

Abstract
BACKGROUND AND PURPOSE
QT prolongation is commonly used as a surrogate marker for Torsade de Pointes (TdP) risk of non-cardiovascular drugs. However, use of this indirect marker often leads to misinterpretation of the realistic TdP risk, as tested compounds may cause QT prolongation without evoking TdP in humans. A negative electro-mechanical (E-M) window has recently been proposed as an alternative risk marker for TdP in a canine LQT1 model. Here, we evaluated the E-M window in anaesthetized guinea pigs as a screening marker for TdP in humans.
EXPERIMENTAL APPROACH
The effects of various reference drugs and changes in body temperature on the E-M window were assessed in instrumented guinea pigs. The E-M window was defined as the delay between the duration of the electrical (QT interval) and mechanical (QLVPend) systole.
KEY RESULTS
Drugs with known TdP liability (quinidine, haloperidol, domperidone, terfenadine, thioridazine and dofetilide), but not those with no TdP risk in humans (salbutamol and diltiazem) consistently decreased the E-M window. Interestingly, drugs with known clinical QT prolongation, but with low risk for TdP (amiodarone, moxifloxacin and ciprofloxacin) did not decrease the E-M window. Furthermore, the E-M window was minimally affected by changes in heart rate or body temperature.
CONCLUSIONS AND IMPLICATIONS
A decreased E-M window was consistently observed with drugs already known to have high TdP risk, but not with drugs with low or no TdP risk. These results suggest that the E-M window in anaesthetized guinea pigs is a risk marker for TdP in humans.
Keywords: guinea pig, electro-mechanical window, E-M window, QT interval, Torsade de Pointes, TdP, proarrhythmia, drug induced QT prolongation, ICH S7B, anaesthetized, safety pharmacology
Introduction
Delayed ventricular repolarization – whether by congenital defects such as the long QT syndrome (LQTS) (Vohra, 2007) or by the effects of drugs that block potassium channels, such as terfenadine or dofetilide – is associated with a high incidence of Torsade de Pointes (TdP) and sudden cardiac death. The incidence of TdP with cardiac proarrhythmic drugs is 1–8% (Darpo, 2001) while the incidence with non-cardiac drugs is considerably lower (approximately 1 in 100 000). Because of its infrequent occurrence, TdP is typically not seen in clinical trials. As such, QT interval prolongation has become a surrogate marker of TdP risk and is commonly used as a preclinical and clinical biomarker for proarrhythmic activity of non-cardiovascular drugs (Cavero and Crumb, 2005).
Currently, QT prolongation is a major safety issue in drug development and a dedicated regulatory guideline (ICH S7B) describes strategies for identification of drug candidates affecting cardiac repolarization. As no single assay is sufficient to predict the risk of TdP in humans, the ICH S7B document recommends an integrated approach including at least an in vitro assessment of inhibition of the rapidly activating delayed rectifier potassium current (IKr) and an in vivo QT assay.
In line with the ICH S7B guideline, frontloading of cardiovascular safety data, that is, screening for TdP liability early on in drug development, has become common practice. In this context, guinea pigs have received a great deal of attention with regard to evaluation of effects of newly discovered drugs on the QT interval at an early stage. Indeed, the anaesthetized guinea pig model is a useful tool for ‘first in line’in vivo cardiovascular safety assessment due to the limited amounts of test compound required, the similarity of cardiac ionic currents between guinea pigs and humans (Terrar et al., 2007), and the ease of manipulation of guinea pigs. Over the last years, many reference drugs have been evaluated in the anaesthetized guinea pig model and the predictive value of this model for QT prolongation in humans is well documented (Testai et al., 2004; Kagstrom et al., 2007; Yao et al., 2008).
Although the current strategies based on identification of drug-induced QT prolongation in animal models (ICH S7B) and its clinical counterpart – the thorough QT study (ICH E14) – have been relatively successful in the attrition of unsafe drug candidates, this approach inherently generates a number of false positive results due to the lack of direct association between the surrogate marker (QT interval) and the final endpoint (TdP risk). Indeed, the magnitude of QTc prolongation does not correlate with the TdP risk in humans; and several drugs prolong the QT interval without causing TdP in humans (Darpo, 2001; Hoffmann and Warner, 2006; Hondeghem, 2006).
Over the last years, a number of new risk markers and models for TdP risk evaluation have been proposed, illustrating the urgent need of the pharmaceutical industry to develop more accurate markers for TdP risk in humans. The current state-of-the-art of risk markers and proarrhythmia models are comprehensively described elsewhere (Lawrence et al., 2005; Hoffmann and Warner, 2006; Gintant, 2008). The electro-mechanical (E-M) window has recently been proposed as one of these alternative risk markers for TdP in humans.
The current work aimed to evaluate the E-M window in anaesthetized guinea pigs as a new risk marker for TdP. The E-M window is defined as the delay between the electrical (QT interval) and mechanical (QLVPend) systole (E-M window = QLVPend interval – QT interval, Figure 1). Despite intensive studies on E-M coupling in humans since the 1980s, the concept of disturbed E-M coupling as a risk marker for TdP was only recently introduced in safety pharmacology (van der Linde et al., 2010). In healthy individuals, the duration of the electrical systole (QT interval) is shorter than, but closely parallels, the duration of mechanical systole (measured indirectly by the time between the Q wave and the second heart sound; QS2) throughout the normal range of resting heart rates (Boudoulas et al., 1981b). Interestingly, changes in autonomic tone (De Caprio et al., 1984) or high circulating catecholamine levels (Boudoulas et al., 1981a) were reported to invert the normal QT/QS2 ratio. Furthermore, the QT/QS2 ratio was found to be a useful indicator for several cardiovascular diseases (Boudoulas et al., 1982; Airaksinen et al., 1984; Chambers and Ward, 1987). Moreover, patients with the Romano-Ward inherited LQTS were found to have an altered QT/QS2 ratio (Vincent et al., 1991). Recently, van der Linde et al. (2010) showed that in a canine LQT1 model, an inverted (negative) E-M window was a prerequisite for the initiation of TdP. A negative E-M window is a highly proarrhythmic condition due to the fact that under these conditions there is a mismatch of electrical and mechanical activities. This provides a situation where arrhythmias can ensue, most likely because Ca2+ can continue to enter the cardiac myocytes favouring the occurrence of Ca2+ overload, leading to both early and delayed afterdepolarizations as well as aftercontractions which may eventually lead to TdP. Restoration of the E-M window by atenolol, a β-adrenoceptor antagonist, or verapamil, an L-type Ca2+ channel blocker, prevented the induction of TdP by IKs blockade in combination with β-adrenoceptor stimulation (van der Linde et al., 2010).
Figure 1.
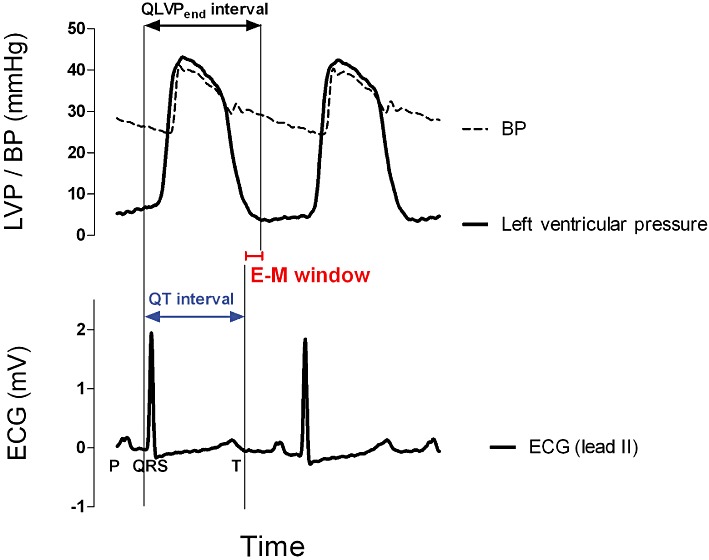
The Wiggers diagram illustrates the synchronization of the electrical (ECG) and mechanical (left ventricular pressure, LVP) activity of the cardiac muscle in anaesthetized guinea pigs. The E-M window is defined as the delay between the duration of the electrical (QT interval) and mechanical (QLVPend interval) systole. E-M window = QLVPend interval – QT interval. BP, blood pressure.
Based on these observations, we hypothesized that a negative E-M window in the anaesthetized guinea pig model might be a (LQT1) risk marker of TdP in humans. Implementation of such a new risk marker for TdP in an early screening platform, like the anaesthetized guinea pig model, would mean a significant breakthrough in cardiovascular safety pharmacology. In an attempt to determine the predictive value of the E-M window in anaesthetized guinea pigs, the effects of various pharmacological agents with different torsadogenic profiles (high, low and no TdP risk) were assessed.
Methods
Animal use and care
The studies were approved by the Ethical Committee of Vito (Mol, Belgium) and conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institute of Health (NIH Publication no. 85–23, revised 1996). Female Dunkin Hartley guinea pigs (weight 380–540 g) were purchased from Charles Rivers (L'Arbresle, France). Animals arrived at the animal facilities at least 1 week before the study and were housed in groups (up to five animals per cage; >3500 cm2 per cage) with access to food and water ad libitum.
Surgical procedure
Animals were anaesthetized by i.p. administration of sodium pentobarbital (60 mg·kg−1). A tracheotomy was performed and the guinea pigs were mechanically ventilated with room air (model ‘683’ respirator, Harvard Apparatus, Les Ulis, France) using a stroke volume of 10 mL·kg−1 at a rate of 60 strokes min−1. The tracheal tube was connected to a pressure transducer to monitor the pulmonary inflation pressure throughout the experiment. Animals were placed on an electrical heat pad at 37°C (small operating table, Harvard Apparatus). Both jugular veins were cannulated for i.v. drug administration (right jugular vein) and continuous infusion of pentobarbital (6 mg·h−1) to maintain anaesthesia (left jugular vein). An open-lumen catheter was inserted in the left carotid artery for blood pressure (BP) measurement and collection of arterial blood samples. A tip catheter (SPR-249, Millar Instruments, Houston, TX, USA) was positioned in the left ventricle for measurement of left ventricular pressure (LVP; accessed via the right carotid artery). Four needle electrodes were placed subcutaneously to record the ECG. Pressure and ECG signals were relayed to a bio-amplifier (EMKA Technologies, Paris, France) with output to a data acquisition system (IOX, EMKA Technologies). Raw data were captured at sampling rates of 1000 Hz (ECG and LVP) or 500 Hz (BP). After being instrumented, animals were allowed to stabilize for a period of at least 20 min.
Experimental protocol
The effect of various reference drugs on cardio-electrophysiological and cardio-haemodynamic parameters was assessed in anaesthetized guinea pigs. Guinea pigs received consecutively five increasing doses of the test compounds. All compounds were infused i.v. at a rate of 1 mL·kg−1 over a 10 min infusion period with 5 min intervals between consecutive doses. Animals in the separate vehicle group received infusions of equal volumes of saline. At the end of the vehicle experiments, dofetilide was infused over a 1 min period as positive control.
In addition, a separate set of experiments (n = 3) was performed to assess the effect of temperature (range 34–38°C) on the cardiovascular parameters. After stabilization, the heating pad was switched off and temperature (measured via a rectal probe) and associated cardiovascular parameters were monitored during the cooling phase.
Data analysis
Analysis of raw data was performed using ECG-Auto software (version 2.5.1.35, EMKA Technologies). ECG signals (lead II) were automatically analysed based on shape recognition linked to a user-defined library of ECG waveforms. The QTcB interval was calculated for every single beat by using the previous RR interval. The end of the ventricular relaxation (LVPend point) was defined by applying a threshold (−100 mmHg·s−1) on the first derivative of the relaxation part of the LVP signal. The RR interval, PQ interval, QRS interval, QT interval, QTcB interval, QLVPend interval and the E-M window were calculated beat by beat. The accuracy of the analysis was reviewed by manual over-reading. For each minute-interval, median values were calculated and used for reporting.
The QT interval, the QLVPend interval and the E-M window were reported in actual units (ms), whereas for QTcB the changes from baseline in percentage were calculated. The parameters 60 s prior to the first drug administration were defined as baseline values (t = 0 min). The results from individual animals were pooled by treatment group and shown in graphs as mean ± SEM. Changes from baseline (in actual units) were analysed by a one-way anova test with Bonferroni post hoc test.
Materials
Clinical reference substances were purchased as dry powder. Quinidine, haloperidol, domperidone, terfenadine and diltiazem were purchased from Tocris Bioscience (Bristol, UK). Moxifloxacin, ciprofloxacin, thioridazine, isoprenaline and salbutamol were purchased from Sigma Aldrich (Bornem, Belgium). The dry powder (taking into account a correction factor for the different molecular weights of various salt forms) was dissolved in a vehicle consisting of physiological saline solution. Vehicles of terfenadine and domperidone contained 10% hydroxypropyl-β-cyclodextrin to increase solubility. Amiodarone was purchased in a clinically available formulation (Cordarone® 150 mg per 3 mL, Sanofi Aventis, Diegem, Belgium) and was diluted with saline.
Results
A range of pharmacological agents were administered i.v. to instrumented and anaesthetized guinea pigs. Following drug administration, the duration of the QT interval and the QLVPend interval was continuously monitored. In addition, the E-M window, defined as the delay between the electrical and mechanical systole (E-M window = QLVPend interval – QT interval) was calculated beat by beat (Figure 1).
Drugs with well-documented TdP risk
Drugs associated with prolongation of the QT interval and with well-documented TdP risk in humans [quinidine (n = 4), haloperidol (n = 3), domperidone (n = 4), terfenadine (n = 4), thioridazine (n = 5) and dofetilide (positive control, n = 4)] all showed dose-dependent prolongation of the QT and QTcB interval (Figures 2 and 3). The QLVPend interval was not prolonged to the same extent as the QT interval resulting in a dose-dependent decrease of the E-M window. Administration of quinidine, haloperidol and domperidone eventually led to a negative E-M window (Figure 3), whereas terfenadine and thioridazine decreased the E-M window, but failed to make this parameter negative (Table 1). Interestingly, quinidine decreased the E-M window, but a slight restoration of the E-M window between consecutive doses was observed, which was not observed for the QTcB interval. Terfenadine caused marked bradycardia and hypotension at the highest dose (10 mg·kg−1), thereby generating less reliable results at this dose.
Figure 2.
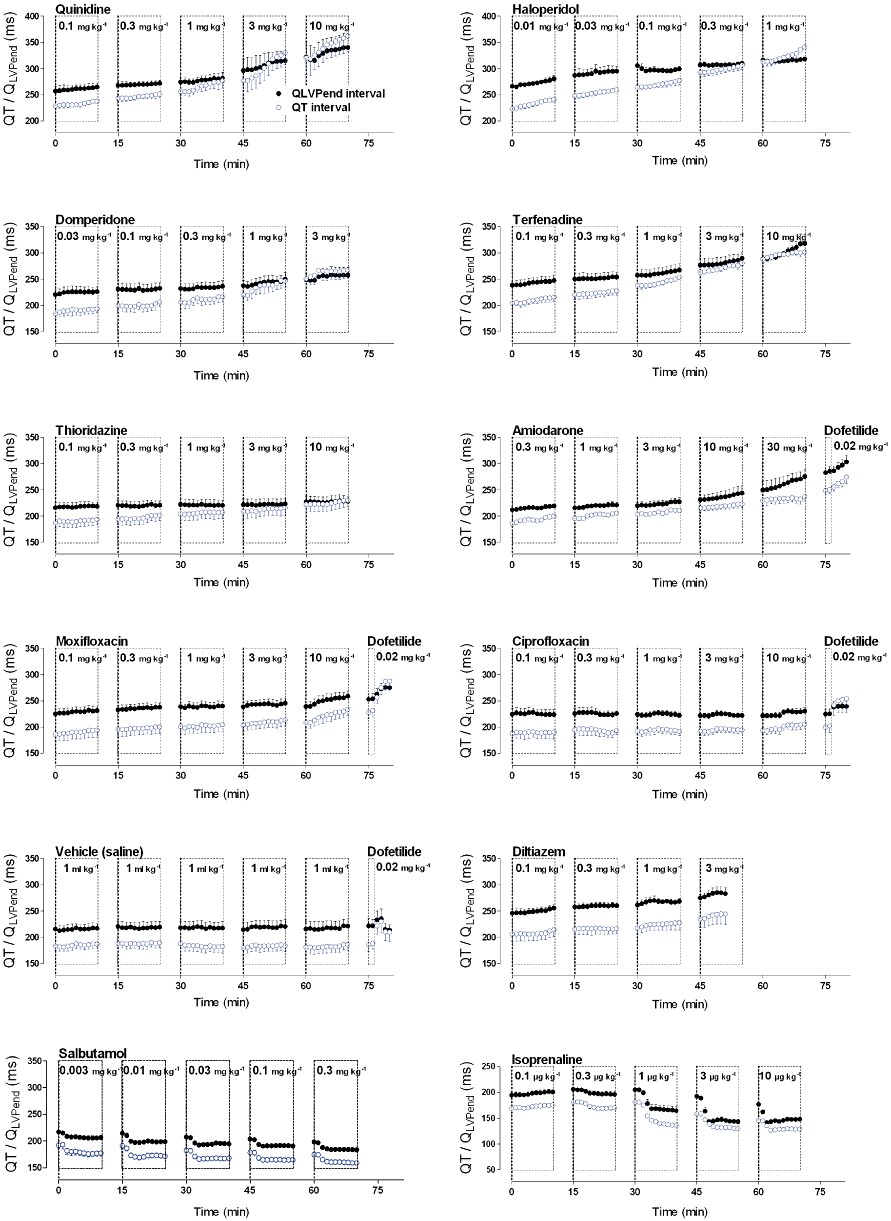
Graphs show the effect of various pharmacological agents on the electro-mechanical coupling in anaesthetized guinea pigs. Drugs with high TdP risk (quinidine, haloperidol, domperidone, terfenadine, dofetilide and thioridazine) induced unequal changes of the QT interval versus the QLVPend interval, whereas drugs with low (amiodarone, moxifloxacin and ciprofloxacin), or no TdP risk (vehicle, salbutamol and diltiazem) induced parallel changes of the QT interval and LVPend interval. Finally, isoprenaline showed different kinetics of its effect on the QT and QLVPend interval leading to a transient decrease of the E-M window. Plots show mean ± SEM.
Figure 3.
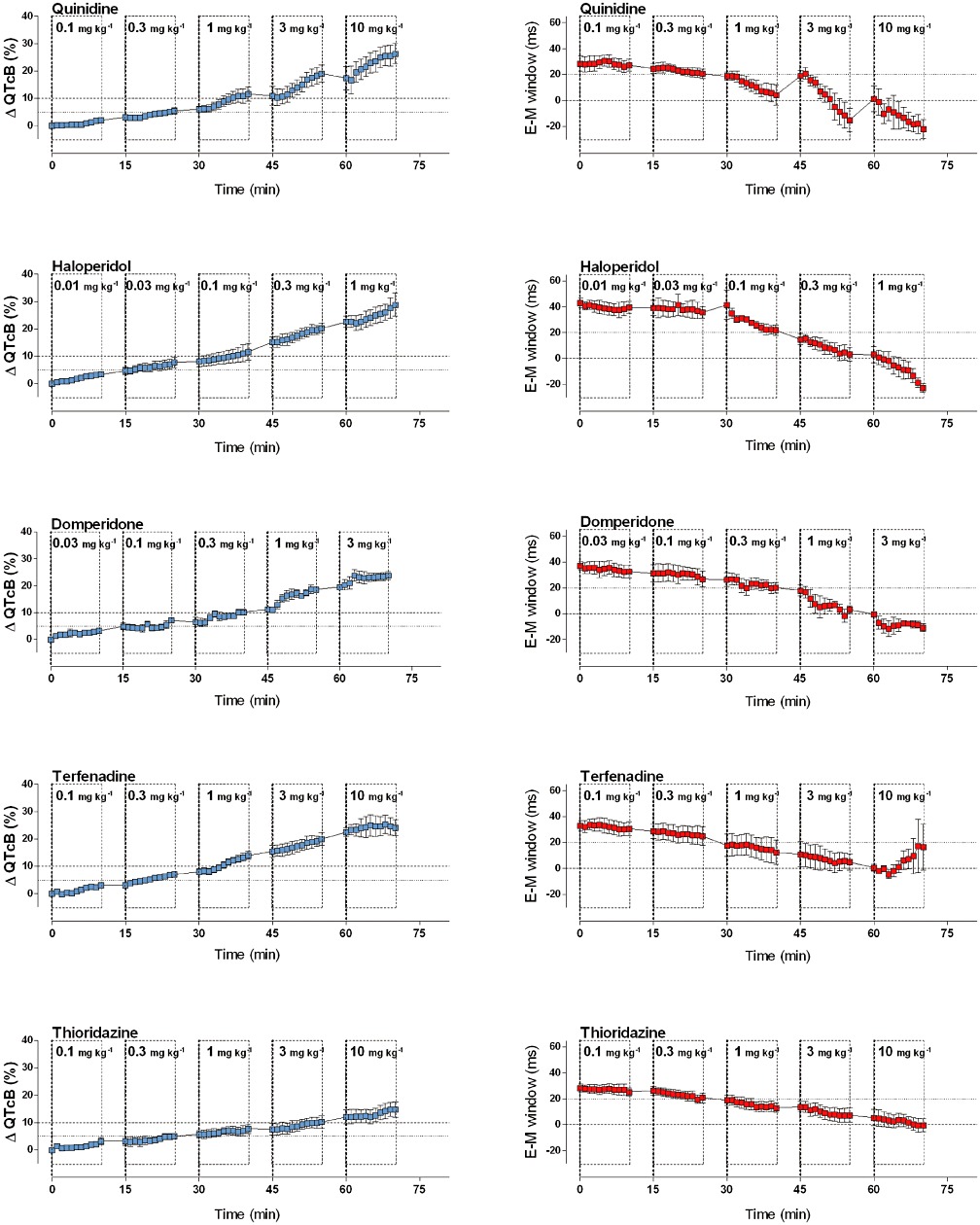
Graphs show the effects of drugs with documented TdP risk on the commonly used surrogate marker for TdP risk (QTcB, % change from baseline; in blue) and on the E-M window (ms; in red). Quinidine, haloperidol, domperidone, terfenadine and thioridazine prolonged the QTcB interval and consistently decreased the E-M window. Graphs show mean ± SEM.
Table 1.
Effects of a range of clinically used drugs on the E-M window and on the QTcB interval in anaesthetized guinea pigs
| Treatment | Baseline (actual units) | Dose 1 | Dose 2 | Dose 3 | Dose 4 | Dose 5 | |
|---|---|---|---|---|---|---|---|
| 10 min | 25 min | 40 min | 55 min | 70 min | |||
| Quinidine | E-M window | 28.4 ± 5.9 | −1.2 ± 1.7 | −7.7 ± 5.7 | −24.2 ± 8.8 | −43.6 ± 9.9** | −50.4 ± 8.9** |
| n = 4 | QTcB | 403.0 ± 6.2 | 7.9 ± 0.6 | 21.6 ± 4.3 | 46.5 ± 10.1* | 76.5 ± 11.4*** | 105.9 ± 14.2*** |
| Haloperidol | E-M window | 43.2 ± 4.0 | −3.6 ± 0.7 | −7.4 ± 2.2 | −21.3 ± 7.8 | −39.8 ± 7.7** | −66.0 ± 7.5*** |
| n = 3 | QTcB | 407.4 ± 6.8 | 13.7 ± 2.7 | 31.4 ± 7.2 | 47.2 ± 11.8* | 82.5 ± 6.6*** | 117.2 ± 15.8*** |
| Domperidone | E-M window | 37.3 ± 3.4 | −4.5 ± 1.7 | −10.5 ± 2.9 | −17.0 ± 3.8 | −34.0 ± 4.6*** | −48.0 ± 5.6*** |
| n = 4 | QTcB | 369.3 ± 10.0 | 11.8 ± 2.8 | 26.2 ± 3.9 | 37.5 ± 4.3*** | 68.9 ± 4.8*** | 88.3 ± 7.1*** |
| Terfenadine | E-M window | 32.8 ± 4.0 | −2.3 ± 2.1 | −7.9 ± 3.8 | −20.4 ± 5.3 | −28.0 ± 2.2 | −12.6 ± 17.9 |
| n = 4 | QTcB | 385.9 ± 5.2 | 10.6 ± 2.4 | 25.5 ± 2.1* | 54.5 ± 3.7*** | 79.8 ± 6.1*** | 92.5 ± 9.7*** |
| Thioridazine | E-M window | 28.2 ± 3.0 | −3.0 ± 1.3 | −7.4 ± 2.0 | −15.2 ± 3.9 | −20.9 ± 6.8* | −28.6 ± 6.8** |
| n = 5 | QTcB | 364.9 ± 11.6 | 11.5 ± 4.2 | 18.0 ± 2.9 | 27.6 ± 6.0* | 36.9 ± 7.8** | 53.1 ± 9.3*** |
| Vehicle | E-M window | 27.7 ± 9.2 | −1.7 ± 0.7 | −2.3 ± 0.9 | −3.2 ± 0.4 | −1.0 ± 2.6 | −2.3 ± 1.8 |
| n = 4 | QTcB | 365.3 ± 9.1 | 4.6 ± 2.2 | 7.5 ± 3.5 | −0.9 ± 7.2 | 0.7 ± 7.9 | 3.0 ± 7.9 |
| Diltiazem | E-M window | 40.2 ± 6.0 | 0.8 ± 4.9 | 3.4 ± 11.0 | 0.5 ± 16.4 | ||
| n = 4 | QTcB | 382.9 ± 17.2 | 7.1 ± 2.9 | 0.7 ± 8.9 | 5.6 ± 14.4 | ||
| Salbutamol | E-M window | 25.0 ± 5.3 | 4.4 ± 3.2 | 2.6 ± 2.7 | 2.2 ± 2.9 | 1.6 ± 3.4 | 0.1 ± 2.6 |
| n = 5 | QTcB | 372.1 ± 6.2 | −8.6 ± 4.4 | −15.7 ± 3.9 | −21.6 ± 6.9 | −27.3 ± 5.5** | −36.5 ± 5.8*** |
| Isoprenaline | E-M window | 25.0 ± 3.2 | 0.3 ± 1.4 | 1.6 ± 2.8 | 3.0 ± 5.1 | −19.5 ± 1.0** | −19.0 ± 4.1** |
| n = 4 | QTcB | 352.3 ± 3.8 | 6.6 ± 0.9 | 5.5 ± 4.4 | −41.4 ± 2.8*** | −38.1 ± 6.1*** | −44.2 ± 7.7*** |
| Amiodarone | E-M window | 26.3 ± 2.3 | −6.3 ± 1.9 | −10.5 ± 1.8 | −9.5 ± 2.2 | −6.0 ± 4.4 | 12 ± 2.4 |
| n = 4 | QTcB | 373.1 ± 2.3 | 13.6 ± 3.6 | 19.9 ± 3.2 | 26.1 ± 5.9* | 37.5 ± 7.7** | 38.2 ± 6.0** |
| Moxifloxacin | E-M window | 39.0 ± 5.0 | −2.0 ± 0.9 | −1.0 ± 2.8 | −3.0 ± 2.4 | −6.8 ± 2.8 | −13.5 ± 3.3* |
| n = 4 | QTcB | 364.6 ± 12.9 | 10.6 ± 1.1 | 18.8 ± 0.6 | 25.4 ± 1.7* | 33.6 ± 4.9** | 52.0 ± 6.4*** |
| Ciprofloxacin | E-M window | 37.5 ± 3.8 | −4.3 ± 1.3 | −4.5 ± 1.9 | −7.0 ± 2.0 | −9.8 ± 1.3 | −12.4 ± 4.0 |
| n = 4 | QTcB | 367.5 ± 13.7 | 4.0 ± 1.0 | 9.7 ± 9.6 | 8.5 ± 9.3 | 12.5 ± 10.5 | 22.0 ± 10.5 |
Baseline and changes from baseline (both in actual units, ms) for the E-M window and the QTcB interval after administration of a range of clinical reference drugs. Data show mean and SEM at the end of the drug infusion period. For isoprenaline (doses 4 and 5), the maximal effect (as shown in the table) was observed, respectively, 4 and 3 min after the start of the infusion. A one-way anova with Bonferroni post hoc test was performed on the ‘change from baseline’ values.
P < 0.05,
P < 0.01,
P < 0.001.
Negative controls (no TdP risk)
I.v. administration of vehicle (saline, n = 4) did not affect the E-M window or the QTcB interval (Figures 2 and 4). Administration of the positive control (dofetilide) caused a negative E-M window and prolongation of the QTcB interval, illustrating the validity of the assay. In addition, two clinical drugs with no TdP risk were tested: salbutamol (n = 5) and diltiazem (n = 4). Both drugs failed to affect the E-M window. Salbutamol caused a drastic increase in heart rate (data not shown), thereby significantly decreasing the QT and QLVPend interval. The QTcB interval was shortened by salbutamol illustrating the limitations of Bazett's formula for heart rate correction, whereas the E-M window was hardly influenced by the marked changes in heart rate. Diltiazem caused a decrease of heart rate (data not shown) and eventually cardiovascular collapse at 3 mg·kg−1 and animals died at the highest dose 10 mg·kg−1. However, the E-M window was not altered by diltiazem administration.
Figure 4.
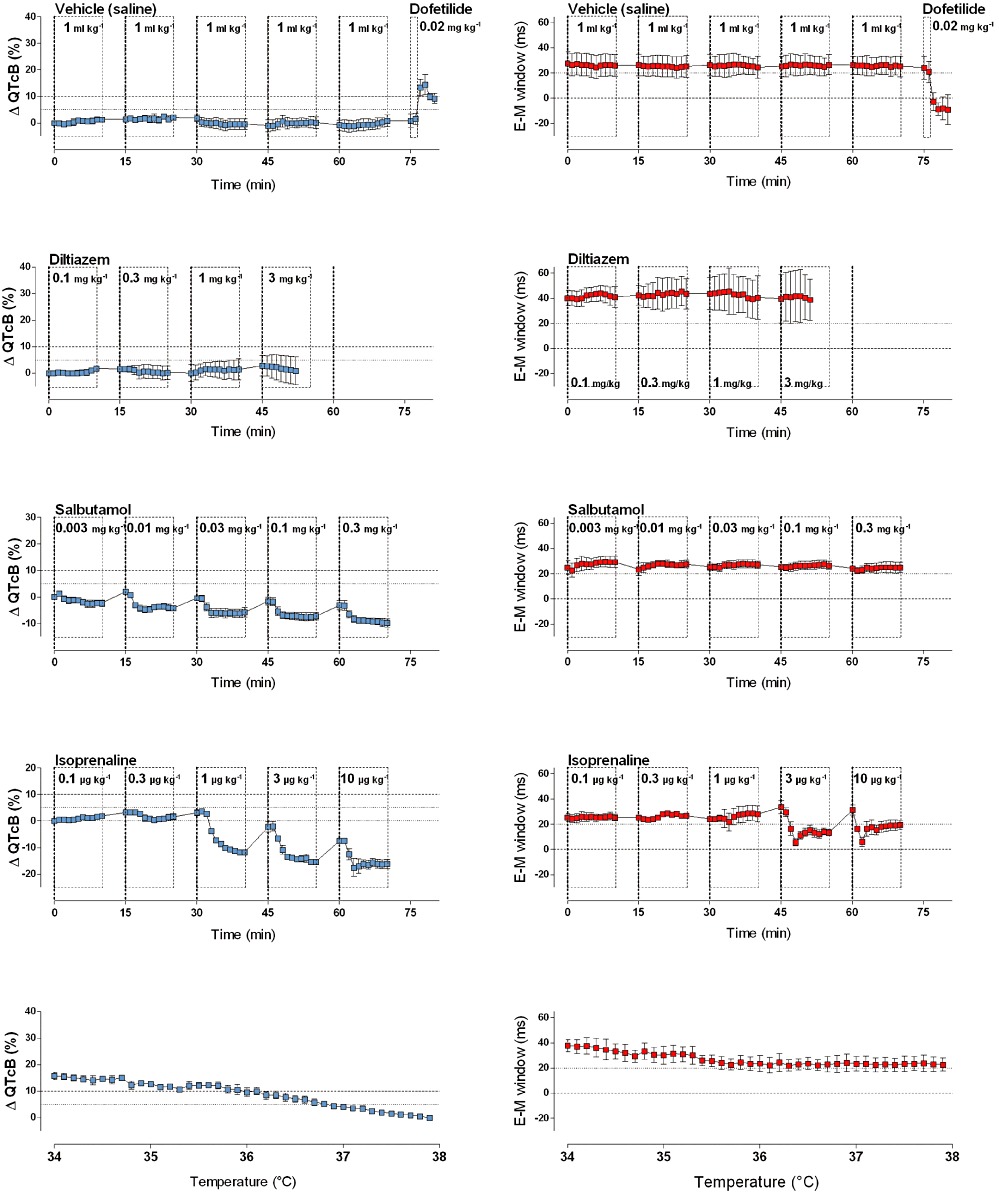
Plots show the effects of vehicle, of drugs with no TdP risk (diltiazem, salbutamol), of isoprenaline and of changes in body temperature (lowest graphs) on the commonly used surrogate marker for TdP risk (QTcB, % change from baseline; in blue) and on the E-M window (ms; in red). The vehicle did not cause QTcB prolongation or shortening of the E-M window. Salbutamol and diltiazem, respectively, increased and decreased heart rate (data not shown), but did not affect the E-M window. Isoprenaline transiently reduced the E-M window due to the different kinetics of its effect on the QT and QLVPend interval. Finally, cooling of anaesthetized animals from 38°C to 34°C prolonged the QTcB interval (−14.6 ms·°C−1), but had no major effect on the duration of the E-M window. Graphs show mean ± SEM.
Amiodarone, moxifloxacin and ciprofloxacin
Amiodarone (n = 4) and moxifloxacin (n = 4) showed remarkable results (Figures 2 and 5). Both drugs caused a significant prolongation of the QTcB interval of 12% and 14% respectively. However, as the QLVPend interval prolonged to the same (moxifloxacin) or greater (amiodarone) extent, no negative E-M window was observed after administration of these compounds. Furthermore, dofetilide failed to evoke a negative E-M window following amiodarone administration. Ciprofloxacin (n = 4) showed a tendency for QT prolongation (5% increase) at the highest dose, but the E-M window was not affected by ciprofloxacin administration.
Figure 5.
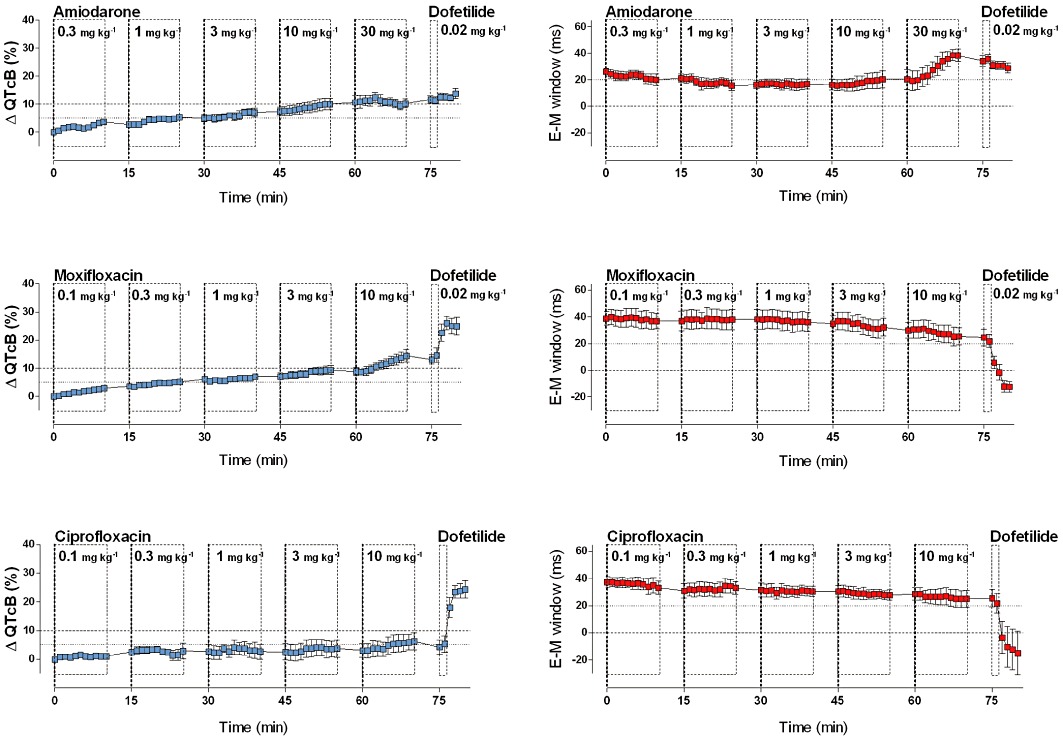
Graphs show the effects of amiodarone and two antibiotics (moxifloxacin and ciprofloxacin) on the commonly used surrogate marker for TdP risk (QTcB, % change from baseline; in blue) and on the E-M window (ms; in red). Amiodarone increased the QTcB interval, but did not decrease the E-M window; in fact, it significantly increased the E-M window and prevented induction of a negative E-M window by dofetilide. Moxifloxacin and ciprofloxacin hardly affected the E-M window despite prolongation of the QTcB interval. Graphs show mean ± SEM.
Isoprenaline
Isoprenaline (n = 4) showed a shortening in the QT interval, as would be expected due to the chronotropic effects of this compound. At the same time, a decrease of the QLVPend interval was observed, which was not completely synchronized with the effects on QT. This led to a transient decrease in the E-M window at the highest concentrations (Figures 2 and 4).
Effect of temperature
The effect of temperature on the electro-mechanical coupling was investigated in a different set of experiments (n = 3, Figure 4). Cooling of the animals from 38°C to 34°C caused increases of the QT interval, the QTcB interval (slope: −14.6 ms·°C−1) and the QLVPend interval. The E-M window remained stable during cooling; and even slightly increased when cooling below 35.5°C.
Occurrence of arrhythmias
None of the reference drugs evoked episodes of TdP. Isolated ventricular premature contractions (VPCs) and an atrial premature contraction were observed in 8 and 1 out of 45 animals respectively. Most of these VPCs were observed during replacement of the catheters and were not observed in later experiments where the catheters were appropriately fixed. Right bundle branch block was reported after administration of diltiazem (1 of 4), terfenadine (4 of 4), haloperidol (1 of 3), thioridazine (2 of 5) and amiodarone (1 of 4). Furthermore, atrio-ventricular block was observed after treatment with diltiazem (3 of 4 animals), terfenadine (1 of 4), haloperidol (1 of 3) and dofetilide (2 of 4). Finally, diltiazem caused complete cardiovascular collapse in all animals (starting from 3 mg·kg−1), eventually leading to death.
Discussion
The anaesthetized guinea pig model and the E-M window
Over the last years, the anaesthetized guinea pig model has received a great deal of attention as a useful tool for early in vivo cardiovascular safety screening. Guinea pigs have a cardiac electrophysiology that largely resembles that of humans and the small size of the animals favours the use of this model for cardiovascular safety screening. Several anaesthesia regimens have been described for assessing QT prolongation in guinea pigs (Hamlin et al., 2003; Hauser et al., 2005; Sakaguchi et al., 2005), with the pentobarbital anaesthetized model being the most common and best documented (Testai et al., 2004; Kagstrom et al., 2007; Tabo et al., 2007; Yao et al., 2008). In addition to the ECG-derived information (QT interval, QRS interval and PQ interval), the anaesthetized guinea pig model provides information on haemodynamics (heart rate, BP, QA interval), cardiac performance (left ventricular pressure) and on the induction of spontaneous arrhythmias making it a versatile cardiovascular screening model (Figure 6). Recently, the E-M window has been proposed as a novel risk marker for TdP in a LQT1 dog model (van der Linde et al., 2010). The present work provides the first documented report of the E-M window as a general risk marker for TdP within an early screening model.
Figure 6.
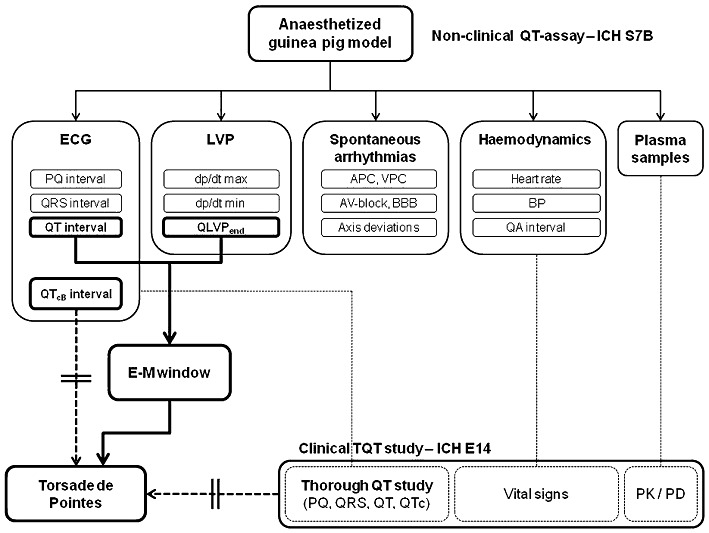
Figure shows a schematic overview of the anaesthetized guinea pig model as a versatile early cardiovascular safety screening tool. At present, both preclinical (ICH S7B) and clinical (ICH E14) studies use the QTc interval as a surrogate marker for TdP risk in humans. The E-M window provides a new risk marker for TdP risk in humans that might eventually be implemented in clinical research. APC, atrial premature contraction; BBB, bundle branch block (left or right); VPC, ventricular premature contraction.
Drugs with high TdP risk are selectively flagged
Drugs with known high TdP risk (Redfern et al., 2003; De Bruin et al., 2005) consistently decreased the E-M window. Quinidine, domperidone, haloperidol and dofetilide caused a negative E-M window that might form a substrate for the development of TdP. Due to a large repolarization reserve in guinea pigs (Lu et al., 2001; Zicha et al., 2003), IKr blockade by itself did not elicit TdP in the anaesthetized guinea pig model.
The results of terfenadine and thioridazine are less clear. Despite a substantial decrease of the E-M window, terfenadine and thioridazine did not cause negative E-M windows on their own. This might suggest that additional challenges to the repolarization reserve are required to trigger TdP with these agents. Terfenadine, for example, has been shown to be a multi-ion channel blocker, affecting both inward and outward currents, including the L-type calcium current (ICaL) and the slowly activating delayed rectifier potassium current (IKr), which could be an explanation for the absence of a negative E-M window with this compound (Ming and Nordin, 1995). Interestingly, terfenadine did not trigger TdP in a phenylephrine-sensitized rabbit model, whereas halofantrine and clofilium did lead to TdP (Batey and Coker, 2002).
In the anaesthetized guinea pig model, a 5% increase in the QTcB interval can be accurately detected (Guns et al., 2010) and a 10% prolongation of the QTcB interval should be classified as important QT prolongation. Similarly, based on our current experience, we would suggest that a shortening of the E-M window to below 20 ms warrants further evaluation, whereas a negative E-M window points directly to high TdP risk in humans. Drugs with no TdP risk (nor the vehicle) had no effect on the E-M window demonstrating the selectivity of the new risk marker.
Amiodarone
The multi-target agent amiodarone has extremely complex electropharmacological actions, with effects on multiple ion channels as well as β-adrenoceptors (Kodama et al., 1997), and is an interesting agent in the context of QT prolongation and TdP. Despite causing a modest prolongation of the QT interval, it is an effective anti-arrhythmic drug with a relatively low potential for TdP. Amiodarone has been found safe in patients who had experienced TdP while taking other drugs (Mattioni et al., 1989; Hohnloser et al., 1994) and can be used to treat TdP (Lazzara, 1989), although it has various off-target side effects which can limit chronic administration in a large cohort of patients. Furthermore, amiodarone showed relatively low potential for TdP in the canine chronic AV-block model, after both chronic (van Opstal et al., 2001) and acute dosing (Yoshida et al., 2002) and also in the Screenit rabbit model (Shah and Hondeghem, 2005). In line with these reports, acute dosing of amiodarone did not elicit a negative E-M window in our experiments. Moreover, amiodarone increased the E-M window in anaesthetized guinea pigs and prevented inversion of the E-M window by dofetilide. A similar protective effect of amiodarone has been described in a setting of sotalol-induced arrhythmias in dogs (Merot et al., 1999). The mechanism of this protective effect might be the conservation of a positive E-M window, most likely due to the alterations in Ca2+ handling, although a reduction in heterogeneity of cardiac repolarization has been attributed to amiodarone as well (Drouin et al., 1998).
Moxifloxacin and ciprofloxacin
Fluoroquinolone antibiotics are all associated with some degree of QT prolongation (Patmore et al., 2000; Lu et al., 2007), but the different members of this therapeutic class have different torsadogenic profiles (DiMasi and Paquette, 2004). Grepafloxacin (withdrawn from the market in 1999) and sparfloxacin have a high risk for induction of TdP in humans (Ball, 2000), whereas ciprofloxacin and moxifloxacin have a low risk for TdP in humans. In agreement with the use of moxifloxacin as a positive control in thorough QT studies (Bloomfield et al., 2008), and consistent with published pre-clinical data (Yao et al., 2008; Van Deuren et al., 2009), moxifloxacin prolonged the QTcB interval in the present study. Despite considerably prolonging the QTcB interval, moxifloxacin only caused a minor decrease in the E-M window in anaesthetized guinea pigs, suggesting a minimal risk for TdP at the maximal dose used in this study (10 mg·kg−1). In line with these results, moxifloxacin (8 mg·kg−1) did not produce episodes of TdP in a canine chronic AV-block model (Thomsen et al., 2006). Whether higher doses of moxifloxacin would further compromise the E-M window is not known, but pharmacokinetics of moxifloxacin (400 mg, oral administration, once daily) are well controlled, which could explain why only a few cases of moxifloxacin-related TdP have been reported. For ciprofloxacin, no effect on the E-M window was observed. This favourable safety outcome in the anaesthetized guinea pig model is compatible with the frequent use of ciprofloxacin in clinical practice as a safe antibiotic.
Taken together, the E-M window appears to classify drugs with high and low TdP risk correctly and might be a valuable addition to the classical QTc interval screening.
Isoprenaline
Isoprenaline transiently reduced the E-M window due to the different kinetics of its effect on the QT and QLVPend interval. The decrease of the E-M window by isoprenaline might explain the torsadogenic activity of isoprenaline in the LQT1 dog model (with compromised IKs current) (van der Linde et al., 2010). However, in anaesthetized guinea pigs, combined administration of IKr- (quinidine 10 mg·kg−1) and IKs-blockers (JNJ303 2 mg·kg−1) – resulting in large negative E-M windows (less than −50 ms) – did not result in spontaneous or β-adrenoceptor (isoprenaline 0.0125 mg·kg−1)-induced TdP (data not shown). As such, no direct association between a negative E-M window and initiation of TdP could currently be shown in guinea pigs. However, in the canine LQT1 model, the necessity of a negative E-M window for the onset of TdP has been demonstrated (van der Linde et al., 2010).
Effect of heart rate and body temperature on the E-M window
The duration of the QT interval is dependent on several factors including heart rate, autonomic influences (Magnano et al., 2002) and body temperature (van der Linde et al., 2008). Different correction formulas have been applied in an attempt to discriminate between heart rate-dependent and heart rate-independent changes of the QT interval. In anaesthetized guinea pigs, Bazett's formula is frequently applied to correct for heart rate (Hauser et al., 2005; Yao et al., 2008), and this correction has been shown to be the most effective in this model (Hamlin et al., 2003). The shortening of the QTcB interval with salbutamol in the present study, however, illustrates the limitations of Bazett's formula. Interestingly, the E-M window hardly changed with salbutamol despite drastic increases in heart rate. Similarly, significant decreases in heart rate associated with diltiazem did not affect the E-M window either.
The effect of temperature on the duration of the QT interval has been studied in detail in anaesthetized dogs (van der Linde et al., 2008). A similar temperature dependency of the QT interval was observed in anaesthetized guinea pigs: −14.6 ms·°C−1 (QTcB) in anaesthetized guinea pigs versus −14 ms·°C−1 (QTcVDW) in anaesthetized dogs. However, the E-M window remained remarkably stable during cooling and even tended to increase slightly when cooling below 35.5°C.
Taken together, the E-M window is, in contrast to the QTcB interval, a robust parameter that is minimally affected by changes in heart rate or body temperature.
Limitations of the study
No bio-analysis of plasma samples was performed, and so results could not be presented as x-folds of therapeutic concentrations and consequently no safety margins could be determined. However, data on plasma concentrations and QTcB prolongation of reference drugs in anaesthetized guinea pigs and dogs have been published (Redfern et al., 2003; Yao et al., 2008; Van Deuren et al., 2009) that could be useful to put our results in perspective. The QTcB prolongations evoked by reference compounds observed in the current study are consistent with these previous reports.
Conclusions
The present work evaluated, for the first time, the E-M window in anaesthetized guinea pigs as a general risk marker for TdP in humans. A decreased E-M window was consistently observed with drugs with high TdP risk, but not with drugs with low or no TdP risk. Furthermore, the E-M window was found to be a robust marker, minimally affected by changes in heart rate or body temperature. These results suggest that the E-M window in anaesthetized guinea pigs is an alternative risk marker for TdP in humans that complements the traditional QT interval screening.
Acknowledgments
The authors wish to thank Hanne Daems for her excellent technical assistance during the execution of the experiments. This work was financially supported by the Institute for Encouragement of Innovation by Science and Technology in Flanders (kmo-programma van het agentschap voor Innovatie door Wetenschap en Technologie, IWT).
Glossary
- E-M
window, electro-mechanical window
- ICaL
L-type calcium current
- IKr
rapidly activating delayed rectifier potassium current
- IKs
slowly activating delayed rectifier potassium current
- LQTS
long QT syndrome
- QTcB
the heart rate corrected QT interval based on Bazett's formula
- TdP
Torsade de Pointes
Conflict of interest
The authors are employees of Bio-Plus Services, a contract research organization in cardiovascular safety pharmacology.
References
- Airaksinen J, Ikaheimo M, Kaila J, Linnaluoto M, Takkunen J. Systolic time intervals and the QT-QS2 interval in young female diabetics. Ann Clin Res. 1984;16:188–191. [PubMed] [Google Scholar]
- Ball P. Quinolone-induced QT interval prolongation: a not-so-unexpected class effect. J Antimicrob Chemother. 2000;45:557–559. doi: 10.1093/jac/45.5.557. [DOI] [PubMed] [Google Scholar]
- Batey AJ, Coker SJ. Proarrhythmic potential of halofantrine, terfenadine and clofilium in a modified in vivo model of torsade de pointes. Br J Pharmacol. 2002;135:1003–1012. doi: 10.1038/sj.bjp.0704550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloomfield DM, Kost JT, Ghosh K, Hreniuk D, Hickey LA, Guitierrez MJ, et al. The effect of moxifloxacin on QTc and implications for the design of thorough QT studies. Clin Pharmacol Ther. 2008;84:475–480. doi: 10.1038/clpt.2008.33. [DOI] [PubMed] [Google Scholar]
- Boudoulas H, Geleris P, Lewis RP, Leier CV. Effect of increased adrenergic activity on the relationship between electrical and mechanical systole. Circulation. 1981a;64:28–33. doi: 10.1161/01.cir.64.1.28. [DOI] [PubMed] [Google Scholar]
- Boudoulas H, Geleris P, Lewis RP, Rittgers SE. Linear relationship between electrical systole, mechanical systole, and heart rate. Chest. 1981b;80:613–617. doi: 10.1378/chest.80.5.613. [DOI] [PubMed] [Google Scholar]
- Boudoulas H, Sohn YH, O'Neill W, Brown R, Weissler AM. The QT greater than QS2 syndrome: a new mortality risk indicator in coronary artery disease. Am J Cardiol. 1982;50:1229–1235. doi: 10.1016/0002-9149(82)90454-4. [DOI] [PubMed] [Google Scholar]
- Cavero I, Crumb W. ICH S7B draft guideline on the non-clinical strategy for testing delayed cardiac repolarisation risk of drugs: a critical analysis. Expert Opin Drug Saf. 2005;4:509–530. doi: 10.1517/14740338.4.3.509. [DOI] [PubMed] [Google Scholar]
- Chambers JB, Ward DE. The QT and QS2 intervals in patients with mitral leaflet prolapse. Am Heart J. 1987;114:355–361. doi: 10.1016/0002-8703(87)90503-5. [DOI] [PubMed] [Google Scholar]
- Darpo B. Spectrum of drugs prolonging QT interval and theincidence of torsades de pointes. Eur Heart J. 2001;3:K70–K80. [Google Scholar]
- De Bruin ML, Pettersson M, Meyboom RH, Hoes AW, Leufkens HG. Anti-HERG activity and the risk of drug-induced arrhythmias and sudden death. Eur Heart J. 2005;26:590–597. doi: 10.1093/eurheartj/ehi092. [DOI] [PubMed] [Google Scholar]
- De Caprio L, Ferro G, Cuomo S, Volpe M, Artialo D, De luca N, et al. QT/QS2 ratio as an index of autonomic tone changes. Am J Cardiol. 1984;53:818–822. doi: 10.1016/0002-9149(84)90411-9. [DOI] [PubMed] [Google Scholar]
- DiMasi JA, Paquette C. The economics of follow-on drug research and development: trends in entry rates and the timing of development. Pharmacoeconomics. 2004;22:1–14. doi: 10.2165/00019053-200422002-00002. [DOI] [PubMed] [Google Scholar]
- Drouin E, Lande G, Charpentier F. Amiodarone reduces transmural heterogeneity of repolarization in the human heart. J Am Coll Cardiol. 1998;32:1063–1067. doi: 10.1016/s0735-1097(98)00330-1. [DOI] [PubMed] [Google Scholar]
- Gintant GA. Preclinical Torsades-de-Pointes screens: advantages and limitations of surrogate and direct approaches in evaluating proarrhythmic risk. Pharmacol Ther. 2008;119:199–209. doi: 10.1016/j.pharmthera.2008.04.010. [DOI] [PubMed] [Google Scholar]
- Guns P-J, Teisman A, Van Ammel K, Towart R, Straetemans R, Bult H, et al. Can retrospective analysis of preclinical cardiovascular safety data improve predictivity of future results? J Pharmacol Toxicol Methods. 2010;62:e29–e30. [Google Scholar]
- Hamlin RL, Kijtawornrat A, Keene BW, Hamlin DM. QT and RR intervals in conscious and anesthetized guinea pigs with highly varying RR intervals and given QTc-lengthening test articles. Toxicol Sci. 2003;76:437–442. doi: 10.1093/toxsci/kfg254. [DOI] [PubMed] [Google Scholar]
- Hauser DS, Stade M, Schmidt A, Hanauer G. Cardiovascular parameters in anaesthetized guinea pigs: a safety pharmacology screening model. J Pharmacol Toxicol Methods. 2005;52:106–114. doi: 10.1016/j.vascn.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Hoffmann P, Warner B. Are hERG channel inhibition and QT interval prolongation all there is in drug-induced torsadogenesis? A review of emerging trends. J Pharmacol Toxicol Methods. 2006;53:87–105. doi: 10.1016/j.vascn.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Hohnloser SH, Klingenheben T, Singh BN. Amiodarone-associated proarrhythmic effects. A review with special reference to torsade de pointes tachycardia. Ann Intern Med. 1994;121:529–535. doi: 10.7326/0003-4819-121-7-199410010-00009. [DOI] [PubMed] [Google Scholar]
- Hondeghem LM. Thorough QT/QTc not so thorough: removes torsadogenic predictors from the T-wave, incriminates safe drugs, and misses profibrillatory drugs. J Cardiovasc Electrophysiol. 2006;17:337–340. doi: 10.1111/j.1540-8167.2006.00347.x. [DOI] [PubMed] [Google Scholar]
- Kagstrom J, Sjogren EL, Ericson AC. Evaluation of the guinea pig monophasic action potential (MAP) assay in predicting drug-induced delay of ventricular repolarisation using 12 clinically documented drugs. J Pharmacol Toxicol Methods. 2007;56:186–193. doi: 10.1016/j.vascn.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Kodama I, Kamiya K, Toyama J. Cellular electropharmacology of amiodarone. Cardiovasc Res. 1997;35:13–29. doi: 10.1016/s0008-6363(97)00114-4. [DOI] [PubMed] [Google Scholar]
- Lawrence CL, Pollard CE, Hammond TG, Valentin JP. Nonclinical proarrhythmia models: predicting Torsades de Pointes. J Pharmacol Toxicol Methods. 2005;52:46–59. doi: 10.1016/j.vascn.2005.04.011. [DOI] [PubMed] [Google Scholar]
- Lazzara R. Amiodarone and torsade de pointes. Ann Intern Med. 1989;111:549–551. doi: 10.7326/0003-4819-111-7-549. [DOI] [PubMed] [Google Scholar]
- van der Linde HJ, Van Deuren B, Teisman A, Towart R, Gallacher DJ. The effect of changes in core body temperature on the QT interval in beagle dogs: a previously ignored phenomenon, with a method for correction. Br J Pharmacol. 2008;154:1474–1481. doi: 10.1038/bjp.2008.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Linde HJ, Van Deuren B, Somers Y, Loenders B, Towart R, Gallacher DJ. The electro-mechanical window: a risk marker for Torsade de Pointes in a canine model of drug induced arrhythmias. Br J Pharmacol. 2010;161:1444–1454. doi: 10.1111/j.1476-5381.2010.00934.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu HR, Vlaminckx E, Van de Water A, Rohrbacher J, Hermans A, Gallacher DJ. In-vitro experimental models for the risk assessment of antibiotic-induced QT prolongation. Eur J Pharmacol. 2007;577:222–232. doi: 10.1016/j.ejphar.2007.07.070. [DOI] [PubMed] [Google Scholar]
- Lu Z, Kamiya K, Opthof T, Yasui K, Kodama I. Density and kinetics of I(Kr) and I(Ks) in guinea pig and rabbit ventricular myocytes explain different efficacy of I(Ks) blockade at high heart rate in guinea pig and rabbit: implications for arrhythmogenesis in humans. Circulation. 2001;104:951–956. doi: 10.1161/hc3401.093151. [DOI] [PubMed] [Google Scholar]
- Magnano AR, Holleran S, Ramakrishnan R, Reiffel JA, Bloomfield DM. Autonomic nervous system influences on QT interval in normal subjects. J Am Coll Cardiol. 2002;39:1820–1826. doi: 10.1016/s0735-1097(02)01852-1. [DOI] [PubMed] [Google Scholar]
- Mattioni TA, Zheutlin TA, Sarmiento JJ, Parker M, Lesch M, Kehoe RF. Amiodarone in patients with previous drug-mediated torsade de pointes. Long-term safety and efficacy. Ann Intern Med. 1989;111:574–580. doi: 10.7326/0003-4819-111-7-574. [DOI] [PubMed] [Google Scholar]
- Merot J, Charpentier F, Poirier JM, Coutris G, Weissenburger J. Effects of chronic treatment by amiodarone on transmural heterogeneity of canine ventricular repolarization in vivo: interactions with acute sotalol. Cardiovasc Res. 1999;44:303–314. doi: 10.1016/s0008-6363(99)00232-1. [DOI] [PubMed] [Google Scholar]
- Ming Z, Nordin C. Terfenadine blocks time-dependent Ca2+, Na+, and K+ channels in guinea pig ventricular myocytes. J Cardiovasc Pharmacol. 1995;26:761–769. doi: 10.1097/00005344-199511000-00013. [DOI] [PubMed] [Google Scholar]
- van Opstal JM, Schoenmakers M, Verduyn SC, de Groot SH, Leunissen JD, van Der Hulst FF, et al. Chronic amiodarone evokes no torsade de pointes arrhythmias despite QT lengthening in an animal model of acquired long-QT syndrome. Circulation. 2001;104:2722–2727. doi: 10.1161/hc4701.099579. [DOI] [PubMed] [Google Scholar]
- Patmore L, Fraser S, Mair D, Templeton A. Effects of sparfloxacin, grepafloxacin, moxifloxacin, and ciprofloxacin on cardiac action potential duration. Eur J Pharmacol. 2000;406:449–452. doi: 10.1016/s0014-2999(00)00694-4. [DOI] [PubMed] [Google Scholar]
- Redfern WS, Carlsson L, Davis AS, Lynch WG, MacKenzie I, Palethorpe S, et al. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc Res. 2003;58:32–45. doi: 10.1016/s0008-6363(02)00846-5. [DOI] [PubMed] [Google Scholar]
- Sakaguchi Y, Sugiyama A, Takao S, Akie Y, Takahara A, Hashimoto K. Halothane sensitizes the guinea-pig heart to pharmacological IKr blockade: comparison with urethane anesthesia. J Pharmacol Sci. 2005;99:185–190. doi: 10.1254/jphs.fp0050295. [DOI] [PubMed] [Google Scholar]
- Shah RR, Hondeghem LM. Refining detection of drug-induced proarrhythmia: QT interval and TRIaD. Heart Rhythm. 2005;2:758–772. doi: 10.1016/j.hrthm.2005.03.023. [DOI] [PubMed] [Google Scholar]
- Tabo M, Kimura K, Ito S. Monophasic action potential in anaesthetized guinea pigs as a biomarker for prediction of liability for drug-induced delayed ventricular repolarization. J Pharmacol Toxicol Methods. 2007;55:254–261. doi: 10.1016/j.vascn.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Terrar DA, Wilson CM, Graham SG, Bryant SM, Heath BM. Comparison of guinea-pig ventricular myocytes and dog Purkinje fibres for in vitro assessment of drug-induced delayed repolarization. J Pharmacol Toxicol Methods. 2007;56:171–185. doi: 10.1016/j.vascn.2007.04.005. [DOI] [PubMed] [Google Scholar]
- Testai L, Calderone V, Salvadori A, Breschi MC, Nieri P, Martinotti E. QT prolongation in anaesthetized guinea-pigs: an experimental approach for preliminary screening of torsadogenicity of drugs and drug candidates. J Appl Toxicol. 2004;24:217–222. doi: 10.1002/jat.975. [DOI] [PubMed] [Google Scholar]
- Thomsen MB, Beekman JD, Attevelt NJ, Takahara A, Sugiyama A, Chiba K, et al. No proarrhythmic properties of the antibiotics moxifloxacin or azithromycin in anaesthetized dogs with chronic-AV block. Br J Pharmacol. 2006;149:1039–1048. doi: 10.1038/sj.bjp.0706900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Deuren B, Van Ammel K, Somers Y, Cools F, Straetemans R, van der Linde HJ, et al. The fentanyl/etomidate-anaesthetised beagle (FEAB) dog: a versatile in vivo model in cardiovascular safety research. J Pharmacol Toxicol Methods. 2009;60:11–23. doi: 10.1016/j.vascn.2009.04.195. [DOI] [PubMed] [Google Scholar]
- Vincent GM, Jaiswal D, Timothy KW. Effects of exercise on heart rate, QT, QTc and QT/QS2 in the Romano-Ward inherited long QT syndrome. Am J Cardiol. 1991;68:498–503. doi: 10.1016/0002-9149(91)90785-j. [DOI] [PubMed] [Google Scholar]
- Vohra J. The long QT syndrome. Heart Lung Circ. 2007;16(Suppl. 3):S5–12. doi: 10.1016/j.hlc.2007.05.008. [DOI] [PubMed] [Google Scholar]
- Yao X, Anderson DL, Ross SA, Lang DG, Desai BZ, Cooper DC, et al. Predicting QT prolongation in humans during early drug development using hERG inhibition and an anaesthetized guinea-pig model. Br J Pharmacol. 2008;154:1446–1456. doi: 10.1038/bjp.2008.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Sugiyama A, Satoh Y, Ishida Y, Yoneyama M, Kugiyama K, et al. Comparison of the in vivo electrophysiological and proarrhythmic effects of amiodarone with those of a selective class III drug, sematilide, using a canine chronic atrioventricular block model. Circ J. 2002;66:758–762. doi: 10.1253/circj.66.758. [DOI] [PubMed] [Google Scholar]
- Zicha S, Moss I, Allen B, Varro A, Papp J, Dumaine R, et al. Molecular basis of species-specific expression of repolarizing K+ currents in the heart. Am J Physiol Heart Circ Physiol. 2003;285:H1641–H1649. doi: 10.1152/ajpheart.00346.2003. [DOI] [PubMed] [Google Scholar]