective expression of signalling components involved in regulating cAMP levels, for example, by activating GPCRs that increase cAMP formation, inhibiting GPCRs that decrease cAMP formation or inhibiting PDEs that hydrolyse cAMP. Results showing that an AC6 adenovirus or fibroblast-targeted AC6 enhances GPCR-promoted cAMP formation and antifibrotic activity (Swaney et al., 2005; Liu et al., 2010) imply that efforts to enhance cAMP levels in fibroblasts may have antifibrotic effects in vivo. Preliminary data indicate that fibroblasts selectively express certain GPCRs that regulate cAMP formation (Insel et al., 2012a; the 2009 BJP Guide to Receptors and Channels, which is available for free download at http://www3.interscience.wiley.com/journal/122684220/issue) and particular isoforms of cAMP–PDE (data not shown) that might be targeted as ways to prevent, blunt or perhaps even reverse fibrosis. Previous data have shown that targeting cAMP-PDEs, in particular PDE4 and PDE1, by family-specific inhibitors and siRNA can inhibit fibroblast activation, EMT and collagen synthesis (Dunkern et al., 2007; Kolosionek et al., 2009; Sachs et al., 2007; Miller et al., 2011). Approaches that increase cAMP levels, such as GPCRs and/or PDE inhibitors in fibroblasts, perhaps combined with Epac or PKA activators, may be novel strategies for the treatment of tissue fibrosis (Leask, 2010; Kisseleva and Brenner, 2011). The GPCR/cAMP/Epac pathway offers intriguing possibilities for new pharmacological approaches directed at achieving antifibrosis in vivo.
Acknowledgments
We thank Dr Richard Bundey for initial studies in our laboratory on this topic and Mary Alice Kiisel for assistance in preparation of the manuscript. Work described here was supported by grants from the National Institutes of Health and the Ellison Medical Foundation.
Glossary
- CF
cardiac fibroblast
- ECM
extracellular matrix
- Epac
Exchange protein activated by cyclic AMP
- 8-Me-cAMP
8-CPT-2′-O-Me-cAMP
- N6-cAMP
N6-Benzoyl-cAMP
- NSAIDs
non-steroidal anti-inflammatory drugs
- α-SMA
α-smooth muscle actin
Conflict of interest
None.
References
- Armulik A, Genové G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21:193–215. doi: 10.1016/j.devcel.2011.07.001. [DOI] [PubMed] [Google Scholar]
- Basoni C, Nobles M, Grimshaw A, Desgranges C, Davies D, Perretti M, et al. Inhibitory control of TGF-beta1 on the activation of Rap1, CD11b, and transendothelial migration of leukocytes. FASEB J. 2005;19:822–824. doi: 10.1096/fj.04-3085fje. [DOI] [PubMed] [Google Scholar]
- Bauman KA, Wettlaufer SH, Okunishi K, Vannella KM, Stoolman JS, Huang SK, et al. The antifibrotic effects of plasminogen activation occur via prostaglandin E2 synthesis in humans and mice. J Clin Invest. 2010;120:1950–1960. doi: 10.1172/JCI38369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black SA, Jr, Palamakumbura AH, Stan M, Trackman PC. Tissue-specific mechanisms for CCN2/CTGF persistent in fibrotic gingiva: interactions between cAMP and MAPK signaling pathways and prostaglandin E2-EP3 receptor mediated activation of the c-JUN N-terminal kinase. J Biol Chem. 2007;282:15416–15429. doi: 10.1074/jbc.M610432200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos JL. Epac proteins: multi-purpose cAMP targets. Trends Biochem Sci. 2006;31:680–686. doi: 10.1016/j.tibs.2006.10.002. [DOI] [PubMed] [Google Scholar]
- Brack AS, Conboy MJ, Roy S, Lee M, Kuo CJ, Keller C, et al. Increased Wnt signaling during agin alters muscle stem cell fate and increases fibrosis. Science. 2007;317:807–810. doi: 10.1126/science.1144090. [DOI] [PubMed] [Google Scholar]
- Breckler M, Berthouze M, Laurent AC, Crozatier B, Morel E, Lezoualc'h F. Rap-linked cAMP signaling Epac proteins: compartmentation, functioning and disease implications. Cell Signal. 2011;23:1257–1266. doi: 10.1016/j.cellsig.2011.03.007. [DOI] [PubMed] [Google Scholar]
- Brown L, Murray F, Zang L, Kipps T, Insel PA. Epacl, an anti-apoptotic protein, is up-regulated in chronic lymphocytic leukemia B cells. FASEB J. 2011;25:1090.1. [Google Scholar]
- Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331:1559–1564. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- Chan EC, Dusting GJ, Guo N, Peshavariya HM, Taylor CJ, Dilley R, et al. Prostacyclin receptor suppresses cardiac fibrosis: role of CREB phosphorylation. J Mol Cell Cardiol. 2010;49:176–185. doi: 10.1016/j.yjmcc.2010.04.006. [DOI] [PubMed] [Google Scholar]
- Chen W, Frangogiannis NG. The role of inflammatory and fibrogenic pathways in heart failure associated with aging. Heart Fail Rev. 2010;5:415–422. doi: 10.1007/s10741-010-9161-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho ME, Kopp JB. Pirfenidone: an anti-fibrotic therapy for progressive kidney disease. Expert Opin Investig Drugs. 2010;19:275–283. doi: 10.1517/13543780903501539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy RM, Zheng P, O'Mahony M, Izmirly P, Zavadil J, Gardner L, et al. Role of hypoxia and cAMP in the transdifferentiation of human fetal cardiac fibroblasts: implications for progression to scarring in autoimmune-associated congenital heart block. Arthritis Rheum. 2007;56:4120–4131. doi: 10.1002/art.23061. [DOI] [PubMed] [Google Scholar]
- Cufí S, Vazquez-Martin A, Oliveras-Ferraros C, Martin-Castillo B, Joven J, Menendez JA. Metformin against TGF-β-induced epithelial-to-mesenchymal transition (EMT): from cancer stem cells to aging-induced fibrosis. Cell Cycle. 2010;9:4461–4468. doi: 10.4161/cc.9.22.14048. [DOI] [PubMed] [Google Scholar]
- D'Souza KM, Malhotra R, Philip JL, Staron ML, Theccanat T, Jeevanandam V, et al. G protein-coupled receptor kinase-2 is a novel regulator of collagen synthesis in adult human cardiac fibroblasts. J Biol Chem. 2011;286:15507–15516. doi: 10.1074/jbc.M111.218263. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Dunkern TR, Feurstein D, Rossi GA, Sabatini F, Hatzelmann A. Inhibition of TGF-beta induced lung fibroblast to myofibroblast conversion by phosphodiesterase inhibiting drugs and activators of soluble guanylyl cyclase. Eur J Pharmacol. 2007;572:12–22. doi: 10.1016/j.ejphar.2007.06.036. [DOI] [PubMed] [Google Scholar]
- Dussaule JC, Guerrot D, Huby AC, Chadjichristos C, Shweke N, Boffa J, et al. The role of cell plasticity in progression and reversal of renal fibrosis. Int J Exp Pathol. 2011;92:151–157. doi: 10.1111/j.1365-2613.2011.00760.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloerich M, Bos JL. Epac: defining a new mechanism for cAMP action. Annu Rev Pharmacol Toxicol. 2010;50:355–375. doi: 10.1146/annurev.pharmtox.010909.105714. [DOI] [PubMed] [Google Scholar]
- Grandoch M, Roscioni SS, Schmidt M. The role of Epac proteins, novel cAMP mediators, in the regulation of immune, lung and neuronal function. Br J Pharmacol. 2010;159:265–284. doi: 10.1111/j.1476-5381.2009.00458.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieb G, Steffens G, Pallua N, Bernhagen J, Bucala R. Circulating fibrobcytes-biology and mechanisms in wound healing and scar formation. Int Rev Cell Mol Biol. 2011;291:1–19. doi: 10.1016/B978-0-12-386035-4.00001-X. [DOI] [PubMed] [Google Scholar]
- Heusinger-Ribeiro J, Eberlein M, Wahab NA, Goppelt-Struebe M. Expression of connective tissue growth factor in human renal fibroblasts: regulatory roles of RhoA and cAMP. J Am Soc Nephrol. 2001;12:1853–1861. doi: 10.1681/ASN.V1291853. [DOI] [PubMed] [Google Scholar]
- Hinz B. The myofibroblast: paradigm for a mechanically active cell. J Biomech. 2010;43:146–155. doi: 10.1016/j.jbiomech.2009.09.020. [DOI] [PubMed] [Google Scholar]
- Holz GG, Chepurny OG, Schwede F. Epac-selective cAMP analogs: new tools with which to evaluate the signal transduction properties of cAMP-regulated guanine nucleotide exchange factors. Cell Signal. 2008;20:10–20. doi: 10.1016/j.cellsig.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SK, Wettlaufer SH, Chung J, Peters-Golden M. Prostaglandin E2 inhibits specific lung fibroblast functions via selective action of PKA and Epac-1. Am J Respir Cell Mol Biol. 2008;39:482–489. doi: 10.1165/rcmb.2008-0080OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SK, White ES, Wettlaufer SH, Grifka H, Hogaboam CM, Thannickal VJ, et al. Prostaglandin E(2) induces fibroblast apoptosis by modulating multiple survival pathways. FASEB J. 2009;23:4317–4326. doi: 10.1096/fj.08-128801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphreys BD, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, et al. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol. 2010;176:85–97. doi: 10.2353/ajpath.2010.090517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel PA, Bourne HR, Coffino P, Tomkins GM. Cyclic AMP-dependent protein kinase: pivotal role in regulation of enzyme induction and growth. Science. 1975;190:896–898. doi: 10.1126/science.171770. [DOI] [PubMed] [Google Scholar]
- Insel PA, Snead A, Murray F, Zhang L, Yokouchi H, Katakia T, et al. GPCR expression in tissues and cells: are the optimal receptors being used as drug targets? Br J Pharmacol. 2012a;165:1613–1616. doi: 10.1111/j.1476-5381.2011.01434.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel PA, Zhang L, Murray F, Yokoushi H, Zambon AC. Cyclic AMP is both a pro-apoptotic and anti-apoptotic second messenger. Acta Physiol (Oxf) 2012b;204:277–287. doi: 10.1111/j.1748-1716.2011.02273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim A, Son M, Kim KI, Yang Y, Song EY, Lee HG, et al. Elevation of intracellular cyclic AMP inhibits NF-κB-mediated thymosin β4 expression in melanoma cells. Exp Cell Res. 2009;315:3325–3335. doi: 10.1016/j.yexcr.2009.05.024. [DOI] [PubMed] [Google Scholar]
- Kisseleva T, Brenner DA. Anti-fibrogenic strategies and the regression of fibrosis. Best Pract Res Clin Gastroenterol. 2011;25:305–317. doi: 10.1016/j.bpg.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klijn K, Slivka SR, Bell K, Insel PA. Renal alpha1-adrenergic receptor subtypes: MDCK-D1 cell, but not rat cortical membranes, possess a single population of receptors. Mol Pharmacol. 1991;39:407–413. [PubMed] [Google Scholar]
- Kohyama T, Yamauchi Y, Takizawa H, Itakura S, Kamitani S, Desaki M, et al. Procaterol inhibits lung fibroblast migration. Inflammation. 2009;32:387–392. doi: 10.1007/s10753-009-9147-x. [DOI] [PubMed] [Google Scholar]
- Kolosionek E, Savai R, Ghofrani HA, Weissmann N, Guenther A, Grimminger F, et al. Expression and activity of phosphodiesterase isoforms during epithelial mesenchymal transition: the role of phosphodiesterase 4. Mol Biol Cell. 2009;20:4751–4765. doi: 10.1091/mbc.E09-01-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kothapalli D, Hayashi N, Grotendorst GR. Inhibition of TGF-beta-stimulated CTGF gene expression and anchorage-independent growth by cAMP identifies a CTGF-dependent restriction point in the cell cycle. FASEB J. 1998;12:1151–1161. doi: 10.1096/fasebj.12.12.1151. [DOI] [PubMed] [Google Scholar]
- Krenning G, Zeisberg EM, Kalluri R. The origin of fibroblasts and mechanism of cardiac fibrosis. J Cell Physiol. 2010;225:631–637. doi: 10.1002/jcp.22322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriz W, Kaissling B, Le Hir M. Epithelial-mesenchymal transition (EMT) in kidney fibrosis: fact or fantasy. J Clin Invest. 2011;121:468–474. doi: 10.1172/JCI44595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakatta EG. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part II: cellular and molecular clues to heart and arterial aging. Circulation. 2003;107:490–497. doi: 10.1161/01.cir.0000048894.99865.02. [DOI] [PubMed] [Google Scholar]
- Leask A. Potential therapeutic targets for cardiac fibrosis: TGFβ, angiotensin, endothelin, CCN2 and PDGF, partners in fibroblast activation. Circ Res. 2010;106:1675–1680. doi: 10.1161/CIRCRESAHA.110.217737. [DOI] [PubMed] [Google Scholar]
- Li Y, Zhu X, Zeng Y, Wang J, Zhang X, Ding YQ, et al. FML2 enhances invation of colorectal carcinoma by inducing epithelial-mesenchymal transition. Mol Cancer Res. 2010;8:1579–1590. doi: 10.1158/1541-7786.MCR-10-0081. [DOI] [PubMed] [Google Scholar]
- Liu X, Ostrom RS, Insel PA. cAMP-elevating agents and adenylyl cyclase overexpression promote an antifibrotic phenotype in pulmonary fibroblasts. Am J Physiol Cell Physiol. 2004;286:C1089–C1099. doi: 10.1152/ajpcell.00461.2003. [DOI] [PubMed] [Google Scholar]
- Liu X, Sun SQ, Ostrom RS. Fibrotic lung fibroblasts show blunted inhibitiion by cAMP due to deficient cAMP response element-binding protein phosphorylation. J Pharmacol Exp Ther. 2005;315:678–687. doi: 10.1124/jpet.105.090324. [DOI] [PubMed] [Google Scholar]
- Liu X, Sun SQ, Hassid A, Ostrom RS. cAMP inhibits transforming growth factor-beta-stimulated collagen synthesis via inhbition of extracellular signal-regulated kinase 1/2 and Smad signaling in cardiac fibroblasts. Mol Pharmacol. 2006;70:1992–2003. doi: 10.1124/mol.106.028951. [DOI] [PubMed] [Google Scholar]
- Liu X, Thangavel M, Sun SQ, Kaminsky J, Mahautmr P, Stitham J, et al. Adenylyl cyclase type 6 overexpression selectively enhances beta-adrenergic and prostacyclin receptor-mediated inhibition of cardiac fibroblast function because of colocalization in lipid rafts. Naunyn Schmiedebergs Arch Pharmacol. 2008;377:359–369. doi: 10.1007/s00210-007-0196-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Li F, Sun SQ, Thangavel M, Kaminsky J, Balazs L, et al. Fibroblast-specific expression of AC6 enhances beta-adrenergic and prostacyclin signaling and blunts bleomycin-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2010;298:L819–L829. doi: 10.1152/ajplung.00429.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacPherson MR, Molina P, Souchelnytskyi S, Wernstedt C, Martin-Pérez J, Portillo F, et al. Phosphorylation of serine 11 and serine 92 as new positive regulators of human Snail1 function: potential involvement of casein kinase-2 and the cAMP-activated kinase protein kinase A. Mol Biol Cell. 2010;21:244–253. doi: 10.1091/mbc.E09-06-0504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marian AJ. Experimental therapies in hypertrophic cardiomyopathy. J Cardiovasc Transl Res. 2009;2:483–492. doi: 10.1007/s12265-009-9132-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maskrey BH, Megson IL, Whitfield PD, Rossi AG. Mechanisms of resolution of inflammation: a focus on cardiovascular diseas. Arterioscler Thromb Vasc Biol. 2011;31:1001–1006. doi: 10.1161/ATVBAHA.110.213850. [DOI] [PubMed] [Google Scholar]
- Masszi A, Kapus A. Smaddening complexity: the role of Smad3 in epithelial-myofibroblast transition. Cells Tissues Organs. 2011;193:41–52. doi: 10.1159/000320180. [DOI] [PubMed] [Google Scholar]
- Masszi A, Speight P, Charbonney E, Ladyga M, Nakano H, Sz?szi K, et al. Fate-determining mechanisms in epithelial-myofibroblast transition: major inhibitory role for Smad3. J Cell Biol. 2010;188:383–399. doi: 10.1083/jcb.200906155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier KE, Snavely MD, Brown SL, Brown JH, Insel PA. alpha 1- and beta 2-adrenergic receptor expression in the Madin-Darby canine kidney epithelial cell line. J Cell Biol. 1983;97:405–415. doi: 10.1083/jcb.97.2.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meran S, Steadman R. Fibroblasts and myofibroblasts in renal fibrosis. Int J Exp Pathol. 2011;92:158–167. doi: 10.1111/j.1365-2613.2011.00764.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CL, Cai Y, Oikawa M, Thomas T, Dostmann WR, Zaccolo M, et al. Cyclic nucleotide phosphodiesterase 1A: a key regulator of cardiac fibroblast activation and extracellular matrix remodeling in the heart. Basic Res Cardiol. 2011;106:1023–1039. doi: 10.1007/s00395-011-0228-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironov SL, Skorova EY, Kügler S. Epac-mediated cAMP signaling in the mouse model of Rett Syndrome. Neuropharmacology. 2011;60:869–877. doi: 10.1016/j.neuropharm.2011.01.002. [DOI] [PubMed] [Google Scholar]
- Okunishi K, Sisson TH, Huang SK, Hogaboam CM, Simon RH, Peters-Golden M. Plasmin overcomes resistance to prostaglandin E2 in fibrotic lung fibroblasts by reorganizing protein kinase A signaling. J Biol Chem. 2011;286:32231–32243. doi: 10.1074/jbc.M111.235606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Österreicher CH, Penz-Österreicher M, Grivennikov SI, Guma M, Koltsova EK, Datz C, et al. Fibroblast-specific protein 1 identifies an inflammatory subpopulation of macrophages in the liver. Proc Natl Acad Sci U S A. 2011;108:308–313. doi: 10.1073/pnas.1017547108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh A, Sandulache VC, Lieb AS, Dohar JE, Hebda PA. Differential regulation of free-floating collagen gel contraction by human fetal and adult dermal fibroblasts in response to prostaglandin E2 mediated by EP2/cAMP-dependent mechanism. Wound Repair Regen. 2007;15:390–398. doi: 10.1111/j.1524-475X.2007.00241.x. [DOI] [PubMed] [Google Scholar]
- Parekh A, Sandulache VC, Singh T, Cetin S, Sacks MS, Dohar JE, et al. Prostaglandin E2 differentially regulates contraction and structural reorganization of anchored collagen gels by human adult and fetal dermal fibroblasts. Wound Repair Regen. 2009;17:88–98. doi: 10.1111/j.1524-475X.2008.00445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SH, Choi MJ, Song IK, Choi SY, Nam JO, Kim CD, et al. Erythropoietin decreases renal fibrosis in mice with uretral obstruction: role of inhibiting TGF-beta-induced epithelial-to-mesenchymal transition. J Am Soc Nephrol. 2007;18:1497–1507. doi: 10.1681/ASN.2005080866. [DOI] [PubMed] [Google Scholar]
- Pinzani M, Macias-Barragan J. Update on the pathophysiology of liver fibrosis. Expert Rev Gastroenterol Hepatol. 2010;4:459–472. doi: 10.1586/egh.10.47. [DOI] [PubMed] [Google Scholar]
- Quaggin SE, Kapus A. Scar wars: mapping the fate of epithelial-mesenchymal-myofibroblast transition. Kidney Int. 2011;80:41–50. doi: 10.1038/ki.2011.77. [DOI] [PubMed] [Google Scholar]
- Ramachandran P, Iredale JP. Reversibility of liver fibrosis. Ann Hepatol. 2009;8:283–291. [PubMed] [Google Scholar]
- Sachs BD, Baillie GS, McCall JR, Passino MA, Schachtrup C, Wallace DA, et al. P75 neurotrophin receptor regulates tissue fibrosis through inhibition of plasminogen activation via a PDE4/cAMP/PKA pathway. J Cell Biol. 2007;177:1119–1132. doi: 10.1083/jcb.200701040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandbo N, Kregel S, Taurin S, Bhorade S, Dulin NO. Critical role of serum response factor in pulmonary myofibroblast differentiation induced by TGF-beta. Am J Respir Cell Mol Biol. 2009;41:332–338. doi: 10.1165/rcmb.2008-0288OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandulache VC, Parekh A, Li-Korotky HS, Dohar JE. Prostaglandin E2 differentially modulates human fetal and adult dermal fibroblast migration and contraction: implication for wound healing. Wound Repair Regen. 2006;14:633–643. doi: 10.1111/j.1743-6109.2006.00156.x. [DOI] [PubMed] [Google Scholar]
- Sandulache VC, Parekh A, Li-Korotky H, Dohar JE, Hebda PA. Prostaglandin E2 inhibition of keloid fibroblast migration, contraction and transforming growth factor (TGF)-beta1-induced collagen synthesis. Wound Repair Regen. 2007;15:122–133. doi: 10.1111/j.1524-475X.2006.00193.x. [DOI] [PubMed] [Google Scholar]
- Schiller M, Verrecchia F, Mauviel A. Cyclic adenosine 3′,5′-monophosphate-elevating agents inhibit transforming growth factor-beta-induced SMAD3/4-dependent transcription via a protein kinase A-dependent mechanism. Oncogene. 2003;22:8881–8890. doi: 10.1038/sj.onc.1206871. [DOI] [PubMed] [Google Scholar]
- Schrimpf C, Duffield JS. Mechanisms of fibrosis: the role of the pericyte. Curr Opin Nephrol Hypertens. 2011;20:397–405. doi: 10.1097/MNH.0b013e328344c3d4. [DOI] [PubMed] [Google Scholar]
- Selige J, Tenor H, Hatzelmann A, Dunkern T. Cytokine-dependent balance of mitogenic effects in primary human lung fibroblasts related to cyclic AMP signaling and phosphodiesterase 4 inhibition. J Cell Physiol. 2010;223:317–326. doi: 10.1002/jcp.22037. [DOI] [PubMed] [Google Scholar]
- Serhan CN. Novel lipid mediators and resolution mechanisms in acute inflammation: to resolve or not? Am J Pathol. 2010;177:1576–1591. doi: 10.2353/ajpath.2010.100322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokman G, Qin Y, Genieser HG, Schwede F, de Heer E, Bos JL, et al. Epac-Rap signaling reduces cellular stress and ischemia-induced kidney failure. J Am Soc Nephrol. 2011;22:859–872. doi: 10.1681/ASN.2010040423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumm CL, Wettlaufer SH, Jancar S, Peters-Golden M. Airway remodeling in murine astham correlates with a defect in PGE2 synthesis by lung fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2011;301:L636–L644. doi: 10.1152/ajplung.00158.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaney JS, Roth DM, Olson ER, Naugle JE, Meszaros JG, Insel PA. Inhibition of cardiac myofibroblast formation and collagen synthesis by activation and overexpression of adenylyl cyclase. Proc Natl Acad Sci U S A. 2005;102:437–442. doi: 10.1073/pnas.0408704102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togo S, Liu X, Wang X, Sugiura H, Kamio K, Kawasaki S, et al. PDE4 inhibitors roflumilast and rolipram augment PGE1 inhibition of TGF beta1-stimulated fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2009;296:L959–L969. doi: 10.1152/ajplung.00508.2007. [DOI] [PubMed] [Google Scholar]
- Wallace WA, Fitch PM, Simpson AJ, Howie SE. Inflamation-associated remodeling and fibrosis in the lung- a process and an end point. Int J Exp Pathol. 2007;88:103–110. doi: 10.1111/j.1365-2613.2006.00515.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windmeier C, Gressner AM. Pharmacological aspects of pentoxifylline with emphasis on its inhibitory actions on hepatic fibrogenesis. Gen Pharmacol. 1997;29:181–196. doi: 10.1016/s0306-3623(96)00314-x. [DOI] [PubMed] [Google Scholar]
- Wynn TA. Integrating mechanisms of pulmonary fibrosis. J Exp Med. 2011;208:1339–1350. doi: 10.1084/jem.20110551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing D, Bonanno JA. Effect of cAMP on TGF1-induced corneal keratocyte-myofibroblast transformation. Invest Ophthalmol Vis Sci. 2009;50:626–633. doi: 10.1167/iovs.08-2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Lee CJ, Zhang L, Sans MD, Simeone DM. Regulation of transforming growth factor beta-induced responses by protein kinase A in pancreatic acinar cells. Am J Physiol Gastrointest Liver Physiol. 2008;295:G170–G178. doi: 10.1152/ajpgi.00492.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates CC, Bodnar R, Wells A. Matrix control of scarring. Cell Mol Life Sci. 2011;68:1871–1881. doi: 10.1007/s00018-011-0663-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama U, Patel HH, Lai NC, Aroonsakool N, Roth DM, Insel PA. The cyclic AMP effector Epac integrates pro-and anti-fibrotic signals. Proc Natl Acad Sci U S A. 2008;105:6386–6391. doi: 10.1073/pnas.0801490105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Prado GN, Schreiber B, Polgar P, Polgar P, Taylor L. Role of prostaglandin E(2) EP receptors and cAMP in the expression of connective tissue growth factor. Arch Biochem Biophys. 2002;404:302–308. doi: 10.1016/s0003-9861(02)00276-x. [DOI] [PubMed] [Google Scholar]
- Zhang A, Dong Z, Yang T. Prostaglandin D2 inhibits TGF-beta1-induced epithelial-to-mesenchymal transition in MDCK cells. Am J Physiol Renal Physiol. 2006a;291:F1332–F1342. doi: 10.1152/ajprenal.00131.2006. [DOI] [PubMed] [Google Scholar]
- Zhang A, Wang MH, Dong Z, Yang T. Prostaglandin E2 is a potent inhibitor of epithelial-to-mesenchymal transition: interaction with hepatocyte growth factor. Am J Physiol Renal Physiol. 2006b;291:F1323–F1331. doi: 10.1152/ajprenal.00480.2005. [DOI] [PubMed] [Google Scholar]
- Zhang L, Yun H, Murray F, Lu R, Wang L, Hook V, et al. Cytotoxic T lymphocyte antigen-2 alpha induces apoptosis of murine T-lymphoma cells and cardiac fibroblasts and is regulated by cAMP/PKA. Cell Signal. 2011;23:1611–1616. doi: 10.1016/j.cellsig.2011.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]