

Abstract
GABAA receptors are ligand-gated chloride channels composed of five subunits that can belong to different subunit classes. The existence of 19 different subunits gives rise to a multiplicity of GABAA receptor subtypes with distinct subunit composition; regional, cellular and subcellular distribution; and pharmacology. Most of these receptors are composed of two α, two β and one γ2 subunits. GABAA receptors are the site of action of a variety of pharmacologically and clinically important drugs, such as benzodiazepines, barbiturates, neuroactive steroids, anaesthetics and convulsants. Whereas GABA acts at the two extracellular β+α− interfaces of GABAA receptors, the allosteric modulatory benzodiazepines interact with the extracellular α+γ2− interface. In contrast, barbiturates, neuroactive steroids and anaesthetics seem to interact with solvent accessible pockets in the transmembrane domain. Several benzodiazepine site ligands have been identified that selectively interact with GABAA receptor subtypes containing α2βγ2, α3βγ2 or α5βγ2 subunits. This indicates that the different α subunit types present in these receptors convey sufficient structural differences to the benzodiazepine binding site to allow specific interaction with certain benzodiazepine site ligands. Recently, a novel drug binding site was identified at the α+β− interface. This binding site is homologous to the benzodiazepine binding site at the α+γ2− interface and is thus also strongly influenced by the type of α subunit present in the receptor. Drugs interacting with this binding site cannot directly activate but only allosterically modulate GABAA receptors. The possible importance of such drugs addressing a spectrum of receptor subtypes completely different from that of benzodiazepines is discussed.
Keywords: GABAA receptors, structure, pharmacology, receptor subtypes, allosteric binding sites, benzodiazepines, barbiturates, steroids, anesthetics, CGS 9895
GABAA receptors are the major inhibitory transmitter receptors in the brain. They are ligand-gated chloride channels composed of five subunits that can belong to different subunit classes. The existence of six α, three β, three γ, one δ, one ε, one θ, one π and three ρ subunits gives rise to an enormous diversity of GABAA receptor subtypes with different subunit composition and different pharmacological properties (Olsen and Sieghart, 2008; Sieghart, 1995). The majority of GABAA receptors, however, are composed of two α, two β and one γ subunit (Olsen and Sieghart, 2008). GABAA receptors are the site of action of a variety of pharmacologically and clinically important drugs such as benzodiazepines, barbiturates, neuroactive steroids, anaesthetics and convulsants, which allosterically modulate GABA-induced currents (Sieghart, 1995). Due to the action of these drugs, it is now clear that GABAA receptors are modulating anxiety, the excitability of the brain, muscle tonus, vigilance, circadian rhythms, learning and memory (Sieghart, 1995). Here, we are briefly reviewing what is known on the structure and pharmacology of GABAA receptors and their heterogeneity. We then shortly discuss some drugs that selectively interact with some of the GABAA receptor subtypes and finally describe the identification and importance of drugs mediating their effects via a novel drug binding site at the α+β− interface of GABAA receptors.
Structure and pharmacology of GABAA receptors
Binding sites for GABA and for some allosterically modulating drugs have already been identified on these receptors (Olsen and Sieghart, 2008). Thus, in the extracellular part of a GABAA receptor composed of 2α, 2β and one γ subunit, the two GABA binding sites are located at the two β+α− interfaces (Smith and Olsen, 1995), and the high-affinity benzodiazepine binding site is located at the α+γ− interface (Sigel and Buhr, 1997; Ernst et al., 2003) (Figure 1A). Interestingly, benzodiazepine site ligands, in contrast to GABA or GABA agonists, cannot directly activate GABAA receptors and thus only allosterically modulate GABA-induced currents. This might be explained by a less efficient transduction of benzodiazepine-induced conformational changes to the channel as compared with GABA, or by the existence of only a single high-affinity benzodiazepine binding site at the α+γ− interface, which alone is not able to directly activate the channel in the absence of GABA. For some benzodiazepines, additional binding sites have been described at GABAA receptors (Hauser et al., 1997; Walters et al., 2000; Hanchar et al., 2006; Baur et al., 2008), but in many cases, these benzodiazepine site ligands exhibit a quite low affinity for these additional binding sites, and no systematic study on a possible direct activation of GABAA receptor associated channels by such high concentrations of benzodiazepine site ligands has been performed so far.
Figure 1.
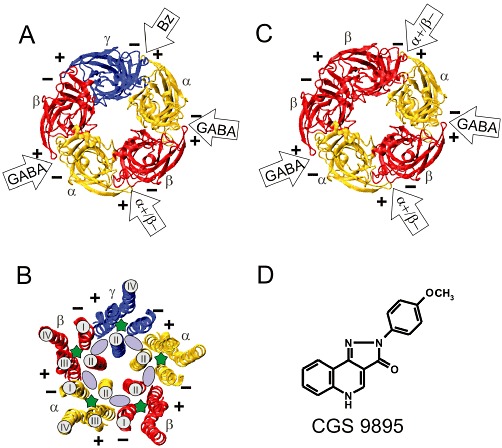
Model structures of the extracellular and transmembrane domains of GABAA receptors and chemical structure of CGS 9895. (A) Extracellular domain or (B) transmembrane domain of a GABAA receptor composed of one γ, two α and two β subunits. (C) Extracellular domain of a GABAA receptor composed of two α and three β subunits. (A, B, C) The structures are shown in ribbon representation and are viewed from the extracellular side. The plus (+) and the minus (−) side of each subunit is indicated. The two GABA sites are located at the β+α− interfaces, and the benzodiazepine (BZ) binding site is located at the α+γ− interface. (B) Solvent accessible space contained in the transmembrane domain of GABAA receptor models. The intra-subunit pockets are located within the four helix transmembrane domains and are marked with a green asterisk. Only helix II is marked in all subunits. Helices I–IV are only marked for a single β and an α subunit. The inter-subunit pockets are indicated by a light purple ellipse and are located between helices II and III of one subunit (+ side) and helices II and I of a neighbouring subunit (− side). (D) Chemical structure of CGS 9895.
In contrast to the allosterically modulating benzodiazepines, steroids, inhalation anaesthetics, i.v. anaesthetics or barbiturates exhibit two different actions. At low concentrations, they enhance GABA-induced currents, and at higher concentrations, they are able to directly elicit GABAA receptor-mediated currents in the absence of GABA (Sieghart, 1995). These compounds, thus, presumably interact with at least two binding sites at GABAA receptors. A recent modelling study indicated the existence of multiple solvent accessible pockets within the transmembrane domain of GABAA receptors (Ernst et al., 2005). Such pockets are present within each four helix bundle of the five GABAA receptor subunits (intra-subunit pockets, marked with a green asterisk in Figure 1B) as well as at the five interfaces between the four helix bundles of two neighbouring subunits (inter-subunit pockets, marked with a purple ellipse in Figure 1B). Mutagenesis studies indicated that neuroactive steroids might have at least two different binding sites within the transmembrane domain of GABAA receptors: one site seems to be located within the four helical transmembrane domains of α subunits mediating allosteric modulation of receptors at low steroid concentrations. Another binding site seems to be located at the β+α− interface below the GABA binding pocket in the transmembrane domain and mediates direct activation of GABAA receptors at high steroid concentrations (Hosie et al., 2006; 2009). Additional binding sites might exist for the inhibitory sulphated steroids (Hosie et al., 2007).
Other mutagenesis studies have indicated that inhalation anaesthetics might elicit their action by binding to the intra-subunit pocket of α subunits (Mihic et al., 1997; Jenkins et al., 2001; Yamakura et al., 2001), whereas i.v. anaesthetics might interact with the intra-subunit pocket of β subunits (Belelli et al., 1997; Krasowski et al., 2001; Bali and Akabas, 2004) or at the transmembrane interface of β and α subunits (Olsen and Sieghart, 2008; 2009). Mutagenesis studies in transmembrane domains, however, are difficult to interpret, because amino acid residues in the transmembrane domain apparently important for drug action might be either located close to the respective binding site or/and be important for the transduction of drug effects. In addition, in the absence of a crystal structure of GABAA receptors, an unequivocal assignment of amino acid residues to an intra- or inter-subunit pocket is not possible. A slightly different conformation of the transmembrane helix to which the respective amino acid residue contributes could result in its pointing into the intra-subunit or the inter-subunit pocket (Figure 1B). Finally, the exact movements and possible rotations of the transmembrane helices during opening of the channel or allosteric modulation of the receptor are not known, thus again making the assignment of amino acid residues to the one or the other pocket type difficult.
In any case, using a photo-incorporable etomidate derivative, a single type of inter-subunit binding pocket was identified for both the allosteric modulation as well as the direct opening of the channel by etomidate. This pocket seemed to be located within the transmembrane domain of GABAA receptors at the β+α− interface (Li et al., 2006). Photo-incorporation of etomidate at this interface seemed to be competitively inhibited by isoflurane (Li et al., 2010), suggesting that at least this inhalation anaesthetic (also) interacts with the β+α− interface. Photo-incorporation of etomidate, however, seemed to be allosterically inhibited by neurosteroids, barbiturates and propofol (for discussion, see Olsen and Li, 2011), indicating that these drugs do not bind to the transmembrane β+α− interface, or at least bind to a site not overlapping with the etomidate binding site. An allosteric interaction of etomidate and propofol seems to be supported also by another study (Richardson et al., 2007). In addition, a recent crystal structure from a bacterial homologue (Gloeobacter violaceus) of the pentameric ligand-gated ion channels indicated that propofol (and desflurane) is binding to the intra-subunit transmembrane pockets of this receptor (Nury et al., 2011), again supporting an allosteric interaction of propofol (intra-subunit pocket) and etomidate (inter-subunit pocket). The additional evidence for the existence of linking channels between inter-subunit and intra-subunit pockets (Nury et al., 2011; Figure 1C), together with the high flexibility of TM3 indicates, that the respective structures may not form well-defined pockets. Pockets may either communicate or even fuse with each other or disappear in certain conformational states of the protein. This dynamical view could well resolve some of the discrepancies concerning anaesthetic binding sites.
In addition to the intra- and inter-subunit pockets in the transmembrane domain of GABAA receptors, a variety of convulsant drugs, such as picrotoxinin and other ‘cage convulsants’ seem to block the chloride channel of GABAA receptors by binding to a site within the channel formed by the second transmembrane domains of the five subunits (Figure 1B) (Olsen, 2006; Sedelnikova et al., 2006; Bali and Akabas, 2007). The existence of at least 16 solvent accessible spaces within the same receptor composed of two α, two β and one γ subunits, that all might act as drug binding sites, explains the extremely complex pharmacology of GABAA receptors and also the difficulty to unequivocally identify the individual binding sites for these drugs. The two intra-subunit α or β subunit pockets or the two inter-subunit β+α− pockets not necessarily have the same structure due to the influence of different surrounding subunits and might thus exhibit a differential interaction with different ligands. In addition, since each binding step of a pharmacologically active compound elicits or stabilizes a different conformation of the receptor, it might also change the properties of a binding site at a homologous binding pocket, thus complicating analysis of the effects observed. Drugs acting for instance via the ‘same’ binding site in an intra-subunit transmembrane pocket of an α or a β subunit, due to the existence of two such pockets in a receptor could nevertheless show allosteric interactions. Such considerations might also explain some of the discrepant results discussed above on the location of binding sites in intra- or inter-subunit pockets and their apparent allosteric interactions. In addition, they also provide an explanation for the requirement of different concentrations of a drug for eliciting the allosteric modulatory or the directly activating effects of drugs, even if the drug apparently interacts with only a single type of binding site at GABAA receptors as discussed for etomidate (Li et al., 2010; Olsen and Li, 2011).
Multiplicity of GABAA receptor subtypes
GABAA receptor subunits exhibit a distinct but overlapping regional distribution in the brain. At the single cell level, there are cells expressing only a few GABAA receptor subunits, and others expressing most, if not all, of these subunits (Wisden et al., 1992; Pirker et al., 2000), giving rise to a multiplicity of these receptors. So far, however, only a limited number of receptor subtypes have been unequivocally identified (Olsen and Sieghart, 2008). Nevertheless, individual receptor subtypes often have a quite specific regional, cellular and subcellular distribution. Receptors composed of α1βγ2, α2βγ2, α3βγ2 are located synaptically, although due to the small proportion of cell surface occupied by synapses, even these receptors might be located predominantly extrasynaptically (Kasugai et al., 2010). In contrast, receptors composed of α5βγ2, or α4βδ and α6βδ subunits might predominantly or exclusively be located extrasynaptically (Nusser et al., 1998; Brunig et al., 2002; Crestani et al., 2002; Farrant and Nusser, 2005). Thus, extrasynaptic receptors seem to be much more important for drug action than previously thought. Considering the fact that usually only low GABA concentrations are present in the extrasynaptic space, drugs interacting with these receptors will more strongly modulate the current amplitude of GABA-induced currents than at synaptic receptors, where GABA often more or less saturates the receptors on synaptic transmission and where allosteric modulators will predominantly act via a prolongation of the current decay (Farrant and Nusser, 2005). Currently, we don't have much information on the regional, cellular and subcellular localization of receptors composed of αβ subunits (Bencsits et al., 1999; Mortensen and Smart, 2006; Sinkkonen et al., 2004) or those containing ε, π, θ or ρ subunits (Hedblom and Kirkness, 1997; Bonnert et al., 1999; Enz and Cutting, 1999; Bormann, 2000; Moragues et al., 2000; 2002). But it is clear that these receptor subtypes, although not very abundant in the brain, exhibit a quite specific regional and cellular distribution.
A distinct regional, cellular and subcellular distribution of receptor subtypes also suggests a distinct function. This conclusion was supported by transgenic knock-in mice (Rudolph et al., 1999; McKernan et al., 2000). GABAA receptors of mice containing a H101R mutation within the α1 subunit no longer can be modulated by diazepam. Thus, all diazepam actions mediated via GABAA receptors containing the α1 subunit are absent in these mice and thus can be identified by comparison of diazepam effects in wild-type mice. Studies investigating these point mutated and wild-type mice indicated that GABAA receptors containing α1 subunits seem to be involved in the sedative, anticonvulsant and anterograde amnestic actions of diazepam (Rudolph et al., 1999; McKernan et al., 2000). Similar experiments performed with mice containing the homologous point mutation in the α2 subunit indicated that receptors containing α2 subunits seem to primarily mediate the anxiolytic, and partially also the muscle relaxant effects of diazepam (Low et al., 2000), and, in addition, seem to mediate the analgesic action of local diazepam in the spinal cord (Knabl et al., 2008). Receptors containing the α3 subunit seem to mediate the anti-absence effects of clonazepam, as indicated by the respective point-mutated mouse (Sohal et al., 2003), and the α3 global knock-out mice displayed a hyper-dopaminergic phenotype relevant for GABAergic control of psychotic-like symptoms (Yee et al., 2005). Under conditions of higher receptor occupancy, α3-containing receptors also mediate anxiolytic actions (Dias et al., 2005; Yee et al., 2005). In contrast, receptors containing the α5 subunit seem to mediate the effects of diazepam on learning and memory, shown by improved spatial memory in mice with knock-out of α5 subunits (Collinson et al., 2002), and improved trace fear conditioning in mice with a partial deficit of α5 GABAA receptors in hippocampus (Crestani et al., 2002). These and other studies for the first time indicated a possible function of specific GABAA receptor subtypes in the rodent brain.
Some benzodiazepine site ligands can distinguish between different receptor subtypes
All these conclusions were supported by studies using receptor subtype-selective drugs. Drugs presumably interacting with the transmembrane domain of GABAA receptors, such as barbiturates, steroids or anaesthetics, exhibit no or only a weak subtype selectivity for receptors composed of α1–6βγ2 subunits (Sieghart and Ernst, 2005). Steroids seem to preferentially modulate receptors containing the δ subunit (Stell et al., 2003; Hosie et al., 2009; Olsen and Sieghart, 2009). Similarly, classical benzodiazepines, such as diazepam, predominantly and with comparable affinity and efficacy interact with GABAA receptors composed of α1βγ2, α2βγ2, α3βγ2 and α5βγ2 subunits (Sieghart, 1995; Hevers and Luddens, 1998). The type of β subunit present in these receptors seems not to significantly modulate benzodiazepine action. Benzodiazepine site ligands also interact with receptors containing γ1 or γ3 instead of γ2 subunits, but their affinity and efficacy at these receptors usually is lower than that for γ2-containing receptors (Sieghart, 1995; Hevers and Luddens, 1998; Khom et al., 2006). Imidazobenzodiazepines in addition interact with receptors composed of α4βγ2 and α6βγ2 subunits.
In the last years, however, subtype-selective drugs interacting with the high-affinity benzodiazepine binding site of GABAA receptors were developed and could be used to investigate the proposed selective functions of GABAA receptors containing different α subunits. Thus, drugs more or less selectively enhancing GABA-induced currents in GABAA receptors containing α2 or α3 subunits [TPA023 (Atack et al., 2006), HZ166 (Rivas et al., 2009; Di Lio et al., 2011), SL651498 (Griebel et al., 2003)], exhibited anxiolytic properties and were active against neuropathic pain. Interestingly, drugs selectively interacting with α3-containing receptors [TP003 (Dias et al., 2005) or α3IA (Atack et al., 2005)], too, exhibited anxiolytic or anxiogenic properties, respectively, supporting the conclusion that α3-containing receptors are also involved in controlling fear and anxiety. And negative allosteric modulators on GABAA receptors containing α5 subunits, such as α5IA (Dawson et al., 2006; Buettelmann et al., 2009), PWZ029 (Savic et al., 2008), or RO4938581 (Achermann et al., 2009), improved learning and memory. Recent work thus indicates that it will be possible to develop benzodiazepine site ligands that can selectively address αβγ2 receptors containing a specific α subunit. Since there are six different α and three different γ subunits, it might be possible to develop selective ligands for 18 different GABAA receptor subtypes.
A novel drug binding site at the α+β− interface
In addition to the benzodiazepine binding site located at the extracellular α+γ− interface and the two GABA binding sites located at the β+α− interface, GABAA receptors composed of two α, two β and one γ subunit contain two other extracellular interfaces, the α+β− and the γ+β− interfaces (Figure 1A). These interfaces so far have not been systematically investigated as candidates for a possible drug binding site. The α+β− interface is similar to the high-affinity benzodiazepine binding site formed at the α+γ− interface. It also contains one of the six α+sides that together with the γ2− side determine the receptor subtype-specific benzodiazepine pockets, and on combining six different α and three different β subunits, a total of 18 different α+β− interfaces can be formed. Therefore, in a recent study, we systematically investigated the α+β− interface for the presence of a possible drug binding site (Ramerstorfer et al., 2011). We reasoned that due to the similarity of the binding sites, a binding site at the α+β− interface might accommodate at least some of the benzodiazepine site ligands. To identify drugs possibly mediating some of their effects via the α+β− interface and to avoid interaction with the benzodiazepine binding site, we used GABAA receptors composed of α1 and β3 subunits only (Figure 1C). Such receptors are assumed to be composed of three β and two α subunits (Tretter et al., 1997; Farrar et al., 1999; Baumann et al., 2001) and should thus have two β3+α1− interfaces (GABA binding sites), two α1+β3− interfaces and one β3+β3− interface, but no high-affinity benzodiazepine binding site. In a screening of >100 benzodiazepine site ligands from different structural classes, the anxiolytic pyrazoloquinoline 2-p-methoxyphenylpyrazolo[4,3-c]quinolin-3(5H)-one [CGS 9895, Figure 1D (Yokoyama et al., 1982; Bennett, 1987)] at 1 and 10 µM concentration was able to strongly enhance GABA-induced currents in recombinant α1β3 receptors expressed in Xenopus oocytes but did not directly elicit a chloride current in the absence of GABA. Since this compound in radioligand displacement studies has been demonstrated previously to exhibit a low nanomolar affinity for the high-affinity benzodiazepine binding site of GABAA receptors (Yokoyama et al., 1982), we compared its action at α1β3 and α1β3γ2 receptors. CGS 9895 did not modulate GABA-induced currents at nanomolar concentrations in α1β3γ2 receptors but at micromolar concentrations elicited a comparable current enhancement in α1β3 and α1β3γ2 receptors (Ramerstorfer et al., 2011). The effect of CGS 9895 on α1β3γ2 receptors in contrast to that of diazepam could not be inhibited by the benzodiazepine site antagonist Ro15-1788, and 50 nM CGS 9895, a concentration that completely saturates the high affinity benzodiazepine binding site of GABAA receptors but does not stimulate GABA-induced currents, was able to completely inhibit the effects of diazepam on α1β3γ2 receptors (Ramerstorfer et al., 2011). These data indicate that CGS 9895 is an antagonist at the high-affinity benzodiazepine binding site at nM concentration and stimulates the receptor at µM concentrations via a second binding site that is also present at α1β3 receptors.
This binding site was then identified by applying a steric hindrance approach. For that, several amino acid residues at the α1+, γ2− or β3− side, which presumably line the pocket at the α1+γ2− or the α1+β3− interface, were selected by using our homology model of GABAA receptors as a guide (Ernst et al., 2003) and were individually mutated to cysteines (Figure 2). Only mutations that produced no or only small changes in the GABA induced currents of the receptors, and did not significantly change the potency and efficacy of diazepam or CGS 9895 for stimulating these receptors, were further used for our experiments. Wild-type and mutated receptors were then exposed to 2 mM MTSEA-biotin that covalently binds to the introduced cysteines and prevents access of drugs to the respective binding site. GABA-elicited currents as well as their potentiation by diazepam or CGS 9895 were then measured before and after MTSEA-biotin exposure.
Figure 2.

Model structure indicating amino acid residues important for binding of CGS 9895 into the α+β− interface. The structural model of the α+ side and the β− side is based on the glutamate bound structure of the glutamate gated chloride channel (Hibbs and Gouaux, 2011), PDB entry 3RIF. This new template shares the highest sequence similarity with GABAA receptors of all available homologous crystal structures. Modeller was used for modelling, VMD for visualization. (http://salilab.org/modeller/; http://www.ks.uiuc.edu/Research/vmd/) Residues used in steric hindrance experiments are shown in yellow label, and those affecting ligand potency or efficacy upon mutagenesis are shown in gray labels. All shown residues were individually mutated into cysteines. For α1S204C, α1V211C and β3Q64C the effect of CGS9895 on the mutant receptor was nearly unchanged, and cysteine modification with MTSEA-biotin drastically reduced ligand efficacy (Ramerstorfer et al., 2011). The mutations α1H101C and α1F99C induced a strong reduction of the ligand effect. Mutations α1S205C, α1T206C and β3A45C caused an increased potency of CGS 9895. These data strongly indicate that the binding site is structurally similar to the benzodiazepine binding site as identical or homologous residues are involved in ligand effects.
Results indicated that it was possible to selectively block the effects of diazepam but not of CGS 9895 by preventing access to the benzodiazepine binding pocket via covalently modified cysteine mutations at the γ2− side and to selectively inhibit the effects of CGS 9895 but not of diazepam by preventing access to the α1+β3− pocket via covalently modified cysteine mutations at the β3− side. In addition, blocking access of CGS 9895 to the α1+β3− pocket by covalently modifying cysteine mutations at the α1+as well as β3− sides in α1β3 receptors completely abolished the actions of this drug on these receptors (Ramerstorfer et al., 2011). These data strongly suggested that CGS 9895 exerts its action by binding to the α1+β3− interface. The inhibition was not caused by a general inactivation of GABAA receptors, because the GABA-enhancing effect of other ligands of the GABAA receptors was not influenced by MTSEA-biotin. Other experiments indicated that the CGS 9895 effect was dependent on the α and β subunit types forming the interface. CGS 9895 thus represents the first prototype of drugs mediating benzodiazepine-like modulatory effects via a defined binding site at the extracellular α1+β3− interface of GABAA receptors (Figure 2).
Many other compounds have been demonstrated to allosterically modulate GABA-induced currents in αβ receptors (Sieghart, 1995) and thus could produce their effects via the extracellular α1+β3− interface. Some of these, such as the above-mentioned barbiturates, steroids and anaesthetics, seem to interact with binding sites probably located in the transmembrane domain of these receptors. For other compounds, the situation is not as clear. Thus, flurazepam has been shown to interact with at least two different binding sites in α1β2γ2 receptors (Walters et al., 2000): it not only enhanced GABA-induced currents via the high-affinity benzodiazepine binding site but at concentrations >10 µM also inhibited its own stimulation by interacting with the α1+β2− interface (Baur et al., 2008; Sigel and Luscher, 2011). However, flurazepam neither positively, nor negatively, modulated GABA-induced currents via this binding site when the classical benzodiazepine binding site at the α1+γ2− interface of αβγ receptors was replaced by a flurazepam-insensitive α6+γ2− interface in concatenated receptors (Baur et al., 2008), possibly indicating that it acts as a neutral modulator (antagonist) at this site in the absence of a high-affinity benzodiazepine binding site and thus possibly also in α1β3 receptors. We thus investigated whether the low-affinity flurazepam binding site is identical with the low-affinity CGS 9895 binding site located at the extracellular α1+β3− interface. In α1β3 receptors, flurazepam at 250 µM concentrations was able to partially inhibit the GABA-stimulatory effect of 10 µM CGS 9895, possibly supporting the conclusion that flurazepam is able to inhibit the action of CGS 9895 via the same binding site (Ramerstorfer et al., 2011). In other experiments, however, it was demonstrated that at the same concentration flurazepam negatively influenced GABA-induced currents in α1β3 receptors, and that this effect could not be blocked by steric hindrance under conditions where the effect of CGS 9895 was completely abolished. This clearly indicates that the negative effect of flurazepam at α1β3 receptors was not mediated via the extracellular α1+β3− interface. It is possible, however, that the effect of flurazepam on α1β3 receptors represented a third effect unrelated to the low potency inhibitory effect of flurazepam on its own action observed by Baur et al. (2008). Whether the low potency self-inhibitory effect of flurazepam is mediated via the extracellular α1+β2− interface of α1β2γ2 receptors can only be answered by steric hindrance experiments in appropriately mutated concatenated receptors.
Some other compounds interacting with αβ receptors have already been excluded as possible ligands for the CGS 9895 binding site. Thus, the additional GABA-potentiating effect at 100 µM diazepam in α1βγ2 receptors (Walters et al., 2000) or the effect of 10 µM ROD 188 (Thomet et al., 2000) on α1β3 receptors could not be inhibited by the steric hindrance approach under conditions where the effects of CGS 9895 were completely abolished (Ramerstorfer et al., 2011). Further experiments have to clarify, whether the effects of for instance Valerenic acid (Khom et al., 2010; Kopp et al., 2010) or of some flavonoids (Hanrahan et al., 2011) on α1β3 receptors are mediated via the CGS 9895 binding site.
Although CGS 9895 interacts with the α+β− site with a relatively low potency, this compound can now be used as a screening tool for the identification of other possible candidates interacting with this binding site. This approach is especially important for the identification of possible antagonists at this binding site (see above). In the absence of a direct effect of such compounds at GABAA receptors, they only can be detected by their inhibition of the effects of CGS 9895. In addition, using the cysteine mutations introduced into the α+β− pocket and their modification with MTSEA-biotin, compounds that cause a positive or negative modulation of GABAA receptors via this binding site can now be identified.
Potential importance of drugs interacting with the extracellular α+β− interface
Drugs interacting with the α+β− interface, irrespective of the type of the α or β subunit, should be able to modulate all αβ, αβγ, αβδ, αβε, αβπ, and αβθ receptors. Due to their modulation of a larger number of receptor types, they should exhibit a much broader action than benzodiazepine site ligands, which only interact with αβγ2 receptors and to a lesser extent with αβγ1 or αβγ3 receptors. Drugs interacting with the α+β− interface might thus also exhibit some novel effects so far not observed with benzodiazepine site ligands. In addition, the broader action of such drugs might be especially important for the treatment of epilepsy. By interacting with a larger number of receptor subtypes, CGS 9895 type drugs have more possibilities to enhance GABAergic inhibition and thus might be able to better reduce over-excitation in neurons that cannot be sufficiently modulated by benzodiazepines. Although benzodiazepines are excellent anticonvulsants, they rapidly loose this property on repeated application, making them largely unsuitable for a long-term treatment of epilepsy. This might be due to a benzodiazepine-induced uncoupling of the GABA and benzodiazepine site of GABAA receptors (Ali and Olsen, 2001). Such a mechanism presumably would not be possible for drugs interacting via the α+β− pocket, since α and β subunits are also essential for forming the GABA binding site. In addition, even after drug mediated down-regulation of αβγ receptors and/or change in subunit composition, the remaining receptors still would be modulated by these drugs. It thus can be expected that drugs interacting with the α+β− pocket also would be suitable for a long-term treatment of epilepsy. Since these drugs cannot directly activate GABAA receptors, they should have low toxicity similar to benzodiazepines, and by enhancing only ongoing GABAergic transmission, they should be specifically active in brain areas with exaggerated excitatory and thus also enhanced GABAergic activity.
Nevertheless, some of these drugs are also able to distinguish between different receptor subtypes depending on the exact α and β subunit type forming their binding site (Ramerstorfer et al., 2011). Different α and β subunits have a distinct regional and cellular distribution (Wisden et al., 1992; Pirker et al., 2000), and it was demonstrated that not only benzodiazepine site ligands selectively interacting with receptors containing a certain α subunit (see above) but also drugs selectively interacting with receptors containing β2 or β3 subunits exhibit quite specific actions (Wingrove et al., 1994; Hill-Venning et al., 1997; Pirker et al., 2000; Jurd et al., 2003; Reynolds et al., 2003; Rudolph and Antkowiak, 2004; Olsen and Sieghart, 2008). So far, only a few compounds, such as salicylidene salicylhydrazide (Thompson et al., 2004) or some fragrant dioxine derivatives (Sergeeva et al., 2010), have been identified that preferentially interact with GABAA receptors containing a β1 subunit. In posterior hypothalamic neurons such receptors might be involved in inducing sleep (Sergeeva et al., 2010) (Yanovsky et al., 2011). Other experiments have indicated that the hypnotic effect of diazepam is not mediated by GABAA receptors containing α1 but by receptors containing α2, α3 or α5 subunits (Tobler et al., 2001). Drugs selectively activating these β1 subunit-containing GABAA receptors might thus become more selective sleep-inducing agents, and the development of drugs that cannot interact with these receptors might be a good strategy to reduce sleep-inducing side effects of anxiolytics.
Of course, drugs interacting with an interface composed of αx+ and βy−, would modulate αxβy, αxβyγ, αxβyδ, αxβyε, αxβyπ or αxβyθ in a similar manner. But such a set of receptors cannot selectively be addressed so far, and their modulation might elicit novel and interesting effects. Especially, receptors composed of less abundant α subunits, such as α2, α3, α4, α5 or α6, combined with any one of the β subunit types, or receptors containing δ, ε, π or θ subunits, not only have a distinct regional and cellular distribution but might also be involved in quite specific behavioural effects, that then could be specifically modulated by selective α+β− drugs. The importance of such a novel pharmacology of drugs interacting with the interface of specific α and β subunits will be comparable with that of benzodiazepines interacting with the interface of specific α and γ subunits. The overall effects of the two types of drugs, however, will be different, because they not only address a subgroup of common receptors (containing the same α, β and γ subunit types), but overall different receptor populations [all receptors containing the same α and γ subunit types (β subunits don't significantly influence the effects of benzodiazepine site ligands) (Sieghart, 1995; Hevers and Luddens, 1998) versus all receptors containing the same α and β subunit types in the absence or presence of γ, δ, ε, π,or θ subunits. Subtype-selective drugs interacting with the α+β− interface in contrast to benzodiazepine site ligands thus also have the potential to distinguish between αβγ2 receptor subtypes containing the same α but different β subunits. And both types of drugs will have a comparably low toxicity because they are not able to directly activate the respective receptors in the absence of GABAergic transmission. The development of drugs interacting with α+β− binding sites of GABAA receptors will thus increase the spectrum of GABAA receptor subtypes that can be addressed experimentally and clinically and will thus have a substantial therapeutic potential. Of course, the question remains whether results from experimental animals can be extrapolated to man. Actually, in several cases, drugs that were non-sedating anxiolytics in experimental animals caused sedation in humans (Atack, 2003; Basile et al., 2004; Atack et al., 2011b). Recently, however, it was demonstrated that an anxiolytic drug exhibiting no effect at all on α1βγ2 receptors behaved as a non-sedating anxiolytic in animal and man (Atack et al., 2011a). Thus, avoiding modulation of α1βγ2 GABAA receptors altogether might be one solution for avoiding sedative effects of drugs. But avoiding the specific α1βxγ2 receptor subtype mediating the sedative effect might be an alternative possibility. A more thorough knowledge on the regional and cellular distribution of GABAA receptor subtypes in the human brain and their function in modulating behaviour will facilitate the rational development of drugs with more selective actions.
Acknowledgments
Financial support by Project P19653 of the Austrian Science Fund (to ME) and by the FP7 project HEALTH-F4-2008-202088 (Neurocypres; to WS) is gratefully acknowledged. CGS 9895 was a gift from Novartis, formerly Ciba-Geigy.
Glossary
- CGS 9895
pyrazoloquinoline 2-p-methoxyphenylpyrazolo[4,3-c]quinolin-3(5H)-one
- GABAA receptors
GABA type A receptors
- MTSEA-biotin
N-Biotinylaminoethyl methanethiosulphonate
Conflict of interest
The authors state no conflict of interest.
References
- Achermann G, Ballard TM, Blasco F, Broutin PE, Buttelmann B, Fischer H, et al. Discovery of the imidazo[1,5-a][1,2,4]-triazolo[1,5-d][1,4]benzodiazepine scaffold as a novel, potent and selective GABA(A) alpha5 inverse agonist series. Bioorg Med Chem Lett. 2009;19:5746–5752. doi: 10.1016/j.bmcl.2009.07.153. [DOI] [PubMed] [Google Scholar]
- Ali NJ, Olsen RW. Chronic benzodiazepine treatment of cells expressing recombinant GABA(A) receptors uncouples allosteric binding: studies on possible mechanisms. J Neurochem. 2001;79:1100–1108. doi: 10.1046/j.1471-4159.2001.00664.x. [DOI] [PubMed] [Google Scholar]
- Atack JR. Anxioselective compounds acting at the GABA(A) receptor benzodiazepine binding site. Curr Drug Targets CNS Neurol Disord. 2003;2:213–232. doi: 10.2174/1568007033482841. [DOI] [PubMed] [Google Scholar]
- Atack JR, Hutson PH, Collinson N, Marshall G, Bentley G, Moyes C, et al. Anxiogenic properties of an inverse agonist selective for alpha3 subunit-containing GABA A receptors. Br J Pharmacol. 2005;144:357–366. doi: 10.1038/sj.bjp.0706056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atack JR, Wafford KA, Tye SJ, Cook SM, Sohal B, Pike A, et al. TPA023 [7-(1,1-dimethylethyl)-6-(2-ethyl-2H-1,2,4-triazol-3-ylmethoxy)-3-(2-fluorophenyl)-1,2,4-triazolo[4,3-b]pyridazine], an agonist selective for alpha2- and alpha3-containing GABAA receptors, is a nonsedating anxiolytic in rodents and primates. J Pharmacol Exp Ther. 2006;316:410–422. doi: 10.1124/jpet.105.089920. [DOI] [PubMed] [Google Scholar]
- Atack JR, Hallett DJ, Tye S, Wafford KA, Ryan C, Sanabria-Bohorquez SM, et al. Preclinical and clinical pharmacology of TPA023B, a GABAA receptor α2/α3 subtype-selective partial agonist. J Psychopharmacol. 2011a;25:329–344. doi: 10.1177/0269881109354928. [DOI] [PubMed] [Google Scholar]
- Atack JR, Wafford KA, Street LJ, Dawson GR, Tye S, Van Laere K, et al. MRK-409 (MK-0343), a GABAA receptor subtype-selective partial agonist, is a non-sedating anxiolytic in preclinical species but causes sedation in humans. J Psychopharmacol. 2011b;25:314–328. doi: 10.1177/0269881109354927. [DOI] [PubMed] [Google Scholar]
- Bali M, Akabas MH. Defining the propofol binding site location on the GABAA receptor. Mol Pharmacol. 2004;65:68–76. doi: 10.1124/mol.65.1.68. [DOI] [PubMed] [Google Scholar]
- Bali M, Akabas MH. The location of a closed channel gate in the GABAA receptor channel. J Gen Physiol. 2007;129:145–159. doi: 10.1085/jgp.200609639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basile AS, Lippa AS, Skolnick P. Anxioselective anxiolytics: can less be more? Eur J Pharmacol. 2004;500:441–451. doi: 10.1016/j.ejphar.2004.07.043. [DOI] [PubMed] [Google Scholar]
- Baumann SW, Baur R, Sigel E. Subunit arrangement of gamma-aminobutyric acid type A receptors. J Biol Chem. 2001;276:36275–36280. doi: 10.1074/jbc.M105240200. [DOI] [PubMed] [Google Scholar]
- Baur R, Tan KR, Luscher BP, Gonthier A, Goeldner M, Sigel E. Covalent modification of GABAA receptor isoforms by a diazepam analogue provides evidence for a novel benzodiazepine binding site that prevents modulation by these drugs. J Neurochem. 2008;106:2353–2363. doi: 10.1111/j.1471-4159.2008.05574.x. [DOI] [PubMed] [Google Scholar]
- Belelli D, Lambert JJ, Peters JA, Wafford K, Whiting PJ. The interaction of the general anesthetic etomidate with the gamma-aminobutyric acid type A receptor is influenced by a single amino acid. Proc Natl Acad Sci U S A. 1997;94:11031–11036. doi: 10.1073/pnas.94.20.11031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bencsits E, Ebert V, Tretter V, Sieghart W. A significant part of native gamma-aminobutyric acid A receptors containing alpha 4 subunits do not contain gamma or delta subunits. J Biol Chem. 1999;274:19613–19616. doi: 10.1074/jbc.274.28.19613. [DOI] [PubMed] [Google Scholar]
- Bennett DA. Pharmacology of the pyrazolo-type compounds: agonist, antagonist and inverse agonist actions. Physiol Behav. 1987;41:241–245. doi: 10.1016/0031-9384(87)90360-x. [DOI] [PubMed] [Google Scholar]
- Bonnert TP, McKernan RM, Farrar S, le Bourdelles B, Heavens RP, Smith DW, et al. Theta, a novel gamma-aminobutyric acid type A receptor subunit. PNAS. 1999;96:9891–9896. doi: 10.1073/pnas.96.17.9891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bormann J. The ‘ABC’ of GABA receptors. Trends Pharmacol Sci. 2000;21:16–19. doi: 10.1016/s0165-6147(99)01413-3. [DOI] [PubMed] [Google Scholar]
- Brunig I, Scotti E, Sidler C, Fritschy J-M. Intact sorting, targeting, and clustering of gamma-aminobutyric acid A receptor subtypes in hippocampal neurons in vitro. J Comp Neurol. 2002;443:43–55. doi: 10.1002/cne.10102. [DOI] [PubMed] [Google Scholar]
- Buettelmann B, Ballard TM, Gasser R, Fischer H, Hernandez MC, Knoflach F, et al. Imidazo[1,5-a][1,2,4]-triazolo[1,5-d][1,4]benzodiazepines as potent and highly selective GABAA alpha5 inverse agonists with potential for the treatment of cognitive dysfunction. Bioorg Med Chem Lett. 2009;19:5958–5961. doi: 10.1016/j.bmcl.2009.08.027. [DOI] [PubMed] [Google Scholar]
- Collinson N, Kuenzi FM, Jarolimek W, Maubach KA, Cothliff R, Sur C, et al. Enhanced learning and memory and altered GABAergic synaptic transmission in mice lacking the alpha 5 subunit of the GABAA receptor. J Neurosci. 2002;22:5572–5580. doi: 10.1523/JNEUROSCI.22-13-05572.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crestani F, Keist R, Fritschy J-M, Benke D, Vogt K, Prut L, et al. Trace fear conditioning involves hippocampal alpha5 GABA(A) receptors. Proc Natl Acad Sci U S A. 2002;99:8980–8985. doi: 10.1073/pnas.142288699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson GR, Maubach KA, Collinson N, Cobain M, Everitt BJ, MacLeod AM, et al. An inverse agonist selective for alpha5 subunit-containing GABAA receptors enhances cognition. J Pharmacol Exp Ther. 2006;316:1335–1345. doi: 10.1124/jpet.105.092320. [DOI] [PubMed] [Google Scholar]
- Di Lio A, Benke D, Besson M, Desmeules J, Daali Y, Wang ZJ, et al. HZ166, a novel GABA(A) receptor subtype-selective benzodiazepine site ligand, is antihyperalgesic in mouse models of inflammatory and neuropathic pain. Neuropharmacology. 2011;60:626–632. doi: 10.1016/j.neuropharm.2010.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias R, Sheppard WF, Fradley RL, Garrett EM, Stanley JL, Tye SJ, et al. Evidence for a significant role of alpha 3-containing GABAA receptors in mediating the anxiolytic effects of benzodiazepines. J Neurosci. 2005;25:10682–10688. doi: 10.1523/JNEUROSCI.1166-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enz R, Cutting GR. GABAC receptor rho subunits are heterogeneously expressed in the human CNS and form homo- and heterooligomers with distinct physical properties. Eur J Neurosci. 1999;11:41–50. doi: 10.1046/j.1460-9568.1999.00423.x. [DOI] [PubMed] [Google Scholar]
- Ernst M, Brauchart D, Boresch S, Sieghart W. Comparative modeling of GABAA receptors: limits, insights, future developments. Neuroscience. 2003;119:933–943. doi: 10.1016/s0306-4522(03)00288-4. [DOI] [PubMed] [Google Scholar]
- Ernst M, Bruckner S, Boresch S, Sieghart W. Comparative models of GABAA receptor extracellular and transmembrane domains: important insights in pharmacology and function. Mol Pharmacol. 2005;68:1291–1300. doi: 10.1124/mol.105.015982. [DOI] [PubMed] [Google Scholar]
- Farrant M, Nusser Z. Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat Rev Neurosci. 2005;6:215–229. doi: 10.1038/nrn1625. [DOI] [PubMed] [Google Scholar]
- Farrar SJ, Whiting PJ, Bonnert TP, McKernan RM. Stoichiometry of a ligand-gated ion channel determined by fluorescence energy transfer. J Biol Chem. 1999;274:10100–10104. doi: 10.1074/jbc.274.15.10100. [DOI] [PubMed] [Google Scholar]
- Griebel G, Perrault G, Simiand J, Cohen C, Granger P, Depoortere H, et al. SL651498, a GABAA receptor agonist with subtype-selective efficacy, as a potential treatment for generalized anxiety disorder and muscle spasms. CNS Drug Rev. 2003;9:3–20. doi: 10.1111/j.1527-3458.2003.tb00241.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanchar HJ, Chutsrinopkun P, Meera P, Supavilai P, Sieghart W, Wallner M, et al. Ethanol potently and competitively inhibits binding of the alcohol antagonist Ro15-4513 to alpha4/6beta3delta GABAA receptors. Proc Natl Acad Sci U S A. 2006;103:8546–8551. doi: 10.1073/pnas.0509903103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanrahan JR, Chebib M, Johnston GA. Flavonoid modulation of GABA(A) receptors. Br J Pharmacol. 2011;163:234–245. doi: 10.1111/j.1476-5381.2011.01228.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser CA, Wetzel CH, Berning B, Gerner FM, Rupprecht R. Flunitrazepam has an inverse agonistic effect on recombinant alpha6beta2gamma2-GABAA receptors via a flunitrazepam-binding site. J Biol Chem. 1997;272:11723–11727. doi: 10.1074/jbc.272.18.11723. [DOI] [PubMed] [Google Scholar]
- Hedblom E, Kirkness EF. A novel class of GABAA receptor subunit in tissues of the reproductive system. J Biol Chem. 1997;272:15346–15350. doi: 10.1074/jbc.272.24.15346. [DOI] [PubMed] [Google Scholar]
- Hevers W, Luddens H. The diversity of GABAA receptors. Pharmacological and electrophysiological properties of GABAA channel subtypes. Mol Neurobiol. 1998;18:35–86. doi: 10.1007/BF02741459. [DOI] [PubMed] [Google Scholar]
- Hibbs RE, Gouaux E. Principles of activation and permeation in an anion-selective Cys-loop receptor. Nature. 2011;474:54–60. doi: 10.1038/nature10139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill-Venning C, Belelli D, Peters JA, Lambert JJ. Subunit-dependent interaction of the general anaesthetic etomidate with the gamma-aminobutyric acid type A receptor. Br J Pharmacol. 1997;120:749–756. doi: 10.1038/sj.bjp.0700927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosie AM, Wilkins ME, da Silva HM, Smart TG. Endogenous neurosteroids regulate GABAA receptors through two discrete transmembrane sites. Nature. 2006;444:486–489. doi: 10.1038/nature05324. [DOI] [PubMed] [Google Scholar]
- Hosie AM, Wilkins ME, Smart TG. Neurosteroid binding sites on GABA(A) receptors. Pharmacol Ther. 2007;116:7–19. doi: 10.1016/j.pharmthera.2007.03.011. [DOI] [PubMed] [Google Scholar]
- Hosie AM, Clarke L, da Silva H, Smart TG. Conserved site for neurosteroid modulation of GABA A receptors. Neuropharmacology. 2009;56:149–154. doi: 10.1016/j.neuropharm.2008.07.050. [DOI] [PubMed] [Google Scholar]
- Jenkins A, Greenblatt EP, Faulkner HJ, Bertaccini E, Light A, Lin A, et al. Evidence for a common binding cavity for three general anesthetics within the GABAA receptor. J Neurosci. 2001;21:RC136. doi: 10.1523/JNEUROSCI.21-06-j0002.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurd R, Arras M, Lambert S, Drexler B, Siegwart R, Crestani F, et al. General anesthetic actions in vivo strongly attenuated by a point mutation in the GABA(A) receptor beta3 subunit. FASEB J. 2003;17:250–252. doi: 10.1096/fj.02-0611fje. [DOI] [PubMed] [Google Scholar]
- Kasugai Y, Swinny JD, Roberts JD, Dalezios Y, Fukazawa Y, Sieghart W, et al. Quantitative localisation of synaptic and extrasynaptic GABAA receptor subunits on hippocampal pyramidal cells by freeze-fracture replica immunolabelling. Eur J Neurosci. 2010;32:1868–1888. doi: 10.1111/j.1460-9568.2010.07473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khom S, Baburin I, Timin EN, Hohaus A, Sieghart W, Hering S. Pharmacological properties of GABAA receptors containing gamma1 subunits. Mol Pharmacol. 2006;69:640–649. doi: 10.1124/mol.105.017236. [DOI] [PubMed] [Google Scholar]
- Khom S, Strommer B, Ramharter J, Schwarz T, Schwarzer C, Erker T, et al. Valerenic acid derivatives as novel subunit-selective GABAA receptor ligands – in vitro and in vivo characterization. Br J Pharmacol. 2010;161:65–78. doi: 10.1111/j.1476-5381.2010.00865.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knabl J, Witschi R, Hosl K, Reinold H, Zeilhofer UB, Ahmadi S, et al. Reversal of pathological pain through specific spinal GABAA receptor subtypes. Nature. 2008;451:330–334. doi: 10.1038/nature06493. [DOI] [PubMed] [Google Scholar]
- Kopp S, Baur R, Sigel E, Mohler H, Altmann KH. Highly potent modulation of GABA(A) receptors by valerenic acid derivatives. ChemMedChem. 2010;5:678–681. doi: 10.1002/cmdc.201000062. [DOI] [PubMed] [Google Scholar]
- Krasowski MD, Nishikawa K, Nikolaeva N, Lin A, Harrison NL. Methionine 286 in transmembrane domain 3 of the GABAA receptor beta subunit controls a binding cavity for propofol and other alkylphenol general anesthetics. Neuropharmacology. 2001;41:952–964. doi: 10.1016/s0028-3908(01)00141-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GD, Chiara DC, Sawyer GW, Husain SS, Olsen RW, Cohen JB. Identification of a GABAA receptor anesthetic binding site at subunit interfaces by photolabeling with an etomidate analog. J Neurosci. 2006;26:11599–11605. doi: 10.1523/JNEUROSCI.3467-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GD, Chiara DC, Cohen JB, Olsen RW. Numerous classes of general anesthetics inhibit etomidate binding to gamma-aminobutyric acid type A (GABAA) receptors. J Biol Chem. 2010;285:8615–8620. doi: 10.1074/jbc.M109.074708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low K, Crestani F, Keist R, Benke D, Brunig I, Benson JA, et al. Molecular and neuronal substrate for the selective attenuation of anxiety. Science. 2000;290:131–134. doi: 10.1126/science.290.5489.131. [DOI] [PubMed] [Google Scholar]
- McKernan RM, Rosahl TW, Reynolds DS, Sur C, Wafford KA, Atack JR, et al. Sedative but not anxiolytic properties of benzodiazepines are mediated by the GABA(A) receptor alpha1 subtype. Nat Neurosci. 2000;3:587–592. doi: 10.1038/75761. [DOI] [PubMed] [Google Scholar]
- Mihic SJ, Ye Q, Wick MJ, Koltchine VV, Krasowski MD, Finn SE, et al. Sites of alcohol and volatile anaesthetic action on GABA(A) and glycine receptors. Nature. 1997;389:385–389. doi: 10.1038/38738. [DOI] [PubMed] [Google Scholar]
- Moragues N, Ciofi P, Lafon P, Odessa MF, Tramu G, Garret M. cDNA cloning and expression of a gamma-aminobutyric acid A receptor epsilon-subunit in rat brain. Eur J Neurosci. 2000;12:4318–4330. [PubMed] [Google Scholar]
- Moragues N, Ciofi P, Tramu G, Garret M. Localisation of GABA(A) receptor epsilon-subunit in cholinergic and aminergic neurones and evidence for co-distribution with the theta-subunit in rat brain. Neuroscience. 2002;111:657–669. doi: 10.1016/s0306-4522(02)00033-7. [DOI] [PubMed] [Google Scholar]
- Mortensen M, Smart TG. Extrasynaptic alphabeta subunit GABAA receptors on rat hippocampal pyramidal neurons. J Physiol. 2006;577:841–856. doi: 10.1113/jphysiol.2006.117952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nury H, Van Renterghem C, Weng Y, Tran A, Baaden M, Dufresne V, et al. X-ray structures of general anaesthetics bound to a pentameric ligand-gated ion channel. Nature. 2011;469:428–431. doi: 10.1038/nature09647. [DOI] [PubMed] [Google Scholar]
- Nusser Z, Sieghart W, Somogyi P. Segregation of different GABAA receptors to synaptic and extrasynaptic membranes of cerebellar granule cells. J Neurosci. 1998;18:1693–1703. doi: 10.1523/JNEUROSCI.18-05-01693.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen RW. Picrotoxin-like channel blockers of GABAA receptors. Proc Natl Acad Sci U S A. 2006;103:6081–6082. doi: 10.1073/pnas.0601121103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen RW, Li GD. GABA(A) receptors as molecular targets of general anesthetics: identification of binding sites provides clues to allosteric modulation. Can J Anaesth. 2011;58:206–215. doi: 10.1007/s12630-010-9429-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen RW, Sieghart W. International Union of Pharmacology. LXX. Subtypes of gamma-aminobutyric acid(A) receptors: classification on the basis of subunit composition, pharmacology, and function. Update. Pharmacol Rev. 2008;60:243–260. doi: 10.1124/pr.108.00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen RW, Sieghart W. GABA A receptors: subtypes provide diversity of function and pharmacology. Neuropharmacology. 2009;56:141–148. doi: 10.1016/j.neuropharm.2008.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirker S, Schwarzer C, Wieselthaler A, Sieghart W, Sperk G. GABA(A) receptors: immunocytochemical distribution of 13 subunits in the adult rat brain. Neuroscience. 2000;101:815–850. doi: 10.1016/s0306-4522(00)00442-5. [DOI] [PubMed] [Google Scholar]
- Ramerstorfer J, Furtmuller R, Sarto-Jackson I, Varagic Z, Sieghart W, Ernst M. The GABAA receptor alpha+beta- interface: a novel target for subtype selective drugs. J Neurosci. 2011;31:870–877. doi: 10.1523/JNEUROSCI.5012-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds DS, Rosahl TW, Cirone J, O'Meara GF, Haythornthwaite A, Newman RJ, et al. Sedation and anesthesia mediated by distinct GABA(A) receptor isoforms. J Neurosci. 2003;23:8608–8617. doi: 10.1523/JNEUROSCI.23-24-08608.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson JE, Garcia PS, O'Toole KK, Derry JM, Bell SV, Jenkins A. A conserved tyrosine in the beta2 subunit M4 segment is a determinant of gamma-aminobutyric acid type A receptor sensitivity to propofol. Anesthesiology. 2007;107:412–418. doi: 10.1097/01.anes.0000278875.36639.2c. [DOI] [PubMed] [Google Scholar]
- Rivas FM, Stables JP, Murphree L, Edwankar RV, Edwankar CR, Huang S, et al. Antiseizure activity of novel gamma-aminobutyric acid (A) receptor subtype-selective benzodiazepine analogues in mice and rat models. J Med Chem. 2009;52:1795–1798. doi: 10.1021/jm801652d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph U, Antkowiak B. Molecular and neuronal substrates for general anaesthetics. Nat Rev Neurosci. 2004;5:709–720. doi: 10.1038/nrn1496. [DOI] [PubMed] [Google Scholar]
- Rudolph U, Crestani F, Benke D, Brunig I, Benson JA, Fritschy JM, et al. Benzodiazepine actions mediated by specific gamma-aminobutyric acid(A) receptor subtypes. Nature. 1999;401:796–800. doi: 10.1038/44579. [DOI] [PubMed] [Google Scholar]
- Savic MM, Clayton T, Furtmuller R, Gavrilovic I, Samardzic J, Savic S, et al. PWZ-029, a compound with moderate inverse agonist functional selectivity at GABA(A) receptors containing alpha5 subunits, improves passive, but not active, avoidance learning in rats. Brain Res. 2008;1208:150–159. doi: 10.1016/j.brainres.2008.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedelnikova A, Erkkila BE, Harris H, Zakharkin SO, Weiss DS. Stoichiometry of a pore mutation that abolishes picrotoxin-mediated antagonism of the GABAA receptor. J Physiol. 2006;577:569–577. doi: 10.1113/jphysiol.2006.120287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergeeva OA, Kletke O, Kragler A, Poppek A, Fleischer W, Schubring SR, et al. Fragrant dioxane derivatives identify beta1-subunit-containing GABAA receptors. J Biol Chem. 2010;285:23985–23993. doi: 10.1074/jbc.M110.103309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieghart W. Structure and pharmacology of γ-aminobutyric acidA receptor subtypes. Pharmacol Rev. 1995;47:181–234. [PubMed] [Google Scholar]
- Sieghart W, Ernst M. Heterogeneity of GABAA receptors: revived interest in the development of subtype-selective drugs. Curr Med Chem – Central Nervous System Agents. 2005;5:217–242. [Google Scholar]
- Sigel E, Buhr A. The benzodiazepine binding site of GABAA receptors. Trends Pharmacol Sci. 1997;18:425–429. doi: 10.1016/s0165-6147(97)01118-8. [DOI] [PubMed] [Google Scholar]
- Sigel E, Luscher BP. A closer look at the high affinity benzodiazepine binding site on GABAA receptors. Curr Top Med Chem. 2011;11:241–246. doi: 10.2174/156802611794863562. [DOI] [PubMed] [Google Scholar]
- Sinkkonen ST, Luscher B, Luddens H, Korpi ER. Autoradiographic imaging of altered synaptic alphabetagamma2 and extrasynaptic alphabeta GABAA receptors in a genetic mouse model of anxiety. Neurochem Int. 2004;44:539–547. doi: 10.1016/j.neuint.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Smith GB, Olsen RW. Functional domains of GABAA receptors. Trends Pharmacol Sci. 1995;16:162–168. doi: 10.1016/s0165-6147(00)89009-4. [DOI] [PubMed] [Google Scholar]
- Sohal VS, Keist R, Rudolph U, Huguenard JR. Dynamic GABA(A) receptor subtype-specific modulation of the synchrony and duration of thalamic oscillations. J Neurosci. 2003;23:3649–3657. doi: 10.1523/JNEUROSCI.23-09-03649.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stell BM, Brickley SG, Tang CY, Farrant M, Mody I. Neuroactive steroids reduce neuronal excitability by selectively enhancing tonic inhibition mediated by delta subunit-containing GABAA receptors. Proc Natl Acad Sci U S A. 2003;100:14439–14444. doi: 10.1073/pnas.2435457100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomet U, Baur R, Razet R, Dodd RH, Furtmuller R, Sieghart W, et al. A novel positive allosteric modulator of the GABA(A) receptor: the action of (+)-ROD188. Br J Pharmacol. 2000;131:843–850. doi: 10.1038/sj.bjp.0703558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SA, Wheat L, Brown NA, Wingrove PB, Pillai GV, Whiting PJ, et al. Salicylidene salicylhydrazide, a selective inhibitor of beta 1-containing GABAA receptors. Br J Pharmacol. 2004;142:97–106. doi: 10.1038/sj.bjp.0705689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobler I, Kopp C, Deboer T, Rudolph U. Diazepam-induced changes in sleep: role of the alpha 1 GABA(A) receptor subtype. Proc Natl Acad Sci U S A. 2001;98:6464–6469. doi: 10.1073/pnas.111055398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tretter V, Ehya N, Fuchs K, Sieghart W. Stoichiometry and assembly of a recombinant GABAA receptor subtype. J Neurosci. 1997;17:2728–2737. doi: 10.1523/JNEUROSCI.17-08-02728.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters RJ, Hadley SH, Morris KD, Amin J. Benzodiazepines act on GABAA receptors via two distinct and separable mechanisms. Nat Neurosci. 2000;3:1274–1281. doi: 10.1038/81800. [DOI] [PubMed] [Google Scholar]
- Wingrove PB, Wafford KA, Bain C, Whiting PJ. The modulatory action of loreclezole at the gamma-aminobutyric acid type A receptor is determined by a single amino acid in the beta 2 and beta 3 subunit. Proc Natl Acad Sci U S A. 1994;91:4569–4573. doi: 10.1073/pnas.91.10.4569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisden W, Laurie DJ, Monyer H, Seeburg PH. The distribution of 13 GABAA receptor subunit mRNAs in the rat brain. I. Telencephalon, diencephalon, mesencephalon. J Neurosci. 1992;12:1040–1062. doi: 10.1523/JNEUROSCI.12-03-01040.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakura T, Bertaccini E, Trudell JR, Harris RA. Anesthetics and ion channels: molecular models and sites of action. Annu Rev Pharmacol Toxicol. 2001;41:23–51. doi: 10.1146/annurev.pharmtox.41.1.23. [DOI] [PubMed] [Google Scholar]
- Yanovsky Y, Schubring S, Fleischer W, Gisselmann G, Zhu XR, Lübbert H, et al. GABAA receptors involved in sleep and anaesthesia: β1- versus β3-containing assemblies. Pfügers Arch. 2011 doi: 10.1007/s00424-011-0988-4. PMID: 21735059. [DOI] [PubMed] [Google Scholar]
- Yee BK, Keist R, von Boehmer L, Studer R, Benke D, Hagenbuch N, et al. A schizophrenia-related sensorimotor deficit links alpha 3-containing GABAA receptors to a dopamine hyperfunction. Proc Natl Acad Sci U S A. 2005;102:17154–17159. doi: 10.1073/pnas.0508752102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama N, Ritter B, Neubert AD. 2-Arylpyrazolo[4,3-c]quinolin-3-ones: novel agonist, partial agonist, and antagonist of benzodiazepines. J Med Chem. 1982;25:337–339. doi: 10.1021/jm00346a002. [DOI] [PubMed] [Google Scholar]