

Abstract
In contrast to the tree of life (TOF) theory, species are mosaics of gene sequences with different origins. Observations of the extensive lateral sequence transfers in all organisms have demonstrated that the genomes of all life forms are collections of genes with different evolutionary histories that cannot be represented by a single TOF. Moreover, genes themselves commonly have several origins due to recombination. The human genome is not free from recombination events, so it is a mosaic like other organisms' genomes. Recent studies have demonstrated evidence for the integration of parasitic DNA into the human genome. Lateral transfer events have been accepted as major contributors of genome evolution in free-living bacteria. Furthermore, the accumulation of genomic sequence data provides evidence for extended genetic exchanges in intracellular bacteria and suggests that such events constitute an agent that promotes and maintains all bacterial species. Archaea and viruses also form chimeras containing primarily bacterial but also eukaryotic sequences. In addition to lateral transfers, orphan genes are indicative of the fact that gene creation is a permanent and unsettled phenomenon. Currently, a rhizome may more adequately represent the multiplicity and de novo creation of a genome. We wanted to confirm that the term “rhizome” in evolutionary biology applies to the entire cellular life history. This view of evolution should resemble a clump of roots representing the multiple origins of the repertoires of the genes of each species.
Keywords: rhizome, genealogic tree, horizontal sequence transfer, recombination, orphan genes
Speciation
The definition of a species established by Mayr (Mayr, 1957), known as the “Biological Species Concept,” postulates that species are groups of interbreeding natural populations that are reproductively isolated from other such groups. Therefore, sympatric speciation can occur only between closely related species, in which gene exchange is possible, whereas reproductive isolation is a process that can only occur if geographical separation partially or completely impedes gene flow. This definition establishes allopatric speciation as the norm (Mayr, 1957). Ecological changes or natural disasters lead to organismal isolation and bottlenecks, and therefore, to speciation. To this end, the main characteristic of speciation in eukaryotes is geographic isolation or allopatry, which restricts the capacity to exchange genes (Via, 2009); this feature is also observed in specialized bacteria (Doolittle and Papke, 2006).
However, a species is not a stable entity because species are continuously created and transformed. Due to their previous sympatric lifestyle, in which genetic exchanges were not limited, genomes are mosaics of sequences with different origins (Georgiades et al., 2011a,b). Indeed, the observations of extensive lateral sequence transfers in all organisms demonstrated that the genomes of all life forms are collections of genes with different evolutionary histories that cannot be represented by a single tree of life (TOL) (Koonin, 2009).
Many alternatives have been proposed to represent all the gene exchange events in organisms, such as networks (Kunin et al., 2005; Dagan et al., 2008; Halary et al., 2010; Beauregard-Racine et al., 2011; Kloesges et al., 2011; Popa et al., 2011), forests (Lopez and Bapteste, 2009) and bushes (Gould, 1987).
We sought to confirm that the term “rhizome” in evolutionary biology is the most suitable descriptor and to demonstrate that the entire history of cellular life is a rhizome (Raoult, 2010a; Merhej et al., 2011; Koonin, 2012).
Lateral inheritance
Lateral gene transfers (LGT) have been considered to be marginal phenomena, important under specific circumstances, which could be discarded in the study of organismal evolution (Koonin and Wolf, 2008). However, recent extensive comparisons of multiple whole genome sequences have revealed a vast and surprising variability in gene content, even among closely related-species (Mira et al., 2001; Berg and Kurland, 2002; Konstantinidis and Tiedje, 2004; Koonin and Wolf, 2008; Georgiades et al., 2011a,b). Comparative genomics revealed that most genes are susceptible to LGT (Sorek et al., 2007), although the tendency to undergo LGT is highly variable among genes (Nakamura et al., 2004; Cohen et al., 2008, 2011; Hao and Golding, 2008). LGT affects different classes of genes to different extents, but no single gene is completely immune to LGT (Koonin and Wolf, 2008). LGT may be mediated by the inheritance of a plasmid, the integration of a lysogenic phage into a chromosome, or by the insertion of a linear fragment into a chromosome (Kaper and Hacker, 1999). Complete genome sequences highlight the confounding effects of lateral transfers in reconstructing the history of organismal evolution (Koonin et al., 1997, 2000; Doolittle, 1999; Nelson et al., 1999; Boucher et al., 2001; Gogarten et al., 2002; Zhaxybayeva and Gogarten, 2002). A practical necessity for detecting LGT is a species tree that depicts the phylogeny of the compared organisms in their entirety. Indeed, the most common practice used to detect LGT involves identifying the reliable discrepancies between the topologies of a gene tree and a species tree (Merhej et al., 2011). The species tree concept was validated by comparing the phylogenetic trees of sets of several hundred single-copy clusters of orthologous groups (COGs) from well-characterized, widespread bacterial groups, such as alphaproteobacteria, gamma-proteobacteria or Bacillus-Clostridium (Koonin and Wolf, 2008). However, LGT is a misleading definition because lateral inheritance may refer to sequences and not to entire or single genes (Chan et al., 2009, 2011; Merhej et al., 2011).
Lateral sequence transfers in eukaryotes and the human rhizome
Two elements participate in eukaryotic genetic change. The first is sexuality. In 1889, August Weismann proposed that sex evolved because of the advantage in creating variation among siblings (Weismann, 1889). Therefore, natural selection favors the parents who can produce a variety of offspring. A similar hypothesis was proposed by Darwin in his “Origin of Species” (Darwin, 1859), which states that all sexually reproducing organisms are derived from a common, single-celled eukaryotic ancestor. Many protists reproduce sexually, as do multicellular plants, animals, and fungi. A few species, such as Bdelloidea and some parthenocarpic plants have secondarily lost this feature (Letunic and Bork, 2006). Some species, such as arthropods, can reproduce sexually and asexually or undergo parthenogenesis, which is the development of embryos in the absence of male fertilization. Wolbachia are known to induce female parthenogenesis in infected arthropods (Renvoisé et al., 2011).
The second mechanism of genetic exchange in eukaryotes is lateral sequence transfer by infection or recombination. Sequence transfers from bacteria to eukaryotes have occurred due to the ancestral symbiotic events that led to the establishment of plant and animal lineages (Thomas and Greub, 2010). Amoebae have also played a significant role as a melting pot for genetic exchange (Ogata, 2006; Moliner et al., 2010; Raoult and Boyer, 2010). A dramatic lateral sequence transfer has been reported: nearly the entire Wolbachia genome was observed to be integrated into the host genome. Comparative genomic studies support the existence of progressive sequence transfers from Wolbachia to arthropods, insects, and nematodes (Dunning Hotopp et al., 2007; Nikoh et al., 2008; McNulty et al., 2010). In particular, Drosophila ananassae harbors the entire genome of a Wolbachia species (Callaway, 2007).
The human genome does not escape such mixtures and also is a mosaic. Evidence from a recent study supports the integration of parasitic DNA into the human genome. The authors revealed that Trypanosoma cruzi sequences were integrated into the genomes of patients from five families from different Brazilian ecosystems. An investigation of the role of saliva in the transmission of human herpesvirus 6 (HHV6) revealed that all nine submandibular glands and one of four parotid glands harbored the HHV6 genome and expressed the corresponding proteins (Fox et al., 1990). The consequences of parasite DNA acquisition, the vertical inheritance of integrated DNA and its subsequent drift may contribute to the ongoing genetic diversity and speciation in the human population (Hecht et al., 2010), which is indicative of a chimera (Raoult, 2011).
Genome mixing has also occurred between archaic hominins and modern humans. Indeed, evidence for a notable presence of a Neanderthal-derived X chromosome segment among all contemporary human populations outside Africa has been presented (Yotova et al., 2011). Another archaic hominin was recently discovered in the Denisovan cave in Siberia. This group is derived from a hominin migration out of Africa distinct from the Neanderthal ancestors and modern humans, as suggested by the highly divergent morphological features. Evidence suggests that this archaic human lived close in time and space with Neanderthals and modern humans and that its genome contributed 4–6% of its genetic material to the genomes of the present-day Malaysians (Krause et al., 2010; Reich et al., 2010), whereas Neanderthals contributed approximately 1–4% of their genetic material to modern Europeans (Abi-Rached et al., 2011). Furthermore, a recent analysis of the highly polymorphic human leukocyte antigen (HLA) class I revealed how modern humans acquired the HLA-B*73 allele in West Asia through mixing with Denisovans (Abi-Rached et al., 2011). All these surprising data suggest that the genetic constitution of modern humans may be the outcome of a mosaic of lineages from different times and geographic origins (Yotova et al., 2011), as defined by our vision of the rhizome (Raoult, 2011) (Figure 1).
Figure 1.

The human rhizome. The current human genome harbors genes acquired vertically, predominantly from Cro-Magnon ancestors but also from Denisovans and Neanderthals. In addition to the Cro-Magnon genes, Europeans possess a few Denisovan genes, Africans possess a small and approximately similar amount of Denisovan and Neanderthal genes, and Malaysians possess few Neanderthal genes.
The mitochondrial rhizome
Mitochondrial evolution has recently been demonstrated to constitute a rhizome (Georgiades and Raoult, 2011). The results of this study suggest that the origins of mitochondrial genes are not limited to Rickettsiales and that their creation did not occur in a single event but through multiple successive events. Contrary to what has been believed until now, recent evidence strongly suggests that mitochondria do not have a single common ancestor (Emelyanov, 2001) but likely numerous ancestors, including proto-Rickettsiales, proto-Rhizobiales, proto-alphaproteobacteria and current alphaproteobacterial species. The mosaisism of the mitochondrial genome is also discussed by Esser et al. (2007), however, in our study, the use of four different types of mitochondria from four different organisms (protozoa, yeast, louse, human) revealed that the mitochondria of different organisms comprise different elements, while the analysis of the louse multi-chromosomal mitochondrion (Shao et al., 2009) showed that the mitochondria creation model is not fixed: mitochondria do not have a stable or unique form, and thus their evolution cannot be the same. We conclude from these results that the TOL is not sufficient to explain the chimeric structure of mitochondrial genomes and that their evolution should be represented as a rhizome as well.
Lateral sequence transfers in bacteria and their rhizome
Lateral inheritance has been accepted as a major contributor of genome evolution in free-living bacteria since the Lederberg experiments on pneumococcus (Lederberg et al., 1951). Acquired genes play a major role in bacterial diversification by supplying previously unavailable traits, which permit the rapid exploitation of new environments (Ochman et al., 2005). Selfish and mobile genetic elements, such as virophages, plasmids and transposons, are the primary vehicles for lateral transfers among prokaryotes (Pace et al., 2008). The insertion sequences (IS) that were initially identified in the Escherichia coli galactose operon are also examples (Shapiro, 1969).
Conversely, the absence of evidence for lateral transfers has long been considered to be a fundamental characteristic of the genomic evolution in obligate intracellular bacteria. The isolated lifestyles of obligate intracellular bacteria were thought to reduce gene acquisition opportunities. Genetic exchanges have been judged to be negligible in these species because their intracellular environment is relatively constant and does not select for the genetic diversity promoted by more challenging environments (Moreno, 1998; Blanc et al., 2007a). These early views have progressively changed with the accumulation of genomic data, which have provided evidence for extended genetic exchanges in intracellular bacteria (Bordenstein and Reznikoff, 2005). Therefore, lateral sequence transfer is now viewed as an agent that promotes and maintains all bacterial species. Bacteria can acquire genes by several means, including conjugal gene transfer, phage-mediated insertion and native DNA from outside sources (Ochman et al., 2005). The process of gene loss has substantially contributed to the differences in the gene contents between the modern Rickettsia species. Additionally, the Typhus Group (TG) genomes were predominantly shaped by the reductive evolution from the ancestral Rickettsia genome.
However, the R. bellii genome includes numerous genes related to amoebal symbionts, perhaps due to ancient gene exchanges between an R. bellii ancestor and other amoebal hosts (Ogata, 2006). In a recent study, we provided evidence for lateral transfers between Rickettsiales species and other bacteria or other organisms. R. canadensis acquired genes from gamma-proteobacteria, Bacteroidetes, R. akari, and R. felis acquired genes from gamma- and delta-proteobacteria, and SFG species acquired genes from Bacteroidetes and Cyanobacteria, whereas the R. belli genome contained genes of a eukaryotic origin (Georgiades et al., 2011a). Finally, the plasmids in R. felis may have been acquired via conjugation (Ogata et al., 2005). The R. felis genome is actually a collection of genes and parts of genes with diverse evolutionary histories. The apparent horizontal transfer of different sized DNA segments corresponding to genes in various functional categories has been detected, indicating that any segment of DNA may be horizontally transferred (Merhej et al., 2011). Other Rickettsiales, such as Orientia spp., also acquired genes by LGT from Cyanobacteria, protists, and viruses (Georgiades et al., 2011a). Wolbachia spp. also have highly recombinogenic genomes, as their sympatric lifestyle enables recombination among the intracellular bacterial community (Klasson et al., 2009). Although lateral transfers were thought to be rare phenomenon in obligate intracellular bacteria (Audic et al., 2007), the “mobilome,” composed of mobile genetic elements, promotes horizontal genetic fluidity in Rickettsia species and has likely shaped the evolution of these genomes (Merhej and Raoult, 2011). Bacteria of the Legionella genus can also grow in amoebae, although a clear demonstration of intra-amoebal growth remains lacking for most of them (Thomas and Greub, 2010). Degtyar et al. demonstrated that the distribution of effector-encoding genes is highly variable in Legionella species: most genes of eukaryotic origin are present in different Legionella species, whereas others are specific to L. pneumophila (Degtyar et al., 2009). In another recent study, 557 laterally transferred genes were observed in L. pneumophila. Most of the transferred genes are part of the metabolism functional category. An exchange of genetic material with a common amoeba host most likely explains the multiple phylogenetic origins of a significant fraction of the Legionella genes (Coscolla et al., 2011). Other bacteria have also acquired genes through lateral transfers. Lawrence and Ochman (Lawrence and Ochman, 1997) proposed that at least 15% of the E. coli genome is atypical and may have arisen by recent lateral inheritance, while the diversity within the species E. coli and the overlap in gene content suggests a continuum rather than sharp species borders in the group of Enterobacteriaceae (Lukjancenko et al., 2010). Nelson et al. (Nelson et al., 1999) concluded that almost 25% of the Thermotoga maritime genes are most closely related to archaeal genes and have a history of gene transfer between these lineages (Gogarten et al., 2002). Firmicutes, Bacteroidetes, and gamma-proteobacteria species were observed to possess genes of Rickettsiales origin (Georgiades et al., 2011a). Approximately half of the species-specific genes in Streptococcus species have been proposed to be acquired by lateral transfer from diverse backgrounds. Specifically, multiple lateral sequence transfer events occurred during polyclonal infections among the nasopharyngeal Streptococcus pneumoniae strains recovered from a child suffering from chronic upper respiratory and middle-ear infections (Hiller et al., 2010). Finally, recent evidence has revealed the presence of horizontally transferred fragments of the human long interspersed nuclear element L1 in the genome of the strictly human pathogen Neisseria gonorrhoeae (Andersson and Seifert, 2011). Generally, it has been demonstrated that at least 81 ± 15% of the genes in each studied genome were involved in LGT at some point of their history even thought they can be vertically inherited after acquisition (Dagan et al., 2008). Taken together, these data suggest that the rhizome hypothesis is well suited for describing bacteria (Merhej et al., 2011) (Figure 2).
Figure 2.

The rhizome of bacteria—the Rickettsiales paradigm. Rickettsiales harbor genes from various bacterial species, protists, and viruses.
Lateral sequence transfers in archaea and their rhizome
Many Archaea inhabit extreme environments similar to those in which life originated. Although Archaea members may be seen as evolutionary relics of the earliest life forms, none of the organisms living today are primitive. All extant life forms are modern organisms that are well adapted to their ecological niches. Numerous authors have observed many horizontally transferred genes in Archaea, confirming that lateral sequence transfer is a wide-ranging phenomenon (Koonin and Galperin, 1997; Aravind et al., 1998). Koonin and Galperin (1997) observed large fractions of genes of bacterial or eukaryotic origins in Archaea genomes, suggesting a chimeric origin for Archaea. The percentages of horizontally transferred sequences in bacteria and Archaea are similar, ranging from 5% in Methanococcus jannaschii to 14% in Aeropyrum pernix (Garcia-Vallvé et al., 2000). Massive gene exchanges between the extremely thermophilic Archaea and the hyperthermophilic Aquifex have been suggested. Previous studies revealed that the Aquifex genome is a chimera that shares a large component with the Archaea genome in addition to the core gene set shared with the rest of the bacteria. Bacterial hyperthermophily has likely evolved secondarily within moderately thermophilic bacteria by the continuous acquisition of thermotolerance genes from pre-adapted hyperthermophiles, namely the Archaea (Aravind et al., 1998). Finally, M. jannaschii protein sequences revealed that the number of proteins that are more similar to bacterial homologs significantly exceeds the number of those that are closer to eukaryotic homologs. The prevalence of the genes of bacterial origin in archaeal genomes suggests a genetic basis for the prokaryotic phenotype (Koonin and Galperin, 1997).
Lateral sequence transfers in viruses and their rhizome
Viral genomes are not without lateral sequence transfers. A recently discovered virophage, Sputnik, was considered to be a vehicle that mediated lateral transfers between giant viruses (La Scola et al., 2008). The Acanthamoeba polyphaga mimivirus has unique features, including the presence of dsDNA, which was previously undocumented in viruses. Phylogenetic analyses identified a large number of bacterial homologs, suggesting an acquisition by lateral inheritance. Most of these genes were related to the orthologs in bacterial species, such as L. pneumophila, that are known to grow within amoebae. Mimiviruses likely acquired these genes from degraded or live bacteria sharing the same environment, particularly within amoebae (Moliner et al., 2010). In mammalian Herpesvirus, 141 putative transferred genes were identified, of which 91 were from gamma-herpesvirus, 42 from beta-herpesvirus, and 8 from alpha-herpesvirus, suggesting that gene acquisition in gamma-herpesvirus was more active than in the others (Figure 3). Although the functions of most of the putative transferred genes remain uncharacterized, many genes have been predicted to encode glycoproteins or membrane proteins (Fu et al., 2008). Furthermore, genomic analyses revealed that approximately 13% of the Herpesvirus proteins have clear sequence similarity to the human genome. The human homologs present in a large proportion of herpesvirus genomes, such as DNA polymerase and uracil-DNA glycosylase, are likely to have been acquired from a distant host by an ancestral Herpesvirus. Generally, gamma-herpesvirus genomes are particularly rich in genes that have a human counterpart (Holzerlandt et al., 2002).
Figure 3.
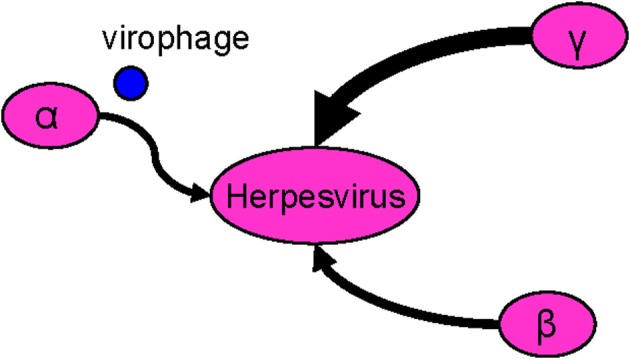
The viral rhizome. The mammalian herpesvirus harbors genes from the α, β, and γ herpesvirus. Gene exchange often occurs by virophages.
Not only does the gene repertoire exhibit a substantial plasticity, but nearly all genes of all organisms, including Prokaryotes, Eukaryotes, Archaea and Viruses, have been exchanged or recombined at some point in time (Raoult, 2010b). Therefore, all life forms are mosaics that are part of the rhizome of life.
Intragenic recombination
Other than lateral sequence transfer and recombination between various organisms, the origin of some genes cannot be simply explained because of the occurrence of intragenic recombination (Raoult, 2010a). Different varying sites recombine within the same gene locus, and intragenic recombination can generate new allelic variation at a locus. New gene products with potentially new properties will then arise (Watt, 1972). Chimeric gene sequences that could result from recombination between divergent alleles have been detected in several species (Kelly et al., 2009). A recent R. felis study revealed random transfers of DNA sequences in the R. felis genomes that occurred independently of gene function or sequence length. The apparent horizontal transfer of DNA segments with different sizes has been detected, thus indicating that any segment of DNA may be laterally transferred (Merhej et al., 2011).
Staphylococcus aureus expresses several proteases, including the thermolysin-like metalloprotease aureolysin. Sequence analyses revealed that the aur gene is present in two distinct types of related sequences and that it is very polymorphic. The gene trees constructed from aur and concatenated multilocus sequence typing (MLST) genes revealed several putative assortative recombination events, such as exchanges of the entire aur gene between divergent S. aureus lineages. Evidence for intragenic recombination events, such as exchanges of internal aur segments across aur genes, was also observed (Sabat et al., 2008). Moreover, results from Nicotiana illustrated that intragenic recombination may be a relatively common occurrence and can provide evidence for distinct parental contributions within diploid species of likely reticulate origin (Kelly et al., 2009). Bluetongue virus (BTV) is the pathogen that causes Bluetongue disease, which threatens sheep, deer, cattle, and goats. Mosaic viral genes have been observed to share a similar recombination event. Recombination can occur in BTVs and likely plays a role in the evolution and genetic diversity of the virus (He et al., 2010). However, the recombination rate of BTV is much lower than that of positive-sense RNA viruses, such as the foot and mouth disease virus, in which 10–20% of the viral genomes undergo recombination during a single replication cycle (Alejska et al., 2001). Intragenic recombination between two different existing alleles in a population can create new alleles. The role of this process in maintaining variation in a natural population has been investigated through the assumptions that one gene consists of two sites, each of which can mutate to an infinite number of unique alleles (Kimura and Crow, 1964; Strobeck and Morgan, 1978; Morgan and Strobeck, 1979). This process, like lateral sequence transfer, represents a challenge for the reconstruction of the evolutionary relationships between species because they cannot be represented adequately with classic trees (Kelly et al., 2009).
Orphan or newly created genes
Orphan genes constitute a class of lineage-specific genes that are not homologous to the sequences of other species (Fischer and Eisenberg, 1999). They typically encode small proteins and have high non-synonymous substitution rates, but their functions remain unknown (Domazet-Loso and Tautz, 2003; Daubin and Ochman, 2004). Recently, a classification of ORFans has been proposed, dividing ORFans into singletons (unique predicted genes with no significant homolog), multipletons (orphan with one or more paralogs in its residing genome but none in other genomes), and lineage ORFans (orphan with homologs among a given taxonomic rank and none outside) (Boyer et al., 2010). Comparative analyses have revealed that new genes appear frequently in genomes and that useful genes can be retained, whereas purposeless genes are often removed from the genome. The current methods for identifying the newly acquired genes can be divided into three categories: compositional analysis, detection of phylogenetic anomalies, and comparison of genome content (Ochman et al., 2000; Nakamura et al., 2004; Gogarten and Townsend, 2005; Kuo and Ochman, 2009). Compositional analysis is the only method that does not rely on comparing sequences from multiple genomes. In short, this approach compares the sequence features, such as nucleotide composition and codon usage, of the genes within a genome. A direct genome alignment is the most frequently used approach for identifying new genes, in which the presence and absence of genes among the organisms is examined. This method entails comparing multiple related genomes and can provide the most direct evidence of genes that are gained or lost from a lineage (Kuo and Ochman, 2009). Each newly sequenced genome contains a significant number of ORFans (Toll-Riera et al., 2009). For example, in the 60 fully sequenced microbial genomes, 14% of the genes are species-specific orphans (Siew and Fischer, 2003), whereas 18% of the Drosophila genes are restricted to the Drosophila group (Zhang et al., 2007; Zhou et al., 2008). However, the origin of the orphan genes remains elusive (Merkeev and Mironov, 2008). One scenario proposes that they were derived from gene duplication events, in which one copy accumulated enough sequence changes so that the ancestral similarity is no longer detectable (Domazet-Loso and Tautz, 2003). Such ORFans were recently proposed to represent genes of viral or plasmid origin (Rocha et al., 2006), and some may correspond to genuine new genes formed de novo through diverse mechanisms of gene evolution (Boyer et al., 2010). This proposed mechanism has significantly impacted the process by which novel genes in mammals form, specifically in primates, in which 5.5% of orphan genes could have originated de novo from non-coding genomic regions (Toll-Riera et al., 2009). The formation of novel genes has also been described in Drosophila (Begun et al., 2006; Levine et al., 2006; Zhou et al., 2008) and Saccharomyces cerevisiae (Cai et al., 2008). To characterize the genetic basis of evolution and development in insects, the genomes of insect-specific proteomes were analyzed. The characterization of the proteomes-based on genome sequences provides a rapid method to approximate and update the putative proteomes as genome sequences become available. Using this approach, 50 insect-specific proteins were isolated, and many have been supported by experimental studies. The proteins-related to stress and immune responses constitute an extensive fraction of the proteins characterized in the insect-specific proteome. The presence of numerous insect-specific olfaction and cuticle development proteins underscores the significance of communication and adaptation to the environment during insect evolution (Zhang et al., 2007).
In a recent study by our laboratory, a small number of gene sequences in Rickettsia species were identified that did not match any database and may have resulted from de novo creation (Georgiades et al., 2011a). Indeed, 17 Rickettsia gene sequences do not possess homologs in the non-redundant (NR) database. The Ka/Ks ratio revealed that 15 of these sequences were either non-functional or had gained functionality later on. The probability of pseudogenization or a viral origin of these genes should not be excluded, however, because these genes were not found in the regions with traces of active or ancient integrated extra-chromosomal elements, we strongly believe that they are novel genes (Georgiades et al., 2011a).
A study reconstructing the gene content of ancestral archaeal and proteobacterial genomes demonstrated that Archaea exhibit an abnormally high number of genesis events, particularly Aeropyrum pernix. The estimates for gene genesis also reveal at least 240 genes that originated at the branch leading to the Archaea. For Proteobacteria, this number was at least 320. Such genes can be considered typical of a taxon because they are unique and widespread within it (Snel et al., 2002).
Finally, the formation of new genes has been suggested to be essential to the viability of an organism. In the case of Drosophila, 59 de novo genes were observed to be as vitally essential as the old genes. The lethal phenotypes caused by the knockout of new genes suggested that the genes created de novo may integrate a vital pathway by interacting with existing genes, and this co-evolution may cause the new gene to become indispensable (Chen et al., 2010). Recently, an attempt to estimate the fraction of acquired genes that become dispensable and the features of those that are retained was made by examining the distribution of genes along an evolutionary lineage. Based on the numbers of the recent arrivals present in the E. coli K12 genome compared with the number of ancestral genes maintained by all members of this species, only 10–15% of the acquired genes were retained (Ochman and Davalos, 2006). Moreover, by establishing the origins of the genes acquired during the diversification of E. coli and Shigella, new genes that have distant homologs in other bacteria were acquired much more frequently but not retained as often as the acquired genes with no identifiable homologs (van Passel et al., 2008). These results suggest that genuinely novel genes, i.e., those that never conferred a function in a cellular genome, are more likely to persist in bacterial genomes (Kuo and Ochman, 2009).
In summary, gene creation is a continuous and unsettled phenomenon that is supported by the discovery of new genes that are permanently generated and are identified more frequently (Boyer et al., 2010; Raoult, 2010a). Genes created de novo illustrate the fact that nature is creative and not parsimonious. A rhizome is the most suitable representation for orphans; newly created genes should be represented as emerging roots on the rhizome.
The tree versus the rhizome of life
The TOL, initially elaborated by Darwin 150 years ago (Darwin, 1859), remains a biological fact for many supporters. For those who question it, the TOL is nothing more than a scientific hypothesis not yet proven to be true. Darwin's theory reflects the religious and social beliefs of its time: the existence of one common ancestor at the origin of each branch is a direct resurgence of the Adam and Eve creation theory (Raoult, 2010b). The origin of life is a biblical definition (Penny, 2011) that reflects the “hierarchical natural order” originating from a single ancestor (Doolittle, 1999). Indeed, this hypothesis could be falsified and is based on the mixed message that classifications should be constructed as hierarchies because evolution is a branching process, and a hierarchical classification is a proof of a branching evolution (Doolittle and Bapteste, 2007). Nevertheless, the traditional biological explanations have mostly relied on the construction of a genealogical tree that describes lineages diverging from a last common ancestor. Such a hierarchy provides a dichotomous topology that structures biodiversity in the most informative way (Schliep et al., 2011). However, the robustness and universal scope of such tree-based evolutionary explanations has been recently questioned more frequently (Doolittle, 1999, 2009a,b; Bapteste et al., 2009; Dagan and Martin, 2009; Ragan and Beiko, 2009). For example, the ubiquity of lateral sequence transfers suggests that the TOL does not adequately represent prokaryote evolution (Bapteste and Boucher, 2008; Koonin and Wolf, 2008). Bacterial classification implies that bacteria have a phylogeny and that the taxonomic system that functions adequately for the metazoans is also meaningful for the microbial world. Further, such a hierarchy and classification do not consider the numerous lateral sequence transfer events observed in all organisms. Up to 30% of the intra-species genome-to-genome variation in gene content results from lateral inheritance and gene loss. In some species, the pan-genome appears unlimited (Doolittle and Bapteste, 2007).
Human beings are complex ecosystems comprising more bacteria and viruses than eukaryotic cells in their mucosa, particularly in the gut. The human genome is a mosaic of genes with eukaryotic, bacterial, and viral origins. Bacteria harbor genes that originated from eukaryotic, viral, and archaeal organisms. Giant viruses also have chimeric genomes of different origins (Raoult, 2010a). Finally, genome analyses have revealed high proportions of newly generated ORFan genes (Fraser et al., 2000). An increasing number of scientists now argue that Darwin's TOL is best seen as an approximation to describe some parts of the living world but is less adequate elsewhere (e.g., viruses and prokaryotes). However, it is currently more complicated to place the root on the TOL (Ragan et al., 2009). A genealogic tree is a more adequate representation (Figure 4).
Figure 4.
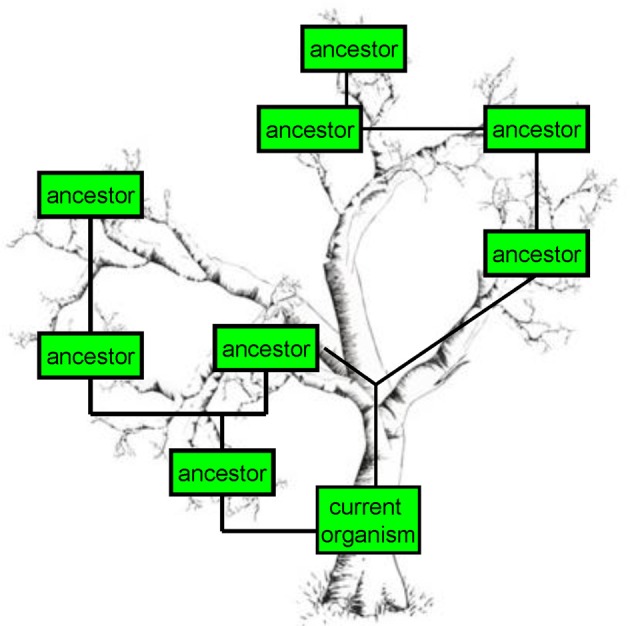
Genealogic tree. A genealogic tree sample that harbors organisms with many ancestors closely or distantly related.
The theories of multiplicity and de novo creation need to be integrated in a post-Darwinian concept of the living species. When all these features are considered, the evolution of species seems more like a rhizome (Raoult, 2010a). A rhizome is a descriptive and epistemological model that does not organize the elements by a hierarchical coordination line, including a base and a root constituting the starting point of branches according to the well-known model of the Porphyrian tree (Eco, 1984). Instead, it is a model in which each element can affect or influence all the others (Deleuze and Guittari, 1972). A rhizome has a predominantly semiotic perspective. It is not easy to perceive things from the middle or from the top to the bottom. In contrast to trees, a rhizome connects any point with any other, and all of the features do not necessarily correspond to features of the same nature (Deleuze and Guittari, 1972). Consequently, this view of evolution considers the occurrence of multiplicities as emerging species grow from the rhizome with gene repertoires of various origins that allow for the multiplication and perpetuation of the species. Even the human genome is, and should be, viewed as a mosaic with eukaryotic, bacterial, archaeal, and viral genes that comprise an ecosystem that drives a network of gene exchange (Figure 5).
Figure 5.

The human genome as a mosaic and an ecosystem. Everything is a rhizome, in which lateral transfers occur between and among bacteria, viruses, archaea, and eukaryotes mediated by plasmids, virophages, and archaeophages. The modern human genome comprises vertically inherited elements from ancestors and from laterally inherited elements from other eukaryotes, bacteria, and viruses. The arrows identify the lateral sequence transfers, and the colorful arrows indicate the transfer events toward humans.
Conclusion
Darwin's TOL presents a single common ancestor on the root and different species on the major branches that separate and continuously diverge. However, the history of life cannot be attributed to a single ancestral species that yielded descendants that have adapted to their environment and developed into various species completely distinct and different from each other in such a short time. Furthermore, new species are continuously created and are not necessarily derived from other species (Raoult, 2010b). Furthermore, recent and massive gene transfer events have been identified in all living organisms. A rhizome could more adequately represent the multiplicities of genomes and their de novo creations. Emerging species grow from the rhizome with gene repertoires of various origins that allow for the multiplication of species under permissive environmental conditions (Raoult, 2010a). This view of evolution should resemble a clump of roots representing the multiple origins of the genetic repertoire of each species (Halary et al., 2010).
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Abi-Rached L., Jobin M. J., Kulkarni S., McWhinnie A., Dalva K., Gragert L., Babrzadeh F., Gharizabeh B., Luo M., Plummer F. A., Kimani J., Carrington M., Middleton D., Rajalingam R., Beksac M., Marsh S. G. E., Maiers M., Guethlein L. A., Tavoulakis S., Little A.-M., Green R. E., Norman P. J., Parham P. (2011). The shaping of modern human immune systems by multiregional admixture with archaic humans. Science 334, 89–94 10.1126/science.1209202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alejska M., Kurzyniska-Kokorniak A., Broda M., Kierzek R., Figlerowicz M. (2001). How RNA viruses exchange their genetic material. Acta Biochim. Pol. 48, 391–407 [PubMed] [Google Scholar]
- Andersson M. T., Seifert S. H. (2011). Opportunity and means: horizontal gene transfer from the human host to a bacterial pathogen. MBio 2, e00005–e00011 10.1128/mBio.00005-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind L., Tatusov R. L., Wolf Y. I., Walker D. R., Koonin E. V. (1998). Evidence for massive gene exchange between archaeal and bacterial hyperthermophiles. Trends Genet. 14, 442–444 10.1016/S0168-9525(98)01553-4 [DOI] [PubMed] [Google Scholar]
- Audic S., Robert C., Campagna B., Parinello H., Claverie J. M., Raoult D., Drancourt M. (2007). Genome analysis of Minibacterium massiliensis highlights the convergent evolution of water-living bacteria. PLoS Genet. 3:e138 10.1371/journal.pgen.0030138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bapteste E., O'Malley M. A., Beiko R. G., Ereshefsky M., Gogarten J. P., Franklin-Hall L., Lapointe F. J., Dupré J., Dagan T., Boucher Y., Martin W. (2009). Prokaryotic evolution and the tree of life are two different things. Biol. Direct 4, 34 10.1186/1745-6150-4-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bapteste E., Boucher Y. (2008). Lateral gene transfer challenges principles of microbial systematics. Trends Microbiol. 16, 200–207 10.1016/j.tim.2008.02.005 [DOI] [PubMed] [Google Scholar]
- Beauregard-Racine J., Bicep C., Schliep K., Lopez P., Lapointe F. J., Bapteste E. (2011). Of woods and webs: possible alternatives to the tree of life for studying genomic fluidity in Escherichia coli. Biol. Direct 6, 39 10.1186/1745-6150-6-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begun D. J., Lindfors H. A., Thompson M. E., Holloway A. K. (2006). Recently evolved genes identified from Drosophila yakuba and D. erecta accessory gland expressed sequence tags. Genetics 172, 1675–1681 10.1534/genetics.105.050336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg O. G., Kurland C. G. (2002). Evolution of microbial genomes: sequence acquisition and loss. Mol. Biol. Evol. 19, 2265–2276 [DOI] [PubMed] [Google Scholar]
- Blanc G., Ogata H., Robert C., Audic S., Claverie J. M., Raoult D. (2007a). Lateral gene transfer between obligate intracellular bacteria: evidence from the Rickettsia massiliae genome. Genome Res. 17, 1657–1664 10.1101/gr.6742107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordenstein S. R., Reznikoff W. S. (2005). Mobile DNA in obligate intracellular bacteria. Nat. Rev. Microbiol. 3, 688–699 10.1038/nrmicro1233 [DOI] [PubMed] [Google Scholar]
- Boucher Y., Nesbo C. L., Doolittle W. F. (2001). Microbial genomes: dealing with diversity. Curr. Opin. Microbiol. 4, 285–289 10.1016/S1369-5274(00)00204-6 [DOI] [PubMed] [Google Scholar]
- Boyer M., Madoui M. A., Gimenez G., La Scola B., Raoult D. (2010). Phylogenetic and phyletic studies of informational genes in genomes highlight existence of a 4th domain of life including giant viruses. Plos One 5:e15530 10.1371/journal.pone.0015530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J., Zhao R., Jiang H., Wang W. (2008). De novo origination of a new protein-coding gene in Saccharomyces cerevisiae. Genetics 179, 487–496 10.1534/genetics.107.084491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaway E. (2007). Genomes within genomes. Nature 449, 6 10.1038/449006b [DOI] [PubMed] [Google Scholar]
- Chan C. X., Beiko R. G., Darling A. E., Ragan M. A. (2009). Lateral transfer of genes and gene fragments in prokaryotes. Genome Biol. Evol. 1, 429–438 10.1093/gbe/evp044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan C. X., Beiko R. G., Ragan M. A. (2011). Lateral transfer of genes and gene fragments in Staphylococcus extends beyond mobile elements. J. Bacteriol. 193, 3964–3977 10.1128/JB.01524-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S., Zhang Y. E., Long M. (2010). New genes in Drosophila quickly become essential. Science 330, 1682–1685 10.1126/science.1196380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen O., Gophna U., Pupko T. (2011). The complexity hypothesis revisited: connectivity rather than function constitutes a barrier to HGT. Mol. Biol. Evol. 28, 1481–1489 10.1093/molbev/msq333 [DOI] [PubMed] [Google Scholar]
- Cohen O., Rubinstein N. D., Stern A., Gophna U., Pupko T. (2008). A likelihood framework to analyse phyletic patterns. Philos. Trans. R. Soc. Lond. B Biol. Sci. 363, 3903–3911 10.1098/rstb.2008.0177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coscolla M., Comas I., Gonzales-Candelas F. (2011). Quantifying non-vertical inheritance in the evolution of Legionella pneumophila. Mol. Biol. Evol. 28, 285–1001 10.1093/molbev/msq278 [DOI] [PubMed] [Google Scholar]
- Dagan T., Artzy-Randrup Y., Martin W. (2008). Modular networks and cumulative impact of lateral transfer in prokaryote genome evolution. Proc. Natl. Acad. Sci. U.S.A. 105, 10039–10044 10.1073/pnas.0800679105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagan T., Martin W. (2009). Getting a better picture of microbial evolution en route to a network of genomes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 364, 2187–2196 10.1098/rstb.2009.0040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darwin C. (1859). The Origin of Species. London: Murray [Google Scholar]
- Daubin V., Ochman H. (2004). Bacterial genomes as new gene homes: the genealogy of ORFans in E. coli. Genome Res. 14, 1036–1042 10.1101/gr.2231904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degtyar E., Zusman T., Ehrlich M., Segal G. (2009). A Legionella effector acquired from protozoa is involved in sphingolipids metabolism and is targeted to the host cell mitochondria. Cell. Microbiol. 11, 1219–1235 10.1111/j.1462-5822.2009.01328.x [DOI] [PubMed] [Google Scholar]
- Deleuze G., Guittari F. (1972). Rhizome: Introduction. Paris, France: Ed de Minuit [Google Scholar]
- Domazet-Loso T., Tautz D. (2003). An evolutionary analysis of orphan genes in Drosophila. Genome Res. 13, 2213–2219 10.1101/gr.1311003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolittle W. F. (1999). Phylogeneticc classification and the universal tree. Science 284, 2124–2129 [DOI] [PubMed] [Google Scholar]
- Doolittle W. F. (2009a). Eradicating typological thinking in prokaryotic systematics and evolution. Cold Spring Harb. Symp. Quant. Biol. 74, 197–204 10.1101/sqb.2009.74.002 [DOI] [PubMed] [Google Scholar]
- Doolittle W. F. (2009b). The practice of classification and the theory of evolution, and what the demise of Charles Darwin's tree of life hypothesis means for both of them. Philos. Trans. R. Soc. Lond. B Biol. Sci. 364, 2221–2228 10.1098/rstb.2009.0032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolittle W. F., Bapteste E. (2007). Pattern plurarism and the tree of life hypothesis. Proc. Natl. Acad. Sci. U.S.A. 104, 2043–2049 10.1073/pnas.0610699104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolittle W. F., Papke R. T. (2006). Genomics and the bacterial species problem. Genome Biol. 7, 116–123 10.1186/gb-2006-7-9-116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunning Hotopp J. C., Clark M. E., Oliveira D. C. S. G., Foster J. M., Fischer P., Munoz Torres M. C., Giebel J. D., Kumar N., Ishmael N., Wang S., Ingram J., Nene R. V., Shepard J., Tomkins J., Richard S., Spiro D. J., Ghedin E., Slatko B. E., Tettelin H., Werren J. H. (2007). Widespread lateral gene transfer from intracellular bacteria to multicellular eukaryotes. Science 317, 1753–1756 10.1126/science.1142490 [DOI] [PubMed] [Google Scholar]
- Eco U. (1984). “Dictionary vs. encyclopedia,” in Semiotics and the Philosophy of Language, (Bloomington, USA: Indiana University Press; ), 46–87 [Google Scholar]
- Emelyanov V. V. (2001). Rickettsiaceae, Rickettsia-like endo- symbionts and the origin of mitochondria. Biosci. Rep. 21, 1–17 [DOI] [PubMed] [Google Scholar]
- Esser C., Martin W., Dagan T. (2007). The origin of mitochondria in light of a fluid prokaryotic chromosome model. Biol. Lett. 3, 180–184 10.1098/rsbl.2006.0582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer D., Eisenberg D. (1999). Finding families for genomic ORFans. Bioinformatics 15, 759–762 10.1093/bioinformatics/15.9.759 [DOI] [PubMed] [Google Scholar]
- Fox J. D., Briggs M., Ward P. A., Tedder R. S. (1990). Human herpesvirus 6 in salivary glands. Lancet 336, 590–593 10.1016/0140-6736(90)93392-3 [DOI] [PubMed] [Google Scholar]
- Fraser C. M., Eisen J. A., Salzberg S. L. (2000). Microbial genome sequencing. Nature 406, 799–803 10.1038/35021244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu M., Denga R., Wang J., Wang X. (2008). Detection and analysis of horizontal gene transfer in herpesvirus. Virus Res. 131, 65–76 10.1016/j.virusres.2007.08.009 [DOI] [PubMed] [Google Scholar]
- Garcia-Vallvé S., Romeu A., Palau J. (2000). Horizontal gene transfer in bacterial and archaeal complete genomes. Genome Res. 10, 1719–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiades K., Merhej V., El Karkouri K., Raoult D., Pontarotti P. (2011a). Gene gain and loss events in Rickettsia and Orientia species. Biol. Direct 6, 6 10.1186/1745-6150-6-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiades K., Merhej V., Raoult D. (2011b). The influence of Rickettsiologists on post-modern microbiology. Front. Cell. Infect. Microbiol. 1:8 10.3389/fcimb.2011.00008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiades K., Raoult D. (2011). The rhizome of Reclimonas americana, Homo sapiens, Pediculus humanus and Saccharomyces cerevisiae mitochondria. Biol. Direct 6, 55 10.1186/1745-6150-6-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogarten J. P., Doolittle W. F., Lawrence J. G. (2002). Prokaryotic evolution in light of gene transfer. Mol. Biol. Evol. 19, 2226–2238 [DOI] [PubMed] [Google Scholar]
- Gogarten J. P., Townsend J. P. (2005). Horizontal gene transfer, genome innovation and evolution. Nat. Rev. Microbiol. 3, 679 10.1038/nrmicro1204 [DOI] [PubMed] [Google Scholar]
- Gould S. J. (1987). The empire of the apes. Nat. Hist. 96, 20 [Google Scholar]
- Halary S., Leigh J. W., Cheaib B., Lopez P., Bapteste E. (2010). Network analyses structure genetic diversity in independent genetic worlds. Proc. Natl. Acad. Sci. U.S.A. 107, 127–132 10.1073/pnas.0908978107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao W., Golding G. B. (2008). Uncovering rate variation of lateral gene transfer during bacterial genome evolution. BMC Genomics 9, 235 10.1186/1471-2164-9-235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C. Q., Ding N. Z., He M., Li S. N., Wang X. M., He H. B., Liu X. F., Guo H. C. (2010). Intragenic recombination as a mechanism of genetic diversity in bluetongue virus. J. Virol. 84, 11487–11495 10.1128/JVI.00889-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht M. M., Nitz N., Araujo P. F., Sousa A. O., de Cássia Rosa A., Gomes D. A., Leonardecz D., Teixeira A. R. L. (2010). Inheritance of DNA transferred from American trypanosomes to human hosts. Plos One 5:e9181 10.1371/journal.pone.0009181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller N. L., Ahmed A., Powell E., Martin D. P., Eutsey R., Earl J., Janto B., Boissy R. J., Hogg J., Barbadora K., Sampath R., Lonergan S., Post J. C., Hu F. Z., Ehrlich G. D. (2010). Generation of genetic diversity among Streptococcus pneumoniae strains via horizontal gene transfers during a chronic polyclonal pediatric infection. Plos Pathog. 6:e1001108 10.1371/journal.ppat.1001108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzerlandt R., Orengo C., Kellam P., Alba M. M. (2002). Identification of new herpesvirus gene homologs in the human genome. Genome Res. 12, 1739–1748 10.1101/gr.334302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaper J., Hacker J. (1999). Pathogenicity Islands and Other Mobile Genetic Elements. Washington, DC: ASM Press; 10.1556/AMicr.50.2003.4.1 [DOI] [Google Scholar]
- Kelly L. J., Leitch A. R., Clarkson J. J., Hunter R. B., Knapp S., Chasel M. W. (2009). Intragenic recombination events and evidence for hybrid speciation in Nicotiana (Solanaceae). Mol. Biol. Evol. 27, 781–799 10.1093/molbev/msp267 [DOI] [PubMed] [Google Scholar]
- Kimura M., Crow J. F. (1964). The number of alleles that can be maintained in a finite population. Genetics 49, 725–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klasson L., Westberg J., Sapountzis P., Naslund K., Lutnaes Y., Darby A. C., Veneti Z., Chen L., Braig H. R., Garrett R., Bourtzis K., Andersson S. G. E. (2009). The mosaic genome structure of the Wolbachia wRi strain infecting Drosophila simulans. Proc. Natl. Acad. Sci. U.S.A. 106, 5725–5730 10.1073/pnas.0810753106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloesges T., Popa O., Martin W., Dagan T. (2011). Networks of gene sharing among 329 proteobacterial genomes reveal differences in lateral gene transfer frequency at different phylogenetic depths. Mol. Biol. Evol. 28, 1057–1074 10.1093/molbev/msq297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinidis K. T., Tiedje L. M. (2004). Trends between gene content and genome size in prokaryotic species with larger genomes. Proc. Natl. Acad. Sci. U.S.A. 101, 3160–3165 10.1073/pnas.0308653100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin E. V. (2009). Darwinian evolution in the light of genomics. Nucleic Acids Res. 37, 1011–1034 10.1093/nar/gkp089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin E. V. (2012). “The postmodern state of evolutionary biology,” in The Logic of Chance: The Nature and Origin of Biological Evolution, (Upper Saddle River, NJ: Pearson Education, Inc.), 403–404 [Google Scholar]
- Koonin E. V., Aravind L., Kondrashov A. S. (2000). The impact of comparative genomics on our understanding of evolution. Cell 101, 573–576 [DOI] [PubMed] [Google Scholar]
- Koonin E. V., Mushegian A. R., Galperin M. Y., Walker D. R. (1997). Comparison of archaeal and bacterial genomes: computer analysis of protein sequences predicts novel functions and suggests a chimeric origin of the archaea. Mol. Microbiol. 25, 619–637 [DOI] [PubMed] [Google Scholar]
- Koonin E. V., Galperin M. Y. (1997). Prokaryotic genomes: the emerging paradigm of genome-based microbiology. Curr. Opin. Genet. Dev. 7, 757–763 10.1016/S0959-437X(97)80037-8 [DOI] [PubMed] [Google Scholar]
- Koonin E. V., Wolf Y. I. (2008). Genomics of bacteria and archaea: the emerging dynamic view of the prokaryotic world. Nucleic Acids Res. 36, 6688–6719 10.1093/nar/gkn668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause J., Fu Q., Good J. M., Viola B., Shunkov M. V., Derevianko A. P., Paabo S. (2010). The complete mitochondrial DNA genome of an unknown hominin from southern Siberia. Nature 464, 894–897 10.1038/nature08976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunin V., Goldovsky L., Darzentas N., Ouzounis C. A. (2005). The net of life: reconstructing the microbial phylogenetic network. Genome Res. 15, 954–959 10.1101/gr.3666505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo C. H., Ochman H. (2009). The fate of new bacterial genes. FEMS Microbiol. Rev. 33, 38–43 10.1111/j.1574-6976.2008.00140.x [DOI] [PubMed] [Google Scholar]
- La Scola B., Desnues C., Paguier I., Robert C., Barrassi I., Fournous G., Mechat M., Suzan-Monti M., Forterre P., Koonin E., Raoult D. (2008). The virophage as a unique parasite of the giant mimivirus. Nature 455, 100–104 10.1038/nature07218 [DOI] [PubMed] [Google Scholar]
- Lawrence J. G., Ochman H. (1997). Amelioration of bacterial genomes: rates of change and exchange. J. Mol. Evol. 44, 383–397 [DOI] [PubMed] [Google Scholar]
- Lederberg J., Lederberg E. M., Zinder N. D., Lively E. R. (1951). Recombination analysis of bacterial heredity. Cold Spring Harb. Symp. Quant. Biol. 16, 413–443 [DOI] [PubMed] [Google Scholar]
- Letunic I., Bork P. (2006). Interactive tree of life. Bioinformatics 23, 127–128 10.1093/bioinformatics/btl529 [DOI] [PubMed] [Google Scholar]
- Levine M. T., Jones C. D., Kern A. D., Lindfors H. A., Begun D. J. (2006). Novel genes derived from noncoding DNA in Drosophila melanogaster are frequently X-linked and exhibit testis-biased expression. Proc. Natl. Acad. Sci. U.S.A. 103, 9935–9939 10.1073/pnas.0509809103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez P., Bapteste E. (2009). Molecular phylogeny: reconstructing the forest. C. R. Biol. 332, 171–182 10.1016/j.crvi.2008.07.003 [DOI] [PubMed] [Google Scholar]
- Lukjancenko O., Wassenaar T. M., Ussery D. W. (2010). Comparison of 61 sequenced Escherichia coli Genomes. Microb. Ecol. 60, 708–720 10.1007/s00248-010-9717-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr E. (1957). The Species Problem. Washington, DC: American Association for the Advancement of Science [Google Scholar]
- McNulty S. N., Foster J. M., Mitreva M., Dunning Hotopp J. C., Martin J., Fischer K., Wu B., Davis P. J., Kumar S., Brattig N. W., Slatko B. E., Weil G. J., Fischer P. U. (2010). Endosymbiont DNA in endobacteria-free filarial nematodes indicates ancient horizontal genetic transfer. PLoS One 5:e11029 10.1371/journal.pone.0011029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merhej V., Notredame C., Roeyr-Carenzi M., Pontarotti P., Raoult D. (2011). The rhizome of life: the sympatric Rickettsia felis paradigm demonstrates the random transfer of DNA sequences. Mol. Biol. Evol. 28, 3213–3223 10.1093/molbev/msr239 [DOI] [PubMed] [Google Scholar]
- Merhej V., Raoult D. (2011). Rickettsial evolution in the light of comparative genomics. Biol. Rev. 86, 379–405 10.1111/j.1469-185X.2010.00151.x [DOI] [PubMed] [Google Scholar]
- Merkeev I. V., Mironov A. A. (2008). Orphan genes: function, evolution and composition. Mol. Biol. 42, 127–132 [PubMed] [Google Scholar]
- Mira A., Ochman H., Moran N. A. (2001). Deletional bias and the evolution of bacterial genomes. Trends Genet. 17, 589–596 10.1016/S0168-9525(01)02447-7 [DOI] [PubMed] [Google Scholar]
- Moliner C., Fournier P. E., Raoult D. (2010). Genome analysis of microorganisms living in amoebae reveals a melting pot of evolution. FEMS Microbiol. Rev. 34, 281–294 10.1111/j.1574-6976.2010.00209.x [DOI] [PubMed] [Google Scholar]
- Moreno E. (1998). Genome evolution within the alpha Proteobacteria: why do some bacteria not possess plasmids and others exhibit more than one different chromosome? FEMS Microbiol. Rev. 22, 255–275 [DOI] [PubMed] [Google Scholar]
- Morgan K., Strobeck C. (1979). Is intragenic recombination a factor in the maintenance of genetic variation in natural populations? Nature 277, 383–384 [DOI] [PubMed] [Google Scholar]
- Nakamura Y., Itoh T., Matsuda H., Gojobori T. (2004). Biased biological functions of horizontally transferred genes in prokaryotic genomes. Nat. Genet. 36, 760–766 10.1038/ng1381 [DOI] [PubMed] [Google Scholar]
- Nelson K. E., Clayton R. A., Gill S. R., Gwinn M. L., Dodson R. J., Haft D. H., Hickey E. K., Peterson J. D., Nelson W. C., Ketchum K. A., McDonald L., Utterback T. R., Malek J. A., Linher K. D., Garrett M. M., Steward A. M., Cotton M. D., Pratt M. S., Phillips C. A., Richardson D., Heidelberg J., Sutton G. G., Fleischmann R. D., Eisen J. A., White O., Salzbug S. L., Smith H. O., Venter J. C., Fraser C. M. (1999). Evidence for lateral gene transfer between archaea and bacteria from genome sequence of Thermotoga maritima. Nature 399, 323–329 10.1038/20601 [DOI] [PubMed] [Google Scholar]
- Nikoh N., Tanaka K., Shibata F., Kondo N., Hizume M., Shimada M., Fukatsu T. (2008). Wolbachia genome integrated in an insect chromosome: evolution and fate of laterally transferred endosymbiotic genes. Genome Res. 18, 272–280 10.1101/gr.7144908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman H., Lawrence J. G., Groisman E. A. (2000). Lateral gene transfer and the nature of bacterial innovation. Nature 405, 299–304 10.1038/35012500 [DOI] [PubMed] [Google Scholar]
- Ochman H., Lerat E., Daubin V. (2005). Examining bacterial species under the specter of gene transfer and exchange. Proc. Natl. Acad. Sci. U.S.A. 102, 6595–6599 10.1073/pnas.0502035102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman H., Davalos L. M. (2006). The nature and dynamics of bacterial genomes. Science 311, 1730–1733 10.1126/science.1119966 [DOI] [PubMed] [Google Scholar]
- Ogata H., La Scola B., Audic S., Renesto P., Blanc G., Robert C., Fournier P. E., Claverie J. M., Raoult D. (2006). Genome sequence of Rickettsia bellii illuminates the role of amoebae in gene exchanges between intracellular pathogens. Plos Genet. 5:e76 10.1371/journal.pgen.0020076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata H., Renesto P., Audic S., Robert C., Blanc G., Fournier P. E., Parinello H., Claverie J. M., Raoult D. (2005). The genome sequence of Rickettsia felis identifies the first putative conjugative plasmid in an obligate intracellular parasite. Plos Biol. 3:e248 10.1371/journal.pbio.0030248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace J. K., Gilbert C., Clark M. S., Feschotte C. (2008). Repeated horizontal transfer of DNA transposon in mammals and other tetrapods. Proc. Natl. Acad. Sci. U.S.A. 105, 17023–17028 10.1073/pnas.0806548105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penny D. (2011). Darwin's theory of descent with modification versus the biblical tree of life. PLoS Biol. 9:e1001096 10.1371/journal.pbio.1001096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popa O., Hazkani-Covo E., Landan G., Martin W., Dagan T. (2011). Directed networks reveal genomic barriers and DNA repair bypasses to lateral gene transfer among prokaryotes. Genome Res. 21, 599–609 10.1101/gr.115592.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragan M. A., McInerney J. O., Lake J. A. (2009). The network of life: genome beginnings and evolution. Philos. Trans. R. Soc. Lond. B Biol. Sci. 364, 2169–2175 10.1098/rstb.2009.0046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragan M. A., Beiko R. G. (2009). Lateral genetic transfer: open issues. Philos. Trans. R. Soc. Lond. B Biol. Sci. 364, 2241–2251 10.1098/rstb.2009.0031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raoult D. (2010a). The post-Darwinist rhizome of life. Lancet 375, 104–105 10.1016/S0140-6736(09)61958-9 [DOI] [PubMed] [Google Scholar]
- Raoult D. (2010b). Dépasser Darwin. France: Plon [Google Scholar]
- Raoult D. (2011). A viral grandfather: genomics in 2010 contradict Darwin's vision of evolution. Eur. J. Clin. Microbiol. Infect. Dis. 30, 935–936 10.1007/s10096-010-1151-4 [DOI] [PubMed] [Google Scholar]
- Raoult D., Boyer M. (2010). Amoebae as genitors and reservoirs of giant viruses. Intervirology 53, 321–329 10.1159/000312917 [DOI] [PubMed] [Google Scholar]
- Reich D., Green R. E., Kircher M., Krause J., Patterson N., Durand E. Y., Viola B., Briggs A. W., Stenzel U., Johnson P. L., Maricic T., Good J. M., Marques-Bonet T., Alkan C., Fu Q., Mallick S., Li H., Meyer M., Eichler E. E., Stoneking M., Richards M., Talamo S., Shunkov M. V., Derevianko A. P., Hublin J. J., Kelso J., Slatkin M., Pääbo S. (2010). Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature 468, 1053–1060 10.1038/nature09710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renvoisé A., Merhej V., Georgiades K., Raoult D. (2011). Intracellular Rickettsiales: onsights into manipulators of eukaryotic cells. Trends Mol. Med. 17, 573–583 10.1016/j.molmed.2011.05.009 [DOI] [PubMed] [Google Scholar]
- Rocha M. T. G., Cooper J. E., Smith N. H., Feil E. J. (2006). Comparisons of dN/dS are time dependent for closely related bacterial genomes. J. Theor. Biol. 239, 226–235 10.1016/j.jtbi.2005.08.037 [DOI] [PubMed] [Google Scholar]
- Sabat A. J., Wladyka B., Kosowska-Shick K., Grundmann H., Maarten van Dijl J., Kowal J., Appelbaum P. C., Dubin A., Hryniewicz W. (2008). Polymorphism, genetic exchangeand intragenic recombinationof the aureolysin gene among Staphylococcus aureus strains. BMC Microbiol. 8, 129 10.1186/1471-2180-8-129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schliep K., Lopez P., Lapointe F. J., Bapteste E. (2011). Harvesting evolutionary signals in a forest of prokaryotic gene trees. Mol. Biol. Evol. 28, 1393–1405 10.1093/molbev/msq323 [DOI] [PubMed] [Google Scholar]
- Shao R., Kirkness E. F., Barker S. C. (2009). The single mitochondrial chromosome typical of animals has evolved into 18 mini chromosomes in the human body louse Pediculus humanus. Genome Res. 19, 904–912 10.1101/gr.083188.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro J. A. (1969). Mutation caused by the insertion of genetic laterial into the galactose operon of Escherichia coli. J. Mol. Biol. 40, 93–105 10.1016/0022-2836(69)90298-8 [DOI] [PubMed] [Google Scholar]
- Siew N., Fischer D. (2003). Analysis of singleton ORFans in fully sequenced microbial genomes. Proteins 53, 241–251 10.1002/prot.10423 [DOI] [PubMed] [Google Scholar]
- Snel B., Bork P., Huynen M. A. (2002). Genomes in flux: the evolution of archaeal and proteobacterial gene content. Genome Res. 12, 17–25 10.1101/gr.176501 [DOI] [PubMed] [Google Scholar]
- Sorek R., Zhu Y., Creevey C. J., Francino M. P., Bork P., Rubin E. M. (2007). Genome-wide experimental determination of barriers to horizontal gene transfer. Science 318, 1449–1452 10.1126/science.1147112 [DOI] [PubMed] [Google Scholar]
- Strobeck C., Morgan K. (1978). The effect of intragenic recombination on the number of alleles in a finite population. Genetics 88, 829–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas V., Greub G. (2010). Amoebae/amoebal symbionts genetic transfers: lessons from giant viruses neighbours. Intervirology 53, 254–267 10.1159/000312910 [DOI] [PubMed] [Google Scholar]
- Toll-Riera M., Bosch N., Bellora N., Castelo R., Armengol L., Estivill X., Alba M. M. (2009). Origin of primate orphan genes: a comparative genomics approach. Mol. Biol. Evol. 26, 603–612 10.1093/molbev/msn281 [DOI] [PubMed] [Google Scholar]
- van Passel M. W., Marri P. R., Ochman H. (2008). The emergence and fate of horizontally acquired genes in Escherichia coli. PLoS Comput. Biol. 4:e1000059 10.1371/journal.pcbi.1000059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Via S. (2009). Natural selection in action during speciation. Proc. Natl. Acad. Sci. U.S.A. 106, 9939–9946 10.1073/pnas.0901397106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt B. W. (1972). Intragenic recombination as a source of population genetic variability. Am. Nat. 106, 737–753 [Google Scholar]
- Weismann A. (1889). Essays on Heredity and Kindred Biological Subjects. Oxford, UK: Oxford University Press [Google Scholar]
- Yotova V., Lefebvre J. F., Moreau C., Gbeha E., Hovhannesyan K., Bourgeois S., Bédarida S., Awevedo L., Amorim A., Sarkisian T., Avogbe P., Chabi N., Hama Dicko M., Sabiba Kou' Santa Amouzou E., Sanni A., Roberts-Thomson J., Boettcher B., Scott R. J., Labuda D. (2011). An X-linked haplotype of Neanderthal origin is present among all non-African populations. Mol. Biol. Evol. 28, 1957–1962 10.1093/molbev/msr024 [DOI] [PubMed] [Google Scholar]
- Zhang G., Wang H., Shi J., Wang X., Zheng H., Wong G. K. S., Clark T., Wang W., Wang J., Kang L. (2007). Identification and characterization of insect-specific proteins by genome data analysis. BMC Genomics 8, 93 10.1186/1471-2164-8-93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhaxybayeva O., Gogarten J. P. (2002). Bootstrap, Bayesian probability and maximum likelihood mapping: exploring new tools for comparative genome analyses. BMC Genomics 3, 4 10.1186/1471-2164-3-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q., Zhang G., Zhang Y., Xu S., Zhao R., Zhan Z., Li X., Ding Y., Yang S., Wang W. (2008). On the origin of new genes in Drosophila. Genome Res. 18, 1446–1455 10.1101/gr.076588.108 [DOI] [PMC free article] [PubMed] [Google Scholar]