

Abstract
Hyperpolarization-activated cation channels generate the If current in the heart. In the sino-atrial node (SAN), If is thought to play an essential role in setting the heart rate and mediating its autonomic control. This review focuses on the role of If in pacemaking and non-pacemaking cardiomyocytes and the resulting therapeutic implications. HCN4 represents the principal isoform underlying sino-atrial If, but other isoforms may also be of importance. To examine the functional role of cardiac channels, several mouse mutants, most of them targeting HCN4, have been generated by different groups. Unexpectedly, these lines display greatly different and as yet unexplained phenotypes. We provide an overview about these HCN mutants and suggest an interpretation of the functional significance of If in the SAN in light of these studies. HCN channels are also present in ventricular myocytes, and an up-regulation of If in the hypertrophic and failing heart may contribute to arrhythmogenesis. Inhibition of If by HCN channel blockers is a novel approach in the treatment of cardiac disorders, and ivabradine is approved for treatment of stable angina pectoris. Remarkably, a recent clinical trial assessing this substance in heart failure showed a significantly improved outcome. The mechanism underlying this beneficial effect is not yet clear and might lie beyond heart rate slowing. Thus, the growing knowledge about cardiac HCN channels will undoubtedly promote the development of the promising class of HCN channel blockers.
Keywords: arrhythmia, cardiac hypertrophy, HCN channel blockers, hyperpolarization-activated channels, pacemaking, sino-atrial node, transgenic mice
Introduction
The class of hyperpolarization-activated cyclic nucleotide-gated (HCN) cation channels contains four members termed HCN1-4 (Ludwig et al., 1998, 1999; Santoro et al., 1998, 1999; Ishii et al., 1999). All four isoforms generate a current with characteristics typical for the conductance known as If or Ih. Since this review deals with the role of cardiac HCN channels, the long-established term If for the current in the heart will be used. The subunits vary in their biophysical properties including activation kinetics, voltage dependence of activation, modulation by low molecular weight factors like cAMP and phosphatidylinositol 4,5-bisphosphate (PIP2) and phosphorylation by tyrosine kinases. For example, the activation kinetic between HCN4 (the slowest) and HCN1 (the fastest HCN channel) differs by more than one magnitude (τ of HCN1: 30–300 ms, HCN4: 300 ms – several s). cAMP enhances the activation kinetic and voltage dependence of activation in an isoform-dependent manner. HCN2 and HCN4 are strongly stimulated, whereas HCN1 is barely and HCN3 is not modulated. These isoform-dependent differences are covered by excellent reviews (e.g. Robinson and Siegelbaum, 2003; Frere et al., 2004; Siu et al., 2006; Biel et al., 2009). The channels are sometimes called pacemaker channels, and If is accordingly dubbed as pacemaker current, since the current is thought to play an essential role in setting the heart rate and mediating its autonomic control, for example, by β-adrenergic stimulation. The expression profile of the channels has been studied in the different cardiac compartments indicating a strong enrichment of the channels, most prominently of HCN4, in the sino-atrial (and atrioventricular) node. Nevertheless, various data indicate that besides HCN4, other HCN isoforms may be important for generating native If in the primary pacemaker centre.
Whereas the identity of HCN subunits as If-mediating channels is beyond dispute, the exact role of the channels in the pacemaking mechanism is still controversial. Various transgenic mouse models were generated to examine the functional role of cardiac HCN channels. However, these models demonstrated pronounced differences, and potential reasons for these will be discussed.
The role of HCN channels/If in non-pacemaking cardiomyocytes is less well-defined. Several groups reported the detection of If in ventricular myocytes and an increase of the current in cardiac disease states like hypertrophy and heart failure (Cerbai et al., 1997; Hoppe et al., 1998; Stilli et al., 2001; Fernandez-Velasco et al., 2003; Stillitano et al., 2008). It was repeatedly suggested that the augmented If promotes the generation of ventricular arrhythmias under these pathological conditions, for example, by favouring diastolic depolarizations in myopathic cells. This hypothesis has not been verified yet, but direct evidence for the role of If in working myocardial cells is emerging.
Inhibition of If is a novel approach in the treatment of cardiac disorders based on the idea of producing a heart rate reduction similar to β-blockers or calcium channel blockers. Several HCN channel blockers including ZD7288, zatebradine, cilobradine and ivabradine are available; these substances are also known as ‘specific bradycardic agents’ or ‘sinus node inhibitors’ (Bucchi et al., 2007). The first clinically approved substance from this new class of drugs is ivabradine which is licensed for treatment of stable angina pectoris when β-blockers are not tolerated or display inadequate effectiveness. Remarkably, a recent clinical trial showed a significant beneficial effect of this substance in patients suffering from heart failure (Boehm et al., 2010; Swedberg et al., 2010). The underlying mechanism is controversial and may lie beyond heart rate lowering as discussed below.
Expression of HCN channels in the cardiac pacemaking and conduction system
Interpretation of the partially conflicting results from the various transgenic HCN mutants as well as therapeutic targeting of the channels requires a firm knowledge about the expression pattern of the HCN isoforms. Expression of HCN1, HCN2 and HCN4 has been documented in the pacemaking and conduction system. Undoubtedly, HCN4 constitutes the predominant isoform in the sino-atrial node (SAN) at both transcript and protein level (Ishii et al., 1999; Shi et al., 1999, 2000; Moosmang et al., 2001; Marionneau et al., 2005; Tellez et al., 2006; Liu et al., 2007; Brioschi et al., 2009; Herrmann et al., 2011b). HCN4 mRNA accounts for around 80% of HCN transcripts in rabbit (Shi et al., 1999) and 60% in murine (Herrmann et al., 2011b) SAN. This prevalent expression is in accordance with data at the protein level. In all analysed mammalian species (mouse, rat, rabbit, dog, human), a strong expression of HCN4 in the SAN was observed by Western blot and immunohistochemical analyses (Figure 1) (Zicha et al., 2005; Liu et al., 2007; Herrmann et al., 2007b, 2011b; Brioschi et al., 2009; Chandler et al., 2009). In addition, all three principal cell types in isolated SAN cell preparations, namely spindle, elongated spindle and spider cells (Verheijck et al., 1998), exhibit a clustered expression of HCN4 protein at the plasma membrane (Figure 1). The specificity of this isoform for sino-atrial cells is high, since no HCN4 protein was observed in the surrounding atrial tissue. In addition, a line carrying a knock-in of an inducible Cre recombinase into the endogenous HCN4 locus (HCN4-KiT Cre) demonstrated that the HCN4 promoter is indeed selectively active (Hoesl et al., 2008). These mice were crossed with a conditional transgenic line expressing diphtheria toxin A to create an animal model with controlled deletion of pacemaking cells. In this sensitive genetic system, no HCN4 promoter activity was detected in atrial or ventricular working myocytes (Herrmann et al., 2011a). Taken together, HCN4 represents an excellent, unique and now widely used marker for the detection of cardiac pacemaking cells. This is also true during cardiac development. HCN4 was selectively detected in those regions of the embryonic heart (caudal portion of heart tube, afterwards junction of right atrial appendage with superior vena cava), where pacemaker activity and SAN development have been located (Garcia-Frigola et al., 2003; Stieber et al., 2003).
Figure 1.
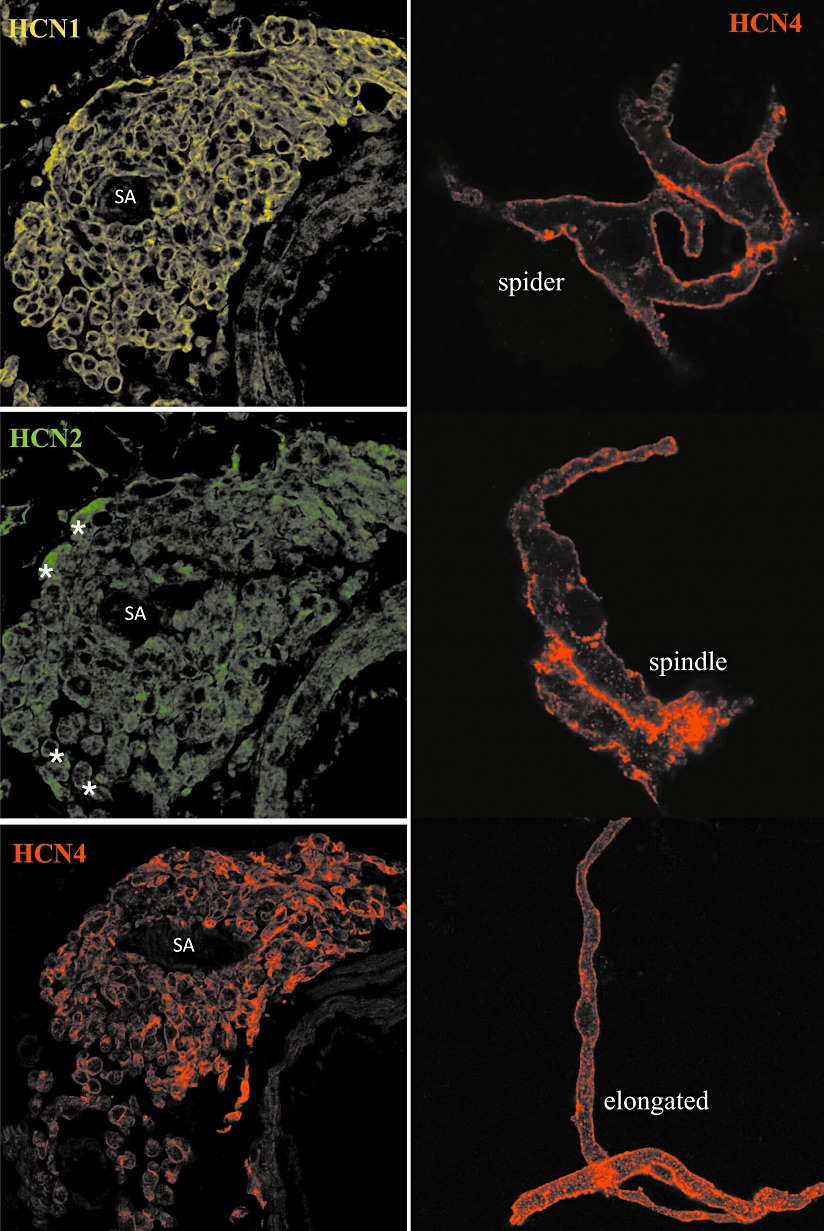
Regional and cellular expression of HCN channel proteins in the sino-atrial node. Left, sections through the SAN region of C57BL/6 mice stained with antibodies directed against HCN1, HCN2 and HCN4 as indicated. * denotes significant HCN2 positive cells. Right, sino-atrial cells isolated from C57BL/6 mice labelled with the HCN4 antibody. Representative examples of spider, spindle and elongated spindle cells are shown. SA, sino-atrial node artery.
There are conflicting reports regarding the expression of other HCN isoforms in the SAN. Several publications reported the presence of low levels of HCN2 transcripts and some, but not all, described a higher expression in the SAN as compared to atrial tissue (Shi et al., 1999; Moosmang et al., 2001; Marionneau et al., 2005; Chandler et al., 2009; Herrmann et al., 2011b). However, other groups, which analysed the same species (mouse, rabbit) could not detect this isoform at the protein level (Liu et al., 2007; Brioschi et al., 2009). A recent study by the authors using HCN2-knockout mice as negative control found a very low expression of this isoform in the SAN, albeit some scattered cells in the periphery of the SAN showed higher HCN2 levels (Herrmann et al., 2011b).
The available data for HCN1 are more divergent. A substantial level of HCN1 transcript in the SAN (∼20–35% of total HCN message) has been reported (Shi et al., 1999; Marionneau et al., 2005; Herrmann et al., 2011b). However, several studies were unable to demonstrate a corresponding HCN1 protein expression (Liu et al., 2007; Brioschi et al., 2009; Baruscotti et al., 2011). We evaluated this issue by developing a sensitive immunostaining procedure and by using sections from HCN1-deficient mice as rigorous negative control (Herrmann et al., 2011b). Unexpectedly, HCN1 showed an identical expression pattern as HCN4, that is, the isoform was clearly present throughout the whole nodal area. At the cellular level, immunostaining was restricted to the plasma membrane of sino-atrial myocytes (Figure 1). Hence, mRNA as well as new protein expression data suggests a significant contribution of HCN1 to cardiac If.
The expression pattern of HCN proteins in the atrioventricular node (AVN) is similar to the SAN (Herrmann et al., 2011b). HCN2 is present in a small number of myocytes, whereas both HCN1 and HCN4 are expressed in the whole AVN. This expression profile changes somewhat in the lower parts of the conduction system. In the Bundle of His, HCN4 is selectively expressed, whereas HCN1, HCN2 and HCN4 are present in the bundle branches and Purkinje fibres.
Transgenic HCN mutants HCN2 knockouts
Over the past years, several genetically engineered HCN mutants were generated in different labs (Table 1). Various targeting strategies were employed ranging from conventional knockouts lacking an isoform in all tissues at any time to single amino acid modifications in a temporally controlled manner altering only defined current properties. We give an overview about the murine HCN mutations and an interpretation of the role of cardiac If in light of these studies. In the first of these mutants, exon 2 and 3 of the HCN2 gene encoding five of the six transmembrane segments including part of the pore were deleted, the mutation results in a frameshift (Ludwig et al., 2003). The cardiac phenotype was analysed in global knockouts as well as in heart-specific mutants. Albeit expression of HCN2 in the SAN is low, the knockout animals displayed a reduction of If density by about 30% and a significant slower activation kinetic of the current. In addition, the maximal diastolic potential (MDP) of sino-atrial cells was slightly (∼5 mV) hyperpolarized as compared to controls. The remaining current and its activation kinetics were increased by cAMP and reached nearly the values of cAMP-stimulated wild-type cells. The knockouts displayed a sinus dysrhythmia which was present at rest and absent during phases of enhanced activity, whereas other electrocardiogram (ECG) parameters including basal and maximal heart rate were normal.
Table 1.
Murine transgenic HCN lines
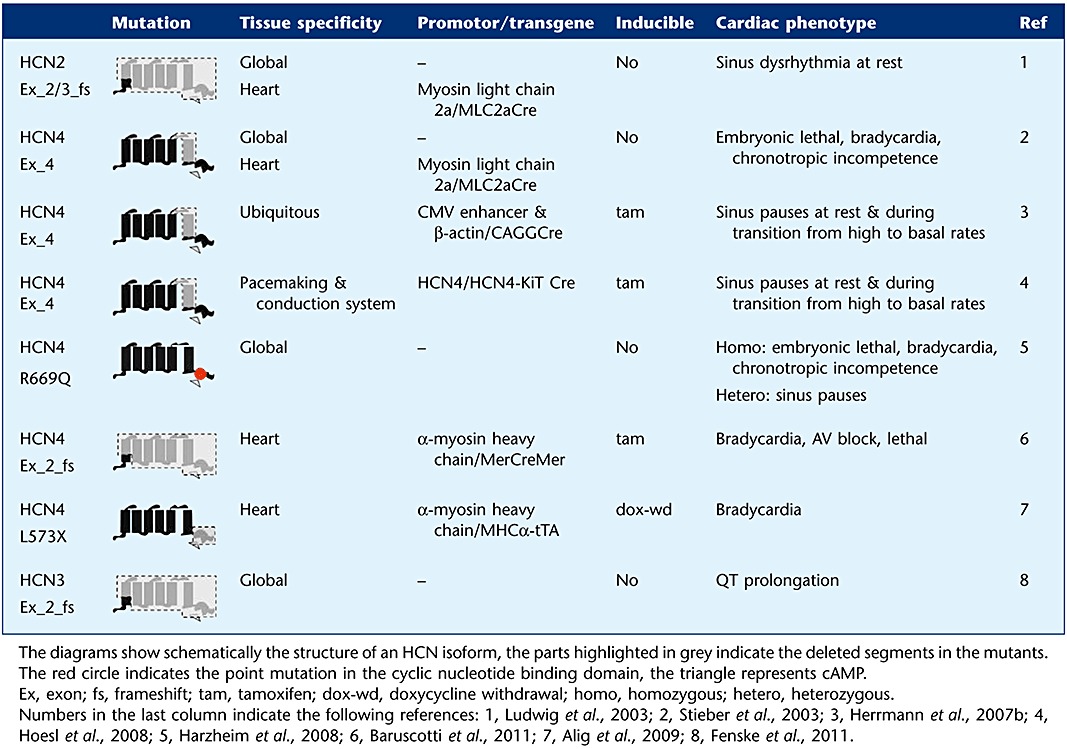 |
The different HCN4 mutants
HCN4 is the predominant isoform in the cardiac pacemaker system regardless of developmental stage or examined species and several independent efforts were made to create HCN4 mutants. A first knockout was created by Cre-mediated excision of exon 4 which results in a non-functional construct lacking the pore (Stieber et al., 2003). Global as well as cardiac-restricted loss of HCN4 led to embryonic death between embryonic days 10.5 and 11.5. At this stage of cardiac development, control myocytes displayed a prominent If which was strongly diminished (∼90%) in knockouts. The remaining current exhibited a much faster activation kinetic and was probably driven by HCN1 (and HCN3). The contraction rate of isolated knockout hearts was markedly reduced by about 40%, and neither heart rate nor action potential frequency could be increased by using isoproterenol or cAMP.
Unexpectedly, another study showed that mice harbouring only a point mutation in the cAMP-binding domain of HCN4 also die in utero (Harzheim et al., 2008). This amino acid exchange (R669Q) abolished cAMP-dependent modulation but did not alter basal channel function. Mice carrying the homozygous mutation died between embryonic days 11 and 12. Similar to global HCN4 knockouts, the homozygous mutants displayed a significant slower heart rate which could not be accelerated by pharmacological stimulation. If activated slower and the voltage dependence of activation was significantly shifted to hyperpolarized potentials. These changes were attributed to the presence of a relatively high basal cAMP level in pacemaker cells.
To overcome the embryonic lethality, we and others generated temporally controlled deletions of HCN4. In our lab, this was achieved by crossing floxed HCN4 with tamoxifen-inducible CMV enhancer/chicken beta-actin-Cre animals. This Cre transgene is ubiquitously expressed driven by a strong promoter, and is strictly dependent on the presence of tamoxifen. The approach led to a complete deletion of HCN4 in all SAN cells (Herrmann et al., 2007b). The ECG was characterized by recurrent sinus pauses; the pauses were more frequent and longer at lower heart rates and during the transition from high to basal frequencies. Remarkably, pauses disappeared during periods of stimulated heart rates (by pharmacological means or exercise). SAN cells showed a drastic reduction of If density by about 75%, whereas the residual If demonstrated a faster activation kinetic. Action potential recordings showed that 90% of the isolated knockout pacemaker cells did not fire spontaneously and displayed a hyperpolarized MDP. Superfusion of cells with isoproterenol rescued this cellular quiescent phenotype. However, the mice showed neither signs of bradycardia nor any impairment in heart rate regulation.
These unexpected results were largely corroborated by two additional mouse studies. One report used floxed HCN4 animals mated with a tamoxifen-inducible Cre transgene knocked into the endogenous HCN4 promoter itself (HCN4-KiT Cre, Hoesl et al., 2008). The HCN4-KiT line allows a temporally controlled deletion of genes exclusively in cells of the cardiac pacemaking and conduction system. The functionality of this line was demonstrated by complete deletion of the HCN4 protein. This approach resulted in an analogous phenotype characterized by sinus pauses and a strongly reduced but significantly faster activating If. Similar observations were made in the report from Harzheim et al. (2008) outlined above. Even though homozygous disruption of the cAMP-binding site resulted in embryonic death, heterozygous R669Q mutants developed normally. ECG recordings revealed that these mutants showed more sinus pauses and more sino-atrial blocks than controls.
Surprisingly, Baruscotti and colleagues described a completely different HCN4 knockout phenotype recently (Baruscotti et al., 2011). This group also used the Cre/loxP system to achieve a heart-specific, tamoxifen-inducible deletion of exon 2 of HCN4 encoding the distal part of the first and the following three transmembrane segments. The deletion produced a frameshift followed by a premature stop after R262; thus only the N-terminal part of HCN4 is translated. Expression of the Cre transgene used (MerCreMer) is controlled by the cardiomyocyte-specific alpha-myosin heavy chain (αMHC) promoter. Immunolabelling of tamoxifen-treated double mutants displayed a robustly reduced but still detectable HCN4 signal in SAN slices as well as in single SAN cells. Mutants developed very rapidly a deep bradycardia and died within 8.5 days after the first tamoxifen injection. The progressively slower heart rate was accompanied by increased prolongation of the PQ interval, which finally resulted in a complete heart block shortly before death. Electrophysiological measurements revealed a robust decline in sino-atrial If density during the treatment procedure. However, neither activation kinetic nor the response to isoproterenol was changed in the residual If. Isoproterenol increased the heart rate in controls and in tamoxifen-treated mutants by about 200 beats per minute, even though the mutants did not reach the same maximal level.
An additional HCN4 mouse study was recently performed by another group (Alig et al., 2009). Mice were generated with a heart-specific and temporally controlled overexpression of a mutated HCN4 (L573X) which abolishes the cAMP-dependent regulation of HCN channels. The mutation was originally described in a human patient with SAN dysfunction. A single base-pair deletion results in a c-terminal-truncated channel lacking the cyclic nucleotide-binding domain and causes cAMP insensitivity of heterologously coexpressed wild-type HCN4 subunits in a dominant-negative manner. Tissue (and time) specific overexpression of the engineered HCN4 protein was controlled by the Tet-Off expression system and the use of the cardiac-specific αMHC promoter, which in this line directed transgene expression predominantly to atrial myocytes including pacemaker cells. The overexpression was temporally repressed by administration of doxycycline. If from mutant SAN cells exhibited similar densities like controls but slower activation kinetics and no response to isoproterenol. The voltage of half-maximal activation was shifted by about −20 mV to hyperpolarized potentials resulting in an If activation range beyond physiologically relevant diastolic membrane potentials. SAN cell automaticity was greatly affected by the transgene expression. Most pacemaker cells were arrhythmic and alternated between spontaneous action potentials and subthreshold membrane potential oscillations or were totally quiescent. Administration of isoproterenol restored this cellular phenotype and induced an acceleration of pacemaking similar to that observed in control cells. However, the SAN cell pacing values with or without isoproterenol were significantly slower compared to that of controls. These observations made at the single-cell level could be transferred to the whole animal. The heart rate of binary transgenic mice was significantly slower and did not reach maximal levels. The If blocker ivabradine reduced heart rate in control mice but not in mutants indicating that the contribution of If to SAN function was eliminated by hHCN4-L573X transgene overexpression.
Potential explanations for the divergent observations in the different adult HCN4 mutants
It is astonishing that the deletion/mutation of one and the same gene leads to fundamentally different results. Taking together the five reports covering adult HCN4 mutations can be classified roughly in two phenotype categories. The first category includes mutants characterized by recurrent sinus pauses (Herrmann et al., 2007b; Harzheim et al., 2008; Hoesl et al., 2008), whereas mutants of the second category exhibit a pronounced bradycardia (Alig et al., 2009; Baruscotti et al., 2011). Considering that different HCN isoforms contribute to sino-atrial If (see later) and that the isoforms possess different kinetic properties, the deletion of one single isoform should result in a change in the kinetics of the residual If. In fact, this prediction was verified only in mutants of category 1 (and the global HCN2 mutant) pointing to a selective deletion of only one isoform in these lines. Mutants of category 2 showed effects which could also be observed by administration of (isoform-unspecific) HCN channel blockers. In the case of the HCN4 mutant described by Alig et al. (2009), it is likely that the dominant-negative effect of the mutated HCN4 protein affects all HCN wild-type isoforms which are expressed in the SAN. This assumption is supported by the fact that the blocker ivabradine has no further impact on SAN automaticity in these mutants. Thus, this study might reflect rather a combined isoform-unspecific HCN repression than a selective repression of HCN4 alone.
The consequences of the HCN4 deletion described by Baruscotti et al. (2011) resemble those of the report from Alig et al. (2009) outlined above. Moreover, this deletion did not result in a kinetic shift of the remaining If (Baruscotti et al., 2011). In fact, the mutation produced a reduction of sino-atrial If mimicking an isoform-unspecific HCN block. Thus, we speculate that besides HCN4, other HCN isoforms might additionally be functionally affected in this line. We cannot provide direct evidence for these considerations, but our interpretation may provide a plausible explanation for the differences found in the various mutants.
HCN1 and HCN3 mutants
The generation of HCN1 knockouts has been described (Nolan et al., 2003, 2004). HCN1-deficient mice display a neuronal phenotype with profound deficits in spatial memory and motor learning. However, a cardiac phenotype of this line has not been reported. Although the cardiac expression of HCN3 channels was frequently questioned and only low levels of HCN3 transcripts could be detected in cardiac myocytes, a ventricular phenotype caused by global deletion of HCN3 has very recently been described (Fenske et al., 2011). The HCN3 locus was disrupted by excision of exon 2. Epicardial myocytes of HCN3 knockouts displayed a reduction of If density by about 30% and a shortening of action potential duration caused by changes during the late repolarization phase. ECG recordings displayed a slight prolongation of the QT interval combined with increased T-wave amplitudes. These alterations were present only at low heart rates. The authors postulated that ventricular If serves as a depolarizing background current and as an antagonist of K+-currents during the late repolarization phase. Loss of HCN3 relieved this antagonistic activity and leads to a shortening of the ventricular action potential waveform.
Implications
Sino-atrial If and HCN subunit composition
HCN4 represents the main sino-atrial subunit followed by a distinct expression of HCN1, whereas HCN2 seems to be expressed only at low levels. Direct evidence for a combined contribution of different isoforms to sino-atrial If came from the various knockout studies, where the loss of HCN4 and also HCN2 caused a significant reduction of sino-atrial If density by about 75% and 30%, respectively. The If reduction in HCN2 knockouts seems astonishingly high compared to the low expression level of this isoform. HCN channels exist as tetramers, and the different isoforms have the ability to co-assemble with each other. Thus, the deletion of HCN2 might result in reduced opportunities for co-assembling, and this may result in a rather strong effect on the formation of functional channels. A significant contribution of HCN1 to sino-atrial If is also likely. This assumption is supported on the one hand by the prominent expression of HCN1 in sino-atrial tissue, and on the other hand by the fact that the combined deletion of HCN2 and HCN4 did not result in a complete sino-atrial If knockout (unpubl. data). Taken together, these results point to a cooperation of all three HCN subunits HCN4, HCN1 and HCN2 in the generation of the pacemaker current in SAN cells.
cAMP modulation is mandatory for the physiological function of sino-atrial If
An unexpected finding was that cardiac HCN channels need to be activated by cyclic nucleotides to fulfil their normal physiological function. With a lack of any cAMP-mediated stimulation, the channels exhibit activation and deactivation characteristics beyond those physiologically observed in pacemaker cells. This was demonstrated by the two cAMP-insensitive mutants described earlier. The first of these mutations (Harzheim et al., 2008) caused in embryonic as well as in adult mice a very similar phenotype as observed in animals with a total disruption of HCN4 (Herrmann et al., 2007b) reflecting the necessity of cAMP to basal HCN4 channel function. The overexpression of the dominant-negative hHCN4-573X protein in the second line (Alig et al., 2009) completely abolished the contribution of If to SAN automaticity also indicating the essential importance of cyclic nucleotide binding for the function of the channels.
The activity of HCN channels is directly modulated by cAMP making them ideal candidates for transducing sympathetic stimulation to accelerated pacing activity. However, all available adult HCN mutants as well as wild types in the presence of HCN channel blockers were able to increase their heart rate upon pharmacological stimulation. This observation seems consequent if one considers that cAMP binding to If channels might rather be an essential precondition for their function under physiological conditions than a modulator of their function. Hence, these data point to cellular mechanisms besides If channels responsible for β-adrenergic heart rate stimulation.
If channels serve as rhythm stabilizer and set the basal rhythm
The knockout studies showed that the contribution of all sino-atrial HCN isoforms is necessary to shield the pacemaker potential from enhanced hyperpolarization and to guarantee a stable heart rhythm. The lack of HCN4 (and with limitations of HCN2) causes a higher propensity for pacemaker rhythm disturbances as apparent by sinus pauses in the ECG. Periods of low heart rates or an enhanced parasympathetic tone increases the magnitude of these abnormalities; hence, a fully functional If seems to be important under these vulnerable conditions (Ludwig et al., 2003; Herrmann et al., 2007b; Harzheim et al., 2008; Hoesl et al., 2008).
There are several indications for a considerable impact of If channels on setting the basal heart rate. First, human mutations in the HCN4 gene are linked to bradycardic conditions (Schulze-Bahr et al., 2003; Milanesi et al., 2006). Second, If channel blockers like ivabradine slow the heart rate in mice and man (Ragueneau et al., 1998; Stieber et al., 2006). Third, the transgenic overexpression of the truncated HCN4-573X construct results in decreased cardiac rates (Alig et al., 2009). Fourth, the heart-specific deletion of HCN4 described by Baruscotti et al. (2011) also produces a deep bradycardia. These studies might share a combined inhibition of several HCN isoforms; either by the use of unselective HCN blockers or by the use of a dominant-negative protein. As explained above, the other studies which clearly resulted in the complete deletion of a single HCN isoform show a destabilized cardiac rhythm, but no signs of bradycardia. Therefore, it seems that only the repression of all relevant HCN isoforms significantly slows the diastolic depolarization and leads to slower heart rates. These slower rates were present at rest and also during exercise or pharmacological stimulation with the consequence that reaching maximal heart rates is not possible without If. In regard to the development of new HCN channel blockers for cardiac use, an isoform-selective blocker which specifically targets one isoform, for example, HCN4, has the theoretical advantage of causing less side effects. However, such a substance might be not effective enough; we suggest that, instead, isoform-unselective blockers have the highest therapeutic potential.
HCN channels in the ventricular working myocardium
Expression of the channels in the working myocardium is highly dependent on the developmental state and the presence of pathological conditions like cardiac hypertrophy or failure. A pronounced If is observed in embryonic and neonatal ventricular myocytes which is strongly down-regulated during postnatal development (Robinson et al., 1997; Yasui et al., 2001). In the adult state, an obvious difference exists in the expression levels between pacemaking and working myocardium, since the amount of HCN transcript in the ventricle is around one magnitude lower compared to the SAN. Nevertheless, If can be detected in ventricular myocytes from animals and humans (Cerbai et al., 1996; Hoppe et al., 1998; Graf et al., 2001; Stillitano et al., 2008). The predominant ventricular isoform is HCN2 representing between 75% and 90% of the total HCN message; the remaining HCN transcript is mainly HCN4 (Shi et al., 1999; Marionneau et al., 2005; Stillitano et al., 2008; Herrmann et al., 2011b). A very recent work found a ventricular phenotype of an HCN3 null mutant (Fenske et al., 2011, see earlier discussion) pointing to a functional expression of this isoform, albeit only low levels of HCN3 mRNA have been detected so far.
A significant increase of ventricular If in a variety of heart diseases including cardiac hypertrophy and failure due to hypertension and ischaemic and dilated cardiomyopathy has been documented (Cerbai et al., 1997; Hoppe et al., 1998; Stilli et al., 2001; Fernandez-Velasco et al., 2003; Stillitano et al., 2008). These studies were performed by using rats and samples from patients undergoing heart transplantation. In addition, preliminary data from our lab show that an up-regulation of ventricular If is also present in mice with ventricular hypertrophy induced by transverse aortic constriction. Based on these observations, it has been suggested that an increase in the depolarizing If current in working myocardium may contribute to the generation of arrhythmias in cardiac disease (Cerbai and Mugelli, 2006; Herrmann et al., 2007a). Recently, a large clinical trial (Systolic Heart Failure Treatment with the If Inhibitor Ivabradine Trial, SHIFT) demonstrated a beneficial effect on heart failure patients, where addition of ivabradine to standard therapy significantly reduced the rates of hospital admissions and cardiovascular death (Boehm et al., 2010; Swedberg et al., 2010). It was proposed that this remarkable effect was accounted for by the heart rate-reducing properties of the drug, that is, by inhibition of If in the SAN. However, this hypothesis was strongly questioned, and suggested that the mechanism responsible for the beneficial effect might well lie beyond heart rate lowering (Teerlink, 2010). Among other mechanisms, it is possible that the beneficial effect of ivabradine in cardiac failure is related, at least in part, to the block of increased HCN channel activity in ventricular myocytes. However, additional studies have to be performed before HCN channels in the ventricular myocardium will be established as new drug targets.
Acknowledgments
Research of the authors was supported by the European Union research project NORMACOR and by the Deutsche Forschungsgemeinschaft.
Glossary
- AVN
atrioventricular node
- ECG
electrocardiogram
- HCN
hyperpolarization-activated cyclic nucleotide-gated cation channel
- MDP
maximal diastolic potential
- SAN
sino-atrial node
Conflicts of interest
None declared.
References
- Alig J, Marger L, Mesirca P, Ehmke H, Mangoni ME, Isbrandt D. Control of heart rate by cAMP sensitivity of HCN channels. Proc Natl Acad Sci U S A. 2009;106:12189–12194. doi: 10.1073/pnas.0810332106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baruscotti M, Bucchi A, Viscomi C, Mandelli G, Consalez G, Gnecchi-Rusconi T, et al. Deep bradycardia and heart block caused by inducible cardiac-specific knockout of the pacemaker channel gene Hcn4. Proc Natl Acad Sci U S A. 2011;108:1705–1710. doi: 10.1073/pnas.1010122108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biel M, Wahl-Schott C, Michalakis S, Zong X. Hyperpolarization-activated cation channels: from genes to function. Physiol Rev. 2009;89:847–885. doi: 10.1152/physrev.00029.2008. [DOI] [PubMed] [Google Scholar]
- Boehm M, Swedberg K, Komajda M, Borer JS, Ford I, Dubost-Brama A, et al. Heart rate as a risk factor in chronic heart failure (SHIFT): the association between heart rate and outcomes in a randomised placebo-controlled trial. Lancet. 2010;376:886–894. doi: 10.1016/S0140-6736(10)61259-7. [DOI] [PubMed] [Google Scholar]
- Brioschi C, Micheloni S, Tellez JO, Pisoni G, Longhi R, Moroni P, et al. Distribution of the pacemaker HCN4 channel mRNA and protein in the rabbit sinoatrial node. J Mol Cell Cardiol. 2009;47:221–227. doi: 10.1016/j.yjmcc.2009.04.009. [DOI] [PubMed] [Google Scholar]
- Bucchi A, Barbuti A, Baruscotti M, DiFrancesco D. Heart rate reduction via selective ‘funny’ channel blockers. Curr Opin Pharmacol. 2007;7:208–213. doi: 10.1016/j.coph.2006.09.005. [DOI] [PubMed] [Google Scholar]
- Cerbai E, Mugelli A. I(f) in non-pacemaker cells: role and pharmacological implications. Pharmacol Res. 2006;53:416–423. doi: 10.1016/j.phrs.2006.03.015. [DOI] [PubMed] [Google Scholar]
- Cerbai E, Barbieri M, Mugelli A. Occurrence and properties of the hyperpolarization-activated current If in ventricular myocytes from normotensive and hypertensive rats during aging. Circulation. 1996;94:1674–1681. doi: 10.1161/01.cir.94.7.1674. [DOI] [PubMed] [Google Scholar]
- Cerbai E, Pino R, Porciatti F, Sani G, Toscano M, Maccherini M, et al. Characterization of the hyperpolarization-activated current, I(f), in ventricular myocytes from human failing heart. Circulation. 1997;95:568–571. doi: 10.1161/01.cir.95.3.568. [DOI] [PubMed] [Google Scholar]
- Chandler NJ, Greener ID, Tellez JO, Inada S, Musa H, Molenaar P, et al. Molecular architecture of the human sinus node: insights into the function of the cardiac pacemaker. Circulation. 2009;119:1562–1575. doi: 10.1161/CIRCULATIONAHA.108.804369. [DOI] [PubMed] [Google Scholar]
- Fenske S, Mader R, Scharr A, Paparizos C, Cao-Ehlker X, Michalakis S, et al. HCN3 contributes to the ventricular action potential waveform in the murine heart. Circ Res. 2011;109:1015–1023. doi: 10.1161/CIRCRESAHA.111.246173. [DOI] [PubMed] [Google Scholar]
- Fernandez-Velasco M, Goren N, Benito G, Blanco-Rivero J, Bosca L, Delgado C. Regional distribution of hyperpolarization-activated current (If) and hyperpolarization-activated cyclic nucleotide-gated channel mRNA expression in ventricular cells from control and hypertrophied rat hearts. J Physiol. 2003;553:395–405. doi: 10.1113/jphysiol.2003.041954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frere SG, Kuisle M, Luthi A. Regulation of recombinant and native hyperpolarization-activated cation channels. Mol Neurobiol. 2004;30:279–305. doi: 10.1385/MN:30:3:279. [DOI] [PubMed] [Google Scholar]
- Garcia-Frigola C, Shi Y, Evans SM. Expression of the hyperpolarization-activated cyclic nucleotide-gated cation channel HCN4 during mouse heart development. Gene Expr Patterns. 2003;3:777–783. doi: 10.1016/s1567-133x(03)00125-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf EM, Heubach JF, Ravens U. The hyperpolarization-activated current If in ventricular myocytes of non-transgenic and beta2-adrenoceptor overexpressing mice. Naunyn Schmiedebergs Arch Pharmacol. 2001;364:131–139. doi: 10.1007/s002100100431. [DOI] [PubMed] [Google Scholar]
- Harzheim D, Pfeiffer KH, Fabritz L, Kremmer E, Buch T, Waisman A, et al. Cardiac pacemaker function of HCN4 channels in mice is confined to embryonic development and requires cyclic AMP. EMBO J. 2008;27:692–703. doi: 10.1038/emboj.2008.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann S, Stieber J, Ludwig A. Pathophysiology of HCN channels. Pflugers Arch. 2007a;454:517–522. doi: 10.1007/s00424-007-0224-4. [DOI] [PubMed] [Google Scholar]
- Herrmann S, Stieber J, Stockl G, Hofmann F, Ludwig A. HCN4 provides a ‘depolarization reserve’ and is not required for heart rate acceleration in mice. EMBO J. 2007b;26:4423–4432. doi: 10.1038/sj.emboj.7601868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann S, Fabritz L, Layh B, Kirchhof P, Ludwig A. Insights into sick sinus syndrome from an inducible mouse model. Cardiovasc Res. 2011a;90:38–48. doi: 10.1093/cvr/cvq390. [DOI] [PubMed] [Google Scholar]
- Herrmann S, Layh B, Ludwig A. Novel insights into the distribution of cardiac HCN channels: an expression study in the mouse heart. J Mol Cell Cardiol. 2011b;51:997–1006. doi: 10.1016/j.yjmcc.2011.09.005. [DOI] [PubMed] [Google Scholar]
- Hoesl E, Stieber J, Herrmann S, Feil S, Tybl E, Hofmann F, et al. Tamoxifen-inducible gene deletion in the cardiac conduction system. J Mol Cell Cardiol. 2008;45:62–69. doi: 10.1016/j.yjmcc.2008.04.008. [DOI] [PubMed] [Google Scholar]
- Hoppe UC, Jansen E, Sudkamp M, Beuckelmann DJ. Hyperpolarization-activated inward current in ventricular myocytes from normal and failing human hearts. Circulation. 1998;97:55–65. doi: 10.1161/01.cir.97.1.55. [DOI] [PubMed] [Google Scholar]
- Ishii TM, Takano M, Xie LH, Noma A, Ohmori H. Molecular characterization of the hyperpolarization-activated cation channel in rabbit heart sinoatrial node. J Biol Chem. 1999;274:12835–12839. doi: 10.1074/jbc.274.18.12835. [DOI] [PubMed] [Google Scholar]
- Liu J, Dobrzynski H, Yanni J, Boyett MR, Lei M. Organisation of the mouse sinoatrial node: structure and expression of HCN channels. Cardiovasc Res. 2007;73:729–738. doi: 10.1016/j.cardiores.2006.11.016. [DOI] [PubMed] [Google Scholar]
- Ludwig A, Zong X, Jeglitsch M, Hofmann F, Biel M. A family of hyperpolarization-activated mammalian cation channels. Nature. 1998;393:587–591. doi: 10.1038/31255. [DOI] [PubMed] [Google Scholar]
- Ludwig A, Zong X, Stieber J, Hullin R, Hofmann F, Biel M. Two pacemaker channels from human heart with profoundly different activation kinetics. EMBO J. 1999;18:2323–2329. doi: 10.1093/emboj/18.9.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig A, Budde T, Stieber J, Moosmang S, Wahl C, Holthoff K, et al. Absence epilepsy and sinus dysrhythmia in mice lacking the pacemaker channel HCN2. EMBO J. 2003;22:216–224. doi: 10.1093/emboj/cdg032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marionneau C, Couette B, Liu J, Li H, Mangoni ME, Nargeot J, et al. Specific pattern of ionic channel gene expression associated with pacemaker activity in the mouse heart. J Physiol. 2005;562:223–234. doi: 10.1113/jphysiol.2004.074047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milanesi R, Baruscotti M, Gnecchi-Ruscone T, DiFrancesco D. Familial sinus bradycardia associated with a mutation in the cardiac pacemaker channel. N Engl J Med. 2006;354:151–157. doi: 10.1056/NEJMoa052475. [DOI] [PubMed] [Google Scholar]
- Moosmang S, Stieber J, Zong X, Biel M, Hofmann F, Ludwig A. Cellular expression and functional characterization of four hyperpolarization-activated pacemaker channels in cardiac and neuronal tissues. Eur J Biochem. 2001;268:1646–1652. doi: 10.1046/j.1432-1327.2001.02036.x. [DOI] [PubMed] [Google Scholar]
- Nolan MF, Malleret G, Lee KH, Gibbs E, Dudman JT, Santoro B, et al. The hyperpolarization-activated HCN1 channel is important for motor learning and neuronal integration by cerebellar Purkinje cells. Cell. 2003;115:551–564. doi: 10.1016/s0092-8674(03)00884-5. [DOI] [PubMed] [Google Scholar]
- Nolan MF, Malleret G, Dudman JT, Buhl DL, Santoro B, Gibbs E, et al. A behavioral role for dendritic integration: HCN1 channels constrain spatial memory and plasticity at inputs to distal dendrites of CA1 pyramidal neurons. Cell. 2004;119:719–732. doi: 10.1016/j.cell.2004.11.020. [DOI] [PubMed] [Google Scholar]
- Ragueneau I, Laveille C, Jochemsen R, Resplandy G, Funck-Brentano C, Jaillon P. Pharmacokinetic-pharmacodynamic modeling of the effects of ivabradine, a direct sinus node inhibitor, on heart rate in healthy volunteers. Clin Pharmacol Ther. 1998;64:192–203. doi: 10.1016/S0009-9236(98)90153-9. [DOI] [PubMed] [Google Scholar]
- Robinson RB, Siegelbaum SA. Hyperpolarization-activated cation currents: from molecules to physiological function. Annu Rev Physiol. 2003;65:453–480. doi: 10.1146/annurev.physiol.65.092101.142734. [DOI] [PubMed] [Google Scholar]
- Robinson RB, Yu H, Chang F, Cohen IS. Developmental change in the voltage-dependence of the pacemaker current, if, in rat ventricle cells. Pflugers Arch. 1997;433:533–535. doi: 10.1007/s004240050309. [DOI] [PubMed] [Google Scholar]
- Santoro B, Liu DT, Yao H, Bartsch D, Kandel ER, Siegelbaum SA, et al. Identification of a gene encoding a hyperpolarization-activated pacemaker channel of brain. Cell. 1998;93:717–729. doi: 10.1016/s0092-8674(00)81434-8. [DOI] [PubMed] [Google Scholar]
- Schulze-Bahr E, Neu A, Friederich P, Kaupp UB, Breithardt G, Pongs O, et al. Pacemaker channel dysfunction in a patient with sinus node disease. J Clin Invest. 2003;111:1537–1545. doi: 10.1172/JCI16387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert R, Scholten A, Gauss R, Mincheva A, Lichter P, Kaupp UB. Molecular characterization of a slowly gating human hyperpolarization-activated channel predominantly expressed in thalamus, heart, and testis. Proc Natl Acad Sci U S A. 1999;96:9391–9396. doi: 10.1073/pnas.96.16.9391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W, Wymore R, Yu H, Wu J, Wymore RT, Pan Z, et al. Distribution and prevalence of hyperpolarization-activated cation channel (HCN) mRNA expression in cardiac tissues. Circ Res. 1999;85:e1–e6. doi: 10.1161/01.res.85.1.e1. [DOI] [PubMed] [Google Scholar]
- Shi W, Yu H, Wu J, Zuckerman J, Wymore R, Dixon J, et al. The distribution and prevalence of HCN isoforms in the canine heart and their relation to the voltage dependence of If. Biophys J. 2000;78((Suppl.)):353A. [Google Scholar]
- Siu CW, Lieu DK, Li RA. HCN-encoded pacemaker channels: from physiology and biophysics to bioengineering. J Membr Biol. 2006;214:115–122. doi: 10.1007/s00232-006-0881-9. [DOI] [PubMed] [Google Scholar]
- Stieber J, Herrmann S, Feil S, Loster J, Feil R, Biel M, et al. The hyperpolarization-activated channel HCN4 is required for the generation of pacemaker action potentials in the embryonic heart. Proc Natl Acad Sci U S A. 2003;100:15235–15240. doi: 10.1073/pnas.2434235100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stieber J, Wieland K, Stockl G, Ludwig A, Hofmann F. Bradycardic and proarrhythmic properties of sinus node inhibitors. Mol Pharmacol. 2006;69:1328–1337. doi: 10.1124/mol.105.020701. [DOI] [PubMed] [Google Scholar]
- Stilli D, Sgoifo A, Macchi E, Zaniboni M, De Iasio S, Cerbai E, et al. Myocardial remodeling and arrhythmogenesis in moderate cardiac hypertrophy in rats. Am J Physiol Heart Circ Physiol. 2001;280:H142–H150. doi: 10.1152/ajpheart.2001.280.1.H142. [DOI] [PubMed] [Google Scholar]
- Stillitano F, Lonardo G, Zicha S, Varro A, Cerbai E, Mugelli A, et al. Molecular basis of funny current (If) in normal and failing human heart. J Mol Cell Cardiol. 2008;45:289–299. doi: 10.1016/j.yjmcc.2008.04.013. [DOI] [PubMed] [Google Scholar]
- Swedberg K, Komajda M, Boehm M, Borer JS, Ford I, Dubost-Brama A, et al. Ivabradine and outcomes in chronic heart failure (SHIFT): a randomised placebo-controlled study. Lancet. 2010;376:875–885. doi: 10.1016/S0140-6736(10)61198-1. [DOI] [PubMed] [Google Scholar]
- Teerlink JR. Ivabradine in heart failure – no paradigm SHIFT … yet. Lancet. 2010;376:847–849. doi: 10.1016/S0140-6736(10)61314-1. [DOI] [PubMed] [Google Scholar]
- Tellez JO, Dobrzynski H, Greener ID, Graham GM, Laing E, Honjo H, et al. Differential expression of ion channel transcripts in atrial muscle and sinoatrial node in rabbit. Circ Res. 2006;99:1384–1393. doi: 10.1161/01.RES.0000251717.98379.69. [DOI] [PubMed] [Google Scholar]
- Verheijck EE, Wessels A, van Ginneken AC, Bourier J, Markman MW, Vermeulen JL, et al. Distribution of atrial and nodal cells within the rabbit sinoatrial node: models of sinoatrial transition. Circulation. 1998;97:1623–1631. doi: 10.1161/01.cir.97.16.1623. [DOI] [PubMed] [Google Scholar]
- Yasui K, Liu W, Opthof T, Kada K, Lee JK, Kamiya K, et al. I(f) current and spontaneous activity in mouse embryonic ventricular myocytes. Circ Res. 2001;88:536–542. doi: 10.1161/01.res.88.5.536. [DOI] [PubMed] [Google Scholar]
- Zicha S, Fernandez-Velasco M, Lonardo G, L'Heureux N, Nattel S. Sinus node dysfunction and hyperpolarization-activated (HCN) channel subunit remodeling in a canine heart failure model. Cardiovasc Res. 2005;66:472–481. doi: 10.1016/j.cardiores.2005.02.011. [DOI] [PubMed] [Google Scholar]