

Abstract
BACKGROUND AND PURPOSE
Vascular endothelial growth factor receptor 2 (VEGFR2) is an attractive therapeutic target for the treatment of diseases such as cancer. Small-molecule VEGFR2 inhibitors of a variety of chemical classes are currently under development or in clinical use. In this study, we describe the de novo design of a new generation pyrazole-based molecule (JK-P3) that targets VEGFR2 kinase activity and angiogenesis.
EXPERIMENTAL APPROACH
JK-P compound series were designed using de novo structure-based identification methods. Compounds were tested in an in vitro VEGFR2 kinase assay. Using primary endothelial cells, JK-P compounds were assessed for their ability to inhibit VEGF-A-stimulated VEGFR2 activation and intracellular signalling. We tested these compounds in cell migration, proliferation and angiogenesis assays.
KEY RESULTS
JK-P3 and JK-P5 were predicted to bind the VEGFR2 kinase domain with high affinity, and both compounds showed pronounced inhibition of endogenous VEGFR2 kinase activity in primary human endothelial cells. Only JK-P3 inhibited VEGF-A-stimulated VEGFR2 activation and intracellular signalling. Interestingly, JK-P3 inhibited endothelial monolayer wound closure and angiogenesis but not endothelial cell proliferation. Both compounds inhibited fibroblast growth factor receptor kinase activity in vitro, but not basic fibroblast growth factor-mediated signalling in endothelial cells.
CONCLUSIONS AND IMPLICATIONS
This is the first report that describes an anti-angiogenic inhibitor based on such a pyrazole core. Using a de novo structure-based identification approach is an attractive method to aid such drug discovery. These results thus provide an important basis for the development of multi-tyrosine kinase inhibitors for clinical use in the near future.
Keywords: VEGFR2, tyrosine kinase inhibitor, structure-based design, endothelium, angiogenesis
Introduction
Angiogenesis is defined as the sprouting of new blood vessels from an existing vascular network (Folkman, 1971). This process is critical in normal physiological development but excessive angiogenesis is a common denominator in a wide range of pathologies, most notably cancer (Hanahan and Weinberg, 2011). During tumour growth, transformed cells secrete a cocktail of pro-angiogenic proteins including vascular endothelial growth factors (VEGFs) and fibroblast growth factors (FGFs) (Carmeliet, 2003; Carmeliet and Jain, 2011). These proteins can stimulate endothelial cell proliferation, migration and vascular remodelling, which contribute to tumour neovascularization. An enriched blood supply provides the tumour with nutrients for further growth and facilitates invasion and metastasis (Carmeliet, 2005). The VEGF gene family encodes soluble secreted cytokines such as VEGF-A, VEGF-B, VEGF-C, VEGF-D and placental growth factor (Ferrara et al., 2003; Latham et al., 2010; Koch et al., 2011). These ligands bind membrane VEGF receptor tyrosine kinases [vascular endothelial growth factor receptor (VEGFR)1-3] where VEGFR2 is a key mediator of VEGF-A-stimulated pro-angiogenic signalling in the endothelium (Olsson et al., 2006; Holmes et al., 2007; Koch et al., 2011). VEGFR2 is a member of the type III receptor tyrosine kinase subfamily comprising a large extracellular domain, a single transmembrane region and a cytoplasmic split tyrosine kinase domain (Holmes et al., 2007; Koch et al., 2011). VEGF-A binding to VEGFR2 promotes receptor dimerization, tyrosine kinase activation and trans-autophosphorylation of specific tyrosine residues within the cytoplasmic domain (Holmes et al., 2007; Koch et al., 2011). An intracellular signalling cascade is initiated, including activation of phospholipase Cγ1 (PLCγ1), c-Akt (PKB) and ERK1/2 (p42/44 MAPK) (Takahashi & Shibuya, 1997; Blanes et al., 2007) leading ultimately to increased expression of pro-angiogenic genes (Liu et al., 2003; Schweighofer et al., 2009).
The dependence of tumour growth and metastasis on a vascular network makes targeting angiogenesis an attractive strategy (Carmeliet and Jain, 2011). Small-molecule VEGFR2 inhibitors (ATP analogues) were some of the first treatments to show anti-angiogenic efficacy with clinical benefits for cancer patients (Jain et al., 2006). Compounds with different chemical core structures have been identified as potent VEGFR2 inhibitors. The anti-cancer drug sunitinib (SU11248; Sutent) belongs to the indolinone family of compounds and has been approved for treatment of renal cell carcinoma (RCC) and imatinib-resistant gastrointestinal stromal tumour (Mendel et al., 2003; Roskoski, 2007). Sorafenib (BAY 43–9006; Nexavar) is a bis-aryl urea, which has been approved for treatment of hepatocellular carcinoma and advanced RCC (Bracarda et al., 2007; Wilhelm et al., 2008). Additional classes of anti-angiogenic agents include anilinophthalazines (e.g. vatalanib/PTK787/ZK 222548), anilinoquinazolines (e.g. ZD6474), isothiazoles (e.g. CP-547632) and pyrimidinyl-indazoles (e.g. pazopanib/Votrient) (Harris et al., 2005; 2008). Despite this variety, certain structural features of inhibitor binding to the VEGFR2 kinase domain are conserved: residues E917 and C919 within the VEGFR2 cytoplasmic domain have been identified as important in inhibitor binding via hydrogen donor and acceptor bonds (Harris et al., 2005; Miyazaki et al., 2005; Latham et al., 2012). Many inhibitors also make contact with an Asp-Phe-Gly (DFG) motif in the kinase domain activation loop (Zhou et al., 2010; Latham et al., 2012).
In the present study, we used structure-based de novo design methods complementary to high-throughput screening to rapidly identify a novel VEGFR2 inhibitor of the pyrazole class of molecules. We further examined the mechanism of action of this compound by analysis of VEGF-A-stimulated VEGFR2 tyrosine kinase activity, intracellular signalling and angiogenesis.
Methods
Reagents
Human umbilical vein endothelial cells (HUVECs) were retrieved from human tissues obtained by local ethical approval from the Leeds Hospitals NHS Trust and cultured as previously described (Howell et al., 2004). Primary human foreskin fibroblasts (pHFF) were a gift from Dr A. Bruns, (University of Leeds, UK). Recombinant human VEGF-A165 was a gift from Genentech Inc. (San Francisco, CA, USA). Recombinant basic fibroblast growth factor (bFGF), EGF and antibody against VEGFR2 extracellular domain were purchased from R&D Systems (Minneapolis, MN, USA). Recombinant insulin-like growth factor 1 (IGF-1) was a gift from Dr Hema Viswambharan (University of Leeds, UK). Phospho-VEGFR2 Y1175, phospho-ERK1/2, phospho-PLCγ1, phospho-Akt, ERK1/2 and Akt antibodies were purchased from Cell Signalling Technology (Danvers, MA, USA). PLCγ1 and patelet-endothelial cell adhesion molecule 1 (PECAM-1) antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA) and horseradish peroxidase (HRP)-conjugated secondary antibodies were from PerBio Sciences (Cramlington, UK). Compound 1, Compound 2, 3,4-dimethoxy-N-(5-phenyl-1H-pyrazol-3-yl)benzamide (JK-P3) and N-[4-(4-methylpiperazin-1-yl)phenyl]-1H-indazole-3-carboxamide (JK-P5) were designed, synthesized and prepared as 10 mM stock solutions in dimethyl sulphoxide (DMSO).
Serial 10-fold dilutions were made in tissue culture medium. All other reagents were obtained from Sigma-Aldrich (Poole, UK) unless otherwise stated.
Structure-based in silico design of VEGFR2 inhibitors
A range of pyrazole-based compounds Compound 1, Compound 2, JK-P3 and JK-P5 were designed using de novo structure-based software, namely SPROUT (SPROUT Software Package, SimBioSys Inc., Toronto, Canada, 2005) (Ali et al., 2005; Boda and Johnson, 2006) and an available crystal structure of VEGFR2 kinase domain [Protein DataBank (PDB) code: 3CJG] (Harris et al., 2008). Using information derived from X-ray crystal structures of receptor tyrosine kinases (e.g. hydrogen bonding capacity of the surrounding amino acid residues present in the active site and presence of hydrophobic regions), SPROUT identified a target region where putative ligands would interact strongly. Molecular fragments were docked at these sites and connected to generate skeletons that satisfied the steric and geometric constraints. Atoms in the skeletons were then substituted to produce molecules that have the required electrostatic and hydrophobic properties. The ligands were ranked and scored to give an estimated pKi. The programme Glide (Schrödinger LLC, New York, NY, USA) (Friesner et al., 2004; 2006; Halgren et al., 2004) was also used to dock these ligands into receptor tyrosine kinase crystal structures and predict compound binding affinity (presented as a Glide score where a lower score represents lower energy and thus greater affinity). The Glide programme searches the positional, orientational and conformational space available to the ligand using a series of hierarchical filters. The programme semi-quantitatively ranks the ability of a ligand to bind to a specified conformation of the protein receptor. The Glide score represents a combined energy of the interaction including energy from charged-charged hydrogen bond motifs and rewards for pi-stacking and pi-cation interactions. Images from Glide software are used in this publication (Figures 2, Supporting Information Figures S1A, S1B, S2A and S2B). The binding mode of compounds within the VEGFR2 kinase domain (with respect to hydrogen bonding) was confirmed to be similar to that of a derivative of pazopanib (N4-methyl-N4-(3-methyl-1H-indazol-6-yl)-N2-[3,4,5-tris(methyoxy)phenyl]-2,4 pyrimidinediamine), the original ligand co-crystallized with VEGFR2 3CJG (Supporting Information Figure S1B and C) (Harris et al., 2008). A full description of the structure–activity relationship of these compounds is currently ongoing.
Figure 2.
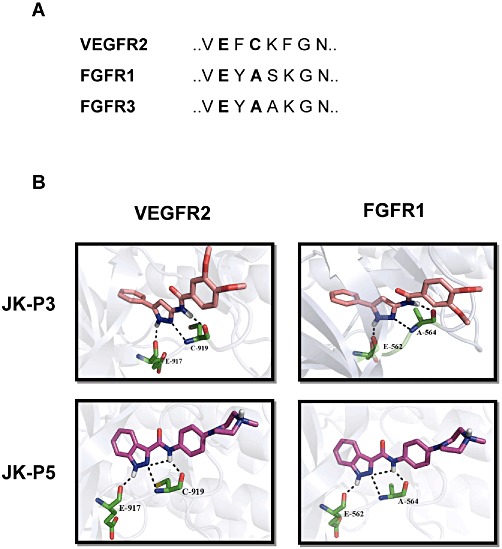
Predicted binding mode of JK-P3 and JK-P5 within the VEGFR2 and FGFR1 tyrosine kinase domains. (A) Amino acid sequence homology within the hinge region of VEGFR2, FGFR1 and FGFR3 kinase domains. Key residues important for JK-P compound binding are highlighted in bold text. (B) JK-P3 and JK-P5 were docked into VEGFR2 and FGFR1 tyrosine kinase domain using Glide programme to predict binding pose and hydrogen bond contacts formed (see Methods section). Pink carbon backbone denotes JK-P3; magenta carbon backbone denotes JK-P5; green carbon backbones denote key amino acid residues in tyrosine kinase domain; black dotted lines denote hydrogen bonds. White, hydrogen; blue, nitrogen; red, oxygen; yellow, sulphur.
33P receptor tyrosine kinase HotSpotSM profiling assay
Full-length recombinant VEGFR2, FGFR1 or FGFR3 were incubated with 10 µM (radio-labelled) [γ33P]-ATP and MgCl2 together with threefold serial dilutions of inhibitors starting at 10, 50 and 100 µM. Inhibition of kinase activity was assessed by measuring the relative reduction of the γ33P signal produced by autophosphorylation events on recombinant receptor (Reaction Biology, Malvern, PA, USA). Ki values were calculated using the Cheng–Prusoff relationship as described by the equation Ki= IC50/(1 + ([ATP]/Km,app)) (Cheng and Prusoff, 1973).
SDS-PAGE and immunoblotting
HUVECs were deprived of serum in MCDB-131 medium (Invitrogen, Amsterdam, Netherlands) containing 0.2% (w/v) BSA for 3 h and pretreated with inhibitors for 1 h before stimulation with 25 ng·mL−1 (0.54 nM) VEGF-A for 7.5 min in the presence of inhibitors, 50 ng·mL−1 (3.1 nM) bFGF, 50 ng·mL−1 (7.8 nM) EGF or 100 ng·mL−1 (13.3 nM) IGF-1 for 10 min in the presence of inhibitors. Cells were lysed in 2% (w/v) SDS in PBS and lysates were boiled and sonicated briefly before the protein content was quantified using a bicinchoninic acid assay. Samples were resuspended in Laemmli buffer and boiled for 5 min before electrophoresis on a denaturing 10% polyacrylamide gel. Proteins were transferred to nitrocellulose membranes and probed with appropriate antibodies. Immunoreactive proteins were visualized by enhanced chemiluminescence using a Fuji LAS-3000 imaging system (Raytek Scientific, Sheffield, UK). Band intensity was quantified by 2D densitometry using AIDA software (Fujifilm, Fuji, Japan).
Scratch wound healing assay
Confluent HUVECs were deprived of serum for 3 h and pretreated with chemical inhibitors for 1 h before a cross-shaped scratch wound was made through the cell monolayer with a 1 mL plastic pipette of 0.9 mm tip width. Wounded cell monolayers were washed with PBS, photographed and stimulated with 25 ng·mL−1 VEGF-A in the presence of inhibitors during a 24 h recovery period, and analysis of wound closure was monitored using digital microscopy. Wound closure was calculated using NIH Image J software and represented as % by ((width before – width after)/width before) × 100.
MTS cell proliferation assay
HUVECs were seeded at 2000 cells per well in 96-well plates, treated with inhibitors for 16 h and incubated with 20 µL CellTiter 96® AQueous One Solution Reagent (Promega, Southampton, UK) for 4 h until sufficient colour change had been reached. The active compound in the reagent 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) is reduced by cellular succinate dehydrogenase to produce a formazan product. Absorbance of formazan at 490 nm was measured.
Fibroblast co-culture assay
pHFF were grown to confluence in a 48-well plate in Dulbecco's modified Eagles medium (DMEM) and then 7 500 HUVECs seeded as a secondary layer in a two-cell co-culture model. After 24 h, the co-culture was incubated in endothelial growth medium supplemented with 25 ng·mL−1 VEGF-A and either DMSO or an appropriate drug for 7 days. The co-cultures were fixed and stained for the endothelial-specific marker PECAM-1 and further with anti-mouse HRP. Tubes were visualized under a light microscope using nickel-enhanced 1,1,diaminobenzidine/urea/hydrogen peroxide development. For immunofluorescence analysis, co-cultures were made on coverslips, and PECAM-1 staining was visualized using AlexaFluor594-conjugated anti-mouse secondary antibody (Invitrogen) followed by examination with a Deltavision wide-field deconvolution microscope equipped with a 60× objective (Applied Precision Inc., Issaguah, WA, USA).
Statistical analysis
Data were analysed using ANOVA with Tukey's post-hoc test using GraphPad Prism software (La Jolla, CA, USA). Significant difference denoted by *P < 0.05, **P < 0.01 or ***P < 0.001.
Results
JK-P compounds are predicted to bind in the ATP binding pocket of VEGFR2 and FGFRs with high affinity
As part of an ongoing research programme to identify novel inhibitors of the VEGFR2 tyrosine kinase, de novo design methods, for example SPROUT and Glide (Boda and Johnson, 2006; Agarwal and Fishwick, 2010), were applied to an available crystal structure of the VEGFR2 cytoplasmic tyrosine kinase domain (PDB code: 3CJG) (Harris et al., 2008). Using these structure-based methods, a series of compounds with a pyrazole core were identified as potential VEGFR2 inhibitors (Figure 1). In an initial in silico screen using the Glide programme, Compound 1 was docked into the VEGFR2 tyrosine kinase domain and was predicted to make two hydrogen bond contacts with the protein (data not shown). Optimization by further molecular modelling led to the identification of its reverse amide, Compound 2 (Figure 1) which had greater predicted binding affinity than Compound 1 and made one extra hydrogen bond contact (data not shown). Refinement of Compound 2 through further iterations of design and synthesis led to the identification of JK-P3 and its benzo-fused indazole derivative, JK-P5 (Figure 1). Both JK-P compounds had improved predicted binding to VEGFR2 with respect to their predecessor molecules. For these compounds, an estimated pKi was calculated using the SPROUT programme (Table 1). Since the tyrosine kinase domain hinge regions of VEGFR2 and related receptors FGFR1 and FGFR3 are highly conserved (Figure 2A), we wished to compare the predicted binding affinity of JK-P3 and JK-P5 to both receptor families (Table 1, Figure 2B). JK-P3 and JK-P5 each made three predicted hydrogen bond contacts in the VEGFR2 ATP-binding pocket hinge region: with the backbone carbonyl of E917, and both the backbone carbonyl and the backbone amino group of C919 (Figure 2B, left panels). The two compounds were predicted to bind the homologous residues in FGFR1 (E562 and A564, respectively) (Figure 2B, right panels) and the same residues in FGFR3 (data not shown). Both compounds exhibited comparatively greater binding affinity to VEGFR2 and FGFR3 (predicted pKi of −8 or less), compared to FGFR1 (predicted pKi of −7) (Table 1). In comparison to JK-P3, JK-P5 was predicted to bind with greater affinity to all three kinases (Table 1), which may be due to an extra predicted intramolecular hydrogen bond contact within this compound (Figure 2). In silico docking studies predicted the orientation and binding mode of JK-P3 and JK-P5 to be similar to that of a pazopanib derivative (the original co-crystallized ligand) (Supporting Information Figure S1). Key hydrogen bond donor and acceptor atoms on the compounds were overlapping (Supporting Information Figure S1B).
Figure 1.
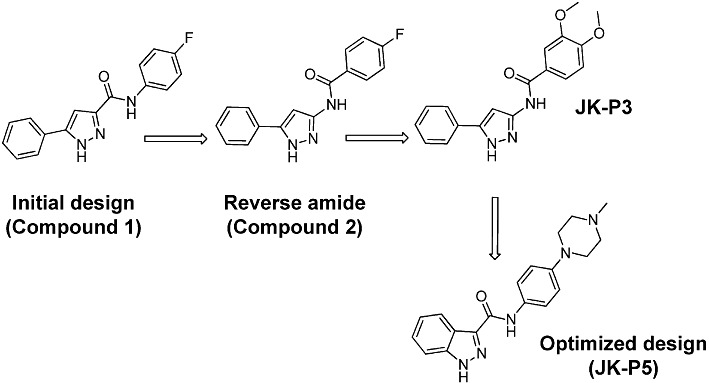
Structure and optimization of JK-P3 and JK-P5 compounds. Key steps in the optimization of JK-P3 pyrazole and JK-P5 indazole, including the predecessor pyrazoles Compound 1 and Compound 2.
Table 1.
Comparison of estimated binding affinities (pKi values) of JK-P compounds to VEGFR2, FGFR1 and FGFR3 kinase domains in silico, as predicted using SPROUT programme (see Methods section)
| Estimated pKi | |||
|---|---|---|---|
| Compound | VEGFR2 | FGFR1 | FGFR3 |
| JK-P3 | −8.44 | −6.94 | −8.71 |
| JK-P5 | −9.16 | −7.29 | −9.01 |
JK-P compounds inhibit the intrinsic tyrosine kinase activity of VEGFR2 and FGFRs
To test the effects of JK-P3 and JK-P5 on the intrinsic tyrosine kinase activity of VEGFR2, FGFR1 and FGFR3, we used an in vitro kinase assay. Both compounds showed dose-dependent inhibition of tyrosine kinase activity using purified recombinant VEGFR2, FGFR1 and FGFR3 (Figure 3), although JK-P3 was clearly a much less potent inhibitor of FGFR1 (Figure 3A). JK-P3 and JK-P5 showed similar inhibitory profiles for VEGFR2, which were not significantly different (Ki∼ 5.4 µM; P < 0.01) (Table 2). Both compounds began to inhibit VEGFR2 kinase activity at a concentration of ∼50 nM (Figure 3). The results of the kinase assay were mostly in keeping with our prediction derived from modelling: JK-P5 was the more potent inhibitor and both compounds in general inhibited FGFR3 > VEGFR2 > FGFR1. However, JK-P5 exhibited comparatively less potent inhibition of VEGFR2 than previously predicted (Tables 1 and 2). It is useful to compare the potency of these compounds with known VEGFR2 inhibitors using this in vitro assay. Both JK-P3 and JK-P5 were more potent than SU5416 in terms of Ki value (JK-P3, 5.65 µM; JK-P5, 5.26 µM; SU5416, 6.41 µM), but were approximately one order of magnitude less potent than sunitinib and PTK787 (sunitinib, 0.42 µM; PTK787, 0.23 µM).
Figure 3.
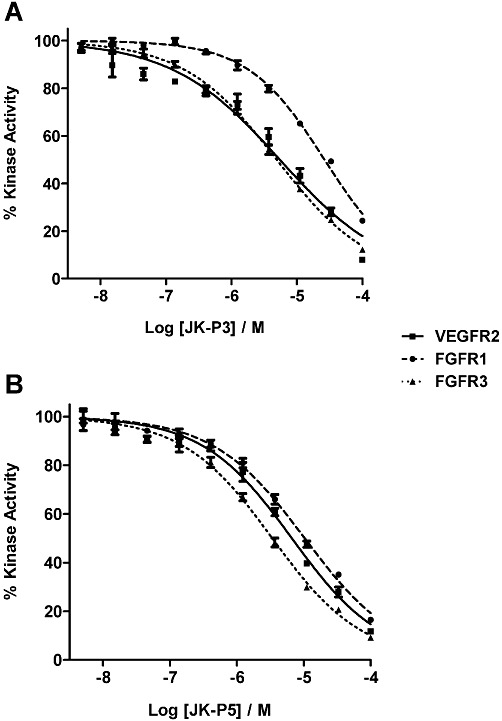
Inhibition of VEGFR2, FGFR1 and FGFR3 tyrosine kinase activity by JK-P compounds. (A) Inhibition of kinase activity by JK-P3 and (B) inhibition of kinase activity by JK-P5. IC50 curves were generated by incubating recombinant receptor with (radio-labelled) [γ33P]-ATP and a concentration range of each inhibitor (5 nM to 100 µM). Inhibition was assessed by measuring relative reduction of the γ33P signal. Data are presented as mean±SEM (n= 3); IC50 values were derived from the curves shown and Ki values using the Cheng–Prusoff equation (see Methods section).
Table 2.
Inhibition of VEGFR2, FGFR1 and FGFR3 tyrosine kinase activity by JK-P compounds
| IC50 value (µM) | Ki value (µM) | |||||
|---|---|---|---|---|---|---|
| Compound | VEGFR2 | FGFR1 | FGFR3 | VEGFR2 | FGFR1 | FGFR3 |
| JK-P3 | 7.83 ± 0.66 | 27.0 ± 0.18 | 5.18 ± 0.30 | 5.65 ± 0.48 | 7.71 ± 0.07 | 3.97 ± 0.23 |
| JK-P5 | 7.28 ± 0.19 | 11.4 ± 0.71 | 3.21 ± 0.15 | 5.26 ± 0.13 | 3.25 ± 0.20 | 2.46 ± 0.11 |
JK-P3 and JK-P5 differentially inhibited the intrinsic tyrosine kinase activity of all three receptors. IC50 curves and Ki values were generated using an in vitro kinase assay (see Methods section).
JK-P3 inhibits VEGF-A-mediated VEGFR2 phosphorylation and downstream signalling, but does not inhibit signalling by other growth factors
The VEGF-A-stimulated VEGFR2 intracellular signalling pathway involves phosphorylation of serine, threonine and tyrosine residues on effector proteins (Bruns et al., 2010; Latham et al., 2012). These include the generation of PLCγ1-pY783, Akt-pS473 and the dual phosphorylated ERK1/2-pT202/pY204. Phosphorylation of these proteins stimulates enzymatic activity and influences endothelial cell migration, proliferation and survival (Koch et al., 2011). To assess the inhibitory efficacy of the two most promising compounds on these pro-angiogenic signalling events, JK-P3 and JK-P5 were assayed by immunoblotting on VEGF-A-stimulated cells (Figure 4). At 10 µM, JK-P3 almost completely inhibited VEGFR2 Y1175 phosphorylation, a key hallmark of VEGFR2 activation that stimulates pro-angiogenic responses by endothelial cells (Figure 4A and B). JK-P3 also inhibited VEGF-A-stimulated PLCγ1, Akt and ERK1/2 phosphorylation (Figure 4A). In contrast, JK-P5 failed to inhibit VEGFR2 phosphorylation in response to a VEGF-A pulse (Figure 4C and D). While JK-P5 also failed to inhibit PLCγ1 phosphorylation, there was partial inhibition of Akt and ERK1/2 phosphorylation (Figure 4C). One possibility is that this phenomenon is due to off-target effects of the JK-P5 compound. Thus, JK-P3 is the more potent inhibitor of VEGF-A-stimulated intracellular signalling in endothelial cells. Since these compounds also inhibit FGFR kinase activity (Figure 3), we tested the ability of JK-P3 and JK-P5 to inhibit intracellular signalling in response to a bFGF pulse. Neither compound inhibited bFGF-stimulated ERK1/2 phosphorylation at concentrations up to 10 µM (Supporting Information Figure S2A). In addition, both compounds failed to inhibit EGF-stimulated Akt and ERK1/2 phosphorylation and IGF-1-stimulated Akt phosphorylation at the same concentration range (Supporting Information Figure S2B and C).
Figure 4.
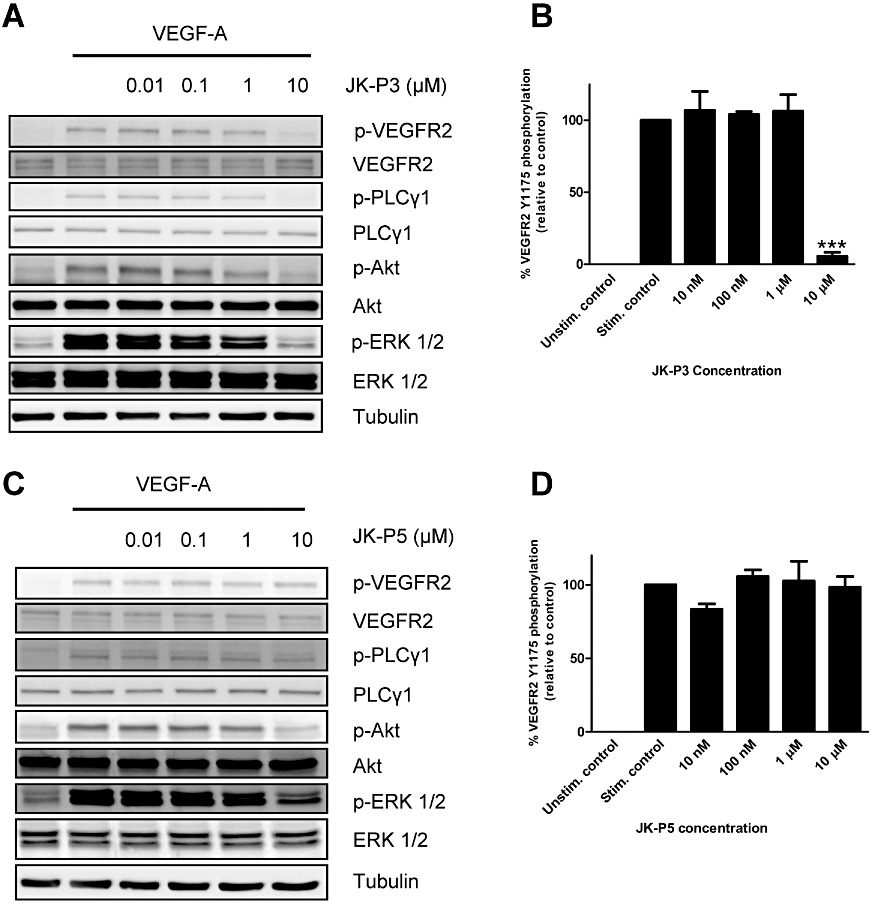
Inhibition of VEGFR2 phosphorylation and intracellular signalling in response to VEGF-A in primary endothelial cells. Cells were treated for 7.5 min with VEGF-A (25 ng·mL−1) in the presence of (A) JK-P3 or (C) JK-P5 at between 10 nM and 10 µM concentration and processed for immunoblotting using anti-phospho VEGFR2 (Y1175), anti-VEGFR2, anti-phospho-PLCγ1 (Y783), anti-PLCγ1, anti-phospho-Akt (S473), anti-Akt, anti-phospho-ERK1/2 (T202/Y204), anti-ERK1/2 and anti-alpha-tubulin antibodies. (B) Quantification of VEGFR2 inhibition by JK-P3. (D) Quantification of VEGFR2 inhibition by JK-P5. Data are presented as mean ± SEM (n= 3). Significant inhibition of VEGFR2 phosphorylation denoted by ***P < 0.001 using anova.
Effects of JK-P compounds on VEGF-A-stimulated endothelial wound healing and cell proliferation
Endothelial cell migration and proliferation are important steps in angiogenesis and key functional outputs of VEGF-A-stimulated intracellular signalling (Bruns et al., 2010; Koch et al., 2011). A simple in vitro model that reproduces early events during angiogenesis is a cell monolayer scratch wound assay. A denuded region was created in a confluent endothelial monolayer, and the migration of cells into the wounded region was monitored over 24 h in the presence of DMSO (control), JK-P3 or JK-P5 (Figure 5). In the presence of exogenous VEGF-A alone, average endothelial wound closure was ∼42% (Figure 5A and B). JK-P3 failed to inhibit VEGF-A-stimulated wound closure at 1 µM, but at 10 µM wound closure was inhibited by ∼90% (Figure 5A). JK-P5 did not significantly inhibit endothelial wound closure at either 1 or 10 µM (Figure 5B). To further test the effects of JK-P3 on endothelial cell proliferation, we used the MTS assay. This assay measures metabolic enzyme activity and is thus a measure of cell viability; however, the absorbance readout correlates directly with cell number (Richardson et al., 1995). Intriguingly, JK-P3 failed to inhibit endothelial cell proliferation at a range of concentrations but paradoxically elicited a small but significant increase in cell proliferation at certain lower concentrations (Figure 5C). JK-P5 also did not inhibit cell proliferation (Figure 5D). These data were confirmed using an alternative cell proliferation assay (bromo-deoxyuridine incorporation), which showed a similar trend (data not shown).
Figure 5.
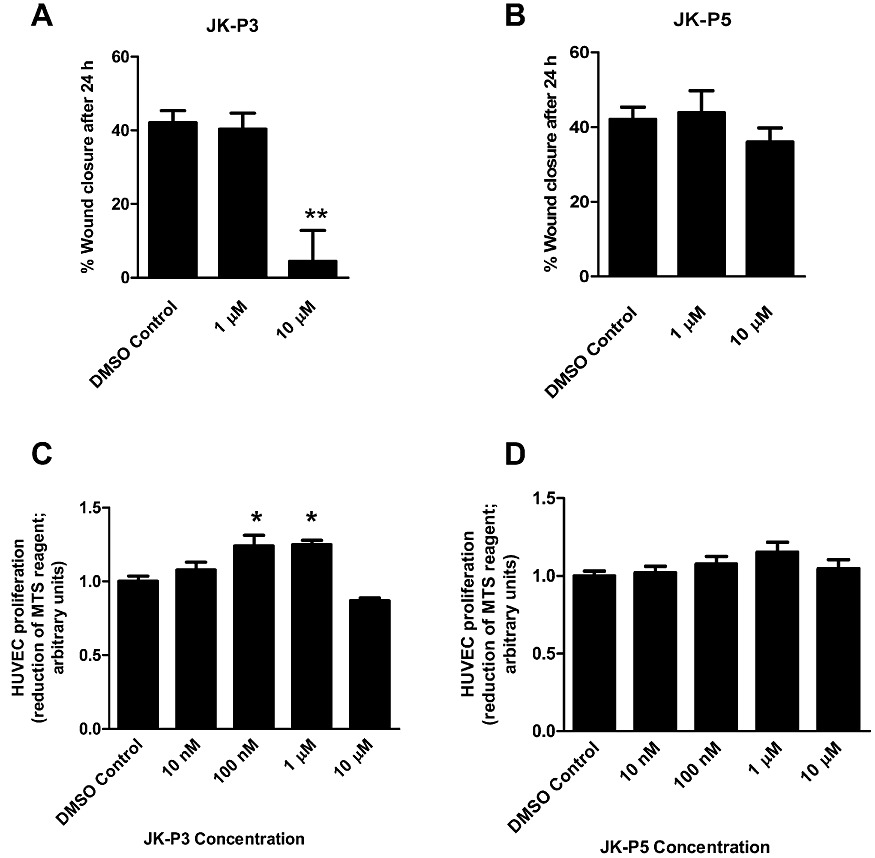
JK-P3 inhibits VEGF-A mediated endothelial wound healing but not cell proliferation. Scratch-wounded cell monolayers were stimulated with VEGF-A (25 ng·mL−1) in serum-free conditions in the presence of 1 µM or 10 µM of (A) JK-P3 or (B) JK-P5. An MTS assay was performed on HUVECs treated with a range of inhibitor concentrations in full growth medium. Effects of (C) JK-P3 and (D) JK-P5 on HUVEC proliferation were measured. Reduction of MTS reagent to formazan product was assessed by measuring absorbance at 490 nm. Data are presented as mean ± SEM (n≥ 5). Significant difference denoted by *P < 0.05 or **P < 0.01 using anova.
JK-P3 inhibits in vitro angiogenesis
During blood vessel sprouting, lumen formation is dependent on the ability of endothelial cells to form into three-dimensional tubular structures (Carmeliet, 2005). In an in vitro model of angiogenesis, endothelial cells incubated in the presence of growth factors and secreted proteins from fibroblasts can elongate and branch to form hollow tubes (Newman et al., 2011). These cellular structures can be examined at low resolution using light microscopy by measuring both the tube length and the number of tubular branch points (Figure 6A). Alternatively, high-resolution microscopy can be used to examine individual cellular phenotypes including intracellular protein localization (Figure 6B). This assay was thus used to examine the effects of novel small-molecule inhibitors on endothelial cell physiology (Figure 6A and B). VEGF-A is a potent stimulus of directional tubule formation and branching and promotes extensive lamellipodial and filopodial projection (Figure 6A and B, first panel). In contrast, bFGF is a less potent stimulus of tubulogenesis in this assay, eliciting ∼50% tubule formation and branching in comparison to VEGF-A (Figure 6C and D); tubules appear narrower and less organized, but many filopodia are still evident (Figure 6A and B, fourth panel). Consistent with our data from the scratch wound assay, JK-P3 failed to significantly inhibit VEGF-A-stimulated endothelial tube formation at 1 µM (Figure 6). However, at 10 µM, JK-P3 almost completely inhibited the ability of endothelial cells to form into elongated hollow tubes in the presence of VEGF-A (Figure 6C) with no evidence of branching (Figure 6D). It is important to note that during treatment with JK-P3 at 10 µM, endothelial cells remain viable in small islands without lamellipodia or filopodia (Figure 6A and B, third and sixth panels). Surprisingly, at a relatively low 1 µM concentration, JK-P3 inhibited bFGF-stimulated tube formation by ∼70% (Figure 6C and D) with evidence of only vestigial lamellipodia (Figure 6B, fifth panel).
Figure 6.
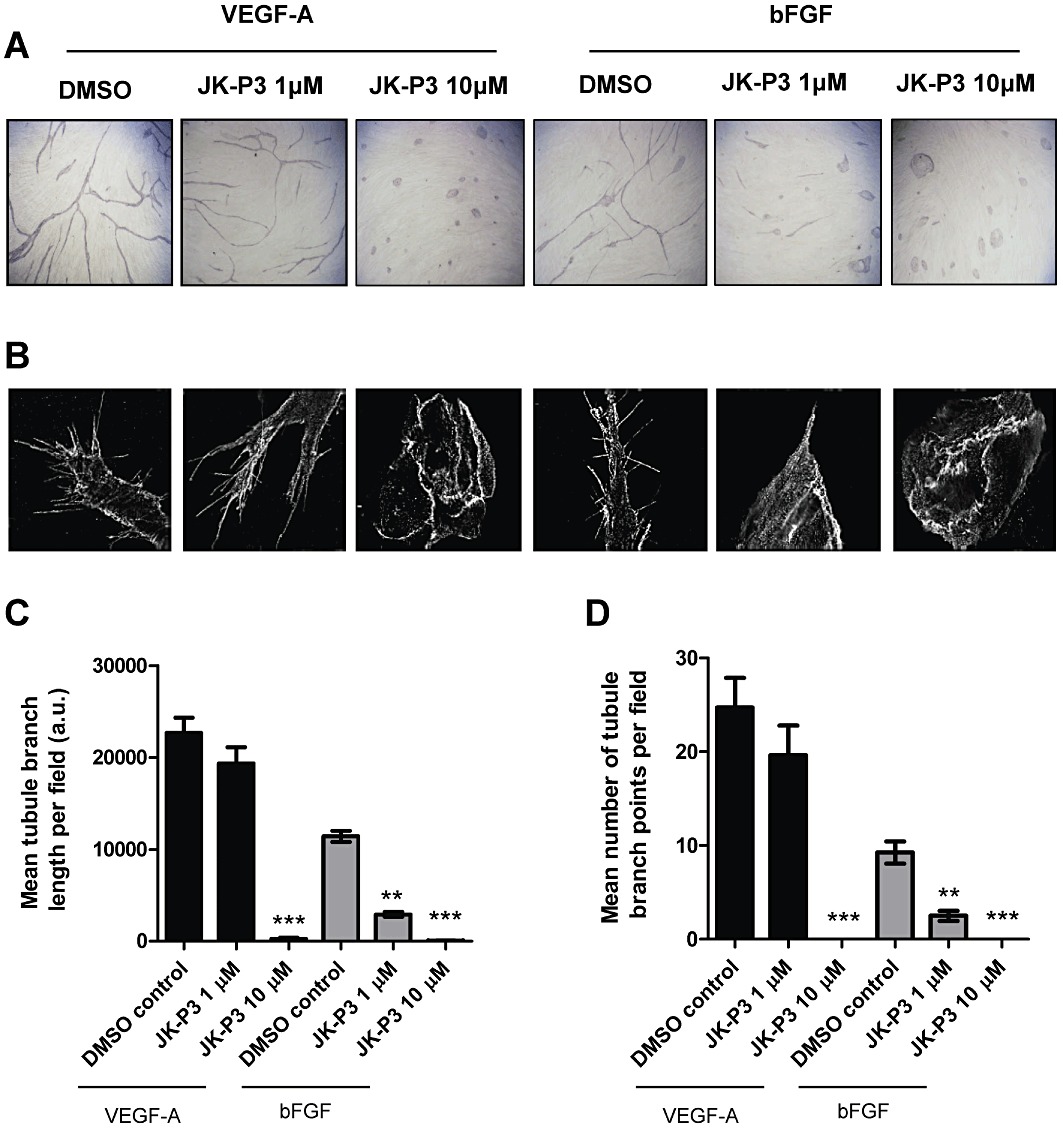
JK-P3 inhibits endothelial tube formation in response to VEGF-A or bFGF. (A) Endothelial tubulogenesis stimulated by the presence of VEGF-A or bFGF in an endothelial-fibroblast co-culture model was evaluated for the effects of JK-P3 over a 7-day period. (B) High-resolution fluorescent images depicting endothelial cell phenotypes and filopodia formation during tubulogenesis in the presence of JK-P3. (C) Quantification of endothelial tube length and (D) quantification of endothelial tube branch point formation. Quantification performed using Image J software. Data are presented as mean± SEM (n≥ 10). Significant difference denoted by **P < 0.01 or ***P < 0.001 using anova.
Discussion
VEGFR2 is an important therapeutic target in the treatment of diseases characterized by excessive angiogenesis, such as cancer (Jain et al., 2006; Carmeliet and Jain, 2011). VEGFR2 inhibitors currently in clinical use include sunitinib, a promiscuous drug that also inhibits the platelet-derived growth factor receptor (PDGFR), c-Kit and Flt-3 kinases (Mendel et al., 2003; Hasinoff and Patel, 2010) and sorafenib, a multi-kinase inhibitor of VEGFRs, PDGFR, Raf and c-Kit (Wilhelm et al., 2008). The advent of structure-based lead optimization has revolutionized the discovery of such drugs (Noble et al., 2004). In the present study, we describe the de novo structure-based identification, design and mechanism of action of a VEGFR2 kinase inhibitor of a novel chemical class, JK-P3.
During de novo design, JK-P3 was predicted to target the VEGFR2, FGFR1 and FGFR3 kinase domains and bind with high affinity. JK-P3 makes hydrogen bond contacts with E917 and C919 of VEGFR2, the same residues as predicted for binding of indolinones such as SU5416 and sunitinib (Latham et al., 2012). Additional residues reported to be involved in inhibitor binding to VEGFR2 include D1046 for anilinophthalazines and E883, N923 and K868 for pyrimidine analogues (Miyazaki et al., 2005; Harris et al., 2008; Latham et al., 2012). In an in vitro kinase assay, JK-P3 inhibited the intrinsic catalytic activity of the VEGFR2, FGFR1 and FGFR3 tyrosine kinases. Particularly noteworthy is that JK-P3 exhibits comparatively greater inhibition of VEGFR2 than FGFR1, a property observed only of more selective VEGFR inhibitors, for example PTK787 (Latham et al., 2012). In primary endothelial cells, JK-P3 inhibited VEGFR2 phosphorylation, activation and downstream signalling in response to VEGF-A treatment, but did not inhibit intracellular signalling in response to other growth factors bFGF and EGF and also IGF-1, a growth factor shown to be important for vascular homeostasis (Imrie et al., 2009). In addition, JK-P3 decreased VEGF-A-stimulated endothelial cell migration in the wound healing assay, a recapitulation of an important early step in angiogenesis. However, JK-P3 failed to inhibit endothelial cell proliferation at an equivalent concentration. This finding is in agreement with other studies where structurally unrelated compounds that target VEGFR2 tyrosine kinase activity have little or no inhibitory effect on cell growth at relatively high micromolar concentrations (Tille et al., 2001; Latham et al., 2012). Finally, we showed that JK-P3 inhibits angiogenesis in vitro in an endothelial tubulogenesis assay in co-culture with fibroblasts. Despite the lack of inhibition of bFGF-mediated signalling by JK-P3, this compound attenuated both VEGF-A and bFGF-stimulated tubulogenesis. While this was unexpected, we must not overlook the contribution of full growth medium (which already contains growth factors) to endothelial tube formation in co-culture. Since endothelial wound closure was inhibited but cell proliferation was unaffected by JK-P3, we propose that the major anti-angiogenic activity of this compound is exerted through inhibition of cell migration. A related compound, JK-P5 was also predicted to target VEGFR2 and FGFRs in a molecular model and showed enhanced inhibitory activity against these receptors in vitro compared to JK-P3. However, JK-P5 failed to inhibit VEGFR2 activation, growth factor-stimulated signalling and VEGF-A-stimulated wound closure in endothelial cells. One possibility is that JK-P5 has structural constraints that prevent it from crossing the plasma membrane bilayer. However, the observed inhibition of Akt and ERK1/2 signalling pathways by JK-P5 in this study would argue otherwise. Recent work by our group has further highlighted the importance of such thorough molecular, biochemical and cell biological characterization of lead compounds with potential therapeutic efficacy (Latham et al., 2012) before conducting animal and clinical studies.
JK-P3 is a potent inhibitor of VEGFR2 activity with a steep curve of inhibition at concentrations between 1 and 10 µM. While this range is comparable to recently identified lead compounds (Zhang et al., 2011), the IC50 values of many clinical and preclinical VEGFR2 inhibitors lie in the nanomolar range (Latham et al., 2012). This leaves considerable scope for optimizing the pyrazole/indazole scaffold as VEGFR2 inhibitors. Indeed, a series of similar indenopyrazoles and related compounds have been identified as inhibitors of VEGFR2 and other protein kinases (Krystof et al., 2006; Dinges et al., 2007; Usui et al., 2008). Inhibitor selectivity against a panel of kinases can be achieved by exploring the size of the gatekeeper residue; for example, in cyclin-dependent kinase 2, a bulky phenylalanine contributes to significant steric hindrance, whereas in VEGFR2, this is replaced with a less hydrophobic valine at the equivalent position (Krystof et al., 2006; Zuccotto et al., 2010).
In addition to VEGFR2, JK-P compounds also show inhibitory activity on FGFR kinases, a related receptor tyrosine kinase subfamily with high sequence homology. The target specificity of tyrosine kinase inhibitors is a key determinant of clinical efficacy (Noble et al., 2004). To date, the most successful VEGF-related therapies have been multi-targeted small-molecule inhibitors such as sorafenib and sunitinib, which have provided the greatest improvement in progression-free survival in cancer patients (Jain et al., 2006). However, off-target inhibition by these drugs is postulated to be the major cause of side effects including cardiotoxicity (Hasinoff and Patel, 2010; Zhang et al., 2011). Thus, maintaining the correct balance of inhibition of a small but select panel of receptors, for example VEGFRs and FGFRs, appears clinically important. Indeed so-called ‘selectively non-selective’ drugs are deemed useful to overcome signalling pathway redundancies (Morphy, 2010). Several dual FGFR-VEGFR tyrosine kinase inhibitors are currently under development, for example, brivanib (Bristol-Myers Squibb, Uxbridge, UK), TKI-258 (Novartis, Basel, Switzerland), Vargatef (Boehringer Ingleheim, Bracknell, UK) and RO438596 (Roche, Basel, Switzerland) (Knights and Cook, 2010).
In summary, the present study not only identifies a novel VEGFR2 inhibitor scaffold but also uses pioneering structure-based design technology to rapidly identify potent small-molecule inhibitors. JK-P3 is a potent inhibitor of the VEGFR2 tyrosine kinase and VEGF-A-stimulated angiogenesis with some inhibitory effect on FGFR kinases in vitro. These results thus provide an important basis for the development of multi-tyrosine kinase inhibitors for clinical use in the near future.
Acknowledgments
This work was supported by a BBSRC-CASE PhD studentship from Pfizer Global Inc. (AML), an ORSAS award (JK) and a Wellcome Trust project grant (SP). We thank Mike Harrison (University of Leeds) and members of the Endothelial Cell Biology Unit including Alex Bruns, Leyuan Bao, Gareth Howell and Adam Odell for their help and advice.
Glossary
- BAY 43-9006 (drug name: sorafenib, trade name: Nexavar)
4-[4-[[4-chloro-3 (trifluoromethyl)phenyl]carbamoylamino]phenoxy]-N-methyl-pyridine-2-carboxamide
- bFGF
basic fibroblast growth factor
- FGFR
fibroblast growth factor receptor
- HUVEC
human umbilical vein endothelial cells
- IGF-1
insulin-like growth factor 1
- pHFF
primary human foreskin fibroblasts
- RCC
renal cell carcinoma
- VEGFR2
vascular endothelial growth factor receptor 2
- SU-11248 (drug name: sunitinib, trade name: Sutent)
N-(2-diethylaminoethyl)-5-[(Z)-(5-fluoro-2-oxo-1H-indol-3-ylidene)methyl]-2,4-dimethyl-1H-pyrrole-3-carboxamide
Conflict of interest
The authors state no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Confirmation of JK-P3 and JK-P5 binding mode within the VEGFR2 tyrosine kinase domain. (A) Docking overlay of JK-P3 and JK-P5 and (B) docking overlay of JK-P3 and co-crystallized pazopanib derivative in VEGFR2 tyrosine kinase domain. (C) Chemical structure of pazopanib derivative. Pink carbon backbone denotes JK-P3; magenta carbon backbone denotes JK-P5; yellow carbon backbone denotes pazopanib derivative. White, hydrogen; blue, nitrogen; red, oxygen.
Figure S2 JK-P3 and JK-P5 do not inhibit bFGF-, EGF- or IGF-1-mediated signalling in endothelial cells at concentrations up to 10 µM. Cells were treated for 10 min with (A) bFGF (50 ng·mL−1) (B) EGF (50 ng·mL−1) or (C) IGF-1 (100 ng·mL−1) in the presence of JK-P3 or JK-P5 at between 10 nM and 10 µM concentration and processed for immunoblotting using anti-phospho-Akt (S473), anti-Akt, anti-phospho-ERK1/2 (T202/Y204), anti-ERK1/2 and anti-alpha-tubulin antibodies.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Agarwal AK, Fishwick CW. Structure-based design of anti-infectives. Ann NY Acad Sci. 2010;1213:20–45. doi: 10.1111/j.1749-6632.2010.05859.x. [DOI] [PubMed] [Google Scholar]
- Ali MA, Bhogal N, Findlay JB, Fishwick CW. The first de novo-designed antagonists of the human NK(2) receptor. J Med Chem. 2005;48:5655–5658. doi: 10.1021/jm050533o. [DOI] [PubMed] [Google Scholar]
- Blanes MG, Oubaha M, Rautureau Y, Gratton JP. Phosphorylation of tyrosine 801 of vascular endothelial growth factor receptor-2 is necessary for Akt-dependent endothelial nitric-oxide synthase activation and nitric oxide release from endothelial cells. J Biol Chem. 2007;282:10660–10669. doi: 10.1074/jbc.M609048200. [DOI] [PubMed] [Google Scholar]
- Boda K, Johnson AP. Molecular complexity analysis of de novo designed ligands. J Med Chem. 2006;49:5869–5879. doi: 10.1021/jm050054p. [DOI] [PubMed] [Google Scholar]
- Bracarda S, Caserta C, Sordini L, Rossi M, Hamzay A, Crino L. Protein kinase inhibitors in the treatment of renal cell carcinoma: sorafenib. Ann Oncol. 2007;18(Suppl. 6):vi22–vi25. doi: 10.1093/annonc/mdm219. [DOI] [PubMed] [Google Scholar]
- Bruns AF, Herbert SP, Odell AF, Jopling HM, Hooper NM, Zachary IC, et al. Ligand-stimulated VEGFR2 signaling is regulated by co-ordinated trafficking and proteolysis. Traffic. 2010;11:161–174. doi: 10.1111/j.1600-0854.2009.01001.x. [DOI] [PubMed] [Google Scholar]
- Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9:653–660. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- Carmeliet P. VEGF as a key mediator of angiogenesis in cancer. Oncology. 2005;69(Suppl. 3):4–10. doi: 10.1159/000088478. [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Dinges J, Albert DH, Arnold LD, Ashworth KL, Akritopoulou-Zanze I, Bousquet PF, et al. 1,4-Dihydroindeno[1,2-c]pyrazoles with acetylenic side chains as novel and potent multitargeted receptor tyrosine kinase inhibitors with low affinity for the hERG ion channel. J Med Chem. 2007;50:2011–2029. doi: 10.1021/jm061223o. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, et al. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Harris PA, Cheung M, Hunter RN, Brown ML, Veal JM, Nolte RT, et al. Discovery and evaluation of 2-anilino-5-aryloxazoles as a novel class of VEGFR2 kinase inhibitors. J Med Chem. 2005;48:1610–1619. doi: 10.1021/jm049538w. [DOI] [PubMed] [Google Scholar]
- Harris PA, Boloor A, Cheung M, Kumar R, Crosby RM, Davis-Ward RG, et al. Discovery of 5-[[4-[(2,3-dimethyl-2H-indazol-6-yl)methylamino]-2-pyrimidinyl]amino]-2-m ethyl-benzenesulfonamide (Pazopanib), a novel and potent vascular endothelial growth factor receptor inhibitor. J Med Chem. 2008;51:4632–4640. doi: 10.1021/jm800566m. [DOI] [PubMed] [Google Scholar]
- Hasinoff BB, Patel D. The lack of target specificity of small molecule anticancer kinase inhibitors is correlated with their ability to damage myocytes in vitro. Toxicol Appl Pharmacol. 2010;249:132–139. doi: 10.1016/j.taap.2010.08.026. [DOI] [PubMed] [Google Scholar]
- Holmes K, Roberts OL, Thomas AM, Cross MJ. Vascular endothelial growth factor receptor-2: structure, function, intracellular signalling and therapeutic inhibition. Cell Signal. 2007;19:2003–2012. doi: 10.1016/j.cellsig.2007.05.013. [DOI] [PubMed] [Google Scholar]
- Howell GJ, Herbert SP, Smith JM, Mittar S, Ewan LC, Mohammed M, et al. Endothelial cell confluence regulates Weibel-Palade body formation. Mol Membr Biol. 2004;21:413–421. doi: 10.1080/09687860400011571. [DOI] [PubMed] [Google Scholar]
- Imrie H, Abbas A, Viswambharan H, Rajwani A, Cubbon RM, Gage M, et al. Vascular insulin-like growth factor-I resistance and diet-induced obesity. Endocrinology. 2009;150:4575–4582. doi: 10.1210/en.2008-1641. [DOI] [PubMed] [Google Scholar]
- Jain RK, Duda DG, Clark JW, Loeffler JS. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat Clin Pract Oncol. 2006;3:24–40. doi: 10.1038/ncponc0403. [DOI] [PubMed] [Google Scholar]
- Knights V, Cook SJ. De-regulated FGF receptors as therapeutic targets in cancer. Pharmacol Ther. 2010;125:105–117. doi: 10.1016/j.pharmthera.2009.10.001. [DOI] [PubMed] [Google Scholar]
- Koch S, Tugues S, Li X, Gualandi L, Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. Biochem J. 2011;437:169–183. doi: 10.1042/BJ20110301. [DOI] [PubMed] [Google Scholar]
- Krystof V, Cankar P, Frysova I, Slouka J, Kontopidis G, Dzubak P, et al. 4-arylazo-3,5-diamino-1H-pyrazole CDK inhibitors: SAR study, crystal structure in complex with CDK2, selectivity, and cellular effects. J Med Chem. 2006;49:6500–6509. doi: 10.1021/jm0605740. [DOI] [PubMed] [Google Scholar]
- Latham AM, Molina-Paris C, Homer-Vanniasinkam S, Ponnambalam S. An integrative model for vascular endothelial growth factor A as a tumour biomarker. Integr Biol (Camb) 2010;2:397–407. doi: 10.1039/c0ib00008f. [DOI] [PubMed] [Google Scholar]
- Latham AM, Bruns AF, Kankanala J, Johnson AP, Fishwick CW, Homer-Vanniasinkam S, et al. Indolinones and anilinophthalazines differentially target VEGF-A and bFGF-mediated responses in primary human endothelial cells. Br J Pharmacol. 2012;165:245–259. doi: 10.1111/j.1476-5381.2011.01545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Jia H, Holmes DI, Stannard A, Zachary I. Vascular endothelial growth factor-regulated gene expression in endothelial cells: KDR-mediated induction of Egr3 and the related nuclear receptors Nur77, Nurr1, and Nor1. Arterioscler Thromb Vasc Biol. 2003;23:2002–2007. doi: 10.1161/01.ATV.0000098644.03153.6F. [DOI] [PubMed] [Google Scholar]
- Mendel DB, Laird AD, Xin X, Louie SG, Christensen JG, Li G, et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003;9:327–337. [PubMed] [Google Scholar]
- Miyazaki Y, Matsunaga S, Tang J, Maeda Y, Nakano M, Philippe RJ, et al. Novel 4-amino-furo[2,3-d]pyrimidines as Tie-2 and VEGFR2 dual inhibitors. Bioorg Med Chem Lett. 2005;15:2203–2207. doi: 10.1016/j.bmcl.2005.03.034. [DOI] [PubMed] [Google Scholar]
- Morphy R. Selectively nonselective kinase inhibition: striking the right balance. J Med Chem. 2010;53:1413–1437. doi: 10.1021/jm901132v. [DOI] [PubMed] [Google Scholar]
- Newman AC, Nakatsu MN, Chou W, Gershon PD, Hughes CC. The requirement for fibroblasts in angiogenesis: fibroblast-derived matrix proteins are essential for endothelial cell lumen formation. Mol Biol Cell. 2011;22:3791–3800. doi: 10.1091/mbc.E11-05-0393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble ME, Endicott JA, Johnson LN. Protein kinase inhibitors: insights into drug design from structure. Science. 2004;303:1800–1805. doi: 10.1126/science.1095920. [DOI] [PubMed] [Google Scholar]
- Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling – in control of vascular function. Nat Rev Mol Cell Biol. 2006;7:359–371. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- Richardson DR, Tran EH, Ponka P. The potential of iron chelators of the pyridoxal isonicotinoyl hydrazone class as effective antiproliferative agents. Blood. 1995;86:4295–4306. [PubMed] [Google Scholar]
- Roskoski R., Jr Sunitinib: a VEGF and PDGF receptor protein kinase and angiogenesis inhibitor. Biochem Biophys Res Commun. 2007;356:323–328. doi: 10.1016/j.bbrc.2007.02.156. [DOI] [PubMed] [Google Scholar]
- Schweighofer B, Testori J, Sturtzel C, Sattler S, Mayer H, Wagner O, et al. The VEGF-induced transcriptional response comprises gene clusters at the crossroad of angiogenesis and inflammation. Thromb Haemost. 2009;102:544–554. doi: 10.1160/TH08-12-0830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Shibuya M. The 230 kDa mature form of KDR/Flk-1 (VEGF receptor-2) activates the PLC-gamma pathway and partially induces mitotic signals in NIH3T3 fibroblasts. Oncogene. 1997;14:2079–2089. doi: 10.1038/sj.onc.1201047. [DOI] [PubMed] [Google Scholar]
- Tille JC, Wood J, Mandriota SJ, Schnell C, Ferrari S, Mestan J, et al. Vascular endothelial growth factor (VEGF) receptor-2 antagonists inhibit. J Pharmacol Exp Ther. 2001;299:1073–1085. [PubMed] [Google Scholar]
- Usui T, Ban HS, Kawada J, Hirokawa T, Nakamura H. Discovery of indenopyrazoles as EGFR and VEGFR-2 tyrosine kinase inhibitors by in silico high-throughput screening. Bioorg Med Chem Lett. 2008;18:285–288. doi: 10.1016/j.bmcl.2007.10.084. [DOI] [PubMed] [Google Scholar]
- Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther. 2008;7:3129–3140. doi: 10.1158/1535-7163.MCT-08-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Cao Z, Tian H, Shen G, Ma Y, Xie H, et al. SKLB1002, a novel potent inhibitor of vascular endothelial growth factor receptor 2 signaling, inhibits angiogenesis and tumor growth in vivo. Clin Cancer Res. 2011;17:4439–4450. doi: 10.1158/1078-0432.CCR-10-3109. [DOI] [PubMed] [Google Scholar]
- Zhou T, Commodore L, Huang WS, Wang Y, Sawyer TK, Shakespeare WC, et al. Structural analysis of DFG-in and DFG-out dual Src-Abl inhibitors sharing a common vinyl purine template. Chem Biol Drug Des. 2010;75:18–28. doi: 10.1111/j.1747-0285.2009.00905.x. [DOI] [PubMed] [Google Scholar]
- Zuccotto F, Ardini E, Casale E, Angiolini M. Through the ‘gatekeeper door’: exploiting the active kinase conformation. J Med Chem. 2010;53:2681–2694. doi: 10.1021/jm901443h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.