

Abstract
Conotoxins (conopeptides) are small disulfide bonded peptides from the venom of marine cone snails. These peptides target a wide variety of membrane receptors, ion channels and transporters, and have enormous potential for a range of pharmaceutical applications. Structurally related ω-conotoxins bind directly to and selectively inhibit neuronal (N)-type voltage-gated calcium channels (VGCCs) of nociceptive primary afferent neurones. Among these, ω-conotoxin MVIIA (Prialt) is approved by the Food and Drug Administration (FDA) as an alternative intrathecal analgesic for the management of chronic intractable pain, particularly in patients refractory to opioids. A series of newly discovered ω-conotoxins from Conus catus, including CVID–F, are potent and selective antagonists of N-type VGCCs. In spinal cord slices, these peptides reversibly inhibit excitatory synaptic transmission between primary afferents and dorsal horn superficial lamina neurones, and in the rat partial sciatic nerve ligation model of neuropathic pain, significantly reduce allodynic behaviour. Another family of conotoxins, the α-conotoxins, are competitive antagonists of mammalian nicotinic acetylcholine receptors (nAChRs). α-Conotoxins Vc1.1 and RgIA possess two disulfide bonds and are currently in development as a treatment for neuropathic pain. It was initially proposed that the primary target of these peptides is the α9α10 neuronal nAChR. Surprisingly, however, α-conotoxins Vc1.1, RgIA and PeIA more potently inhibit N-type VGCC currents via a GABAB GPCR mechanism in rat sensory neurones. This inhibition is largely voltage-independent and involves complex intracellular signalling. Understanding the molecular mechanisms of conotoxin action will lead to new ways to regulate VGCC block and modulation in normal and diseased states of the nervous system.
Keywords: ω-conotoxin, α-conotoxin, voltage-gated calcium channel, GABAB receptor, G protein-coupled receptor, dorsal root ganglion, Xenopus oocyte, HEK293 cell, pain pathway
In recent years, progress in understanding conotoxin genetics and biodiversity and exploring sequence homology within the conotoxin superfamilies has moved rapidly, facilitating the discovery of a series of biologically active conopeptides. Several laboratories have made substantial progress in the identification, separation, synthesis and structural characterization of these peptides (reviewed in Nielsen et al., 2000; Terlau and Olivera, 2004; Schroeder et al., 2005; Jakubowski et al., 2006; Olivera, 2006; Bulaj and Olivera, 2008; Bingham et al., 2010). A number of conopeptides selectively target ion channels, membrane receptors and transporters associated with pain pathways. Among these, N-type voltage-gated calcium channels (VGCCs) play a major role in pain information processing in sensory neurones (Campbell and Meyer, 2006; Zamponi et al., 2009) and represent a validated target for treating chronic and neuropathic pain (Snutch, 2005; Pexton et al., 2011). The N-type VGCC belongs to the group of high voltage-activated calcium channels and is composed of the pore-forming α1B subunit (also known as CaV2.2, which in humans is encoded by the CACNA1B gene) and ancillary cytoplasmic β subunits (CACNB), membrane associated α2δ subunits (CACNA2D) and possibly γ subunits (CACNG) (nomenclature conforms to Alexander et al., 2011). In nociceptive primary afferent neurones, various isoforms of each of these subunits exist, either expressed from similar genes or the result of alternative splicing (for reviews, see Lipscombe et al., 2002; Catterall et al., 2005; Yasuda and Adams, 2007; Catterall, 2011). This review focuses on the effects of ω- and α-conopeptides on N-type VGCC function and evaluates their potential in pain research.
ω-Conotoxins: primary structure and isolation
Typically, ω-conotoxins are composed of 24–30 amino acids, belong to the superfamily of disulfide-rich conopeptides and exhibit a characteristic Cys-residue scaffolding pattern (C-C-CC-C-C) and a protective C-terminal amide, which confers resistance to carboxylase activity (Table 1). Most of the variability in length among ω-conopeptides is in loops 3 and 4. The Cys residues are cross-linked by stabilizing disulfide bridges, resulting in a typical four-loop structure (Terlau and Olivera, 2004). The disulfide-coupled folding and the roles of unique molecular chaperones and enzymes governing post-translational modifications within a single conotoxin sequence are poorly understood. Despite the mechanistic differences in conditions between in vivo and in vitro oxidative folding, correctly folded and biologically active ω-conotoxins can be obtained in a simple oxidative environment (Figure 1) (Bulaj and Olivera, 2008; Bingham et al., 2010).
Table 1.
Amino acid sequence of selected ω-conotoxins from the venom of Conus catus, geographus and magus
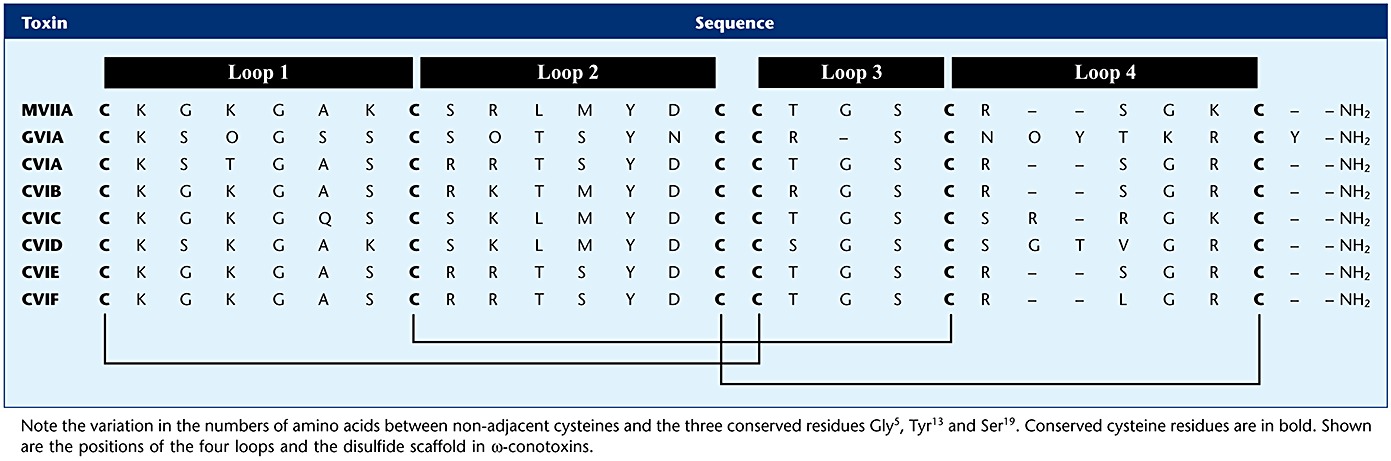 |
Figure 1.
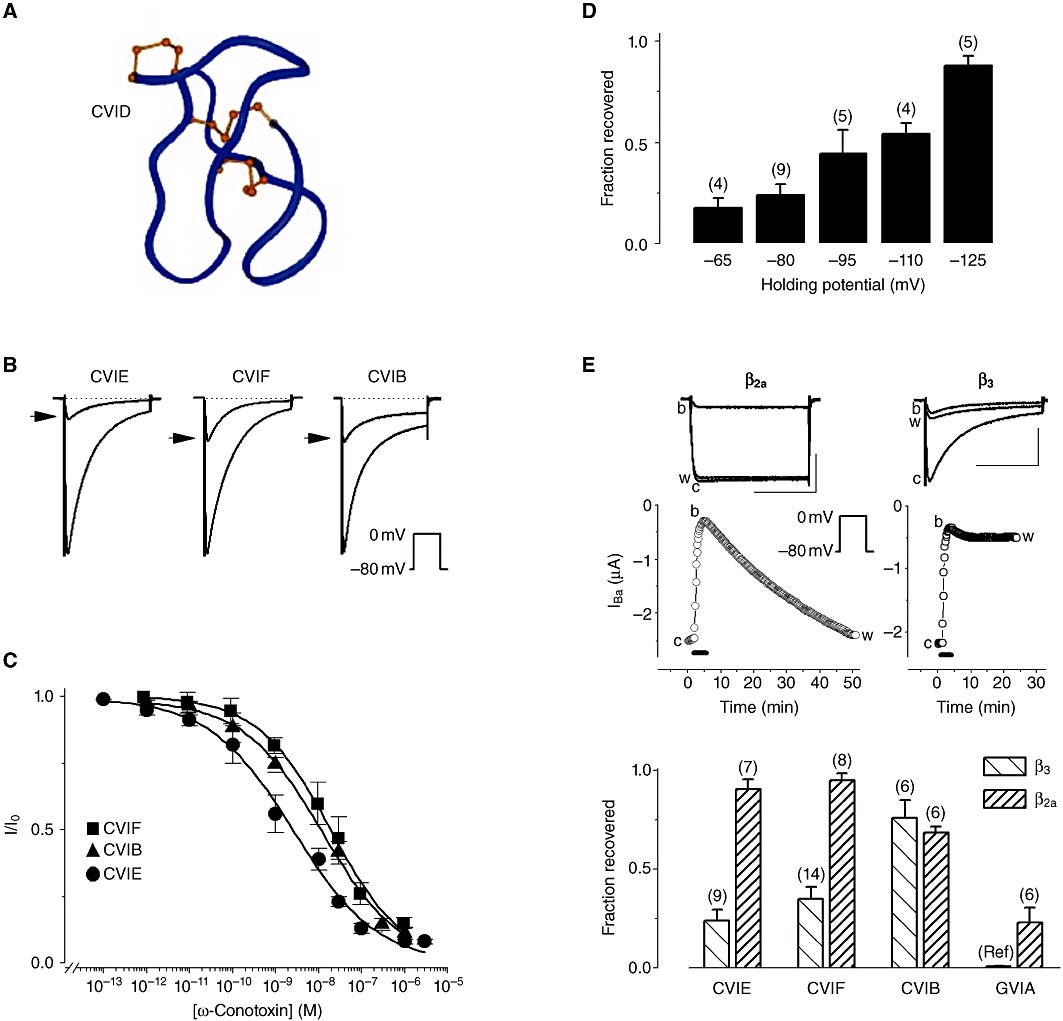
Block of recombinant N-type (Cav2.2) VGCCs by ω-conotoxins in Xenopus oocytes. (A) Three-dimensional structure of ω-conotoxin CVID highlighting the β-bridge and sheets (red arrows), the turns (blue arrows) and the location of disulfide bridges (ball-and-stick) [adapted from Lewis et al. (2000) ]. (B) Representative normalized Ba2+ current traces (IBa) obtained before and after (arrowhead) application of 100 nM ω-conotoxin CVIE, CVIF or CVIB. Inset: voltage protocol. (C) Concentration–response curves for the normalized peak IBa. (D) Recovery from block by ω-conotoxin CVIE (100 nM) is voltage-dependent. IBa traces were evoked by 0.1 Hz, 200 ms step depolarization to 0 mV from the indicated holding potential. (E) Recovery from CVIE block depends on the β subunit: channels with the ‘non-inactivating’β2a auxiliary subunit show full recovery, whereas channels with β3 exhibit weak recovery. Scale bars represent 1 µA and 100 ms. Bottom: reversibility of block after bath application of 100 nM ω-conotoxin CVIE, CVIF, CVIB or GVIA seen with α1B-b/α2δ1/β2a or α1B-b/α2δ1/β3 VGCCs. Data marked by ‘Ref’ are from Mould et al. (2004) and represent recovery from block by 1 nM GVIA. Data in B, C, D and E are modified with permission from Berecki et al. (2010).
The best characterized ω-conotoxins are the N-type VGCC-selective GVIA (Kerr and Yoshikami, 1984) and MVIIA (Olivera et al., 1987), isolated from fish-hunting cone snails. More recently, several novel ω-conotoxins, CVIA-F (Lewis et al., 2000; Adams et al., 2003; Berecki et al., 2010), were identified from the piscivorous Conus catus venom using a PCR-based strategy (see Hillyard et al., 1992). This approach had the advantage of supplying large amounts of synthetic compounds for structure-function analyses, restricting the need for crude material obtained directly from the cone snail's venom duct. To date, CVID, CVIE and CVIF are the most selective ω-conotoxins for N-type VGCCs (Lewis et al., 2000; Berecki et al., 2010).
N-type VGCC determinants of ω-conotoxin binding
ω-Conotoxins contribute to the rapid prey-immobilizing ‘motor cabal’ action of the cone snail venom (Terlau et al., 1996), which require multiple neurotoxins coordinately targeting presynaptic VGCCs and postsynaptic nAChRs and voltage-gated Na+ channels that underlie neuromuscular transmission (Terlau and Olivera, 2004). Calcium influx through VGCCs is necessary for a large number of intracellular events, including the control of neurotransmitter release. The use of ω-conotoxins has been pivotal in both the biochemical and the physiological characterization of VGCCs, primarily due to these agents having very high affinity for VGCC binding sites.
The most commonly used assay to determine the potencies of conotoxins at the N-type VGCC has been radioligand binding (see Nielsen et al., 2000). In such studies, ω-conotoxin affinity for depolarized cells or depolarized membrane fragments is very different from the characteristics of toxin binding to cells exhibiting physiological resting potentials (Table 2). As discussed in more detail below, the most likely reason for this difference is that characteristics of block and/or recovery from block change in accordance with the degree of voltage-dependent inactivation of N-type VGCCs.
Table 2.
ω-Conotoxins that inhibit mammalian N-type VGCCs
| Toxin and species | *Selectivity (VGCC type) | IC50 for N-type VGCCs (cloned/native) | Displacement of 125I-GVIA, IC50 | Mechanism of VGCC block | Reversibility of N-type VGCC block (cloned/native) |
|---|---|---|---|---|---|
| ω-GVIA | N1 | nd/72 nM2 (DRG) | 38 pM3 and §100 pM13 | Pore block4 (complete) | Irreversible in the presence of α1B/α2δ/β1b subunits5/irreversible in spinal cord slice preparation6 |
| Conus geographus | |||||
| ω-MVIIA | N7 | 5 nM8 (X)/200 nM9 (hc) | 55 pM3 | Pore block5 | Poorly reversible in the presence of α1B-b/α2δ1/β3 subunits; irreversible in the presence of α1B/α2δ/β1b subunits5/irreversible in spinal cord slice preparation6 |
| Conus magus | |||||
| ω-MVIIC | P/Q and N10 | nd/3–18 nM11 (SCG) | 7 nM3 and §4 nM13 | Pore block4 (complete) | nd/reversible in SCG neurone11 |
| C. magus | |||||
| ω-MVIID | N12 and P/Q | nd/nd | §100 nM13 | nd | nd/reversible in BC cells13 |
| C. magus | |||||
| ω-RVIA | N14 | nd/nd | §§8.4–10.4 nM15 | nd | nd/nd |
| Conus radiatus | |||||
| ω-TVIA | N16 | nd/nd | 251 pM17 | nd | nd/nd |
| Conus tulipa | |||||
| ω-SVIB | N18 and P/Q | nd/nd | 3.2 nM19 | nd | nd/nd |
| Conus striatus | |||||
| ω-CnVIIA | N20 | nd/nd | 2.7–3.7 pM20 | nd | nd/nd |
| Conus consors | |||||
| ω-CVIA | N3 | nd/nd | 560 pM3 | nd but likely pore block | nd/nd |
| Conus catus | |||||
| ω-CVIB | N3 and P/Q | 33 nM (X)2/23 nM2 (DRG) | 7.7 nM3 | nd but likely pore block | Reversible in the presence of α1B-b/α2δ1/β321 and α1B-b/α2δ1/β2a2 subunits/reversible in spinal cord slice preparation6 |
| C. catus | |||||
| ω-CVIC | N3 and P/Q | nd/nd | 7.6 nM3 | nd but likely pore block | nd/nd |
| C. catus | |||||
| ω-CVID | N3 | 21.8 nM8 (X)/9 nM2 (DRG) | 70 pM3 | nd but likely pore block | Reversible in the absence of α2δ1 subunit8; weakly reversible in the presence of α2δ1 subunit8/irreversible in spinal cord slice preparation6 |
| C. catus | |||||
| ω-SO3 | N22 | nd/160 nM23 (hc) | nd | nd but likely pore block | nd/almost fully reversible in hippocampal neurones23 |
| C. striatus | |||||
| ω-CVIE | N21 | 2.6 nM (X)21/nd | 19 pM21 | nd but likely pore block | Poorly reversible in the presence of α1B-b/α2δ1/β3 subunits21; reversible in the presence of α1B-b/α2δ1/β2a subunits21/poorly reversible in DRG neurone21; reversible in spinal cord slice preparation21 |
| C. catus | |||||
| ω-CVIF | N21 | 19.9 nM21 (X)/nd | 25 pM21 | nd but likely pore block | Poorly reversible in the presence of α1B-b/α2δ1/β3 subunits21; reversible in the presence of α1B-b/α2δ1/β2a subunits21/poorly reversible in DRG neurone21; reversible in spinal cord slice preparation21 |
| C. catus | |||||
| ω-FVIA | N24 | 11.5 nM24 (HEK293 cell)/nd | nd | nd | Reversible in HEK293 cells stably expressing α1B/α2δ/β1b subunits24/nd |
| Conus fulmen |
Selectivity determined from relative potencies to displace 125I-GVIA binding to rat brain membrane.
Displacement of 125I-GVIA binding to bovine adrenal medullary membrane.
Displacement of 125I- GVIA binding in Swiss Webster mouse cortex.
Superscript numbers between parentheses refer to references as follows: (l) Olivera et al., 1984; (2) Motin et al., 2007; (3) Lewis et al., 2000; (4) McDonough et al., 2002; (5) Feng et al., 2003; (6) Motin and Adams, 2008; (7) Olivera et al., 1987; (8) Mould et al., 2004; (9) Wen et al., 2005; (10) Hillyard et al., 1992; (11) McDonough et al., 1996; (12) Monje et al., 1993; (13) Gandia et al., 1997; (14) Olivera et al., 1985; (15) Abbott and Litzinger, 1994; (16) Miljanich and Ramachandran, 1995; (17) Miljanich et al., 1993; (18) Ramilo et al., 1992; (19) Nielsen et al., 2000; (20) Favreau et al., 2001; (21) Berecki et al., 2010; (22) Lu et al., 1999; (23) Wen et al., 2005; (24) Lee et al., 2010.
BC cell, bovine chromaffin cell; DRG, dorsal root ganglion neurone; hc, hippocampal neurone; HEK239 cell, human embryonic kidney cell line; GPCR, G protein-coupled receptor pathway; nACh, nicotinic acetylcholine receptor; nd, not determined (no published information); N, N-type VGCC; P/Q, P/Q-type VGCC; PTX, pertussis toxin; SCG, rat superior cervical ganglion neurone; X, Xenopus oocyte.
N-type VGCCs are relatively heterogeneous in terms of their biophysical properties due to the differences in subunit composition (Yasuda and Adams, 2007), to alternative splicing of the α1B subunit (Lipscombe et al., 2002) or modulation of the α1B subunit by cytosolic proteins, such as G-proteins (Dolphin, 1998). Because of this structural and functional heterogeneity, VGCC sensitivity to various ω-conotoxins can show considerable variation (Table 2). The influence of β and α2δ auxiliary subunits on the ability of ω-conotoxins GVIA, MVIIA, CVID and CVID analogues to block N-type VGCCs was examined using peripheral and central forms of rat Cav2.2 heterologously expressed in Xenopus oocytes (Mould et al., 2004). While the β3 subunit had no significant effect on potency, the α2δ subunit dramatically (∼100-fold) reduced ω-conotoxin affinity at both the central and the peripheral α1B isoforms (Mould et al., 2004; Berecki et al., 2010). This effect of α2δ was not affected by oocyte deglycosylation, which rules out an electrostatic shielding or repulsion effect of the heavily glycosylated extracellular α2 domain to the conotoxin binding site. Given that the α2δ subunit is up-regulated in certain pain states (Luo et al., 2001; Newton et al., 2001), the anti-nociceptive properties of ω-conotoxins might be affected by α2δ subunit expression levels.
In rat parasympathetic ganglia in situ, CVID inhibits evoked neurotransmitter release from preganglionic cholinergic nerve terminals, whereas MVIIA or GVIA produce no effect (Adams et al., 2003). This is most likely due to the presence of a tissue-specific α1B splice variant or auxiliary subunits of the N-type VGCC. Relatively small differences in the sequence of the N-type VGCC α1B subunit can influence ω-conotoxin-channel interaction: Gly(1326) may form a barrier, which controls the access of peptide toxins to their blocking site within the outer vestibule of the channel pore, as well as stabilizing the toxin-channel interaction (Feng et al., 2001b). In the Xenopus expression system, ω-conotoxins CVID and MVIIA have similar potencies to block central (α1B-d) and peripheral (α1B-b) splice variants of the rat N-type VGCCs when co-expressed with rat β3 (Lewis et al., 2000). Intracellular domains of α1B also affect ω-conotoxin affinity. The human N-type VGCC brain variants Cav 2.2 Delta 1 and Delta 2 lack the synaptic protein interaction site and are less sensitive to both GVIA and MVIIA (Kaneko et al., 2002).
The membrane potential can severely affect the severity of ω-conotoxin block and the kinetics of its onset and removal (Nowycky et al., 1985; Neely et al., 1993; Stocker et al., 1997; Feng et al., 2003). Block by ω-conotoxins GVIA, MVIIA and SNX-331, a derivative of MVIIC, is reversible by strong hyperpolarization. This effect of the membrane potential on reversibility of block could be accounted for by a modulated receptor model, in which toxin dissociation from the inactivated state is slower than from the resting state (state-dependent block) (Stocker et al., 1997). Recovery from CVIE and CVIF block also increases with membrane hyperpolarization, indicating that these molecules have a higher affinity for channels in the inactivated state (Figure 1) (Berecki et al., 2010). Contrary to this, hyperpolarization does not significantly enhance the extent of recovery from CVID block (Mould et al., 2004), whereas CVIB block and recovery also appear to be largely voltage-independent (Motin et al., 2007; Berecki et al., 2010).
Several other factors and mechanisms, including N-type VGCC heterogeneity, Ca2+–calmodulin binding (Liang et al., 2003), palmitoylation (Hurley et al., 2000) and steady-state N-type VGCC inactivation, affect the extent of recovery from block by different ω-conotoxins. Auxiliary subunits α2δ and β exert a pronounced regulation of steady-state inactivation characteristics of N-type VGCCs (Scott et al., 1996; Yasuda et al., 2004). Slowly inactivating N-type VGCCs with β2a subunits almost completely recover from CVIE or CVIF block, whereas rapidly inactivating VGCCs containing the β3 subunits exhibit only weak recovery (Figure 1E) (Berecki et al., 2010). Co-expression of α2δ with α1B and β3 subunits reduces recovery from block for CVID but not for MVIIA (Mould et al., 2004). Reversibility of block may be correlated with the effectiveness of these compounds to reverse different painful conditions in vivo and could indicate whether the side effects during the administration of these peptides can be controlled (Wright et al., 2000).
The site and mechanism of block of N-type VGCCs by ω-conotoxins
Although the blocking mechanism by GVIA, MVIIA and MVIIC has been studied in detail, the precise site of interaction of ω-conotoxins with the N-type VGCC is yet to be fully elucidated. At the single channel level, N-type VGCCs exhibit decreased open-state probability in the presence of GVIA, and the Ca2+ binding site (or sites), located in the pore of the channel, allosterically regulates the GVIA binding site (Witcher et al., 1993). Amino acid residues within the S5–H5 region of domain III (located in the vestibule of the α1B subunit of the N-type VGCC) are critical for binding ω-conotoxin GVIA, suggesting that this toxin acts via physical occlusion of the pore (Ellinor et al., 1994). The results of secondary structure prediction methods show a helix–coil–helix structure for this pore region, highly reminiscent of the classical EF hand, but with a weaker Ca2+ binding site than that found in calmodulin and other calcium-binding proteins (Doughty et al., 1998). There is sound electrophysiological evidence for the existence of this ion-permeation pathway-lining, weak Ca2+ binding – EF hand homology motif, at which both Ca2+ and Ba2+ can compete but only Ca2+ can produce an effect on channel gating (Zamponi and Snutch, 1996). An increase in external Ba2+ reduces both the potency and the rate of MVIIC block (Boland et al., 1994; McDonough et al., 1996), and conversely, a decrease in external Ba2+ accelerates the off-rate of GVIA (Liang and Elmslie, 2002). These findings support the idea that toxin molecules compete with Ba2+ bound at the locus of selectivity at the EF hand homology motif on the a1B subunit, immediately adjacent to the ω-conotoxin GVIA binding site.
There are at least two distinct toxin binding sites on the N-type channel and three on the P/Q-type (Cav2.1) channel. MVIIC binding produces complete N- and P/Q-type channel block, but this is prevented by previous partial channel block by ω-agatoxin-IIIA from the venom of the funnel-web spider Agelenopsis aperta, suggesting that MVIIC binds closer to the external mouth of the pore than ω-agatoxin-IIIA. On N-type channels, results are consistent with blockade of the channel pore by GVIA, MVIIA and MVIIC (McDonough et al., 2002; Feng et al., 2003). Although GVIA appears to be an open channel blocker, Yarotskyy and Elmslie (2009) showed that the N-type VGCC gating charge movement is also affected to some extent during the block, and this gating modulation alone inhibited the N-type VGCC current by 50% in a mathematical model.
As previously mentioned, single residues of the N-type VGCC can have a significant impact on toxin-channel interaction (Feng et al., 2001a). Stocker et al. (1997) suggested that, during toxin blockade, preferential interaction of the toxin with the inactivated channel must be accompanied by an externally detectable conformational change near the extracellular pore mouth. It therefore appears that ω-conopeptides exert their biological actions by occluding the ion-conducting pore of VGCCs, with a 1:1 stoichiometry for GVIA (McDonough et al., 2002; Feng et al., 2003). Because CVIA-F are structurally related to GVIA and MVIIA, it is likely that common molecular determinants underlie the block of N-type VGCCs by these ω-conopeptides.
ω-Conotoxin residues affecting the interaction with N-type VGCCs
C. catusω-conotoxins are small, hydrophilic and stable, and are therefore ideally suited for NMR spectroscopy (Daly and Craik, 2009). CVIA-F adopt a common fold, including the anti-parallel triple-stranded β-sheet previously reported in MVIIA, MVIIC and GVIA (Figure 1) (Nielsen et al., 2000). Detailed NMR studies of CVID have revealed the unique orientation of loop 4, which may account for the improved selectivity for the N-type VGCC over MVIIA and GVIA, and the presence of two hydrogen bonds (from the NH protons of Lys10 and Leu11) that enhance the stability of loop 2 (Lewis et al., 2000; Nielsen et al., 2000). Compared with CVID, CVIB lacks the two stabilizing hydrogen bonds between loops 2 and 4 and, like other ω-conotoxins lacking this structural stabilizing feature, it has a disordered loop 2 (Motin et al., 2007).
A series of studies determined the most important ω-conotoxin residues for interaction with VGCCs. The N-terminal amino group (Lampe et al., 1993) and Tyr13 (Kim et al., 1995) are essential for the binding to a number of residues, which partially overlap with the putative EF hand domain on the channel (Ellinor et al., 1994; Feng et al., 2001b; 2003). Homology models of a series of ω-conotoxins exhibit similar structural fold at positions 10, 11 and 13 (Berecki et al., 2010) (see Table 1). The amino acid in position 10 has a secondary effect on binding and affects the extent of ω-conotoxin reversibility (Mould et al., 2004); it is therefore likely that the pharmacophore of ω-conotoxins might be affected by amino acid substitution in this position. There is also evidence that some ω-conopeptides exert allosteric effects on channel structure when they bind (Stocker et al., 1997). The distribution of electrostatic surface properties of ω-conotoxins suggests that the ω-conotoxin–N-type VGCC interactions are dominated by ionic/electrostatic forces (Berecki et al., 2010). The difference in net charge between ω-conotoxins, in addition to differences in structure, can give us clues to explain the difference in potency, selectivity and reversibility among these related peptides.
ω-Conotoxins as neurophysiological tools and potential drug leads for the treatment of pain
The N-type VGCC is revealed as an exciting new target for pain treatment, following the approval of ω-conotoxin MVIIA (Ziconotide or Prialt) by the US Food and Drug Administration and European Medicines Agency for the management of severe chronic pain associated with cancer, acquired immune deficiency syndrome (AIDS) and neuropathies – refractory to other current pain medications. However, due to the wide distribution of N-type VGCCs and the peptidergic nature of MVIIA, current therapy with Prialt is limited to intrathecal delivery and has many side effects (Park and Luo, 2010). In the ascending pain pathways, the pathophysiological roles of N-type VGCCs have been demonstrated by using gene knockout mice or ω-conopeptides (Snutch, 2005; McGivern, 2006). CVIB and CVID-F have been efficiently used to evaluate the contribution of the N-type VGCC component to whole-cell currents in dorsal root ganglia (DRG) neurones (Motin et al., 2007; Berecki et al., 2010) and to characterize excitatory synaptic transmission between primary afferents and dorsal horn superficial lamina neurones. CVID completely and irreversibly inhibits excitatory postsynaptic current (epsc), whereas the epsc amplitude-reducing effect of CVIB and CVIE-F is reversible (Motin and Adams, 2008; Berecki et al., 2010).
During intrathecal application, ω-conopeptides bind to N-type VGCCs of the nociceptive A-δ and C fibres, the cell bodies of which reside in the DRG and project to secondary neurones of the dorsal horn in the spinal cord (Zamponi et al., 2009). N-type VGCC blocker ω-conopeptides are potent inhibitors of nociceptive signalling and therefore represent potential drug leads for the treatment of pain states. Clearly, there is a need for new conotoxins, which may have potential for the treatment of neuropathic pain, depression and various other neurological conditions, as well as exhibiting a more favourable ratio of anti-nociception to side effect profile than MVIIA. CVID-F exhibits the highest N-type over P/Q-type selectivity and could therefore serve as lead structures for novel analgesics. CVID (AM-336, Leconotide) has been tested as a therapeutic agent and has completed phase II clinical trial. Compared with MVIIA, CVID has an improved specificity for N-type VGCCs, better in vitro reversibility, and a more rapid onset and offset of action (Mould et al., 2004); in animal models, intrathecally applied CVID shows improved efficacy (Wright et al., 2000) and fewer cardiovascular side effects (Smith et al., 2002). Similar to CVID, ω-conotoxins CVIE or CVIF completely and reversibly relieve mechanical allodynia, as demonstrated in a nerve injury model of neuropathic pain (Berecki et al., 2010). However, further experiments are required to clarify the anti-nociception to side effect profiles of CVIE-F. Currently, the methods for administering ω-conopeptides as therapeutic agents are limited to intrathecal delivery. Peptides, in general, are unable to cross the blood–brain barrier due to their inherently large size and hydrophilic nature. Therefore, alternative strategies are needed, which would convert active peptides into non-peptidic small molecules and/or would limit their systemic degradation. Schroeder et al. (2004) identified cyclic pentapeptides, which contain residues of loop 2 of CVID and mimic the binding of CVID at the N-type VGCC. A better understanding of conopeptide-VGCC interaction would help in the development of small molecules that selectively and tissue-specifically target N-type VGCCs. Future experiments with structural analogues could result in compounds with stabilized structure, which would permit oral administration. However, development of ω-conotoxin mimetics (through the mimicry of N-type VGCC binding domains) is currently very challenging (Baell et al., 2004). Recently, the cell-specific inhibition of P/Q- and N-type VGCCs in vivo was demonstrated with a tethered-toxin approach, resulting in cell-specific and cell-autonomous silencing of neurotransmission (Auer et al., 2010). This approach could serve as a viable alternative therapy to intrathecal ω-conopeptide delivery.
α-Conotoxins: isolation, structures, targets
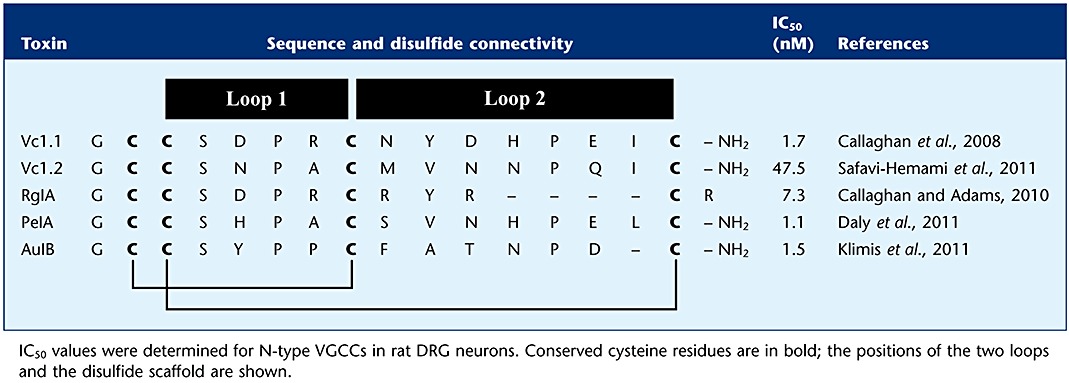
α-Conotoxins have been isolated either by fractionation and purification from cone snail crude venom duct contents or via PCR screens of cDNA sequences derived from Conus venom ducts of fish-, worm- and mollusc-hunting species of Conus (Olivera et al., 1985; Janes, 2005; Kaas et al., 2008). The first α-conotoxins were discovered more than 30 years ago (Spence et al., 1977; Gray et al., 1981) and are currently classified according to their gene superfamily membership and cysteine framework connectivity. The number of amino acids between the C2 and C3 and the C3 and C4 residues further divides the α-conotoxins into five subfamilies: α3/5, α4/3, α4/4, α4/6 and α4/7 (Azam and McIntosh, 2009). The venom repertoire of cone snails undergoes ontogenetic changes, suggestive of alternative functions of peptide toxins during development (Safavi-Hemami et al., 2011). To date, at least 39 mature α-conotoxin sequences of the type I framework are included in the ConoServer database (Kaas et al., 2010). Members of this family typically range in size from 12 to 16 amino acids, exhibit a cysteine framework with two disulfide bonds in C1–C3 and C2–C4 connectivity, and often have an amidated C-terminus (Table 3).
Table 3.
Amino acid sequences of α-conotoxins that inhibit N-type (Cav2.2) calcium channels via GABAB receptor activation
 |
In contrast to the ω-conotoxins that are usually classified as selective antagonists of VGCCs, α-conotoxins are pharmacologically classified as competitive antagonists of muscle and neuronal nAChRs (Adams et al., 1999; Janes, 2005; Bingham et al., 2010). However, a series of recent publications clearly demonstrates that a subset of α-conotoxins indirectly inhibit neuronal N-type VGCCs by acting effectively as agonists of the GABAB GPCR (Figure 2) (Callaghan et al., 2008; Daly et al., 2011; Klimis et al., 2011; Safavi-Hemami et al., 2011). There are also examples of conotoxins that target other GPCRs, including the α1-adrenoreceptor (Sharpe et al., 2001; 2003) and the neurotensin receptor (Craig et al., 1999). GABAB receptors are heterodimers composed of a GABAB1 subunit and the GABAB2 subunit (Figure 3) (Bettler et al., 2004), and their modulation by agonists is well established as a mechanism for producing pain relief (Goudet et al., 2009). Activation of presynaptic GABAB receptors leads to inhibition of N-type calcium channels, which, in turn, results in diminished release of GABA (autoreceptors) and other neurotransmitters (heteroreceptors), whereas postsynaptic GABAB receptor activation is associated with G-protein regulated inward-rectifying potassium (GIRK) channel activation, resulting in hyperpolarization. In the human peripheral and central nervous system, GABAB1 has two major site variants (isoforms), GABAB1a and GABAB1b, which result from alternative transcription initiation and primarily differ by the presence of a pair of sushi repeats (domains) in GABAB1a (Kaupmann et al., 1997), thought to interact with proteins involved in cell–cell adhesion (Bettler et al., 2004). The GABAB1 subunit contains an endoplasmic reticulum (ER) retention domain that is concealed when it forms a dimer with GABAB2, allowing for surface expression (Margeta-Mitrovic et al., 2000), whereas GABAB2 regulates internalization of the receptor (Hannan et al., 2011). Recently, the GABAB heterodimeric receptor has been shown to spontaneously form dimers of heterodimers, or larger complexes in heterologous systems via the interaction of GABAB1 subunits, which leads to negative functional cooperativity between GABAB heterodimers (Comps-Agrar et al., 2011; Kniazeff et al., 2011).
Figure 2.
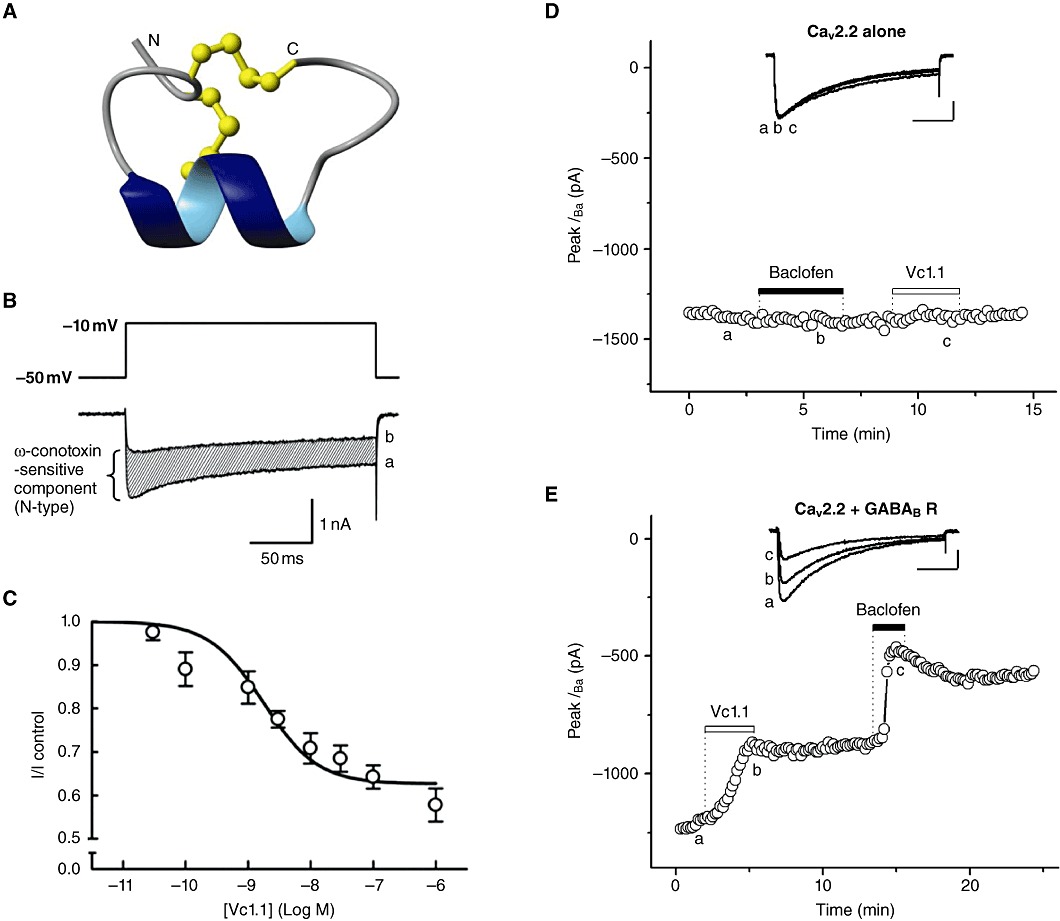
α-Conotoxins inhibit N-type (CaV2.2) VGCCs via the GABAB receptor. (A) Three-dimensional structure of α-conotoxin Vc1.1. Disulfide bonds are shown in ball-and-stick representation and the N and C termini are marked adapted from Clark et al. (2006). (B) Superimposed traces of depolarization-activated whole-cell IBa recorded using 2 mM Ba2+ as the charge carrier, elicited in the absence (a) and presence (b) of 100 nM Vc1.1. Inset: voltage protocol. (C) Concentration–response curve for inhibition of IBa in DRG neurones by Vc1.1; IC50= 1.7 nM. B and C are adapted from Callaghan et al. (2008), with permission. (D) The GABAB receptor mediates Vc1.1 inhibition of N-type (CaV2.2) VGCC currents in transiently transfected HEK-293 cells. Time course of peak IBa evoked with 0.1 Hz, 250 ms depolarizing test pulses from −80 to 10 mV and recorded using 20 mM Ba2+ as the charge carrier; upward deflections represent current inhibition. Baclofen (50 µM) or Vc1.1 (100 nM) does not affect IBa in cells co-transfected only with cDNAs of α1-B, α2δ1 and β1 VGCC subunits (n= 3). Inset: representative IBa traces are shown at the times indicated by lowercase letters. In cells co-transfected with (CaV2.2) VGCC subunits and GABAB1,B2 subunits, Vc1.1 and baclofen (n= 4) (E) inhibit IBa. Scale bars in D and E represent 500 pA and 50 ms (D and E; G. Berecki and D.J. Adams, unpubl. obs.).
Figure 3.
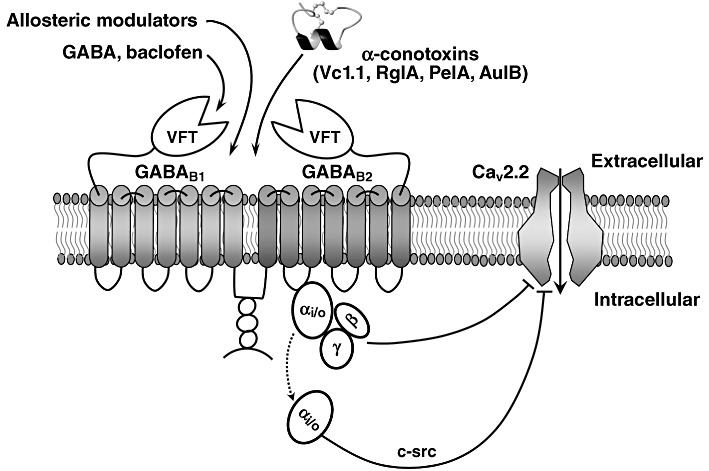
GABAB receptor activation by analgesic α-conotoxins. The highly conserved structural scaffold of the α-conotoxins consists of two disulfide bonds that stabilize a central helical region. GABAB receptor is a heterodimer with two almost identical subunits that are both required for a functional receptor. The GABAB1 subunit is involved in ligand binding and the GABAB2 subunit interacts with the G-protein. The natural ligand of the receptor, GABA, binds to a cleft within the large N-terminal ‘Venus fly-trap (VFT)’ domain of the GABAB1 subunit, triggering a conformational change in the receptor that facilitates interaction with the G-protein by the GABAB2 subunit. GPCR activation leads to dissociation of Gα from Gβγ subunits. A single GPCR can couple to either one or more families of Gα proteins, which activate several downstream effectors (Tedford and Zamponi, 2006). Upon ligand binding, Gβγ subunits function as a dimer to interact with many signalling molecules and with ion channels. The schematically shown pore-forming α1-B subunit of the N-type (Cav2.2) VGCC consists of IV homologous domains linked by cytoplasmic loops (referred to as I-II, II-III and III-IV linkers) and cytoplasmic N- and C-terminal regions.
GPCR stimulation is associated with GDP replacement with GTP and dissociation of the heterotrimeric G protein into Gα-GTP and Gβγ dimers, which then interact with a variety of different effectors (for reviews, see Pierce et al., 2002; Oldham and Hamm, 2006; 2008). Regulators of G-protein signalling proteins accelerate the Gα subunit GTPase activity, thus increasing the rate of G-protein inactivation (McCudden et al., 2005). The activated GPCRs are also phosphorylated by G protein-coupled receptor kinases, resulting in recruitment of β-arrestins to terminate G protein-dependent signalling (Shukla et al., 2010).
N-type VGCC determinants of modulation by GPCRs
The N-type VGCC can be depressed by several neurotransmitters and this modulation constitutes a major mechanism of presynaptic inhibition (Bean, 1989). Two distinct pathways are generally identified in this signalling, between which the voltage-dependent inhibition is the most common form, involving Gβγ binding directly to the I-II linker of the VGCC α1 subunit of the N-type VGCC (Bean, 1989; Hille, 1994; Tedford et al., 2010) (Figure 3). The free Gβγ heterodimers do not only signal exclusively to VGCCs but also modulate downstream effectors such as phospholipase C and adenylyl cyclase. The Gα subunits trigger various intracellular pathways, which may converge on VGCCs to either up-regulate or inhibit their activities (Bernheim et al., 1991; Beech et al., 1992; Delmas et al., 1998; Kammermeier et al., 2000). Because VGCC modulation via second messenger pathways cannot be reversed by strong membrane depolarizations, it is referred to as voltage-independent inhibition. This pathway involves diffusible second messengers and occurs over a slower time course than voltage-dependent inhibition (Luebke and Dunlap, 1994). The voltage-dependent and voltage-independent modulation of neuronal Cav2 channels has been reviewed recently (Tedford and Zamponi, 2006; Brown and Sihra, 2008; Weiss, 2009; Currie, 2010).
As previously mentioned, N-type VGCCs are rather heterogeneous regarding their biophysical properties due to the differences in subunit composition. Alternative splicing markedly expands the functional potential of VGCCs (Gray et al., 2007). In pain pathways, exon 37a not only tailors CaV2.2 channels towards specific roles in pain (Altier et al., 2007) but also alters CaV2.2 channel modulation by GPCRs, acting as a molecular switch linking GPCRs to voltage-independent inhibition of the N-type VGCC (Lipscombe and Raingo, 2007). The 37a splice isoform, which is preferentially expressed in nociceptive neurones (Bell et al., 2004), exhibits both voltage-dependent and voltage-independent inhibition, whereas the 37b isoform exhibits only voltage-dependent inhibition due to the absence of a consensus site for tyrosine kinase phosphorylation in position 1747 (Raingo et al., 2007). In nociceptors of a murine model lacking exon 37a in Cav2.2, spinal morphine analgesia is diminished in vivo due to a decreased activity-independent G protein inhibition of N-type VGCCs (Andrade et al., 2010).
A novel mechanism of N-type VGCC modulation by α-conotoxins
We examined α-conotoxin Vc1.1 (α4/7), a synthetic peptide, against other known targets involved in pain pathways as it had previously been shown that conotoxins may target more than one class of channel or receptor family. For example, pI14a, a conotoxin from the vermivorous cone Conus planorbis, was reported to inhibit both voltage-gated (Kv1.6) and ligand-gated (α3β4, α1β1εδ nAChR) ion channels (Imperial et al., 2006). We found that Vc1.1 and RgIA (α4/3) inhibit N-type VGCC currents in mammalian DRG neurones, whereas vc1a, the native post-translationally modified peptide of Vc1.1, was inactive. The inhibition was indirect and was abolished in the presence of GDPβS, Pertussis toxin (PTX), the selective peptide inhibitor of pp60c-src tyrosine kinase and GABAB receptor antagonists (CGP55845, CGP54626 and phaclofen), whereas selective nAChR, muscarinic AChR, GABAA, α1- and α2-adrenergic, µ-opioid antagonists had no effect on N-type VGCC currents (Callaghan et al., 2008). Inhibition of N-type VGCC currents depended on the frequency and duration of the depolarizing pulse in the presence of Vc1.1, indicating use-dependence of block. The onset of the Vc1.1 effect was slower than that of baclofen and was not reversed by prepulse depolarizations or by toxin washout. It was suggested that this novel type of VGCC inhibition is not a conventional Gβγ-mediated voltage-dependent mechanism but is largely voltage-independent, with the involvement of a PTX-sensitive Gαi/o subfamily and of a c-src kinase (Callaghan et al., 2008). Taken together, these results indicate that analgesic Vc1.1 and Rg1A modulate native N-type CaV2.2 channel currents via a novel mechanism, acting effectively as agonists via G protein-coupled GABAB receptors.
In rat DRG neurones, the presence of GABAB receptor subunits GABAB1 and GABAB2 has been confirmed (Towers et al., 2000) and the GABAB receptor-mediated inhibition of CaV2.2 channels by baclofen has been reported previously (Dolphin and Scott, 1987; Tatebayashi and Ogata, 1992). Neither baclofen nor Vc1.1 mobilized intracellular calcium release in rat DRG neurones, consistent with a Gαo-mediated modulation of N-type VGCCs (Callaghan et al., 2008). In α9 nAChR knockout mice, the inhibition of N-type VGCC currents by Vc1.1 and RgIA was found to be intact, indicating that the α9-containing nAChRs do not mediate the actions of these α-conotoxins (Callaghan and Adams, 2010). Furthermore, we have recently shown that AuIB (α4/6), a selective antagonist of α3β4 nAChRs, inhibits N-type VGCCs via a GABAB receptor-mediated pathway but does not antagonize α9α10 nAChRs (Klimis et al., 2011). PeIA (α4/7), another conotoxin selective for α9α10 nAChRs, inhibits N-type VGCC currents in DRG neurones (Daly et al., 2011). Vc1.2 (α4/7), a recently identified toxin isolated from embryonic Conus victoriae, also inhibits N-type VGCCs; however, it exhibits distinct nAChR selectivity to the adult toxin (Safavi-Hemami et al., 2011). siRNA knockdown of functional GABAB receptor expression significantly reduces the inhibition of N-type VGCC current in response to baclofen, Vc1.1, Rg1A and AuIB in isolated rat DRG neurones (H. Cuny and D.J. Adams, unpubl. obs.). Vc1.1 and RgIA were unable to displace binding of the selective GABAB receptor antagonist [3H]CGP54626 to either human GABAB (1b,2) receptors transiently expressed in HEK293T cells (McIntosh et al., 2009) or rat DRG neurones (S. Nevin and D.J. Adams, unpubl. obs.). These results suggest that Vc1.1 binds to a different ligand binding site than [3H]CGP54626, the competitive antagonist for the GABA binding site on the GABAB1 subunit (see Figure 3). The sequences and IC50 values of α-conotoxins that inhibit N-type calcium channels are shown in Table 3. Generally, α-conopeptides suffer from the disadvantage of short biological half-lives and poor activity when administered orally. Remarkably, synthetic cyclization of Vc1.1 produces an orally active compound with improved in vivo stability, higher potency for GABAB-mediated inhibition of N-type calcium channels and lower potency at α9α10 nAChRs (Clark et al., 2010).
Vc1.1 had no direct effect on recombinant CaV2.1 (P/Q-type), CaV2.3 (R-type) and CaV1.2 (L-type) VGCCs expressed in Xenopus oocytes (Callaghan et al., 2008). However, in approximately two-thirds of oocytes expressing recombinant CaV2.2 channels, application of baclofen inhibited Ba2+ currents, consistent with the previously reported presence of endogenous GABAB receptors in batches of Xenopus oocytes (Yang et al., 2001). In these oocytes, Vc1.1 and Rg1A also inhibited Ba2+ currents through recombinant CaV2.2 channels (see supplemental data in Callaghan et al., 2008). McIntosh et al. (2009) reported that α-conotoxin Vc1.1 does not modulate GIRK (1,4) channels heterologously expressed with GABAB receptors in Xenopus oocytes. Recently, GABAB receptor-mediated modulation of transiently or stably expressed CaV2.2 channels in HEK-293 cells, co-transfected with cDNAs of cloned human GABAB1 and GABAB2 receptor subunits, was successfully reconstituted (Figure 2D,E) (G. Berecki and D.J. Adams, unpubl. obs.). This approach is particularly useful for investigating Cav2.2 channel modulation via GABAB receptors without significant interference of endogenous ion channels.
Anti-allodynic effects of α-conotoxins
Several α-conotoxins with different selectivity for nAChR subtypes have been shown to be effective analgesics in rat models of neuropathic pain (Sandall et al., 2003; Satkunanathan et al., 2005; McIntosh et al., 2009; Klimis et al., 2011). Intramuscular injection of Vc1.1 reversed surgically induced hyperalgesia in rats following chronic constriction injury or partial nerve ligation, with the relief persisting for 1 week post-treatment (Satkunanathan et al., 2005). RgIA also exhibited acute and persistent anti-nociceptive effects in rats following partial nerve ligation (Vincler et al., 2006). Both conopeptides most selectively target α9α10 nAChRs over other nAChR subtypes (Vincler et al., 2006; Nevin et al., 2007), leading to the proposal that antagonism of this receptor is a molecular target for the treatment of neuropathic pain (Vincler et al., 2006). Conversely, the native peptide of Vc1.1, vc1a, has been found to be equipotent at α9α10 nAChRs but inactive in rat neuropathic pain assays (Livett et al., 2006; Nevin et al., 2007), suggesting that an alternative mechanism may underlie the analgesic effects observed with Vc1.1 and RgIA. Recently, Klimis et al. (2011) provided evidence that α9α10 nAChRs are not significantly involved in the pain-relieving actions of α-conotoxins, and that Vc1.1 and AuIB exhibit distinct selectivity profiles for N-type VGCCs and nAChRs. The intramuscular administration of Vc1.1 and AuIB produced a long lasting reversal of allodynia in partial nerve ligation rats. The anti-allodynic effect of Vc1.1 was almost completely antagonized by the selective GABAB inhibitor SCH 50911 in vivo (Klimis et al., 2011). It was concluded that the activation of GABAB receptors by α-conotoxins and subsequent inhibition of N-type VGCCs are the most likely mechanisms mediating these effects, despite their modulation, at lower efficacy, of the α9α10 nAChR.
N-type VGCC inhibition by selective antagonists or via GPCRs produces analgesia in animals and humans (McGivern, 2006; Zamponi et al., 2009). The precise mechanism within the nociceptive pathway responsible for the reversal of allodynia by α-conotoxins is not yet fully understood. In vitro patch-clamp electrophysiological studies in rat spinal cord slices could not identify any modulation of primary afferent synaptic transmission by α-conotoxins Vc1.1 or RgIA (L. Motin and D.J. Adams, unpubl. obs.). This might be due to differences in the signalling mechanisms in DRG cell bodies and their central nerve terminals and/or due to the presence of a Cav2.2 channel splice variant(s) in the presynaptic nerve terminal (Altier et al., 2007; Raingo et al., 2007; Andrade et al., 2010; Gardezi et al., 2010) that is not affected by α-conotoxins.
Conclusion and future directions
Understanding the precise mechanism of action of conotoxins is critical to fully exploit the potential of these peptides to target the N-type VGCCs. Future research will be required to determine the precise binding site(s) for the ω-conotoxins on Cav2.2 and to elucidate the potential of ω-conotoxin CVID-F analogues as pain therapeutics. Similarly, the precise binding site on GABAB receptor subunits for α-conotoxins has to be determined. Specific radiolabelling of the α-conopeptides could help to confirm specific binding to and distribution of GABAB receptors. A more complete understanding of the structure–activity relationships of known and chemically modified α-conotoxins that target GABAB receptors will require further mutational studies (e.g. alanine scanning). This will help in the design of more potent and specific analogues for the GABAB receptor versus nAChR subtypes, including peptide modification to improve stability and oral availability. Further studies aimed at determining the precise signalling pathway and the mechanism of VGCC inhibition by various α-conotoxins via GPCRs will be invaluable for the development of more potent and selective analogues of the GABAB receptor.
Acknowledgments
Our research is supported by grants from the National Health & Medical Research Council and the Australian Research Council (ARC) to DJA. DJA is an ARC Australian Professorial Fellow.
Glossary
- AuIB
α-conotoxin AuIB
- Cav2.1
P/Q-type calcium channel
- Cav2.2
N-type calcium channel
- CVID
ω-conotoxin CVID
- CVIE
ω-conotoxin CVIE
- CVIF
ω-conotoxin CVIF
- DRG
dorsal root ganglion
- epsc
excitatory postsynaptic current
- GABA
γ-aminobutyric acid
- GEF
guanine nucleotide exchange factors
- GIRK
G protein-activated inwardly rectifying potassium channel
- GPCR
G protein-coupled receptor
- GVIA
ω-conotoxin GVIA
- MVIIA
ω-conotoxin MVIIA
- nAChR
nicotinic acetylcholine receptor
- PeIA
α-conotoxin PeIA
- RgIA
α-conotoxin RgIA
- Vc1.1
α-conotoxin Vc1.1
- VGCC
voltage-gated calcium channel
Conflict of interest
None.
References
- Abbott JR, Litzinger MJ. Different ω-conotoxins mark the development of Swiss Webster mouse cortex suggesting N-type voltage sensitive calcium channel subtypes. Int J Dev Neurosci. 1994;12:43–47. doi: 10.1016/0736-5748(94)90094-9. [DOI] [PubMed] [Google Scholar]
- Adams DJ, Alewood PF, Craik DJ, Drinkwater RD, Lewis RJ. Conotoxins and their potential pharmaceutical applications. Drug Dev Res. 1999;46:219–234. [Google Scholar]
- Adams DJ, Smith AB, Schroeder CI, Yasuda T, Lewis RJ. ω-Conotoxin CVID inhibits a pharmacologically distinct voltage-sensitive calcium channel associated with transmitter release from preganglionic nerve terminals. J Biol Chem. 2003;278:4057–4062. doi: 10.1074/jbc.M209969200. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th Edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altier C, Dale CS, Kisilevsky AE, Chapman K, Castiglioni AJ, Matthews EA, et al. Differential role of N-type calcium channel splice isoforms in pain. J Neurosci. 2007;27:6363–6373. doi: 10.1523/JNEUROSCI.0307-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade A, Denome S, Jiang YO, Marangoudakis S, Lipscombe D. Opioid inhibition of N-type Ca2+ channels and spinal analgesia couple to alternative splicing. Nat Neurosci. 2010;13:1249–1256. doi: 10.1038/nn.2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auer S, Stürzebecher AS, Jüttner R, Santos-Torres J, Hanack C, Frahm S, et al. Silencing neurotransmission with membrane-tethered toxins. Nat Methods. 2010;7:229–236. doi: 10.1038/nmeth.1425. [DOI] [PubMed] [Google Scholar]
- Azam L, McIntosh JM. Alpha-conotoxins as pharmacological probes of nicotinic acetylcholine receptors. Acta Pharmacol Sin. 2009;30:771–783. doi: 10.1038/aps.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baell JB, Duggan PJ, Lok YP. ω-Conotoxins and approaches to their non-peptide mimetics. Aust J Chem. 2004;57:179–185. [Google Scholar]
- Bean BP. Neurotransmitter inhibition of neuronal calcium currents by changes in channel voltage dependence. Nature. 1989;340:153–156. doi: 10.1038/340153a0. [DOI] [PubMed] [Google Scholar]
- Beech DJ, Bernheim L, Hille B. Pertussis toxin and voltage dependence distinguish multiple pathways modulating calcium channels of rat sympathetic neurons. Neuron. 1992;8:97–106. doi: 10.1016/0896-6273(92)90111-p. [DOI] [PubMed] [Google Scholar]
- Bell TJ, Thaler C, Castiglioni AJ, Helton TD, Lipscombe D. Cell-specific alternative splicing increases calcium channel current density in the pain pathway. Neuron. 2004;41:127–138. doi: 10.1016/s0896-6273(03)00801-8. [DOI] [PubMed] [Google Scholar]
- Berecki G, Motin L, Haythornthwaite A, Vink S, Bansal P, Drinkwater R, et al. Analgesic ω-conotoxins CVIE and CVIF selectively and voltage-dependently block recombinant and native N-type calcium channels. Mol Pharmacol. 2010;77:139–148. doi: 10.1124/mol.109.058834. [DOI] [PubMed] [Google Scholar]
- Bernheim L, Beech DJ, Hille B. A diffusible second messenger mediates one of the pathways coupling receptors to calcium channels in rat sympathetic neurons. Neuron. 1991;6:859–867. doi: 10.1016/0896-6273(91)90226-p. [DOI] [PubMed] [Google Scholar]
- Bettler B, Kaupmann K, Mosbacher J, Gassmann M. Molecular structure and physiological functions of GABAB receptors. Physiol Rev. 2004;84:835–867. doi: 10.1152/physrev.00036.2003. [DOI] [PubMed] [Google Scholar]
- Bingham JP, Mitsunaga E, Bergeron ZL. Drugs from slugs – past, present and future perspectives of ω-conotoxin research. Chem Biol Interact. 2010;183:1–18. doi: 10.1016/j.cbi.2009.09.021. [DOI] [PubMed] [Google Scholar]
- Boland LM, Morrill JA, Bean BP. ω-Conotoxin block of N-type calcium channels in frog and rat sympathetic neurons. J Neurosci. 1994;14:5011–5027. doi: 10.1523/JNEUROSCI.14-08-05011.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DA, Sihra TS. Presynaptic signaling by heterotrimeric G-proteins. Handb Exp Pharmacol. 2008;184:207–260. doi: 10.1007/978-3-540-74805-2_8. [DOI] [PubMed] [Google Scholar]
- Bulaj G, Olivera BM. Folding of conotoxins: formation of the native disulfide bridges during chemical synthesis and biosynthesis of Conus peptides. Antioxid Redox Signal. 2008;10:141–155. doi: 10.1089/ars.2007.1856. [DOI] [PubMed] [Google Scholar]
- Callaghan B, Adams DJ. Analgesic α-conotoxins Vc1.1 and RgIA inhibit N-type calcium channels in sensory neurons of alpha9 nicotinic receptor knockout mice. Channels (Austin) 2010;4:51–54. doi: 10.4161/chan.4.1.10281. [DOI] [PubMed] [Google Scholar]
- Callaghan B, Haythornthwaite A, Berecki G, Clark RJ, Craik DJ, Adams DJ. Analgesic α-conotoxins Vc1.1 and Rg1A inhibit N-type calcium channels in rat sensory neurons via GABAB receptor activation. J Neurosci. 2008;28:10943–10951. doi: 10.1523/JNEUROSCI.3594-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JN, Meyer RA. Mechanisms of neuropthic pain. Neuron. 2006;52:77–92. doi: 10.1016/j.neuron.2006.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA. Voltage-gated calcium channels. Cold Spring Harb Perspect Biol. 2011;3:a003947. doi: 10.1101/cshperspect.a003947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA, Perez-Reyes E, Snutch TP, Striessnig J. Nomenclature and structure-function relationhships of voltage-gated calcium channels. Pharmacol Rev. 2005;57:411–425. doi: 10.1124/pr.57.4.5. [DOI] [PubMed] [Google Scholar]
- Clark RJ, Fischer H, Nevin ST, Adams DJ, Craik DJ. The synthesis, structural characterization, and receptor specificity of the α-conotoxin Vc1.1. J Biol Chem. 2006;281:23254–23263. doi: 10.1074/jbc.M604550200. [DOI] [PubMed] [Google Scholar]
- Clark RJ, Jensen J, Nevin ST, Callaghan BP, Adams DJ, Craik DJ. The engineering of an orally active conotoxin for the treatment of neuropathic pain. Angew Chem Int Ed Engl. 2010;49:6545–6548. doi: 10.1002/anie.201000620. [DOI] [PubMed] [Google Scholar]
- Comps-Agrar L, Kniazeff J, Nørskov-Lauritsen L, Maurel D, Gassmann M, Gregor N, et al. The oligomeric state sets GABAB receptor signalling efficacy. EMBO J. 2011;30:2336–2349. doi: 10.1038/emboj.2011.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig AG, Norberg T, Griffin D, Hoeger C, Akhtar M, Schmidt K, et al. Contulakin-G, an O-glycosylated invertebrate neurotensin. J Biol Chem. 1999;274:13752–13759. doi: 10.1074/jbc.274.20.13752. [DOI] [PubMed] [Google Scholar]
- Currie KP. G protein modulation of CaV2 voltage-gated calcium channels. Channels (Austin) 2010;4:497–509. doi: 10.4161/chan.4.6.12871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly NL, Craik DJ. Structural studies of conotoxins. IUBMB Life. 2009;61:144–150. doi: 10.1002/iub.158. [DOI] [PubMed] [Google Scholar]
- Daly NL, Callaghan B, Clark RJ, Nevin ST, Adams DJ, Craik DJ. Structure and activity of alpha-conotoxin PeIA at nicotinic acetylcholine receptor subtypes and GABAB receptor-coupled N-type calcium channels. J Biol Chem. 2011;286:10233–10237. doi: 10.1074/jbc.M110.196170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas P, Brown DA, Dayrell M, Abogadie FC, Caulfield MP, Buckley NJ. On the role of endogenous G-protein βγ subunits in N-type Ca2+ current inhibition by neurotransmitters in rat sympathetic neurones. J Physiol. 1998;506:319–329. doi: 10.1111/j.1469-7793.1998.319bw.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin AC. Mechanisms of modulation of voltage-dependent calcium channels by G proteins. J Physiol. 1998;506:3–11. doi: 10.1111/j.1469-7793.1998.003bx.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin AC, Scott RH. Calcium channel currents and their inhibition by (-)-baclofen in rat sensory neurones: modulation by guanine nucleotides. J Physiol. 1987;386:1–17. doi: 10.1113/jphysiol.1987.sp016518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doughty SW, Blaney FE, Orlek BS, Richards WG. A molecular mechanism for toxin block in N-type calcium channels. Protein Eng. 1998;11:95–99. doi: 10.1093/protein/11.2.95. [DOI] [PubMed] [Google Scholar]
- Ellinor PT, Zhang J-F, Horne WA, Tsien RW. Structural determinants of the blockade of N-type calcium channels by a peptide neurotoxin. Nature. 1994;372:272–275. doi: 10.1038/372272a0. [DOI] [PubMed] [Google Scholar]
- Favreau P, Gilles N, Lamthanh H, Bournaud R, Shimahara T, Bouet F, et al. A new ω-conotoxin that targets N-type voltage-sensitive calcium channels with unusual specificity. Biochemistry. 2001;40:14567–14575. doi: 10.1021/bi002871r. [DOI] [PubMed] [Google Scholar]
- Feng ZP, Hamid J, Doering C, Jarvis SE, Bosey GM, Bourinet E, et al. Amino acid residues outside of the pore region contribute to N-type calcium channel permeation. J Biol Chem. 2001a;276:5726–5730. doi: 10.1074/jbc.C000791200. [DOI] [PubMed] [Google Scholar]
- Feng ZP, Hamid J, Doering C, Bosey GM, Snutch TP, Zamponi GW. Residue Gly1326 of the N-type calcium channel alpha 1B subunit controls reversibility of ω-conotoxin GVIA and MVIIA block. J Biol Chem. 2001b;276:15728–15735. doi: 10.1074/jbc.M100406200. [DOI] [PubMed] [Google Scholar]
- Feng ZP, Doering CJ, Winkfein RJ, Beedle AM, Spafford JD, Zamponi GW. Determinants of inhibition of transiently expressed voltage-gated calcium channels by ω-conotoxins GVIA and MVIIA. J Biol Chem. 2003;278:20171–20178. doi: 10.1074/jbc.M300581200. [DOI] [PubMed] [Google Scholar]
- Gandia L, Lara B, Imperial JS, Villarroya M, Albillos A, Maroto R, et al. Analogies and differences between ω-conotoxins MVIIC and MVIID: binding sites and functions in bovine chromaffin cells. Pflugers Arch. 1997;435:55–64. doi: 10.1007/s004240050483. [DOI] [PubMed] [Google Scholar]
- Gardezi SR, Taylor P, Stanley EF. Long C terminal splice variant CaV2.2 identified in presynaptic membrane by mass spectrometric analysis. Channels (Austin) 2010;4:58–62. doi: 10.4161/chan.4.1.10364. [DOI] [PubMed] [Google Scholar]
- Goudet C, Magnaghi V, Landry M, Nagy F, Gereau RW, Pin JP. Metabotropic receptors for glutamate and GABA in pain. Brain Res Brain Res Rev. 2009;60:43–56. doi: 10.1016/j.brainresrev.2008.12.007. [DOI] [PubMed] [Google Scholar]
- Gray AC, Raingo J, Lipscombe D. Neuronal calcium channels: splicing for optimal performance. Cell Calcium. 2007;42:409–417. doi: 10.1016/j.ceca.2007.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray WR, Luque A, Olivera BM, Barrett J, Cruz LJ. Peptide toxins from Conus geographus venom. J Biol Chem. 1981;256:4734–4740. [PubMed] [Google Scholar]
- Hannan S, Wilkins ME, Dehghani-Tafti E, Thomas P, Baddeley SM, Smart TG. γ-Aminobutyric acid type B (GABAB) receptor internalization is regulated by the R2 subunit. J Biol Chem. 2011;286:24324–24335. doi: 10.1074/jbc.M110.220814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. Modulation of ion-channel function by G-protein-coupled receptors. Trends Neurosci. 1994;17:531–536. doi: 10.1016/0166-2236(94)90157-0. [DOI] [PubMed] [Google Scholar]
- Hillyard DR, Monje VD, Mintz IM, Bean BP, Nadasdi L, Ramachandran J, et al. A new Conus peptide ligand for mammalian presynaptic Ca2+ channels. Neuron. 1992;9:69–77. doi: 10.1016/0896-6273(92)90221-x. [DOI] [PubMed] [Google Scholar]
- Hurley JH, Cahill AL, Currie KP, Fox AP. The role of dynamic palmitoylation in Ca2+ channel inactivation. Proc Natl Acad Sci U S A. 2000;97:9293–9298. doi: 10.1073/pnas.160589697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imperial JS, Bansal PS, Alewood PF, Daly NL, Craik DJ, Sporning A, et al. A novel conotoxin inhibitor of Kv1.6 channel and nAChR subtypes defines a new superfamily of conotoxins. Biochemistry. 2006;45:8331–8340. doi: 10.1021/bi060263r. [DOI] [PubMed] [Google Scholar]
- Jakubowski JA, Kelley WP, Sweedler JV. Screening for post-translational modifications in conotoxins using liquid chromatography/mass spectrometry: an important component of conotoxin discovery. Toxicon. 2006;47:688–699. doi: 10.1016/j.toxicon.2006.01.021. [DOI] [PubMed] [Google Scholar]
- Janes RW. α-Conotoxins as selective probes for nicotinic acetylcholine receptor subclasses. Curr Opin Pharmacol. 2005;5:280–292. doi: 10.1016/j.coph.2005.01.013. [DOI] [PubMed] [Google Scholar]
- Kaas Q, Westermann JC, Halai R, Wang CK, Craik DJ. ConoServer, a database for conopeptide sequences and structures. Bioinformatics. 2008;24:445–446. doi: 10.1093/bioinformatics/btm596. [DOI] [PubMed] [Google Scholar]
- Kaas Q, Westermann JC, Craik DJ. Conopeptide characterization and classifications: an analysis using ConoServer. Toxicon. 2010;55:1491–1509. doi: 10.1016/j.toxicon.2010.03.002. [DOI] [PubMed] [Google Scholar]
- Kammermeier PJ, Ruiz-Velasco V, Ikeda SR. A voltage-independent calcium current inhibitory pathway activated by muscarinic agonists in rat sympathetic neurons requires both Gαq/11 and Gβγ. J Neurosci. 2000;20:5623–5629. doi: 10.1523/JNEUROSCI.20-15-05623.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko S, Cooper CB, Nishioka N, Yamasaki H, Suzuki A, Jarvis SE, et al. Identification and characterization of novel human Cav2.2 (α1B) calcium channel variants lacking the synaptic protein interaction site. J Neurosci. 2002;22:82–92. doi: 10.1523/JNEUROSCI.22-01-00082.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaupmann K, Huggel K, Heid J, Flor PJ, Bischoff S, Mickel SJ, et al. Expression cloning of GABAB receptors uncovers similarity to metabotropic glutamate receptors. Nature. 1997;386:239–246. doi: 10.1038/386239a0. [DOI] [PubMed] [Google Scholar]
- Kerr LM, Yoshikami D. A venom peptide with a novel presynaptic blocking action. Nature. 1984;308:282–284. doi: 10.1038/308282a0. [DOI] [PubMed] [Google Scholar]
- Kim JI, Takahashi M, Ohtake A, Wakamiya A, Sato K. Tyr13 is essential for the activity of ω-conotoxin MVIIA and GVIA, specific N-type calcium channel blockers. Biochem Biophys Res Commun. 1995;206:449–454. doi: 10.1006/bbrc.1995.1063. [DOI] [PubMed] [Google Scholar]
- Klimis H, Adams DJ, Callaghan B, Nevin S, Alewood PF, Vaughan CW, et al. A novel mechanism of inhibition of high-voltage activated calcium channels by α-conotoxins contributes to relief of nerve injury-induced neuropathic pain. Pain. 2011;152:259–266. doi: 10.1016/j.pain.2010.09.007. [DOI] [PubMed] [Google Scholar]
- Kniazeff J, Prézeau L, Rondard P, Pin JP, Goudet C. Dimers and beyond: the functional puzzles of class C GPCRs. Pharmacol Ther. 2011;130:9–25. doi: 10.1016/j.pharmthera.2011.01.006. [DOI] [PubMed] [Google Scholar]
- Lampe RA, Lo MMS, Keith RA, Horn MB, McLane MW, Herman JL, et al. Effects of site-specific acetylation on omega-conotoxin GVIA binding and function. Biochemistry. 1993;32:3255–3260. doi: 10.1021/bi00064a007. [DOI] [PubMed] [Google Scholar]
- Lee S, Kim Y, Back SK, Choi HW, Lee JY, Jung HH, et al. Analgesic effect of highly reversible ω-conotoxin FVIA on N type Ca2+ channels. Mol Pain. 2010;6:97. doi: 10.1186/1744-8069-6-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis RJ, Nielsen KJ, Craik DJ, Loughnan ML, Adams DA, Sharpe IA, et al. Novel ω-conotoxins from Conus catus discriminate among neuronal calcium channel subtypes. J Biol Chem. 2000;275:35335–35344. doi: 10.1074/jbc.M002252200. [DOI] [PubMed] [Google Scholar]
- Liang H, Elmslie KS. Rapid and reversible block of N-type calcium channels (CaV 2.2) by ω-conotoxin GVIA in the absence of divalent cations. J Neurosci. 2002;22:8884–8890. doi: 10.1523/JNEUROSCI.22-20-08884.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H, DeMaria CD, Erickson MG, Mori MX, Alseikhan BA, Yue DT. Unified mechanisms of Ca2+ regulation across the Ca2+ channel family. Neuron. 2003;39:951–960. doi: 10.1016/s0896-6273(03)00560-9. [DOI] [PubMed] [Google Scholar]
- Lipscombe D, Raingo J. Alternative splicing matters: N-type calcium channels in nociceptors. Channels (Austin) 2007;1:225–227. doi: 10.4161/chan.4809. [DOI] [PubMed] [Google Scholar]
- Lipscombe D, Pan JQ, Gray AC. Functional diversity in neuronal voltage-gated calcium channels by alternative splicing of Cavα1. Mol Neurobiol. 2002;26:21–44. doi: 10.1385/MN:26:1:021. [DOI] [PubMed] [Google Scholar]
- Livett BG, Sandall DW, Keays D, Down J, Gayler KR, Satkunanathan N, et al. Therapeutic applications of conotoxins that target the neuronal nicotinic acetylcholine receptor. Toxicon. 2006;48:810–829. doi: 10.1016/j.toxicon.2006.07.023. [DOI] [PubMed] [Google Scholar]
- Lu BS, Yu F, Zhao D, Huang PT, Huang CF. Conopeptides from Conus striatus and Conus textile by cDNA cloning. Peptides. 1999;20:1139–1144. doi: 10.1016/s0196-9781(99)00116-3. [DOI] [PubMed] [Google Scholar]
- Luebke JI, Dunlap K. Sensory neuron N-type calcium currents are inhibited by both voltage-dependent and -independent mechanisms. Pflugers Arch. 1994;428:499–507. doi: 10.1007/BF00374571. [DOI] [PubMed] [Google Scholar]
- Luo ZD, Chaplan SR, Higuera ES, Sorkin LS, Stauderman KA, Williams ME, et al. Upregulation of dorsal root ganglion α2δ calcium channel subunit and its correlation with allodynia in spinal nerve-injured rats. J Neurosci. 2001;21:1868–1875. doi: 10.1523/JNEUROSCI.21-06-01868.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margeta-Mitrovic M, Jan YN, Jan LY. A trafficking checkpoint controls GABAB receptor heterodimerization. Neuron. 2000;27:97–106. doi: 10.1016/s0896-6273(00)00012-x. [DOI] [PubMed] [Google Scholar]
- McCudden CR, Hains MD, Kimple RJ, Siderovski DP, Willard FS. G-protein signaling: back to the future. Cell Mol Life Sci. 2005;62:551–577. doi: 10.1007/s00018-004-4462-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonough SI, Swartz KJ, Mintz IM, Boland LM, Bean BP. Inhibition of calcium channels in rat central and peripheral neurons by ω-conotoxin MVIIC. J Neurosci. 1996;76:2612–2623. doi: 10.1523/JNEUROSCI.16-08-02612.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonough SI, Boland LM, Mintz IM, Bean BP. Interactions among toxins that inhibit N-type and P-type calcium channels. J Gen Physiol. 2002;119:313–328. doi: 10.1085/jgp.20028560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGivern JG. Targeting N-type and T-type calcium channels for the treatment of pain. Drug Discov Today. 2006;11:245–253. doi: 10.1016/S1359-6446(05)03662-7. [DOI] [PubMed] [Google Scholar]
- McIntosh JM, Absalom N, Chebib M, Elgoyhen AB, Vincler M. Alpha9 nicotinic acetylcholine receptors and the treatment of pain. Biochem Pharmacol. 2009;78:693–702. doi: 10.1016/j.bcp.2009.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miljanich GP, Ramachandran J. Antagonists of neuronal calcium channels: structure, function, and therapeutic implications. Annu Rev Pharmacol Toxicol. 1995;35:707–734. doi: 10.1146/annurev.pa.35.040195.003423. [DOI] [PubMed] [Google Scholar]
- Miljanich GP, Bowersox SS, Fox JA, Valentino KL, Bitner RS, Yamashiro DH. 1993. Compositions for delayed treatment of ischaemia-related neuronal damage. Publication no. WO 93/10145; International application no. PCT/US92 09766; International Patent Classification, C07K 7/10, A61K37/02.
- Monje VD, Haack JA, Naisbitt SR, Miljanich G, Ramachandran J, Nadasdi L, et al. A new Conus peptide ligand for Ca channel subtypes. Neuropharmacology. 1993;32:1141–1149. doi: 10.1016/0028-3908(93)90008-q. [DOI] [PubMed] [Google Scholar]
- Motin L, Adams DJ. ω-Conotoxin inhibition of excitatory synaptic transmission evoked by dorsal root stimulation in rat superficial dorsal horn. Neuropharmacology. 2008;55:860–864. doi: 10.1016/j.neuropharm.2008.06.049. [DOI] [PubMed] [Google Scholar]
- Motin L, Yasuda T, Schroeder C, Lewis R, Adams D. ω-Conotoxin CVIB differentially inhibits native and recombinant N- and P/Q-type calcium channels. Eur J Neurosci. 2007;25:435–444. doi: 10.1111/j.1460-9568.2006.05299.x. [DOI] [PubMed] [Google Scholar]
- Mould J, Yasuda T, Schroeder CI, Beedle AM, Doering CJ, Zamponi GW, et al. The α2δ auxiliary subunit reduces affinity of ω-conotoxins for recombinant N-type (Cav2.2) calcium channels. J Biol Chem. 2004;279:34705–34714. doi: 10.1074/jbc.M310848200. [DOI] [PubMed] [Google Scholar]
- Neely A, Wei X, Olcese R, Birnbaumer L, Stefani E. Potentiation by the beta subunit of the ratio of the ionic current to the charge movement in the cardiac calcium channel. Science. 1993;262:575–578. doi: 10.1126/science.8211185. [DOI] [PubMed] [Google Scholar]
- Nevin ST, Clark RJ, Klimis H, Christie MJ, Craik DJ, Adams DJ. Are α9α10 nicotinic acetylcholine receptors a pain target for α-conotoxins? Mol Pharmacol. 2007;72:1406–1410. doi: 10.1124/mol.107.040568. [DOI] [PubMed] [Google Scholar]
- Newton RA, Bingham S, Case PC, Sanger G, Lawson SN. Dorsal root ganglion neurons show increased expression of the calcium channel α2δ-1 subunit following partial sciatic nerve injury. Brain Res Mol Brain Res. 2001;95:1–8. doi: 10.1016/s0169-328x(01)00188-7. [DOI] [PubMed] [Google Scholar]
- Nielsen KJ, Schroeder T, Lewis R. Structure-activity relationships of ω-conotoxins at N-type voltage-sensitive calcium channels. J Mol Recognit. 2000;13:55–70. doi: 10.1002/(SICI)1099-1352(200003/04)13:2<55::AID-JMR488>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Nowycky MC, Fox AP, Tsien RW. Three types of neuronal calcium channel with different calcium agonist sensitivity. Nature. 1985;316:440–443. doi: 10.1038/316440a0. [DOI] [PubMed] [Google Scholar]
- Oldham WM, Hamm HE. Structural basis of function in heterotrimeric G proteins. Q Rev Biophys. 2006;39:117–166. doi: 10.1017/S0033583506004306. [DOI] [PubMed] [Google Scholar]
- Oldham WM, Hamm HE. Heterotrimeric G protein activation by G-protein-coupled receptors. Nat Rev Mol Cell Biol. 2008;9:60–71. doi: 10.1038/nrm2299. [DOI] [PubMed] [Google Scholar]
- Olivera BM. Conus peptides: biodiversity-based discovery and exogenomics. J Biol Chem. 2006;281:31173–31177. doi: 10.1074/jbc.R600020200. [DOI] [PubMed] [Google Scholar]
- Olivera BM, McIntosh JM, Cruz LJ, Luque FA, Gray WR. Purification and sequence of a presynaptic peptide toxin from Conus geographus venom. Biochemistry. 1984;23:5087–5090. doi: 10.1021/bi00317a001. [DOI] [PubMed] [Google Scholar]
- Olivera BM, Gray WR, Zeikus R, McIntosh JM, Varga J, Rivier J, et al. Peptide neurotoxins from fish-hunting cone snails. Science. 1985;230:1338–1343. doi: 10.1126/science.4071055. [DOI] [PubMed] [Google Scholar]
- Olivera BM, Cruz LJ, de Santos V, LeCheminant GW, Griffin D, Zeikus R, et al. Neuronal calcium channel antagonists. Discrimination between calcium channel subtypes using ω-conotoxin from Conus magus venom. Biochemistry. 1987;26:2086–2090. doi: 10.1021/bi00382a004. [DOI] [PubMed] [Google Scholar]
- Park J, Luo ZD. Calcium channel functions in pain processing. Channels (Austin) 2010;4:510–517. doi: 10.4161/chan.4.6.12869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pexton T, Moeller-Bertram T, Schilling JM, Wallace MS. Targeting voltage-gated calcium channels for the treatment of neuropathic pain: a review of drug development. Expert Opin Investig Drugs. 2011;20:1277–1284. doi: 10.1517/13543784.2011.600686. [DOI] [PubMed] [Google Scholar]
- Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol. 2002;3:639–650. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- Raingo J, Castiglioni AJ, Lipscombe D. Alternative splicing controls G protein-dependent inhibition of N-type calcium channels in nociceptors. Nat Neurosci. 2007;10:285–292. doi: 10.1038/nn1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramilo CA, Zafaralla GC, Nadasdi L, Hammerland LG, Yoshikami D, Gray WR, et al. Novel α- and ω-conotoxins from Conus striatus venom. Biochemistry. 1992;31:9919–9926. doi: 10.1021/bi00156a009. [DOI] [PubMed] [Google Scholar]
- Safavi-Hemami H, Siero WA, Kuang Z, Williamson NA, Karas JA, Page LR, et al. Embryonic toxin expression in the cone snail Conus victoriae: primed to kill or divergent function? J Biol Chem. 2011;286:22546–22557. doi: 10.1074/jbc.M110.217703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandall DW, Satkunanathan N, Keays DA, Polidano MA, Liping X, Pham V, et al. A novel α-conotoxin identified by gene sequencing is active in suppressing the vascular response to selective stimulation of sensory nerves in vivo. Biochemistry. 2003;42:6904–6911. doi: 10.1021/bi034043e. [DOI] [PubMed] [Google Scholar]
- Satkunanathan N, Livett B, Gayler K, Sandall D, Down J, Khalil Z. α-Conotoxin Vc1.1 alleviates neuropathic pain and accelerates functional recovery of injured neurones. Brain Res. 2005;1059:149–158. doi: 10.1016/j.brainres.2005.08.009. [DOI] [PubMed] [Google Scholar]
- Schroeder CI, Smythe ML, Lewis RJ. Development of small molecules that mimic the binding of ω-conotoxins at the N-type voltage-gated calcium channel. Mol Divers. 2004;8:127–134. doi: 10.1023/b:modi.0000025656.79632.86. [DOI] [PubMed] [Google Scholar]
- Schroeder CI, Lewis RJ, Adams DJ. Block of voltage-gated calcium channels by peptide toxins. In: Zamponi GW, editor. Voltage-Gated Calcium Channels. Georgetown: Eurekah.com and Kluwer Academic/Plenum Publishers; 2005. pp. 294–308. [Google Scholar]
- Scott VE, De Waard M, Liu H, Gurnett CA, Venzke DP, Lennon VA, et al. β subunit heterogeneity in N-type Ca2+ channels. J Biol Chem. 1996;271:3207–3212. doi: 10.1074/jbc.271.6.3207. [DOI] [PubMed] [Google Scholar]
- Sharpe IA, Gehrmann J, Loughnan ML, Thomas L, Adams DA, Atkins A, et al. Two new classes of conopeptides inhibit the α1-adrenoceptor and noradrenaline transporter. Nat Neurosci. 2001;4:902–907. doi: 10.1038/nn0901-902. [DOI] [PubMed] [Google Scholar]
- Sharpe IA, Thomas L, Loughnan M, Motin L, Palant E, Croker DE, et al. Allosteric α1-adrenoceptor antagonism by the conopeptide ρ-TIA. J Biol Chem. 2003;278:34451–34457. doi: 10.1074/jbc.M305410200. [DOI] [PubMed] [Google Scholar]
- Shukla AK, Kim J, Ahn S, Xiao K, Shenoy SK, Liedtke W, et al. Arresting a TRP channel β-arrestin 1 mediates ubiquitination and functional downregulation of TRPV4. J Biol Chem. 2010;285:30115–30125. doi: 10.1074/jbc.M110.141549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MT, Cabot PJ, Ross FB, Robertson AD, Lewis RJ. The novel N-type calcium channel blocker, AM336, produces potent dose-dependent antinociception after intrathecal dosing in rats and inhibits substance P release in rat spinal cord slices. Pain. 2002;96:119–127. doi: 10.1016/s0304-3959(01)00436-5. [DOI] [PubMed] [Google Scholar]
- Snutch TP. Targeting chronic and neuropathic pain: the N-type calcium channel comes of age. NeuroRx. 2005;2:662–670. doi: 10.1602/neurorx.2.4.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spence I, Gillessen D, Gregson RP, Quinn RJ. Characterization of the neurotoxic constituents of Conus geographus (L) venom. Life Sci. 1977;21:1759–1769. doi: 10.1016/0024-3205(77)90156-4. [DOI] [PubMed] [Google Scholar]
- Stocker JW, Nadasdi L, Aldrich RW, Tsien RW. Preferential interaction of ω-conotoxins with inactivated N-type Ca2+ channels. J Neurosci. 1997;17:3002–3013. doi: 10.1523/JNEUROSCI.17-09-03002.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatebayashi H, Ogata N. Kinetic analysis of the GABAB-mediated inhibition of the high-threshold Ca2+ current in cultured rat sensory neurones. J Physiol. 1992;447:391–407. doi: 10.1113/jphysiol.1992.sp019008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedford HW, Zamponi GW. Direct G protein modulation of Cav2 calcium channels. Pharmacol Rev. 2006;58:837–862. doi: 10.1124/pr.58.4.11. [DOI] [PubMed] [Google Scholar]
- Tedford HW, Kisilevsky AE, Vieira LB, Varela D, Chen L, Zamponi GW. Scanning mutagenesis of the I-II loop of the Cav2.2 calcium channel identifies residues arginine 376 and valine 416 as molecular determinants of voltage dependent G protein inhibition. Mol Brain. 2010;3:6. doi: 10.1186/1756-6606-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terlau H, Olivera BM. Conus venoms: a rich source of novel ion channel-targeted peptides. Physiol Rev. 2004;84:41–68. doi: 10.1152/physrev.00020.2003. [DOI] [PubMed] [Google Scholar]
- Terlau H, Shon K, Grilley M, Stocker M, Stuhmer W, Olivera BM. Strategy for rapid immobilization of prey by a fish-hunting cone snail. Nature. 1996;381:148–151. doi: 10.1038/381148a0. [DOI] [PubMed] [Google Scholar]
- Towers S, Princivalle A, Billinton A, Edmunds M, Bettler B, Urban L, et al. GABAB receptor protein and mRNA distribution in rat spinal cord and dorsal root ganglia. Eur J Neurosci. 2000;12:3201–3210. doi: 10.1046/j.1460-9568.2000.00237.x. [DOI] [PubMed] [Google Scholar]
- Vincler M, Wittenauer S, Parker R, Ellison M, Olivera BM, McIntosh JM. Molecular mechanism for analgesia involving specific antagonism of α9α10 nicotinic acetylcholine receptors. Proc Natl Acad Sci U S A. 2006;103:17880–17884. doi: 10.1073/pnas.0608715103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss N. Regulation of N-type calcium channels by G-proteins: multiple pathways to control calcium entry into neurons. Channels (Austin) 2009;3:219–220. doi: 10.4161/chan.3.4.9255. [DOI] [PubMed] [Google Scholar]
- Wen L, Yang S, Qiao HF, Liu ZW, Zhou WX, Zhang YX, et al. SO-3, a new O-superfamily conopeptide derived from Conus striatus, selectively inhibits N-type calcium currents in cultured hippocampal neurons. Br J Pharmacol. 2005;145:728–739. doi: 10.1038/sj.bjp.0706223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witcher DR, DeWaard M, Campbell KP. Characterization of the purified N-type Ca2+ channel and the cation sensitivity of ω-conotoxin GVIA binding. Neuropharmacology. 1993;32:1127–1139. doi: 10.1016/0028-3908(93)90007-p. [DOI] [PubMed] [Google Scholar]
- Wright CE, Robertson AD, Whorlow SL, Angus JA. Cardiovascular and autonomic effects of ω-conotoxins MVIIA and CVID in conscious rabbits and isolated tissue assays. Br J Pharmacol. 2000;131:1325–1336. doi: 10.1038/sj.bjp.0703701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Li ZW, Wei JB. Current responses mediated by endogenous GABAB and GABAC receptors in Xenopus oocytes (in Chinese) Sheng Li Xue Bao. 2001;53:311–315. [PubMed] [Google Scholar]
- Yarotskyy V, Elmslie KS. ω-Conotoxin GVIA alters gating charge movement of N-type (CaV2.2) calcium channels. J Neurophysiol. 2009;101:332–340. doi: 10.1152/jn.91064.2008. [DOI] [PubMed] [Google Scholar]
- Yasuda T, Adams DJ. Voltage-gated calcium channels in nociception. In: Martinac B, editor. Sensing with Ion Channels. Heidelberg: Springer; 2007. pp. 267–298. [Google Scholar]
- Yasuda T, Chen L, Barr W, McRory JE, Lewis RJ, Adams DJ, et al. Auxiliary subunit regulation of high-voltage activated calcium channels expressed in mammalian cells. Eur J Neurosci. 2004;20:1–13. doi: 10.1111/j.1460-9568.2004.03434.x. [DOI] [PubMed] [Google Scholar]
- Zamponi GW, Snutch TP. Evidence for a specific site for modulation of calcium channel activation by external calcium ions. Pflugers Arch. 1996;431:470–472. doi: 10.1007/BF02207290. [DOI] [PubMed] [Google Scholar]
- Zamponi GW, Lewis RJ, Todorovic SM, Arneric SP, Snutch TP. Role of voltage-gated calcium channels in ascending pain pathways. Brain Res Brain Res Rev. 2009;60:84–89. doi: 10.1016/j.brainresrev.2008.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]