

Abstract
BACKGROUND AND PURPOSE
β-Adrenoceptors are expressed in human and experimental animal breast cancer cells. However, the effect of the agonists and antagonists reported on cell proliferation and tumour growth was paradoxical, precluding their utilization as possible adjuvant therapy, mainly in the cases of refractory tumours.
EXPERIMENTAL APPROACH
β-Adrenoceptor expression was analysed by immunofluorescence and RT-PCR. Cell proliferation was assessed by [3H]-thymidine incorporation, tumour growth by measuring with a calliper and ERK 1/2 phosphorylation by Western blotting.
KEY RESULTS
β2-Adrenoceptor expression was confirmed in the mouse and human cells tested. Cell proliferation was increased by adrenaline (by α2-adrenoceptor action) and decreased in every tested cell line by the β-adrenoceptor agonist isoprenaline and the β2-adrenoceptor agonist salbutamol. Isoprenaline and salbutamol reduced tumour growth in every tumour tested (mouse C4-HD and CC4-3-HI and human IBH-4, IBH-6 and MDA-MB-231 cell lines growing as xenografts in nude mice). These effects were reversed by the β-adrenoceptor antagonist propranolol. The α2-adrenoceptor antagonist rauwolscine and the β2-adrenoceptor agonist salbutamol were equally effective in diminishing tumour growth. ERK 1/2 activation analysed in IBH-4 tumours correlated with tumour growth, with the β-adrenoceptor agonists decreasing its activation. Inhibition of ERK 1/2 phosphorylation in vitro was mainly mediated by the PKA pathway.
CONCLUSIONS AND IMPLICATIONS
In our experimental models, the β-adrenoceptor agonists inhibited breast cancer cell proliferation and tumour growth, probably mediated by inhibition of ERK 1/2 phosphorylation. The β-adrenoceptor agonists were as effective as the α2-adrenoceptor antagonist rauwolscine, providing possible novel adjuvant treatments for breast cancer.
Keywords: mammary tumour, salbutamol, isoprenaline, propranolol, rauwolscine, adrenoceptors
Introduction
Breast cancer is by far the most common cancer of women, comprising 23% of all female cancers (Ferlay et al., 2010). In the United States, breast cancer was in 2010 the most important female cancer in incidence, with 28% of female neoplasms, although mortality was of 15%, second after lung and bronchus cancer (Jemal et al., 2011). In the States, in 1987 lung cancer surpassed breast cancer as the leading cause of cancer death in women (Jemal et al., 2011). In the United Kingdom, it has been suggested (Parkin, 2009) that the recent decline in breast cancer incidence observed since 1999 between ages 50 and 59 and since 2003 between ages 60 and 64 is a consequence of the reduced use of hormone replacement therapy.
Clinical studies have suggested that stress, chronic depression and social support might influence cancer onset and progression (Antoni et al., 2009) and β-adrenoceptors could be involved in the mediation of these effects (Antoni et al., 2006; Thaker et al., 2006). The first studies of the adrenergic stimulation and adrenoceptors of the mammary gland involved the normal bovine mammary gland because of their effects on milk production (Roets et al., 1984; Roets et al., 1986; Hammon et al., 1994; Inderwies et al., 2003). β-Adrenoceptors were also characterized in the normal mammary gland (Clegg and Mullaney, 1985; Lavandero et al., 1985) and in mammary tumours of experimental animals (Marchetti et al., 1989; Marchetti et al., 1990; Marchetti and Labrie, 1990; Marchetti et al., 1991). β-Adrenoceptors have since been identified in human breast cancer MDA-MB-231, MCF-7, VHB-1, T47-D and BT-20 cell lines, comparing them with the HBL-100 cells (Vandewalle et al., 1990). A weak correlation (0.05 < P < 0.1) was observed between β-adrenoceptors and progestin receptors but no correlation found between β-adrenoceptors and oestrogen receptors (Draoui et al., 1991).
The paradoxical nature of β-adrenoceptor action in breast cancer cells has recently been reviewed (Luthy et al., 2009). On one hand, β-adrenoceptor agonists inhibited cell proliferation. Thus, MDA-MB-231 human breast cancer cells expressed high β-adrenoceptor levels, and receptor stimulation by an agonist evoked immediate and robust reductions in DNA synthesis (Slotkin et al., 2000). Moreover, stimulation of β2-adrenoceptors induced significant tumour growth suppression and tumour regression in mice bearing established MDA-MB-231 human breast tumours into the mammary fat pads (Carie and Sebti, 2007). On the other hand, inhibition of cell proliferation has been induced by β-adrenoceptor antagonists in oestrogen-responsive (MCF-7, ZR-75-1, MDA-MB-361) and oestrogen non-responsive (MDA-MB-453, MDA-MB-435, MDA-MB-468) cell lines derived from human breast cancers (Cakir et al., 2002). However, theses authors did not assess the possibility that the antagonists behaved as partial agonists. Several clinically used β-adrenoceptor antagonists are agonists at the human β-adrenoceptor. Sometimes these agonist effects were small at the cAMP level, but substantial at the transcriptional level (Baker et al., 2003). Therefore, the reported results may not be contradictory. However, there is a close correlation between progressing, static, and regressing tumours after ovariectomy and β-adrenoceptor concentration in membranes prepared from dimethylbenz(a)anthracene (DMBA)-induced tumours (Marchetti et al., 1989). In the same model, the rapid reduction of the β-adrenoceptor population in the tumour following ovariectomy was accompanied by the well-known regression of tumour growth (Marchetti et al., 1991). Also, some single nucleotide polymorphisms of the β2-adrenoceptor gene have been associated with increased risk of breast cancer (Feigelson et al., 2008).
α2-Adrenoceptors have also been described in the bovine mammary gland, related to milk production (Roets et al., 1986). The number of α-adrenoceptors decreased from the teat to the mammary ducts to the parenchyma (Hammon et al., 1994; Inderwies et al., 2003). Actions at α2-adrenoceptors in human breast cancer cells have been described by our group (Vazquez et al., 1999) and these receptors were characterized in several breast tumour and non-tumour cell lines by RT-PCR, immunocytochemistry and binding studies. These α2-adrenoceptors were associated with increased cell proliferation in vitro (Vazquez et al., 2006) and with increased tumour growth in vivo (Bruzzone et al., 2008).
The aim of the present work was to assess if compounds active at β-adrenoceptors were able to inhibit tumour growth in five tumours from two independent breast cancer models, and if the treatment with a β-adrenoceptor agonist and an α2-adrenoceptor antagonist was equally effective in inhibiting tumour growth in these models.
Methods
Cell culture
The mouse mammary epithelial MC4-L5 (Lanari et al., 2001) cell line was routinely cultured in phenol red-free (Berthois et al., 1986) Dulbecco's modified Eagle's medium (DMEM) : Ham's F12 medium (F12) (1:1) supplemented with heat-inactivated 10% fetal calf serum (FCS), 2 mM glutamine, 100 IU mL−1 penicillin, 100 µg mL−1 streptomycin and 15 mM HEPES. Cells were sub-cultured once weekly after trypsinization (0.25% trypsin – 0.2% EDTA) (Lanari et al., 2001). The medium was changed twice weekly.
The human breast cancer MDA-MB-231 and the IBH-4 and IBH-6 cell lines, developed in our laboratory, were cultured as already described (Vazquez et al., 2004). Briefly, cells were maintained in DMEM : F12 (1:1) medium supplemented with heat-inactivated 10% FCS, 2 mM glutamine, 2 µg mL−1 bovine insulin, 100 IU mL−1 penicillin, 100 µg mL−1 streptomycin and 15 mM HEPES. Cells were grown on plastic, incubated at 37°C in 5% CO2 and sub-cultured at 80% confluence using 0.25% trypsin – 0.025% EDTA.
Preparation of cell suspension for injections
IBH-4, IBH-6 and MDA-MB-231 cells were dispersed with 0.25% trypsin – 0.025% EDTA, washed twice with PBS or medium without serum, concentrations adjusted to 1–2 × 107 viable cells and suspended in 200 µL. The cells were inoculated s.c. in the flank, through a 21 gauge needle tunnelled 1–2 cm to prevent leakage of the cell inoculum (Bruzzone et al., 2009).
Animals
All animal care and experimental procedures complied with institutional guidelines, the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Research, Commission on Life Sciences, National Research Council, 1996), which has been updated and is available online and in print (Institute of Laboratory Animal Research, Commission on Life Sciences, National Research Council, 2011) and the United Kingdom Coordinating Committee on Cancer Research Guidelines for the Welfare of Animals in Experimental Neoplasia (United Kingdom Co-ordinating Committee on Cancer Research U, 1998; Workman et al., 2010). For human breast cell lines growing in vivo, congenitally athymic nude mice on a Swiss background (N : NIH(S)-nu) bred at Bioterio de la Universidad de La Plata, Argentina were used. Balb/c mice from the animal facility of the Instituto de Biología y Medicina Experimental were used for transplantation of the C4-HD and CC4-3-HI mouse mammary tumours. All mice were 8 weeks old sexually mature females, and were housed in ventilated racks, pathogen-free conditions under a 12 h light–dark photoperiod with controlled humidity and temperature (20 ± 2°C). Boxes, bedding, food and water were sterilized. Sterility was maintained during the surgical procedures used for the inoculation of the cells to give rise to solid tumours and for subsequent removal and transplantation of tumours.
Tumours
IBH-4, IBH-6 and MDA-MB-231 human breast cancer cell lines were inoculated in nude mice as already described. Subsequent in vivo passages of the tumours were performed by trocar transplantation. C4-HD and CC4-3-HI tumours were transplanted by trocar in Balb/c mice (Lamb et al., 2005). Tumour growth was monitored every second day. The two major diameters were measured with a calliper and the volume was calculated with the formula: volume = (4 × 3−1) π × minor radius2 × major radius (Bruzzone et al., 2008). Mice were killed and the tumours extracted and frozen at −80°C. The animals were killed when ulceration, distension of covering tissues or cachexia were observed or when tumours attained near 10% body weight.
Treatments
One day after transplantation of the tumour, animals were given daily injections of the drugs. Three different experimental approaches were undertaken: in one, the β-adrenoceptor agonists were compared: the animals were separated in three groups of 10 animals each: (i) one s.c. injected with isoprenaline (1.0 mg kg−1 day−1); (ii) another one s.c. injected with salbutamol (1.2 mg kg−1 day−1); and (iii) the control group received physiological saline. The second approach was the reversal of the effects of β-adrenoceptor activation. For that purpose, the animals were separated in four groups of 10 animals each: (i) the β-adrenoceptor agonist group: s.c. injected salbutamol (1.2 mg kg−1 day−1); (ii) the β-adrenoceptor antagonist group: s.c. injected with propranolol (1.0 mg kg−1 day−1); (iii) the reversal group received salbutamol + propranolol at the stated doses; and (iv) the control group received physiological saline. For the third experimental approach the four groups of 10 animals each one, were: (i) the β-adrenoceptor agonist group: s.c. injected with salbutamol (1.2 mg kg−1 day−1); (ii) the α2-adrenoceptor antagonist group s.c. injected with rauwolscine: 0.5 mg kg−1 day−1; (iii) the combined group: received salbutamol + rauwolscine at the stated doses; and (iv) the control group received physiological saline. The duration of the treatments varied, depending on the animal's condition. Every animal was treated beginning 1 day after the implantation of the tumour and continuing until the mice died. For C4-HD tumours, the duration of the experiments was of 25–27 days, for the CC4-3-HI tumours, the experiments lasted 22 days. Nude mice inoculated with the human breast cancer cell lines were treated for 25 days for IBH-4 cells, 30–35 days for IBH-6 cells and 35–40 days for MDA-MB-231 cells.
Proliferation assays
The MC4-L5 mouse mammary tumour cell line and the human IBH-4, IBH-6 and MDA-MB-231 cell lines were tested for proliferation. Mouse cells were seeded at 7000 cells per well in 96 well plates. They were incubated in phenol red-free DMEM : F12 medium supplemented with 10% FCS, 2 mM glutamine, 100 IU·mL−1 penicillin, 100 µg mL−1 streptomycin, 250 ng mL−1 amphotericin B and 15 mM HEPES. After 24 and 48 h, the medium was changed to a similar one but with FCS replaced by 1% charcoal-stripped FCS and the experiments lasted 48 h for the mouse cells. Human cells were seeded at 5000 cells per well in 2% charcoal-stripped FCS and the incubations lasted 72 h. Different adrenoceptor agonists were added. All solutions of these compounds were prepared in 10 mM ascorbic acid, frozen and diluted immediately before use. [3H]-thymidine at 0.2 µCi per well was added with the last change of medium. After 24 h, cells were harvested in a Nunc Cell Harvester 8 (Nunc, Rochester, NY, USA), and filters were counted in a liquid scintillation counter. The murine cells were trypsinized prior to harvesting.
Immunofluorescence
The cells were cultured in Lab-Tek Chamber Slide System (Nunc, Thermo Fisher Scientific, Rochester, NY, USA) with 10% FCS in phenol red-free DMEM : F12 medium; 1 day before immunocytochemistry the medium was changed to 1% (for mouse cells) or 2% (for human cells) charcoal-stripped FCS medium. The cells were treated with 10% FCS (without heat inactivation) and permeated with 0.1% Triton X-100 in 4% FCS. After overnight incubation at 4°C with the anti-human (which can also be used for the murine receptor) β2-adrenoceptor antibody at a concentration 1:50, and several PBS washes, cells were incubated with secondary donkey anti-rabbit IgG in 4% FCS, conjugated to fluorescein isothiocyanate (FITC). Primary antibody was omitted in the controls. Nuclei were stained with 0.1 µg mL−1 propidium iodine for 1 min, and the cells were mounted with Vectashield H-1000 (Vector Laboratories, Burlingame, CA, USA). Sections were analysed under a Nikon laser confocal microscope (Melville, NY, USA) (Plan Apo 40X/0.95 oil or Apo 60X/1.40 oil).
RT-PCR
When cells attained 80% confluence, they were incubated with medium without serum for additional 48 h. They were washed with PBS and total RNA was extracted by incubation with Tri Reagent (Sigma-Aldrich, St. Louis, MO, USA) for 5 min at room temperature, according to the manufacturer's instructions. Chloroform (0.2 mL) was added per mL of initial extract. These samples were incubated at room temperature for 3 min and centrifuged at 10 464× g for 15 min at 4°C. The aqueous phase was subsequently incubated with cold isopropanol (0.5 mL) for 10 min at room temperature with mild agitation and centrifuged again at 10 464× g for 10 min. The nucleic acid pellet was washed with 75% ethanol, centrifuged at 3360× g for 5 min, resuspended in diethylpyrocarbonate (DEPC) water (Invitrogen, Life Technologies) and the concentration measured with a spectrophotometer. The RNA was reverse transcribed at 42°C for 15 min in 20 µL containing 200 units of Superscript II RNase HK Reverse Transcriptase, 4 mM oligo dT18, 50 mM Tris–HCl pH 8.3, 3 mM MgCl2, 75 mM KCl, 10 mM DTT, 500 mM dNTPs (dATP, dTTP, dCTP, dGTP), 40 units RNase OUT (Recombinant RNase Inhibitor) (Superscript First Strand Synthesis System for RT-PCR, Invitrogen, Carlsbad, CA, USA). The reaction was terminated by heating at 99°C for 5 min and resting for 5 min at 5°C. Also, a parallel incubation with the extract in the absence of reverse transcriptase was performed in order to exclude any contamination with genomic DNA. This control is important in a gene without introns, such as that for the β2-adrenoceptor.
PCR amplification was carried out in the presence of 30 µM of each oligonucleotide primer, 50 mM KCl, 10 mM Tris–HCl pH 8.3, 2 mM MgCl2, 0.2 mM dNTPs (dATP, dTTP, dCTP, dGTP), and 0.05 units·mL−1 Taq DNA polymerase (Taq DNA Polymerase, Invitrogen). Three microlitres of cDNA was amplified in the PCR mix made up to 25 µL with molecular quality water.
An initial denaturation step at 95°C for 2 min was followed by 35 cycles (or 30 cycles for murine β2-adrenoceptor) at 95°C for 35 s, 60°C for 35 s and 72°C for 1 min, followed by a single incubation at 72°C for 8 min. A reaction with water instead of cDNA was performed concurrently as a negative control for the PCR in each PCR to test the reactive purity. The primers used were the following: β2-adrenoceptor: for human cells: forward 5′-GGACTTCCATTGATGTGCTGT-3′ and reverse 5′-GTCAGCAGGCTCTGGTACTTG-3′ (112 bp amplicon), for murine cells: forward 5′-GCCATCCTCATGTCGGGTTAT-3′ and reverse 5′-AGCAGGCTCTGGTACTTGAA-3′ (356 bp amplicon). For glyceraldehyde 3-phosphate dehydrogenase (GAPDH) transcript detection, GAPDH primers forward: 5′-TTCGTCATGGGTGTGACC-3′ and reverse 5′-AGTGAGCTTCCCGTTCAGC-3′, giving a 300 bp PCR product, served as a control for RNA integrity and the RT-PCR process. The PCR products were analysed using electrophoresis on a 2% agarose gel stained with ethidium bromide. A 100 bp DNA ladder was used as a marker to determine the size of the PCR products.
Western blotting for ERK 1/2
For the in vitro experiments, IBH-4 cells were seeded in 6 well plates in 10% FCS supplemented medium. After 12 h the medium was changed to 0% FCS and the cells were arrested for 24 h. The medium was changed again to a medium without serum. After 1–2 h, the adrenoceptor agents or the mimics were added (adrenoceptor agents (1 µM): isoprenaline, propranolol, salbutamol, rauwolscine, dexmedetomidine), 25 µM of the PKA inhibitor H-89, the PKA stimulator 8Br-cAMP (100 µM), 100 µM of the PKA selective activator N6-benzoyladenosine-cAMP (6Bnz) or 100 µM of 8-(4-chlorophenylthio)-2′-O-methyl-cAMP (8CPT) the specific activator of exchange protein directly activated by cAMP (EPAC). The antagonists and inhibitors (rauwolscine, propranolol and H89) were added 20 min before the agonists. The control cells were incubated with medium without FCS. The incubation was performed for 15 min and then the protein extraction buffer (RIPA: 10 mM Tris, 150 mM NaCl, 2 mM sodium vanadate, 1% sodium deoxycholate, 0.1% SDS, 1% Igepal) was added after washing the cells with phosphate saline buffer.
Tumours were homogenized in a Polytron at a 1:4 ratio (tissue : buffer). The buffer was 20 mmol L−1 Tris-HCl (pH 7.4), 1.5 mmol L−1 EDTA, 0.25 mmol L−1 dithiothreitol, 20 mmol L−1 Na2MoO4, and 10% glycerol. Protease inhibitors (0.5 mmol L−1 phenylmethylsulfonyl fluoride, 0.025 mmol L−1 N-Carbobenzyloxy-L-phenylalanyl chloromethyl ketone, 0.025 mmol L−1 tosyl-lysylchloromethane, 0.025 mmol L−1 tosylphenylalanylchloromethane, and 0.025 mmol L−1 Nα-p-Tosyl-L-arginine methyl ester hydrochloride) were added to the buffer immediately before use. The homogenate was sonicated and centrifuged for 20 min at 10 391× g (4°C). The supernatant was stored at −20°C until later use in Western blot assays.
Tumour or cell extracts (50–100 µg total protein per lane) were separated on 10% discontinuous polyacrylamide gels (SDS-PAGE). The proteins were dissolved in sample buffer (6 mM Tris pH 6.8, 2% SDS, 0.002% bromophenol blue, 20% glycerol, 5% mercaptoethanol) and boiled for 5 min. After electrophoresis, they were blotted onto a 0.2 µm nitrocellulose membrane and blocked 1 h at room temperature in 5% skim milk in 0.1% TBST (10 mM Tris, 100 mM NaCl, 0.1% vv−1 Tween 20). The membranes were incubated 1 h at room temperature with antibody to ERK 1 or overnight at 4°C with antibody to phospho-ERK 1/2 (pERK), both at a 1:1000 dilution. Blots were probed with donkey anti-rabbit and sheep anti-mouse IgG horseradish peroxidase linked whole antibody (Amersham Life Science, Arlington Heights, IL, USA; 1:2000 dilutions in 5% skim milk 0.1% TTBS). The luminescent signal was generated with an ECL Western blotting detection solution [2.2 mg luminol (Sigma, St. Louis, MO, USA), 0.33 mg p-coumaric acid (Sigma), 15 µL hydrogen peroxide 30% (Merck, Darmstadt, Germany), 333 µL Tris-HCl 1.5 M pH 8.8, 4.6 mL distilled water], and the blots were exposed to an autoradiographic film (Curix RP1, Agfa, Buenos Aires, Argentina) for 10 s to 5 min. Band intensity was quantified only in unsaturated films. Quantification was performed with ImageJ software.
Statistical analysis
Statistical analysis for the in vitro effect of agonists was performed by anova followed by Dunnet–Kramer test and the effect of the adrenergic compounds on tumour growth and the action of inhibitors or stimulators of ERK 1/2 phosphorylation by anova followed by Tukey–Kramer tests (Dowdy and Wearden, 1983).
Materials
FCS, culture media, antibiotics, trypsin, Superscript First Strand Synthesis System for RT-PCR and Taq DNA Polymerase trypsin were purchased from Invitrogen Life Technologies. Methyl [3H]-thymidine (NET 027E; specific activity: 20 Ci·mmol−1) was from Dupont-New England Nuclear (Boston, MA, USA). Liquid scintillation cocktail was Optiphase ‘Hisafe’ 3 (Wallac, Turku, Finland). Anti-human β2-adrenoceptor rabbit polyclonal antibody (sc-569), Phospho-ERK (pERK; sc-7383) and ERK (ERK 1; sc-94) antibodies were from Santa Cruz Biotechnology. Secondary antibodies were from Amersham (GE Healthcare Argentina S.A., Buenos Aires, Argentina). Vectashield H-1000 was from Vector Laboratories (Burlingame, CA, USA). The cAMP analogues, 8-(4-chlorophenylthio)-2′-O-methyladenosine-3′,5′-cAMP (8-CPT), 6Bnz and 8-bromoadenosine-3′,5′-cAMP (8-Br-cAMP) were purchased from BioLog Life Science Institute (Bremen, Germany). All the other reagents, including isoprenaline, salbutamol, propranolol, rauwolscine-HCl, glutamine, DNAse, luminol, p-coumaric acid, forskolin, EGF and H-89 were from Sigma-Aldrich. Lab-Tek Chamber Slide System was from Nunc.
Drug and receptor nomenclature
Drug and receptor nomenclature conforms to the Guide to Receptors and Channels (Alexander et al., 2011).
Results
Expression of β-adrenoceptors in mammary cell lines of mouse and human origin
The present experiments were undertaken to confirm the expression of β2-adrenoceptors in a mouse mammary tumour cell line and three breast cancer cell lines of human origin. As shown in Figure 1A, mouse mammary tumour MC4-L5 cells express β2-adrenoceptors. These receptors are shown in green, while the nuclei are stained with propidium iodine in red. The control in the absence of primary antibody (Figure 1B) showed no staining. Figure 1C depicts the staining of IBH-4 cells compared with its control (Figure 1D). Figure 1E shows the staining of IBH-6 cells and Figure 1F its control. As a positive control, MDA-MB-231 human breast cancer cells were included (Figure 1G, H for the control), because β2-adrenoceptors are known to be expressed in these cells (Vandewalle et al., 1990). Staining for β2-adrenoceptors was positive in both the plasma membrane and the cytoplasm.
Figure 1.
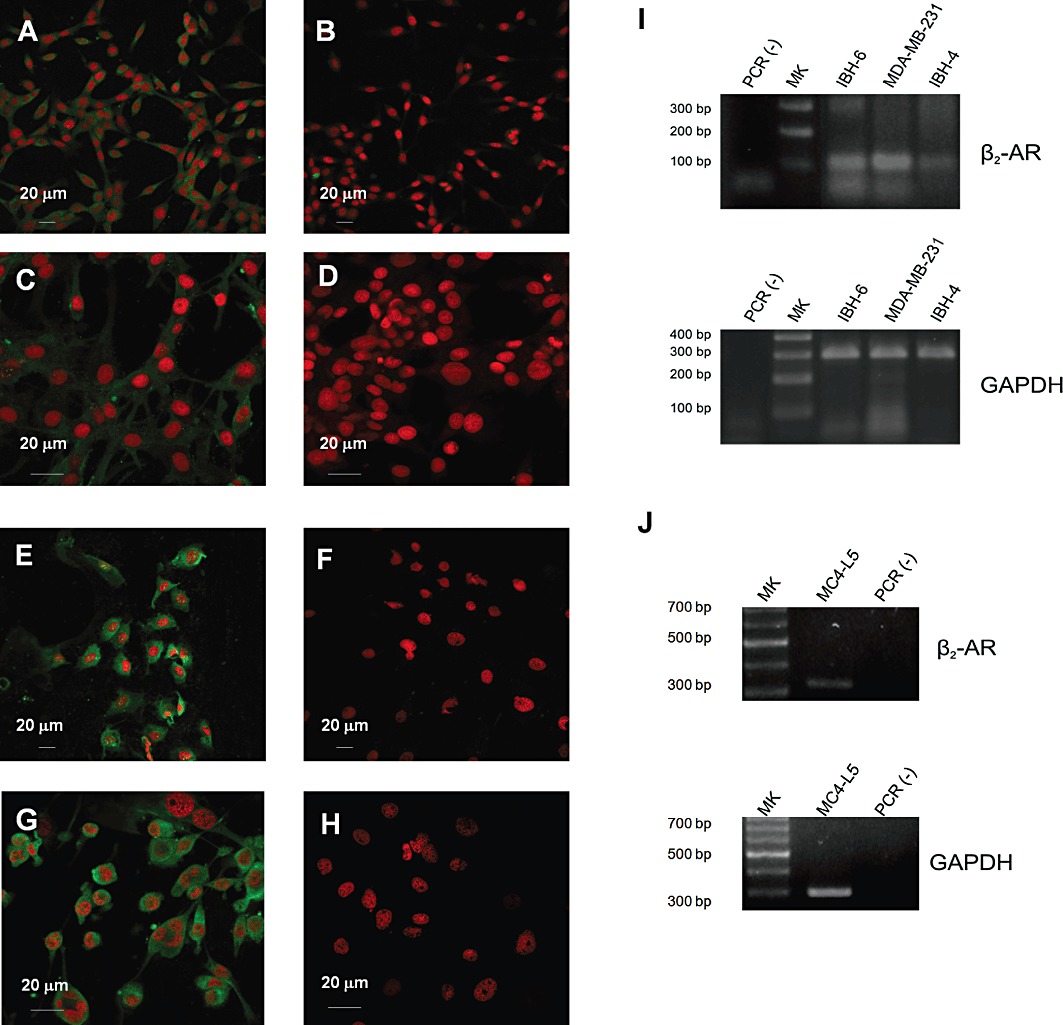
Expression of β2-adrenoceptors in mouse mammary tumour cell line MC4-L5 and the human IBH-4, IBH-6 and MDA-MB-231 cells by immunofluorescence and RT-PCR. (A,B) Mouse mammary tumour cells MC4-L5. (C,D) Human breast cancer cells IBH-4. (E,F) Human breast cancer cells IBH-6. (G,H) Human breast cancer MDA-MB-231 cells. (A,C,E,G) β2-RA. (B,D,F,H) Negative controls incubated without the primary antibody. (A,B,E,F) Photographs taken with a Plan Apo 40X/0.95 oil objective and (C,D,E,F) with an Apo 60X/1.40 oil one. (I) RT-PCR for β2-adrenoceptors (β2-AR) in human IBH-4, IBH-6 and MDA-MB-231 cells. (J) RT-PCR for β2-adrenoceptors in mouse MC4-L5 cells. In every case GAPDH was used as a control for RNA integrity and the RT-PCR process. The negative control was performed in the absence of cDNA.
The expression of these receptors was also confirmed by RT-PCR. As can be seen in Figure 1, all the human (Figure 1I) and murine (Figure 1J) breast cancer cell lines tested expressed β2-adrenoceptors, with IBH-4 cells showing a lighter band for receptor expression as compared with MDA-MB-231. The MC4-L5 murine cells showed a faint but positive band. As the primers are different between human and murine cells, the receptor concentrations cannot be compared.
Cell proliferation is inhibited by β-adrenoceptor agents
Once the expression of β2-adrenoceptors was confirmed in these cell lines, the next step was to analyse the action of the adrenoceptor agents on cell proliferation. Figure 2A shows the effect of adrenaline on the mouse mammary tumour cell line MC4-L5, Figure 2D on the human breast cancer cells IBH-4, Figure 2G on the human breast cancer cells IBH-6 and Figure 2J on the human breast cancer cells MDA-MB-231. In every case, adrenaline caused a significant increase on cell proliferation assessed by [3H]-thymidine incorporation. However, as adrenaline binds to α2, α1 and β-adrenoceptor, this effect could be due to the mitogenic effect of α2-adrenoceptors, already described in breast cancer cells (Vazquez et al., 2006). The MC4-L5 and to a lesser extent, the IBH-4 and MDA-MB-231 cells exhibited biphasic curves, consistent with opposite actions mediated by the α2-adrenoceptor mitogenic effect and a β-adrenoceptor inhibitory effect at higher concentrations. To test this hypothesis, as shown in Figure 2 (Figure 2B for MC4-L5, Figure 2E for IBH-4, Figure 2H for IBH-6 and Figure 2K for MDA-MB-231), every cell line was tested in the presence of adrenaline at the stimulatory concentration in the presence or absence of an excess of the α2-adrenoceptor antagonist rauwolscine (left bars). In every case, significant stimulation of cell proliferation was reversed by the α2-adrenoceptor antagonist. In MC4-L5 and IBH-4 cells, rauwolscine alone showed a significant inhibition of cell proliferation, behaving as an inverse agonist, as previously described (Bruzzone et al., 2011). However, in IBH-6 cells, as previously reported (Bruzzone et al., 2011) and in MDA-MB-231 cells, this antagonist alone exerted no effect in cell proliferation. The cells were also treated in the presence of higher adrenaline concentrations in the presence or absence of the β-adrenoceptor antagonist propranolol (middle bars). The adrenaline concentrations chosen for these incubations were those at which some reversal of the enhancement of cell proliferation was evident in the adrenaline curves. A very significant increase of cell proliferation was evident in every cell line tested, except in IBH-6 (the only showing a non-biphasic adrenaline curve) when the β-adrenoceptor action of adrenaline was reversed by simultaneous incubation with propranolol. The endogenous adrenoceptor agonist adrenaline was tested because of its pivotal role during stress. However, as it binds to several different adrenoceptors, the β2-adrenoceptor agonist salbutamol was also tested for competition with the antagonist propranolol (right bars). In every case significant inhibition of cell proliferation was reversed by propranolol, with no effect of this compound per se.
Figure 2.
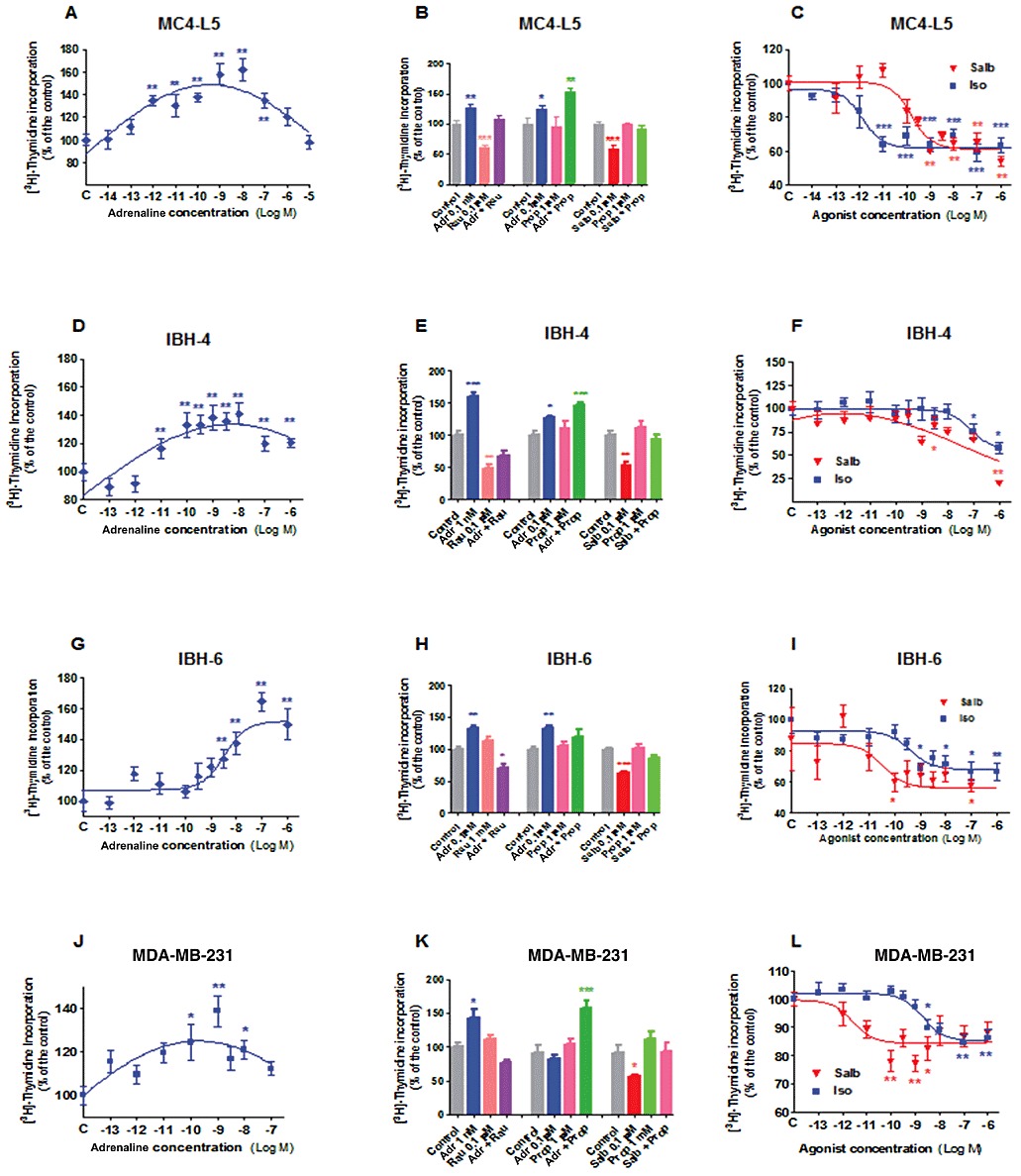
β-adrenoceptor agents and [3H]thymidine incorporation in mouse MC4-L5 and human IBH-4, IBH-6 and MDA-MB-231 cell lines. (A–C) MC4-L5 cells. (D–F) IBH-4 cells. (G–I) IBH-6 cells. (J–L) MDA-MB-231 cells. The cells were incubated with increasing concentrations of adrenaline (Adr; A,D,G,J), the β-adrenoceptor agonist isoprenaline (Iso) and the selective β2-adrenoceptor agonist salbutamol (Salb; C,F,I,L). (B,E,H,K) Cells were incubated with mitogenic concentrations of adrenaline in the presence or absence of the α2-adrenoceptor antagonist rauwolscine (Rau; left bars); higher adrenaline concentrations in the presence or absence of propranolol (middle bars) and with inhibitory concentrations of salbutamol in the presence or absence of propranolol (right bars). The addition of the antagonist was performed 30 min before the agonist. *P < 0.05, **P < 0.01 with respect to control; n = 8 replicates for treatments and 16 for controls; anova followed by Dunnett–Kramer test. The experiments were repeated at least twice with similar results.
The effect of β-adrenoceptor agonists was further analysed. Figure 2C shows the effect of isoprenaline and salbutamol on the mouse mammary tumour cell line MC4-L5, on the human breast cancer cells IBH-4 (Figure 2F), on the human breast cancer cells IBH-6 (Figure 2I) and on the human breast cancer cells MDA-MB-231 (Figure 2L). In every case, the β-adrenoceptor agonists inhibited cell proliferation. The apparent EC50 for isoprenaline on MC4-L5 cells was 1.37 pM, 0.852 nM for the human IBH-4 cells, 0.362 nM for IBH-6 cells and 1.996 nM for MDA-MB-231. The maximum for the Gaussian salbutamol curve for IBH-4 cells was 0.85 pM, and the apparent EC50 for salbutamol on the mouse MC4-L5 cells was 0.165 nM, on the human IBH-6 cells was 23.32 pM and for MDA-MB-231 cells, 2.63 pM. The mouse MC4-L5 cells exhibited a very impressive sensitivity to isoprenaline, whereas in all the human cells the sensitivity was similar for both compounds or higher for salbutamol (MDA-MB-231).
β-adrenoceptor mediated inhibition of tumour growth
Figure 3 depicts the action of the β-adrenoceptor agonist isoprenaline and the specific β2-adrenoceptor agonist salbutamol on tumour growth. Figure 3A shows the action of both agonists on the mouse progestin-dependent tumour C4-HD (Lanari et al., 2009) in the presence of medroxyprogesterone acetate (MPA), Figure 3C on the progestin-independent CC4-3-HI, both growing in Balb/c mice. C4-HD is the parent tumour from MC4-L5 cell line, HD being hormone-dependent, whereas HI signifies hormone-independent. As these tumours were induced by inoculation of MPA, the hormonal status refers to MPA (Lanari et al., 2009). Panels on Figure 3 (Figure 3A, G and I) depict the comparison of isoprenaline and salbutamol in IBH-4, IBH-6 and MDA-MB-231 tumour growth. As a β-adrenoceptor mediated effect has been described for tumours derived from inoculation of MDA-MB-231 cells in nude mice (Carie and Sebti, 2007), these tumours were included as a positive control. As can be seen in Figure 3, in every case the β-adrenoceptor agonist significantly diminished tumour growth, both in mouse tumours growing in Balb/c mice and in human cells growing in nude mice. The reversal of the action by propranolol was also tested (Figure 3B for C4-HD, D for CC4-3-HI, Figure 3F for IBH-4, H for IBH-6 and Figure 3J for MDA-MB-231). Propranolol given alone in IBH-4 tumours was completely ineffective. However in C4-HD, CC4-3-HI and IBH-6 tumour, this antagonist alone showed a curve intermediate between the control and the agonist. In the case of the MDA-MB-231 tumours, the antagonist alone produced an inhibition of growth similar to the agonist. The antagonist was able to reverse the agonist effect in the majority of the tumours.
Figure 3.
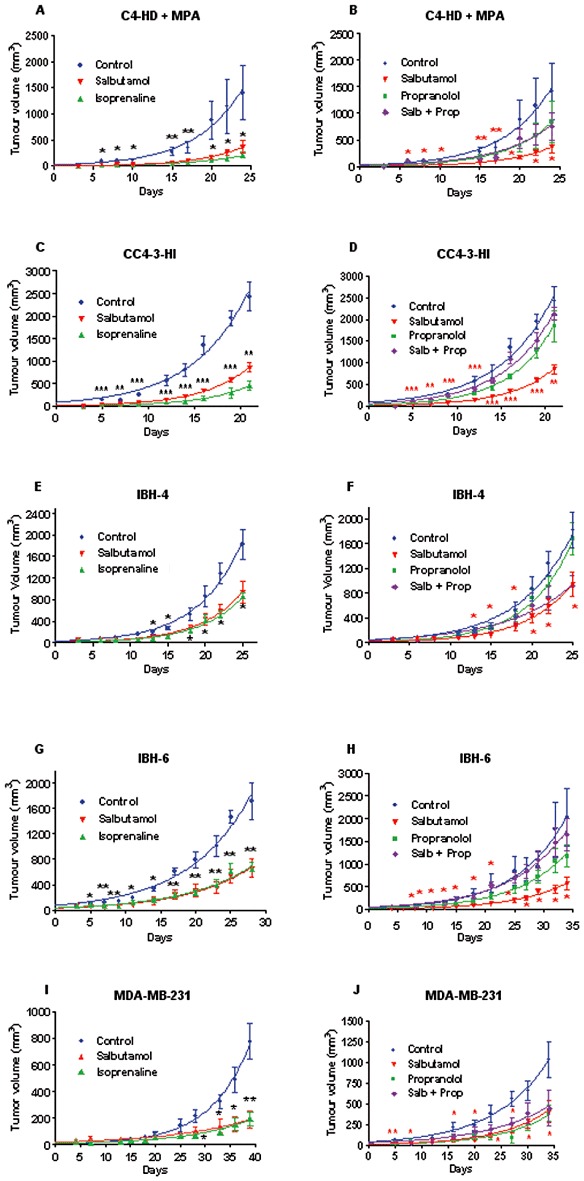
Effect of β-adrenoceptor agents on in vivo tumour growth of the mouse C4-HD and CC4-3-HI and human IBH-4, IBH-6 and MDA-MB-231 tumours. (A,B) Mouse progestin-dependent C4-HD tumour in the presence of MPA depot in Balb/c mice. (C,D) Mouse progestin independent CC4-3-HI in Balb/c mice. (E,F) Human IBH-4 tumour growing in nude mice. (G,H) Human IBH-6 tumour growing in nude mice. (I,J) MDA-MB-231 tumour growing in nude mice. A, C, E, G and I compare the action of isoprenaline and salbutamol. The control group received s.c. daily injections of physiological saline. (B,D,F,H,J) The animals were s.c. daily injected with the following compounds: salbutamol (1.2 mg·kg−1·day−1); propranolol (Prop; 1.0 mg·kg−1·day−1) or (salbutamol + propranolol; Salb + Prop) at the same doses. *P < 0.05, **P < 0.01, with respect to control; n = 10 animals/group; anova followed by Tukey–Kramer test. Each experiment was repeated twice with similar results.
Comparison of α2 and β-adrenoceptor mediated effects on tumour growth
The next set of experiments was undertaken in order to evaluate if the β2-adrenoceptor agonist salbutamol was as efficient as the α2-adrenoceptor antagonist rauwolscine, already described by our group as an inhibitor of tumour growth (Bruzzone et al., 2008; Bruzzone et al., 2011), in diminishing this parameter in the same tumours tested before. As can be seen in Figure 4, in every tumour tested both compounds worked equally well to reduce tumour growth. However, combining them caused no further action on this parameter at the doses tested.
Figure 4.
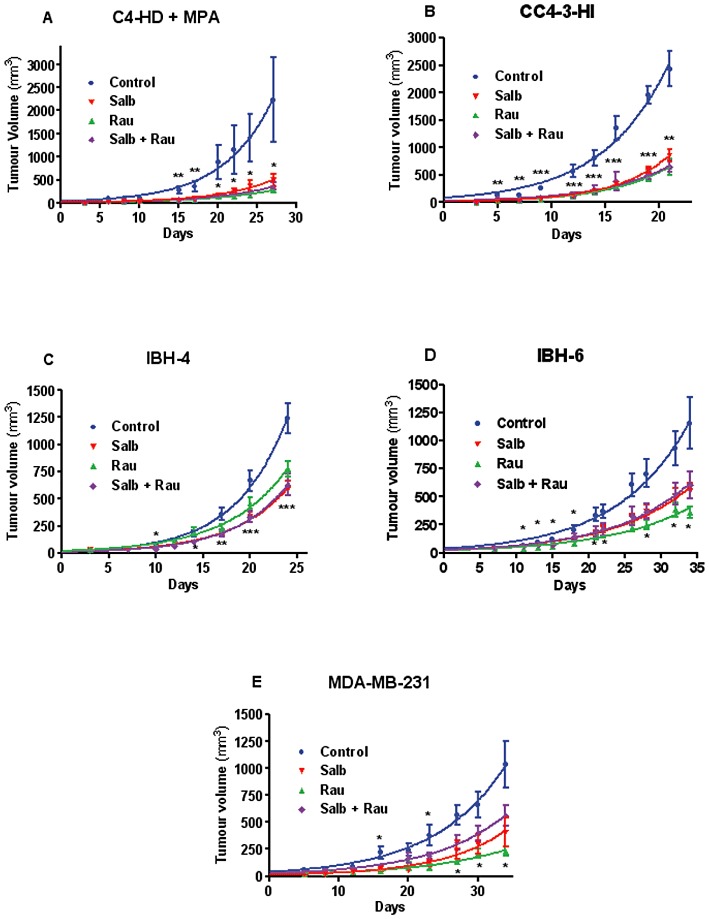
Mouse C4-HD and CC4-3-HI and human IBH-4, IBH-6 and MDA-MB-231 tumour growth in vivo comparing the α2-adrenoceptor antagonist rauwolscine (Rau) and the β2-adrenoceptor agonist salbutamol (Salb). (A) Mouse progestin-dependent C4-HD tumour in the presence of MPA depot in Balb/c mice. (B) Mouse progestin independent CC4-3-HI in Balb/c mice. (C) Human IBH-4 tumour growing in nude mice. (D) Human IBH-6 tumour growing in nude mice. (E) MDA-MB-231 tumour growing in nude mice. The animals were daily injected with the following compounds: salbutamol (1.2 mg kg−1 day−1); rauwolscine: 0.5 mg kg−1 day−1 or (salbutamol + rauwolscine) at the same doses. The control group received physiological saline. *P < 0.05, **P < 0.01; with respect to control; n = 10 animals/group, anova followed by Tukey–Kramer test. Each experiment was repeated twice with similar results.
ERK phosphorylation in the tumours correlated with their effect on tumour growth
A complex signalling pathway for β2-adrenoceptors has been described (Johnson, 2006; Carie and Sebti, 2007) which includes ERK. As the activation of the ERK pathway is sometimes associated with cell proliferation and tumour growth, the phosphorylation of ERK 1/2 was analysed in extracts of IBH-4 tumours, treated with different adrenoceptor agents (Figure 3F and Figure 4C). Figure 5A (a representative Western blot and the mean of the quantification of different experiments) shows phosphorylated ERK 1/2 relative to total ERK in extracts of whole tumours derived from this human breast cancer cell, growing in nude mice. As can be seen in this figure, inhibition of ERK 1/2 phosphorylation in vivo exactly correlated with tumour growth decrease in every treatment.
Figure 5.
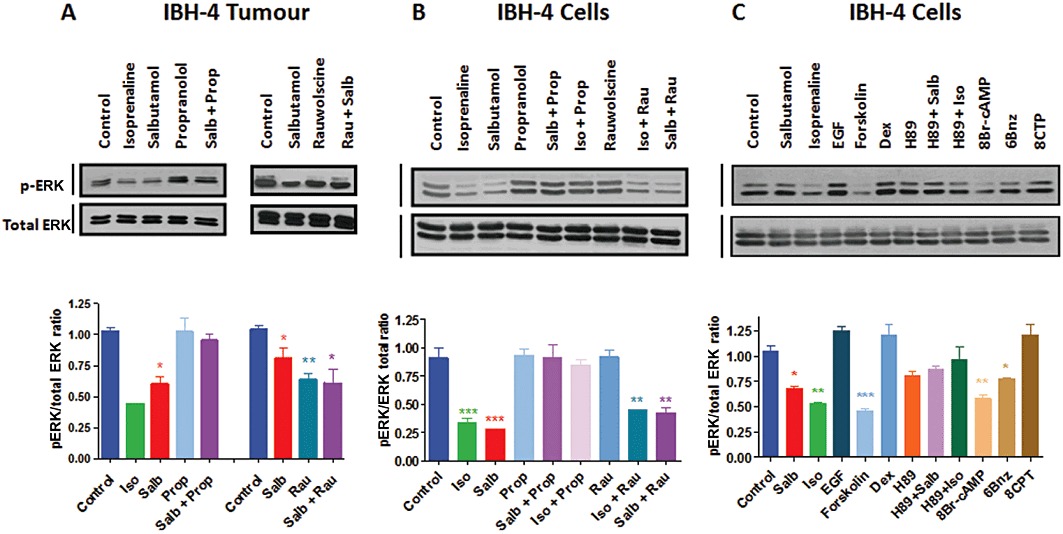
ERK 1/2 phosphorylation in protein extracts from IBH-4 tumours and cells. (A) Whole tumour extracts from IBH-4 tumours were tested for ERK 1/2 phosphorylation by Western blotting with the phospho-ERK (pERK; sc-7383) and ERK (ERK 1; sc-94) antibodies. The results are expressed as the mean ± SEM of three experiments, showing the phospho-ERK 1/2/total ERK 1/2 ratio; a representative sample is shown. (B,C) IBH-4 cells were tested for ERK 1/2 phosphorylation by Western blotting with the same antibodies. The results are expressed as the mean ± SEM of three experiments, stating the phospho-ERK 1/2/total ERK 1/2 ratio and a representative Western blot is shown. The cells were incubated during 15 min in the presence of the following compounds: 1 µM salbutamol (Salb), 1 µM isoprenaline (Iso), 1 µM rauwolscine; 1 µM propranolol, 20 ng mL−1 EGF, 10 µM forskolin, 1 µM dexmedetomidine (Dex), 25 µM of the PKA inhibitor H-89 (with and without salbutamol or isoprenaline), the PKA stimulator 8Br-cAMP (100 µM), 100 µM of the PKA selective activator 6Bnz or 100 µM of the EPAC specific activator (8CPT). *P < 0.05, **P < 0.01, with respect to control; anova followed by Dunnett–Kramer test. The isoprenaline in the in vivo experiment (A) is a single experiment, so no statistics have been performed in this column.
Analysis of signalling pathways involved in the inhibition of ERK phosphorylation
To assess the effects of the adrenoceptor agents on the tumours in vitro, the IBH-4 human breast cancer cell were incubated with the same treatments. As can be seen in Figure 5B,C, incubation with β-adrenoceptor agonists for 15 min caused a significant inhibition of ERK 1/2 phosphorylation, which was reversed by incubation with propranolol. However, no effect on ERK 1/2 phosphorylation was evident in vitro following incubation with rauwolscine. These results suggest that the β-adrenoceptor agonists could exert this effect in tumour ERK 1/2 phosphorylation acting directly on the tumour cells, whereas the effect of the α2-adrenoceptor antagonist rauwolscine could be indirect.
In order to analyse the signalling pathway involved in ERK 1/2 phosphorylation inhibition, IBH-4 cells were incubated in vitro in the presence of the agonists or treated with several mimics or antagonists. The in vitro inhibition of ERK 1/2 phosphorylation (Figure 5C) was similar to that provoked by the adenylyl cyclase activator forskolin or the cAMP analogue 8-Br-cAMP and was reversed by the simultaneous presence of propranolol. The two different pathways activated by cAMP were then studied. The PKA-activating analogue of cAMP (6Bnz) caused a clear inhibition of phosphorylation. The PKA inhibitor H-89 was able to reverse the β-adrenoceptor agonist effect, confirming the implication of PKA in the inhibition of ERK 1/2 phosphorylation. The cAMP pathway mediated by EPAC was also tested. As can be seen in Figure 5, the EPAC-activating analogue of cAMP 8CPT, showed consistent, though not significant, increases in ERK 1/2 phosphorylation in these cells, suggesting that this pathway is not implicated in the inhibition of ERK 1/2 phosphorylation mediated by the β-adrenoceptor agonists. The positive controls EGF and the α2-adrenoceptor agonist dexmedetomidine were also assessed. As these cells have high spontaneous ERK 1/2 phosphorylation and proliferation, it is difficult to cause significant stimulation of this parameter. As a consequence of these signalling studies, the significant decrease of ERK 1/2 phosphorylation caused by β-adrenoceptor agonists seemed to be mediated by PKA stimulation.
Discussion
Expression of β-adrenoceptors in mammary cell lines of mouse and human origin
As stated in the introduction, binding to β-adrenoceptors in the mammary gland has been known since 1985 (Clegg and Mullaney, 1985; Lavandero et al., 1985; Marchetti et al., 1989; Vandewalle et al., 1990; Draoui et al., 1991; Marchetti et al., 1991). The aim of the present investigation was to assess if β-adrenoceptor agonists could be potential adjuvant drugs for breast cancer. To analyse this possibility, the first step was to confirm β-adrenoceptor expression in the cells to be used for in vivo experiments. All cell lines tested showed expression of β2-adrenoceptors. However, this staining was not restricted to the cell membrane but widespread within the cell, and even in the nucleus. The expression of another GPCR, the leukotriene D4 receptor, CysLT1, is increased in colorectal adenocarcinomas and the nuclear localization of this receptor correlated with the proliferative marker Ki-67 (Nielsen et al., 2005). Also, a functional population of intracellular GPCRs coexists with membrane associated receptors (Zhu et al., 2006) and while immediate responses could be associated with the membrane receptors, more long-lasting effects could be caused by the intracellular receptors. Intranuclear receptors have been described (Zhu et al., 2006) for platelet-activating factor, prostaglandins and lysophosphatidic acid in different tissues.
Cell proliferation is inhibited by β-adrenoceptor agents
As there was debate over the activation or inhibition of cell proliferation by β-adrenoceptor agonists, we tested the effect of the natural agonist adrenaline and the β-adrenoceptor agonists, isoprenaline and salbutamol in different cell lines in order to analyse the effect on cell proliferation. The natural agonist adrenaline was included because it is released during both acute and chronic stress and its effects on breast cancer models are therefore relevant. Our data clearly show that in the mouse MC4-L5, and the human IBH-4, IBH-6 and MDA-MB-231 cell lines, the natural agonist adrenaline significantly enhanced cell proliferation, measured by [3H]-thymidine incorporation. However, as adrenaline binds to α1-, α2- and β-adrenoceptors, we hypothesized that this proliferative effect was probably due to α2-adrenoceptor binding. We have previously described in the MC4-L5 and the IBH-6 cell lines, a significant mitogenic effect associated with α2-adrenoceptor binding (Vazquez et al., 2006; Bruzzone et al., 2008). Three cell lines exhibited biphasic curves, consistent with opposing actions mediated by the α2-adrenoceptor mitogenic effect and a β-adrenoceptor inhibitory effect at higher concentrations. The EC50 for the mitogenic action of the α2-adrenoceptor agonists ranged from 20 fM to 80 pM (Vazquez et al., 2006). The incubation of all the cell lines with adrenaline and the specific antagonists confirmed that the stimulation was mediated by α2-adrenoceptors, whereas the decrease in the stimulation was mediated by β-adrenoceptors. The IBH-6 cell line was the only one to show a monophasic curve. The increase in cell proliferation in the presence of both adrenaline and propranolol suggests that when the β-adrenoceptor effect is blocked only the α2-adrenoceptor action is effective. Administration of the α2-adrenoceptor antagonist was always able to reverse adrenaline-induced stimulation of cell proliferation.
We tested then two β-adrenoceptor agonists, isoprenaline and salbutamol, in the same cells. In every case, the agonists inhibited cell proliferation, although the affinity of the effect was rather poor in the case of IBH-4 cells. The maximal inhibitory effect was similar for isoprenaline and for salbutamol in MC4-L5, IBH-4, IBH-6 and MDA-MB-231 cells. However, the sensitivity for these drugs was differed between mouse and human cells. Isoprenaline is an agonist for every β-adrenoceptor, and the expression of other subtypes of β-adrenoceptors have not been assessed in these cells. Then, the possibility of an action of β-adrenoceptors stimulated by this agonist cannot be excluded, mainly in the murine cells, which show an enhanced sensitivity to isoprenaline, compared with the human cells. On the other hand, the most important β-adrenoceptor in human cells is the β2-subtype (Draoui et al., 1991; Badino et al., 1996). Salbutamol and isoprenaline interact with different sites within the β2-adrenoceptor (Granier et al., 2007; Katritch et al., 2009) and polymorphic variants of human β2-adrenoceptors differ in several actions (Insel, 2011). The possibility of different variants in the diverse human cell lines could thus explain the differences in agonist sensitivities. An inhibition of cell proliferation has already been described in MDA-MB-231 human breast cancer cells in culture (Slotkin et al., 2000). β-Adrenoceptor signalling is rather complex and the actions of the agonists comprise a wide range of effects. For example, in the rat skeletal muscle cell line L6, β-adrenoceptor agonists increase 2-deoxy-[3H]-D-glucose uptake via a β2-adrenoceptor-mediated pathway involving both cAMP and PI3K. PKA nevertheless appears to negatively regulate β2-adrenoceptor-mediated glucose uptake (Nevzorova et al., 2006). These effects could affect cell proliferation by regulation of cell metabolism in addition to the other signalling pathways. However, an increase of glucose uptake should increase rather than decrease cell proliferation.
Inhibition of tumour growth mediated by β-adrenoceptors
The aim of these experiments was to analyse the action of β-adrenoceptor agonists on in vivo models of breast cancer. The first model used was a transplantable murine mammary tumour with different sensitivity to MPA (Lanari et al., 2009). As can be seen in Figure 3, the progestin independent CC4-3-HI tumours responded as well as the progestin-sensitive C4-HD tumours to the treatment with β-adrenoceptor agonists. This model has several common features with luminal breast cancer: they are of ductal origin, invasive and metastatic, hormone-responsive, they express oestrogen and progestin receptors and respond to chemotherapy (Lanari et al., 2009). The advantage of this model consists of the intact immune system. The second in vivo model consisted of human breast cancer cell lines IBH-4 and IBH-6 growing in nude mice. These tumours derive from primary human ductal carcinomas. They are able to grow in nude mice without the addition of oestradiol, but they respond to tamoxifen. While IBH-4 metastasizes to the lung, IBH-6 invades the peritoneum (Bruzzone et al., 2009). The advantage of this model is the human origin of the cells and the disadvantage, the need to make them grow in immunodeficient mice. MDA-MB-231 cells growing in nude mice were analysed as positive controls. The tumours of human origin were also sensitive to the inhibitory effects of the β-adrenoceptor agonist.
The doses chosen in the present paper for the in vivo treatment were much lower than the usual ones. For example, the doses of isoprenaline that have been used were 10 mg kg−1 day−1 (Landen et al., 2007) or 200 mg kg−1 day−1 (Carie and Sebti, 2007). Propranolol was used in infants at an initial dose of 0.5 mg kg−1 day−1 and increased to an oral dose of 2 mg kg−1 day−1 (Jadhav and Tolat, 2010). A similar dose of 2 mg kg−1 day−1 has been used in mice (Wang et al., 2008). However, considerably higher doses have also been used (Horinouchi et al., 2007). In preliminary tests the effects on tumour growth of the doses chosen proved sufficient to cause an action, without significant side effects.
In the case of the tumours of human origin, the β-adrenoceptor agonist isoprenaline and the specific β2-adrenoceptor salbutamol similarly inhibited tumour growth. The latter is a drug currently used as a rapid and short acting treatment for asthma (Cazzola and Matera, 2008) and has been classified as a non-catechol partial agonist (Yao et al., 2006). We have already described (Bruzzone et al., 2009) that these cell lines growing in nude mice are insensitive to oestrogen or progestins, but sensitive to tamoxifen inhibition. In MDA-MB-231 cells inoculated into the fat pad of nude mice, the compound ARA-211 induced regression of tumours and potently inhibited tumour growth by acting through β-adrenoceptors (Carie and Sebti, 2007). These authors also stated that isoprenaline was able to inhibit tumour growth without causing regression of tumours (Carie and Sebti, 2007). However, a completely opposite action has been reported associating β-adrenoceptor activation with tumour growth. In tumours induced in rats by DMBA, intact animals with progressing tumours had significantly higher levels of β-adrenoceptors than intact animals with static tumours, and regressing tumours in ovariectomized animals showed a sharp decrease in β-adrenoceptors (Marchetti et al., 1989). The results shown herein with β-adrenoceptor agonists are consistent with the inhibition of tumour growth described for the MDA-MB-231 tumours (Carie and Sebti, 2007). In the present investigation, propranolol in the majority of the cases completely reversed the agonist effect. However, in certain tumours, propranolol administered alone caused a diminution of tumour growth. Although effects on other receptors cannot be excluded, it is possible to ascribe this inhibition of tumour growth as a partial agonist effect, as has already been described mainly at the transcriptional level (Baker et al., 2003). Another possibility is that the in vivo propranolol inhibition of tumour growth could be indirect, as suggested by the complete absence of effect of the drug given alone in the in vitro experiments. This effect could be mediated by the inhibition of macrophage infiltration and crosstalk, inhibiting the prometastatic cell signalling switching in the cancer cells (Powe and Entschladen, 2011). Propranolol is used to treat infantile haemangiomas (Tan et al., 2011) and this compound inhibited in vitro human brain endothelial cells tubulogenesis (Annabi et al., 2009). A similar effect cannot be discounted in this model. Moreover, a very promising effect of β-adrenoceptor antagonists, mainly propranolol, has recently been reported in breast cancer human patients (Powe et al., 2010; Barron et al., 2011; Ganz et al., 2011). Interestingly, the administration of β-adrenoceptor antagonists was associated with improved relapse-free survival but not overall survival among patients with triple-negative breast cancer (Melhem-Bertrandt et al., 2011).
Some important actions described for β-adrenoceptors in different cancer models must be considered in pre-clinical research before considering the possibility of testing adrenoceptor agents in clinical trials. The migration of breast, prostate and colon carcinoma cells is enhanced by the stress-related neurotransmitter noradrenaline in vitro, and this effect can be inhibited by propranolol (Masur et al., 2001; Palm et al., 2006). PC-3 prostate carcinoma cells growing in athymic BALB/c nude mice developed lumbar lymph node metastases at an increased rate with the application of noradrenaline, whereas propranolol inhibited this effect. The growth of the primary tumour was not affected by either treatment (Palm et al., 2006). Nicotine can enhance colon tumour growth (HT-29 cells growing as xenograft in nude mice) partly by stimulation of β-adrenoceptor, preferentially the β2-adrenoceptor, with subsequent stimulation of COX-2, PGE2, and VEGF expression (Wong et al., 2007). Chronic behavioural stress caused higher levels of tissue catecholamines, greater tumour burden and more invasive growth of two different β-adrenoceptor-positive ovarian cancer cell lines (HeyA8 and SKOV3ip1) intraperitoneally inoculated (Thaker et al., 2006). The MDA-MB-231 breast cancer cells injected into the mammary fat pad showed a similar increase of tumour growth. These effects were primarily mediated through activation of β-adrenoceptors and the cAMP–PKA signalling pathway. Ovarian tumours in stressed animals showed markedly increased vascularization and enhanced expression of VEGF, MMP2 and MMP9 (Thaker et al., 2006). All these different effects of adrenaline, noradrenaline or stress, generally mediated by β2-adrenoceptors, must be carefully analysed in pre-clinical studies before considering clinical trials.
Comparison of α2 and β-adrenoceptor action on tumour growth
We have previously demonstrated that the α2-adrenoceptor antagonist/inverse agonist rauwolscine was able to significantly inhibit tumour growth in a model of mouse mammary tumours (Bruzzone et al., 2008) and human breast cancer cells growing in nude mice (Bruzzone et al., 2011) in the absence of agonist treatment. We have carefully analysed the action of this compound elsewhere (Vazquez et al., 2006; Bruzzone et al., 2008; Bruzzone et al., 2011). In summary, although a systemic effect of this compound reversing the stimulatory action of adrenaline or noradrenaline, released by stress in the animal, cannot be discarded as a mechanism of action, in vitro data indicated that at least part of this action could be due to an inverse agonist effect directly on the cells. In the present work, with the objective of finding a possible adjuvant drug for breast cancer, we compared the effect of this α2-adrenoceptor inverse agonist and a β2-adrenoceptor agonist salbutamol in inhibiting tumour growth. Both compounds performed equally well in this parameter. As stated before for the action of the β-adrenoceptor compounds, it is still too soon to think in clinical trials. The action of both β-adrenoceptor agonists and the α2-adrenoceptor antagonist rauwolscine on cell proliferation, apoptosis, invasion, metastasis and differentiation should be carefully investigated before any clinical trial is pursued. At this stage of the investigation it is therefore not possible to describe either of them as more relevant than the other in inhibiting tumour growth.
Several interactions between α2 and β-adrenoceptors have been described (Desai et al., 2004; Desai et al., 2005; Schwencke et al., 2005). We have also previously found an increase of α2-adrenoceptor concentration when the cells were incubated in the presence of isoprenaline (Vazquez et al., 2006). We therefore expected an interaction, to be shown in different growth curves in the simultaneous presence of the α2-adrenoceptor antagonist and the β2-adrenoceptor agonist. However, in the present work the curves of the α2-adrenoceptor antagonist, the β2-adrenoceptor agonist and both compounds together were superimposable, showing no evident interaction of these receptors on tumour growth, at least at the chosen doses.
Adrenoceptors are involved in complex and pleiotropic actions. The α2A-adrenoceptor subtype has very clear hypotensive actions. Stimulation of α2B-adrenoceptors in vascular smooth muscle leads to vasoconstriction, which causes hypertension (Philipp and Hein, 2004).β-Adrenoceptors mediate also the redistribution of blood flow to skeletal muscle during exercise (Figueroa et al., 2009) and are involved in tumour angiogenesis (Chakroborty et al., 2009). However, for the results presented herein, the anti-mitogenic effect seems more relevant than the metabolic and angiogenic actions, because the combined result is a significant reduction of tumour growth in every tumour tested.
ERK phosphorylation
β-Adrenoceptor signalling is complex. As reviewed (Johnson, 2006), although most of the actions of the β2-adrenoceptor seem mediated by Gs proteins and the cAMP-dependent PKA system, these receptors can also couple to Gi proteins, resulting in stimulation of the ERK and p38 MAPK pathways. Activation of this pathway by the β2-adrenoceptor requires receptor phosphorylation by PKA and is mediated by βγ-subunits of the G protein acting as an assembly scaffold for a number of intracellular proteins, including non-receptor tyrosine kinases cSrc, Ras and Raf culminating in the stimulation of MAPKs (Johnson, 2006). Also, β2-adrenoceptor stimulation with an agonist results in a transient pulse of cAMP, GPCR kinase (GRK6)-mediated receptor phosphorylation leading to β-arrestin mediated receptor inactivation and cAMP-dependent kinase-mediated induction of cAMP metabolism by phosphodiesterases (Violin et al., 2008). It has been reported (Houslay and Baillie, 2003) that phosphodiesterase PDE4 isoforms can interact with β-arrestin and thus be recruited to the β2-adrenoceptor in an agonist-dependent fashion. The recruited PDE4 can switch coupling of the β2-adrenoceptor to activation of Gi and hence to ERK. In hepatocellular HepG2 and MHCC97H cancer cells, isoprenaline promoted cell proliferation and transiently ERK 1/2 phosphorylation by EGFR-independent mechanisms (Yuan et al., 2010). Also (Plummer et al., 2004) the expression of mRNA that encodes a G-protein coupled inwardly rectifying potassium channel (GIRK1) has been shown in several human breast cancer cell lines and tissue samples from approximately 40% of primary human breast cancers. The tobacco carcinogen, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, which shows high affinity for β-adrenoceptors, stimulated activation of ERK 1/2 in MDA-MB-453 cells (Plummer et al., 2004).
However, recently Carie and Sebti (Carie and Sebti, 2007) searched for inhibitors of p-ERK 1/2 in the human breast cancer cell line MDA-MB-231. They found a compound, ARA-211, which activated β2-adrenoceptors and suppressed the Raf-1/Mek-1/ERK 1/2 pathway by a cAMP-dependent activation of PKA, but not by the EPAC pathway. Similarly, isoprenaline blocks increases in ERK 1/2 phosphorylation in freshly dispersed rat parotid acinar cells (Soltoff and Hedden, 2010). The inhibitory action of isoprenaline was reproduced by forskolin and mimetics, including one selective for PKA, while EPAC selective activators did not mimic the blockade of ERK by isoprenaline, suggesting that inhibition involved PKA (Soltoff and Hedden, 2010). The stimulation of ERK by phosphorylation induces a series of different actions. As recently reviewed (Kim and Choi, 2010), the ERK signalling pathway plays a role in several steps of tumour progression. ERK phosphorylates several proteins promoting cancer cell migration. The ERK pathway also induces expression of matrix metalloproteinases promoting degradation of extracellular matrix proteins and consequent tumour invasion. ERK 1/2 signalling regulates the activities of pro-apoptotic and anti-apoptotic proteins promoting cancer cell survival. However, the most studied action of ERK 1/2 activation is related to cell proliferation, as recently reviewed (Mebratu and Tesfaigzi, 2009). In the present paper we have analysed the activation of ERK 1/2 by phosphorylation in extracts of the IBH-4 tumours. In this experimental model, ERK inhibition of phosphorylation perfectly matched tumour growth decrease in every treatment.
There is controversy with respect to the effects of β-adrenoceptor agonists on ERK 1/2 phosphorylation. On the one hand, the mechanisms involving the βγ-subunits of the Gi protein, β-arrestin, GIRK1 channel and EGFR transactivation were reported to stimulate ERK 1/2 phosphorylation and thus cell proliferation or tumour growth. On the other hand, PKA stimulation was directly involved in the inhibition of both ERK 1/2 phosphorylation and cell proliferation/tumour growth. A possible explanation for the reported discrepancies on ERK phosphorylation and cell proliferation is that the final result depends on the balance of the stimulating and the inhibiting pathways in each experimental model and conditions.
The results presented herein suggest that the inhibition of ERK 1/2 phosphorylation in this experimental model is mediated by PKA and not by EPAC. These results are consistent with the pathways described for β-adrenoceptor agonists in MDA-MB-231 cells (Carie and Sebti, 2007) and in freshly dispersed rat parotid acinar cells (Soltoff and Hedden, 2010).
In conclusion, our results shown here demonstrated in two independent experimental models of breast cancer that the β-adrenoceptor agonists inhibited breast cancer cell proliferation and tumour growth when growing as xenografts. This effect is probably mediated by ERK 1/2 inhibited phosphorylation. The possibility of inhibiting tumour growth both in hormone-dependent as well as in hormone-independent tumours is promising. The β-adrenoceptor agonist isoprenaline and the β2-adrenoceptor agonist salbutamol (currently in use in humans for the treatment of asthma) were as effective as the α2-adrenoceptor antagonist rauwolscine, providing possible novel therapeutic adjuvant treatments for breast cancer.
Acknowledgments
This work was supported by the Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) (PIP 2004 # 5351 and PIP 2010–2012 # 692), the Agencia Nacional de Promoción Científica y TecnolÓgica (PICT 2004-05-26046) and ‘Fundación Sales’, Argentina. AB and IAL are members of the Research Career, CONICET, Argentina, CPP and LC postdoctoral fellows. The authors thank the skilful technical help from Paola Garcette, Natalia Vasta and Lic. Lara Lapyckyj.
Glossary
- 6Bnz
N6-benzoyl-cAMP
- 8CTP
8-(4-chlorophenylthio)-2′-O-methyl-cAMP
- DMEM
Dulbecco's modified Eagle's medium
- EPAC
exchange protein directly activated by cAMP
- F12
Ham's F12 medium
- FCS
fetal calf serum
Conflicts of interest
None.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th Edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annabi B, Lachambre MP, Plouffe K, Moumdjian R, Beliveau R. Propranolol adrenergic blockade inhibits human brain endothelial cells tubulogenesis and matrix metalloproteinase-9 secretion. Pharmacol Res. 2009;60:438–445. doi: 10.1016/j.phrs.2009.05.005. [DOI] [PubMed] [Google Scholar]
- Antoni MH, Lutgendorf SK, Cole SW, Dhabhar FS, Sephton SE, McDonald PG, et al. The influence of bio-behavioural factors on tumour biology: pathways and mechanisms. Nat Rev Cancer. 2006;6:240–248. doi: 10.1038/nrc1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoni MH, Lechner S, Diaz A, Vargas S, Holley H, Phillips K, et al. Cognitive behavioral stress management effects on psychosocial and physiological adaptation in women undergoing treatment for breast cancer. Brain Behav Immun. 2009;23:580–591. doi: 10.1016/j.bbi.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badino GR, Novelli A, Girardi C, Di Carlo F. Evidence for functional beta-adrenoceptor subtypes in CG-5 breast cancer cell. Pharmacol Res. 1996;33:255–260. doi: 10.1006/phrs.1996.0036. [DOI] [PubMed] [Google Scholar]
- Baker JG, Hall IP, Hill SJ. Agonist and inverse agonist actions of beta-blockers at the human beta 2-adrenoceptor provide evidence for agonist-directed signaling. Mol Pharmacol. 2003;64:1357–1369. doi: 10.1124/mol.64.6.1357. [DOI] [PubMed] [Google Scholar]
- Barron TI, Connolly RM, Sharp L, Bennett K, Visvanathan K. Beta blockers and breast cancer mortality: a population- based study. J Clin Oncol. 2011;29:2635–2644. doi: 10.1200/JCO.2010.33.5422. [DOI] [PubMed] [Google Scholar]
- Berthois Y, Katzenellenbogen JA, Katzenellenbogen BS. Phenol red in tissue culture media is a weak estrogen: implications concerning the study of estrogen-responsive cells in culture. Proc Natl Acad Sci U S A. 1986;83:2496–2500. doi: 10.1073/pnas.83.8.2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruzzone A, Pinero CP, Castillo LF, Sarappa MG, Rojas P, Lanari C, et al. Alpha(2)-Adrenoceptor action on cell proliferation and mammary tumour growth in mice. Br J Pharmacol. 2008;155:494–504. doi: 10.1038/bjp.2008.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruzzone A, Vanzulli SI, Soldati R, Giulianelli S, Lanari C, Luthy IA. Novel human breast cancer cell lines IBH-4, IBH-6, and IBH-7 growing in nude mice. J Cell Physiol. 2009;219:477–484. doi: 10.1002/jcp.21694. [DOI] [PubMed] [Google Scholar]
- Bruzzone A, Pinero CP, Rojas P, Romanato M, Gass H, Lanari C, et al. Alpha(2)-adrenoceptors enhance cell proliferation and mammary tumor growth acting through both the stroma and the tumor cells. Curr Cancer Drug Targets. 2011;11:763–774. doi: 10.2174/156800911796191051. [DOI] [PubMed] [Google Scholar]
- Cakir Y, Plummer HK, III, Tithof PK, Schuller HM. Beta-adrenergic and arachidonic acid-mediated growth regulation of human breast cancer cell lines. Int J Oncol. 2002;21:153–157. [PubMed] [Google Scholar]
- Carie AE, Sebti SM. A chemical biology approach identifies a beta-2 adrenergic receptor agonist that causes human tumor regression by blocking the Raf-1/Mek-1/Erk1/2 pathway. Oncogene. 2007;26:3777–3788. doi: 10.1038/sj.onc.1210172. [DOI] [PubMed] [Google Scholar]
- Cazzola M, Matera MG. Novel long-acting bronchodilators for COPD and asthma. Br J Pharmacol. 2008;155:291–299. doi: 10.1038/bjp.2008.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakroborty D, Sarkar C, Basu B, Dasgupta PS, Basu S. Catecholamines regulate tumor angiogenesis. Cancer Res. 2009;69:3727–3730. doi: 10.1158/0008-5472.CAN-08-4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clegg RA, Mullaney I. Acute change in the cyclic AMP content of rat mammary acini in vitro. Influence of physiological and pharmacological agents. Biochem J. 1985;230:239–246. doi: 10.1042/bj2300239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai AN, Standifer KM, Eikenburg DC. Simultaneous alpha2B- and beta2-adrenoceptor activation sensitizes the alpha2B-adrenoceptor for agonist-induced down-regulation. J Pharmacol Exp Ther. 2004;311:794–802. doi: 10.1124/jpet.104.069674. [DOI] [PubMed] [Google Scholar]
- Desai AN, Standifer KM, Eikenburg DC. Cellular G protein-coupled receptor kinase levels regulate sensitivity of the {alpha}2b-adrenergic receptor to undergo agonist-induced down-regulation. J Pharmacol Exp Ther. 2005;312:767–773. doi: 10.1124/jpet.104.076042. [DOI] [PubMed] [Google Scholar]
- Dowdy S, Wearden S. Statistics for Research. New York: John Wiley & Sons, Inc; 1983. [Google Scholar]
- Draoui A, Vandewalle B, Hornez L, Revillion F, Lefebvre J. Beta-adrenergic receptors in human breast cancer: identification, characterization and correlation with progesterone and estradiol receptors. Anticancer Res. 1991;11:677–680. [PubMed] [Google Scholar]
- Feigelson HS, Teras LR, Diver WR, Tang W, Patel AV, Stevens VL, et al. Genetic variation in candidate obesity genes ADRB2, ADRB3, GHRL, HSD11B1, IRS1, IRS2, and SHC1 and risk for breast cancer in the Cancer Prevention Study II. Breast Cancer Res. 2008;10:R57. doi: 10.1186/bcr2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- Figueroa XF, Poblete I, Fernandez R, Pedemonte C, Cortes V, Huidobro-Toro JP. NO production and eNOS phosphorylation induced by epinephrine through the activation of beta-adrenoceptors. Am J Physiol Heart Circ Physiol. 2009;297:H134–H143. doi: 10.1152/ajpheart.00023.2009. [DOI] [PubMed] [Google Scholar]
- Ganz PA, Habel LA, Weltzien EK, Caan BJ, Cole SW. Examining the influence of beta blockers and ACE inhibitors on the risk for breast cancer recurrence: results from the LACE cohort. Breast Cancer Res Treat. 2011;129:549–556. doi: 10.1007/s10549-011-1505-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granier S, Kim S, Shafer AM, Ratnala VR, Fung JJ, Zare RN, et al. Structure and conformational changes in the C-terminal domain of the beta2-adrenoceptor: insights from fluorescence resonance energy transfer studies. J Biol Chem. 2007;282:13895–13905. doi: 10.1074/jbc.M611904200. [DOI] [PubMed] [Google Scholar]
- Hammon HM, Bruckmaier RM, Honegger UE, Blum JW. Distribution and density of alpha- and beta-adrenergic receptor binding sites in the bovine mammary gland. J Dairy Res. 1994;61:47–57. doi: 10.1017/s0022029900028041. [DOI] [PubMed] [Google Scholar]
- Horinouchi T, Morishima S, Tanaka T, Suzuki F, Tanaka Y, Koike K, et al. Different changes of plasma membrane beta-adrenoceptors in rat heart after chronic administration of propranolol, atenolol and bevantolol. Life Sci. 2007;81:399–404. doi: 10.1016/j.lfs.2007.06.003. [DOI] [PubMed] [Google Scholar]
- Houslay MD, Baillie GS. The role of ERK2 docking and phosphorylation of PDE4 cAMP phosphodiesterase isoforms in mediating cross-talk between the cAMP and ERK signalling pathways. Biochem Soc Trans. 2003;31:1186–1190. doi: 10.1042/bst0311186. [DOI] [PubMed] [Google Scholar]
- Inderwies T, Pfaffl MW, Meyer HH, Blum JW, Bruckmaier RM. Detection and quantification of mRNA expression of alpha- and beta-adrenergic receptor subtypes in the mammary gland of dairy cows. Domest Anim Endocrinol. 2003;24:123–135. doi: 10.1016/s0739-7240(02)00211-4. [DOI] [PubMed] [Google Scholar]
- Insel PA. Beta(2)-Adrenergic receptor polymorphisms and signaling: do variants influence the ‘memory’ of receptor activation? Sci Signal. 2011;4:pe37. doi: 10.1126/scisignal.2002352. [DOI] [PubMed] [Google Scholar]
- Institute of Laboratory Animal Research, Commission on Life Sciences; National Research Council. Guide for the Care and Use of Laboratory Animals. Washington, DC: National Academy Press; 1996. [Google Scholar]
- Institute of Laboratory Animal Research, Commission on Life Sciences; National Research Council. Guide for the Care and Use of Laboratory Animals. Eighth edn. Washington, DC: National Academy Press; 2011. [Google Scholar]
- Jadhav VM, Tolat SN. Dramatic response of propranolol in hemangioma: report of two cases. Indian J Dermatol Venereol Leprol. 2010;76:691–694. doi: 10.4103/0378-6323.72472. [DOI] [PubMed] [Google Scholar]
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- Johnson M. Molecular mechanisms of beta(2)-adrenergic receptor function, response, and regulation. J Allergy Clin Immunol. 2006;117:18–24. doi: 10.1016/j.jaci.2005.11.012. [DOI] [PubMed] [Google Scholar]
- Katritch V, Reynolds KA, Cherezov V, Hanson MA, Roth CB, Yeager M, et al. Analysis of full and partial agonists binding to beta2-adrenergic receptor suggests a role of transmembrane helix V in agonist-specific conformational changes. J Mol Recognit. 2009;22:307–318. doi: 10.1002/jmr.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EK, Choi EJ. Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta. 2010;1802:396–405. doi: 10.1016/j.bbadis.2009.12.009. [DOI] [PubMed] [Google Scholar]
- Lamb CA, Helguero LA, Giulianelli S, Soldati R, Vanzulli SI, Molinolo A, et al. Antisense oligonucleotides targeting the progesterone receptor inhibit hormone-independent breast cancer growth in mice. Breast Cancer Res. 2005;7:R1111–R1121. doi: 10.1186/bcr1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanari C, Luthy I, Lamb CA, Fabris V, Pagano E, Helguero LA, et al. Five novel hormone-responsive cell lines derived from murine mammary ductal carcinomas: in vivo and in vitro effects of estrogens and progestins. Cancer Res. 2001;61:293–302. [PubMed] [Google Scholar]
- Lanari C, Lamb CA, Fabris VT, Helguero LA, Soldati R, Bottino MC, et al. The MPA mouse breast cancer model: evidence for a role of progesterone receptors in breast cancer. Endocr Relat Cancer. 2009;16:333–350. doi: 10.1677/ERC-08-0244. [DOI] [PubMed] [Google Scholar]
- Landen CN, Jr, Lin YG, Armaiz Pena GN, Das PD, Arevalo JM, Kamat AA, et al. Neuroendocrine modulation of signal transducer and activator of transcription-3 in ovarian cancer. Cancer Res. 2007;67:10389–10396. doi: 10.1158/0008-5472.CAN-07-0858. [DOI] [PubMed] [Google Scholar]
- Lavandero S, Donoso E, Sapag-Hagar M. Beta-adrenergic receptors in rat mammary gland. Biochem Pharmacol. 1985;34:2034–2036. doi: 10.1016/0006-2952(85)90330-2. [DOI] [PubMed] [Google Scholar]
- Luthy IA, Bruzzone A, Pinero CP, Castillo LF, Chiesa IJ, Vazquez SM, et al. Adrenoceptors: non-conventional target for breast cancer? Curr Med Chem. 2009;16:1850–1862. doi: 10.2174/092986709788186048. [DOI] [PubMed] [Google Scholar]
- Marchetti B, Labrie F. Hormonal regulation of beta-adrenergic receptors in the rat mammary gland during the estrous cycle and lactation: role of sex steroids and prolactin. Endocrinology. 1990;126:575–581. doi: 10.1210/endo-126-1-575. [DOI] [PubMed] [Google Scholar]
- Marchetti B, Spinola PG, Plante M, Poyet P, Follea N, Pelletier G, et al. Beta-adrenergic receptors in DMBA-induced rat mammary tumors: correlation with progesterone receptor and tumor growth. Breast Cancer Res Treat. 1989;13:251–263. doi: 10.1007/BF02106575. [DOI] [PubMed] [Google Scholar]
- Marchetti B, Fortier MA, Poyet P, Follea N, Pelletier G, Labrie F. Beta-adrenergic receptors in the rat mammary gland during pregnancy and lactation: characterization, distribution, and coupling to adenylate cyclase. Endocrinology. 1990;126:565–574. doi: 10.1210/endo-126-1-565. [DOI] [PubMed] [Google Scholar]
- Marchetti B, Spinola PG, Pelletier G, Labrie F. A potential role for catecholamines in the development and progression of carcinogen-induced mammary tumors: hormonal control of beta-adrenergic receptors and correlation with tumor growth. J Steroid Biochem Mol Biol. 1991;38:307–320. doi: 10.1016/0960-0760(91)90102-b. [DOI] [PubMed] [Google Scholar]
- Masur K, Niggemann B, Zanker KS, Entschladen F. Norepinephrine-induced migration of SW 480 colon carcinoma cells is inhibited by beta-blockers. Cancer Res. 2001;61:2866–2869. [PubMed] [Google Scholar]
- Mebratu Y, Tesfaigzi Y. How ERK1/2 activation controls cell proliferation and cell death: Is subcellular localization the answer? Cell Cycle. 2009;8:1168–1175. doi: 10.4161/cc.8.8.8147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melhem-Bertrandt A, Chavez-Macgregor M, Lei X, Brown EN, Lee RT, Meric-Bernstam F, et al. Beta-blocker use is associated with improved relapse-free survival in patients with triple-negative breast cancer. J Clin Oncol. 2011;29:2645–2652. doi: 10.1200/JCO.2010.33.4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevzorova J, Evans BA, Bengtsson T, Summers RJ. Multiple signalling pathways involved in beta2-adrenoceptor-mediated glucose uptake in rat skeletal muscle cells. Br J Pharmacol. 2006;147:446–454. doi: 10.1038/sj.bjp.0706626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen CK, Campbell JI, Ohd JF, Morgelin M, Riesbeck K, Landberg G, et al. A novel localization of the G-protein-coupled CysLT1 receptor in the nucleus of colorectal adenocarcinoma cells. Cancer Res. 2005;65:732–742. [PubMed] [Google Scholar]
- Palm D, Lang K, Niggemann B, TLt D, Masur K, Zaenker KS, et al. The norepinephrine-driven metastasis development of PC-3 human prostate cancer cells in BALB/c nude mice is inhibited by beta-blockers. Int J Cancer. 2006;118:2744–2749. doi: 10.1002/ijc.21723. [DOI] [PubMed] [Google Scholar]
- Parkin DM. Is the recent fall in incidence of post-menopausal breast cancer in UK related to changes in use of hormone replacement therapy? Eur J Cancer. 2009;45:1649–1653. doi: 10.1016/j.ejca.2009.01.016. [DOI] [PubMed] [Google Scholar]
- Philipp M, Hein L. Adrenergic receptor knockout mice: distinct functions of 9 receptor subtypes. Pharmacol Ther. 2004;101:65–74. doi: 10.1016/j.pharmthera.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Plummer HK, III, Yu Q, Cakir Y, Schuller HM. Expression of inwardly rectifying potassium channels (GIRKs) and beta-adrenergic regulation of breast cancer cell lines. BMC Cancer. 2004;4:93. doi: 10.1186/1471-2407-4-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powe DG, Entschladen F. Targeted therapies: Using beta-blockers to inhibit breast cancer progression. Nat Rev. Clin Oncol. 2011;8:511–512. doi: 10.1038/nrclinonc.2011.123. [DOI] [PubMed] [Google Scholar]
- Powe DG, Voss MJ, Zanker KS, Habashy HO, Green AR, Ellis IO, et al. Beta-blocker drug therapy reduces secondary cancer formation in breast cancer and improves cancer specific survival. Oncotarget. 2010;1:628–638. doi: 10.18632/oncotarget.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roets E, Peeters G, Leysen JE. Identification of beta-adrenoceptors in bovine teat muscles by 3H-dihydroalprenolol binding. Arch Int Pharmacodyn Ther. 1984;270:203–214. [PubMed] [Google Scholar]
- Roets E, Vandeputte-Van Messom G, Peeters G. Relationship between milkability and adrenoceptor concentrations in teat tissue in primiparous cows. J Dairy Sci. 1986;69:3120–3130. doi: 10.3168/jds.s0022-0302(86)80776-7. [DOI] [PubMed] [Google Scholar]
- Schwencke C, Schmeisser A, Weinbrenner C, Braun-Dullaeus RC, Marquetant R, Strasser RH. Transregulation of the alpha2-adrenergic signal transduction pathway by chronic beta-blockade: a novel mechanism for decreased platelet aggregation in patients. J Cardiovasc Pharmacol. 2005;45:253–259. doi: 10.1097/01.fjc.0000154372.03531.e1. [DOI] [PubMed] [Google Scholar]
- Slotkin TA, Zhang J, Dancel R, Garcia SJ, Willis C, Seidler FJ. Beta-adrenoceptor signaling and its control of cell replication in MDA-MB-231 human breast cancer cells. Breast Cancer Res Treat. 2000;60:153–166. doi: 10.1023/a:1006338232150. [DOI] [PubMed] [Google Scholar]
- Soltoff SP, Hedden L. Isoproterenol and cAMP block ERK phosphory0lation and enhance [Ca2+]i increases and oxygen consumption by muscarinic receptor stimulation in rat parotid and submandibular acinar cells. J Biol Chem. 2010;285:13337–13348. doi: 10.1074/jbc.M110.112094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan ST, Itinteang T, Leadbitter P. Low-dose propranolol for infantile haemangioma. J Plast Reconstr Aesthet Surg. 2011;64:292–299. doi: 10.1016/j.bjps.2010.06.010. [DOI] [PubMed] [Google Scholar]
- Thaker PH, Han LY, Kamat AA, Arevalo JM, Takahashi R, Lu C, et al. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat Med. 2006;12:939–944. doi: 10.1038/nm1447. [DOI] [PubMed] [Google Scholar]
- United Kingdom Co-ordinating Committee on Cancer Research U. United Kingdom Co-ordinating Committee on Cancer Research (UKCCCR) Guidelines for the welfare of animals in experimental neoplasia (second edition) Br J Cancer. 1998;77:1–10. doi: 10.1038/bjc.1998.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandewalle B, Revillion F, Lefebvre J. Functional beta-adrenergic receptors in breast cancer cells. J Cancer Res Clin Oncol. 1990;116:303–306. doi: 10.1007/BF01612908. [DOI] [PubMed] [Google Scholar]
- Vazquez SM, Pignataro O, Luthy IA. Alpha2-adrenergic effect on human breast cancer MCF-7 cells. Breast Cancer Res Treat. 1999;55:41–49. doi: 10.1023/a:1006196308001. [DOI] [PubMed] [Google Scholar]
- Vazquez SM, Mladovan A, Garbovesky C, Baldi A, Luthy IA. Three novel hormone-responsive cell lines derived from primary human breast carcinomas: functional characterization. J Cell Physiol. 2004;199:460–469. doi: 10.1002/jcp.10466. [DOI] [PubMed] [Google Scholar]
- Vazquez SM, Mladovan AG, Perez C, Bruzzone A, Baldi A, Luthy IA. Human breast cell lines exhibit functional alpha(2)-adrenoceptors. Cancer Chemother Pharmacol. 2006;58:50–61. doi: 10.1007/s00280-005-0130-4. [DOI] [PubMed] [Google Scholar]
- Violin JD, DiPilato LM, Yildirim N, Elston TC, Zhang J, Lefkowitz RJ. beta2-adrenergic receptor signaling and desensitization elucidated by quantitative modeling of real time cAMP dynamics. J Biol Chem. 2008;283:2949–2961. doi: 10.1074/jbc.M707009200. [DOI] [PubMed] [Google Scholar]
- Wang JF, Min JY, Hampton TG, Amende I, Yan X, Malek S, et al. Clozapine-induced myocarditis: role of catecholamines in a murine model. Eur J Pharmacol. 2008;592:123–127. doi: 10.1016/j.ejphar.2008.06.088. [DOI] [PubMed] [Google Scholar]
- Wong HP, Yu L, Lam EK, Tai EK, Wu WK, Cho CH. Nicotine promotes colon tumor growth and angiogenesis through beta-adrenergic activation. Toxicol Sci. 2007;97:279–287. doi: 10.1093/toxsci/kfm060. [DOI] [PubMed] [Google Scholar]
- Workman P, Aboagye EO, Balkwill F, Balmain A, Bruder G, Chaplin DJ, et al. Guidelines for the welfare and use of animals in cancer research. Br J Cancer. 2010;102:1555–1577. doi: 10.1038/sj.bjc.6605642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao X, Parnot C, Deupi X, Ratnala VR, Swaminath G, Farrens D, et al. Coupling ligand structure to specific conformational switches in the beta2-adrenoceptor. Nat Chem Biol. 2006;2:417–422. doi: 10.1038/nchembio801. [DOI] [PubMed] [Google Scholar]
- Yuan A, Li Z, Li X, Yi S, Wang S, Cai Y, et al. The mitogenic effectors of isoproterenol in human hepatocellular carcinoma cells. Oncol Rep. 2010;23:151–157. [PubMed] [Google Scholar]
- Zhu T, Gobeil F, Vazquez-Tello A, Leduc M, Rihakova L, Bossolasco M, et al. Intracrine signaling through lipid mediators and their cognate nuclear G-protein-coupled receptors: a paradigm based on PGE2, PAF, and LPA1 receptors. Can J Physiol Pharmacol. 2006;84:377–391. doi: 10.1139/y05-147. [DOI] [PubMed] [Google Scholar]