

Abstract
Efficient determination of protein crystal structures requires automated x-ray data analysis. Here, we describe the expert system elves and its use to determine automatically the structure of a 12-kDa protein. Multiwavelength anomalous diffraction analysis of a selenomethionyl derivative was used to image the Asn-16-Ala variant of the GCN4 leucine zipper. In contrast to the parallel, dimeric coiled coil formed by the WT sequence, the mutant unexpectedly formed an antiparallel trimer. This structural switch reveals how avoidance of core cavities at a single site can select the native fold of a protein. All structure calculations, including indexing, data processing, locating heavy atoms, phasing by multiwavelength anomalous diffraction, model building, and refinement, were completed without human intervention. The results demonstrate the feasibility of automated methods for determining high-resolution, x-ray crystal structures of proteins.
Determining the crystal structure of a large molecule is generally a complicated, multistep process that requires considerable time and training to accomplish. Any step can fail, and the manifold computational inputs generally prevent optimization of any step. X-ray structural analysis has been facilitated recently by new experimental methods such as multiwavelength anomalous diffraction (MAD) analysis (1) and by powerful new algorithms for locating heavy atoms, model building, and structural refinement. In particular, the solve and resolve programs have been adopted widely for locating heavy atoms, calculating electron density maps, and modeling (2, 3). The arp/warp program enabled automated model building and refinement (4). Comprehensive program packages such as the CCP4 suite (5) have integrated diverse methods through standardized interfaces and file structures. These methods to speed individual computational steps have created the opportunity to fully automate macromolecular x-ray structure determination.
We developed the expert system elves to automate analysis of crystallographic data without precluding manual control of the process (Fig. 1). elves programs have been used to speed and optimize steps in the determination of many x-ray crystal structures ranging from 8 to 330 kDa in the crystallographic asymmetric unit (au). These structures, solved in several different laboratories, include the human TRAF2/CD40 complex (14), the human papilloma virus E2 protein (15), aspartate transcarbamoylase (16), Escherichia coli primase (17), the tandem bromo domains of human TAF250 (18), the dimerization domain of hepatocyte nuclear factor 1α (19), the human MIA protein (20), the replication initiation factor, DnaA (21), a designed ankyrin repeat motif (22), and the PknB Ser/Thr protein kinase (23). In most of these cases, the elves programs wedger, scaler, phaser, and refmacer (Fig. 1) were used to carry out individual steps or groups of steps under user direction. As an expert system, elves chooses reasonable starting input parameters for each step, optimizes parameters, and marshals several strategies to detect and overcome common failures in the calculations.
Fig. 1.

Levels of automation in elves. elves automates widely used crystallographic software (Lower) that are currently run by means of user-specified scripts. Green wedges indicate the scope of each elves program. Four programs make up the first level of elves automation. wedger processes a single wedge of data by using mosflm (6). scaler performs local scaling (7) and merges multiple wedges of data by using the indicated programs. phaser locates heavy atoms with shelx (8) or rantan (5) and refines and searches for additional heavy atoms with mlphare (9). mlphare and dm (10) are used to calculate phases, and the electron density map is calculated with the CCP4 suite (5). Scripts for o (11), arp/warp (4), refmac (12), and cns (13) are produced. refmacer uses arp/warp and refmac to build and refine a molecular model. These programs are coordinated by the next level of automation, called processer, which also runs solve (2) and arp/warp (4). The elves main program runs processer in the fully automated mode.
Here, we test the ability of elves to determine a protein crystal structure fully automatically. Starting with data frames for three different crystal forms, elves determined the structures of the Asn-16-Ala mutant of the GCN4 leucine zipper. The GCN4 leucine zipper forms a prototypical, parallel, two-helical coiled coil (24). Coiled coils are ropes of two to five helices that perform dynamic oligomerization functions in 3-5% of proteins (25). Understanding how the number of associated helices is determined by coiled-coil sequences has provided insights into the basis of structural uniqueness (26), a general characteristic of proteins that is compromised in protein misfolding diseases. Mutations of Asn-16, a buried polar amino acid, can allow the GCN4 leucine zipper to adopt a mixture of two- and three-helical coiled coils (26-29). In the parallel dimers and trimers formed by hydrophobic replacements of Asn-16, for example, structural specificity was lost because different sets of atoms created similar packing surfaces in alternative folds (27). Strikingly, addition of benzene to the Asn-16-Ala mutant promoted an allosteric switch from a mixture of dimers and trimers to a fully trimeric state (29). The switch in helix number was accompanied by benzene binding to a core hydrophobic cavity present only in the parallel trimer. To better understand the nature of this allosteric switch, we determined the crystal structure of the GCN4 Asn-16-Ala mutant in the absence of benzene. Comparisons with manual calculations demonstrated the accuracy of the structures determined by elves.
Materials and Methods
Synthesis, Purification, and Crystallization. The selenomethionine derivative of GCN4-p1-Asn-16-Ala was synthesized by using solid-phase methods and purified by using reversed-phase HPLC (19). Crystals were grown by vapor diffusion from 1:1 mixtures of 20 mg/ml protein and reservoir solution. Tetragonal crystals (P43212) were grown from 25 mM phosphate, pH 7.0, 12% PEG 1450 (wt/vol), and 400 mM NaBr and frozen in liquid N2 after concentrating the drop ≈2-fold by evaporation. Trigonal crystals (P3121) were grown from 100 mM bis-Tris, pH 7.3. These crystals were transferred slowly into 50 mM phosphate, pH 7.4, 20% 2-methyl-2,4-pentanediol (vol/vol), and 5% PEG 8000 (wt/vol), which allowed flash cooling in liquid N2. A second tetragonal form grew under nearly identical conditions as the trigonal crystals at pH values slightly <7.3. At room temperature, these crystals diffracted x-rays to ≈3-Å resolution and had body-centered symmetry (I41). The symmetry collapsed to P43 upon flash freezing. X-ray data were collected at the Stanford Synchrotron Radiation Laboratory Beam Line 1-5 (Stanford, CA) at 100 K.
X-Ray Structure Determination and Analysis. Each automated structure determination entailed issuing a single command to the elves expert system. elves is a self-configuring shell program that can be installed rapidly on common Unix or Linux systems. elves uses a three-level hierarchy of automation programs that write and optimize scripts for standard crystallographic analysis software (Fig. 1). The individual programs of the elves control hierarchy are packaged within a single text file, and they are deployed as separate scripts when the elves program is run.
Rather than a traditional graphical user interface, a conversational user interface (CUI) was created for the elves programs to translate English language input into values for program variables. The CUI is based on a simple search engine that recognizes crystallographic terms and associates them with command key words and parameter values. The parameter values are then restated succinctly to the user for verification, modeling an English conversation. This verification step makes the CUI more robust than standard, declaration-based interfaces, because it is easier for users to identify incorrect parameters than to input the parameters without error. Lower levels of the hierarchy (Fig. 1) have less abstract interfaces, less automation, and more user options. At the lowest level, users edit standard scripts and pass them to elves for optimization. In this work, elves was directed to accept the computed parameters without user verification.
The levels of abstraction for user input are mirrored by the abstraction of the program outputs. The ultimate product of an elves run is a standardized directory tree containing scripts and output files of all of the programs used in the structure solution. Toward the top of the hierarchy (Fig. 1), elves presents a more distilled version of the underlying program output. Focusing on the key parameters that change the most from project to project makes it easier to evaluate the results. For example, wedger elves reports the most critical parameters refined by mosflm, such as the unit cell and mosaic spread, but less commonly changed parameters such as beam divergence and polarization are reported only in the mosflm script itself. In turn, processer elves reports a summary of the output of wedger elves, such as the image file names and the location of the detailed log file.
elves programs optimize procedures by repeatedly writing and running scripts and examining the output statistics as a function of input parameters. At the end of this process, the best input parameters are used to run the procedure, and the results are passed to the next step. For example, different choices of crystal symmetry are used to merge symmetry-equivalent observations, and the highest symmetry producing the acceptable merging statistics is chosen. Other parameters, such as the crystal unit cell dimensions, are input iteratively into their appropriate refinement programs until the input and output parameter values match. This automated optimization of inputs is faster and more reliable than manually editing scripts, and it reduces the accumulation of errors that can lead to an uninterpretable electron density map.
To manually determine the x-ray structures of the trigonal and tetragonal crystals, data were processed with explicit scripts for mosflm (6). After scaling and merging (7), heavy atoms were located in difference Patterson maps with rsps (5). Minor sites were found in difference Fourier maps, and mlphare (9) was used to refine phases. Using O (11), a helix of the GCN4-pII parallel-trimer structure (30) was docked into the electron density of each of the helices in the MAD-phased map. The structure was refined with refmac (12) and rebuilt in O. Most of the solvent structure was placed by using ARP (4). Twenty-two (trigonal) and 26 (tetragonal) side chains were found in multiple conformations in the experimentally phased maps.
Cavity volumes were calculated with voidoo (31). Structure quality was monitored with procheck (32). Helical and superhelical parameters were calculated by using the program fitcc (Mark Sales, personal communication; http://ucxray.berkeley.edu/~mark/fitcc.html).
Results
The GCN4 Asn-16-Ala mutant produced three different crystal forms. The crystals displayed the symmetry of space groups P3121, P43212, and P43 and diffracted to 1.8-, 1.8-, and 2.7-Å resolution, respectively. Phases for all three crystal forms were determined by MAD analysis (Table 1). Met-2 in the Asn-16-Ala peptide was replaced with selenomethionine by solid-phase peptide synthesis (19). The selenopeptide and the underivitized peptide produced isomorphous crystals in all three forms. The structures were determined automatically by using elves.
Table 1. Comparison of x-ray data collection and refinement statistics for the elves-automated calculations and the manual determinations of the Asn-16-Ala structure.
| Automated structure determination
|
Manual structure determination
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Low | f′ | Mid | f″ | High | Low | f′ | Mid | f″ | High | |
| P3121 | ||||||||||
| Wavelength, Å | 1.0688 | 0.9800 | 0.9795 | 0.9322 | 1.0688 | 0.9800 | 0.9795 | 0.9322 | ||
| Resolution, Å | 1.8 | 1.8 | 1.8 | 1.8 | 1.8 | 1.8 | 1.8 | 1.8 | ||
| Rsym* | 0.064 (0.98) | 0.069 (0.98) | 0.073 (1.07) | 0.083 (1.26) | 0.048 (0.29) | 0.067 (0.83) | 0.062 (0.59) | 0.064 (0.67) | ||
| Completeness, % | 95.6 (76.6) | 99.3 (97.1) | 99.6 (98.4) | 99.2 (97.1) | 93.6 (67.3) | 99.6 (97.5) | 99.4 (96.3) | 96.0 (78.1) | ||
| Multiplicity | 15.4 (5.3) | 19.7 (10.6) | 22.4 (12.0) | 15.7 (12.0) | 8.7 (1.7) | 18.7 (9.4) | 17.5 (5.8) | 10.9 (3.9) | ||
| I/σ(I) | 20.8 (2.9) | 21.1 (4.2) | 21.2 (4.0) | 14.9 (3.5) | 23.5 (2.0) | 28.9 (3.2) | 27.7 (2.9) | 20.6 (2.6) | ||
| Phasing power† | 1.49/0.11 | 0.18/0.27 | -/0.84 | 1.55/0.56 | 1.67/- | -/0.31 | 0.12/1.07 | 1.61/0.7 | ||
| Mean figure of merit‡ | 0.605 (0.897) | 0.540 (0.783) | ||||||||
| Correlation coefficient§ | 0.659 | 0.528 | ||||||||
| Rcryst/Rfree¶ | 0.226/0.357 | 0.202/0.257 | ||||||||
| rmsd bonds, rmsd angles∥ | 0.015 Å, 1.8° | 0.012 Å, 2.0° | ||||||||
| P43212 | ||||||||||
| Wavelength, Å | 1.0688 | 0.9800 | 0.9797 | 0.9795 | 0.9322 | 1.0688 | 0.9800 | 0.9797 | 0.9795 | 0.9322 |
| Resolution, Å | 1.8 | 1.8 | 1.8 | 1.8 | 1.8 | 1.8 | 1.8 | 1.8 | 1.8 | 1.8 |
| Rsym* | 0.082 (0.37) | 0.080 (0.42) | 0.064 (0.56) | 0.081 (0.46) | 0.086 (0.55) | 0.063 (0.41) | 0.069 (0.44) | 0.063 (0.58) | 0.069 (0.47) | 0.079 (0.59) |
| Completeness, % | 89.0 (57.1) | 97.0 (82.7) | 95.2 (75.2) | 97.0 (82.8) | 99.3 (98.2) | 88.3 (55.1) | 96.6 (80.0) | 96.7 (74.8) | 96.7 (80.4) | 99.1 (95.6) |
| Multiplicity | 15.6 (7.8) | 17.4 (11.6) | 11.6 (8.2) | 17.7 (12.0) | 18.0 (14.1) | 15.5 (7.5) | 17.1 (10.8) | 11.3 (7.7) | 17.1 (10.8) | 17.7 (12.8) |
| I/σ(I) | 23.2 (4.9) | 23.0 (5.6) | 19.7 (3.8) | 23.2 (5.7) | 21.5 (5.3) | 30.1 (4.7) | 29.5 (5.5) | 22.0 (3.1) | 30.1 (5.4) | 28.2 (5.2) |
| Phasing power† | 0.94/0.18 | 0.15/0.51 | -/0.64 | 0.27/0.93 | 0.9/0.78 | 1.27/1.03 | 1.03/0.72 | n/a | 1.05/1.19 | 0.63/0.79 |
| Mean figure of merit‡ | 0.515 (0.854) | 0.556 (0.612) | ||||||||
| Correlation coefficient§ | 0.711 | 0.619 | ||||||||
| Rcryst/Rfree¶ | 0.202/0.307 | 0.223/0.241 | ||||||||
| rmsd bonds, rmsd angles∥ | 0.009 Å, 0.9° | 0.013 Å, 2.1° | ||||||||
Values in parentheses refer to the highest resolution shell. The results were tabulated by Table1.com, an Elves utility that examines log files and culls standard statistical values. The differences between the Rsym values of the automated and manual runs were largely caused by the choice of standard deviation correction parameters (SDCORR) in scala (7). Applying the SDCORR parameters from the automated run to the manually reduced data produced nearly identical statistics to the automated run and vice versa. The higher figures of merit and correlation coefficients observed by using the automatically optimized SDCORR parameters suggest that elves produced more realistic estimates that improved the maximum-likelihood phasing and refinement. Na, not available.
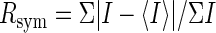 ; I, intensity
; I, intensity
Phasing power (dis/ano) = 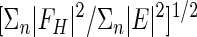 ; FH, calculated heavy atom scattering factor; E, lack of closure error
; FH, calculated heavy atom scattering factor; E, lack of closure error
Mean figure of merit (50- to 1.8-Å resolution) = 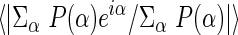 ; α, phase; P(α), phase probability distribution
; α, phase; P(α), phase probability distribution
Correlation coefficient = 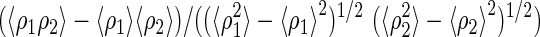 , ρ = electron density map
, ρ = electron density map
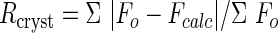 ; Fo, observed structure-factor amplitude; Fcalc, calculated structure-factor amplitude
; Fo, observed structure-factor amplitude; Fcalc, calculated structure-factor amplitude
rmsds from ideal values
elves was launched with a single command containing the location of the diffraction image data and the protein sequence. No other program interfaces were used. For all three structures, the elves main program set up the data processing file system and then handed control to processer elves, which directed the actual program runs and coordinated the parallel use of up to eight computer processors (Fig. 1). wedger elves indexed and processed the x-ray data with mosflm (6). scaler elves scaled the resulting intensities by using the CCP4 program scala (7). phaser elves ran shelx (8) to locate the heavy atom sites, refined the heavy atom parameters with mlphare (9), and did solvent flattening with DM (10). Additional, weak selenium positions corresponding to alternate selenomethionine rotamers were located in difference Fourier maps by the elves program phaser, and these sites were automatically added to the phase calculations. Parameters such as the crystal symmetry and solvent content were determined automatically by performing the scaling, phasing, and model building procedures in each of the six (trigonal) or eight (tetragonal) possible space groups and repeating the solvent flattening run with a series of solvent contents. In each case, the space group and solvent content choices that produced the best scaling (Rmerge), phasing (mean figure of merit), and model (crystallographic R) statistics was selected automatically. The resulting experimental electron density maps (Fig. 2) clearly showed each structure.
Fig. 2.

Experimental, 1.8-Å resolution MAD-phased electron density map (P3121, contoured at 1 σ) produced by elves superimposed on the refined model of the GCN4 Asn-16-Ala leucine-zipper variant. (A) Cross section through the trimer at the level of the Ala-16-Leu-12-Leu-12 layer. (B) Cross section through the trimer at the level of the Leu-12-Ala-16-Ala-16 layer.
For the two high-resolution cases, processer elves ran arp/warp (4) to build and refine molecular models (Fig. 3). The models produced by elves had R/Rfree values of 0.202/0.307, for the tetragonal crystals and 0.226/0.357 for the trigonal form. These models had excellent stereochemistry and they compared favorably with models built and refined manually by using the programs O (11) and refmac (12) (Table 1). The main features apparent in the hand-built models that were not in the automatically determined structures were 22-26 residues assigned to multiple conformers and five additional, relatively disordered residues at the C termini.
Fig. 3.

The Asn-16-Ala variant of the GCN4 leucine zipper forms an antiparallel, trimeric coiled coil. (A) Stereo ribbon diagram of the overall structure. Each helix is colored in increasingly cool colors from the amino to the carboxyl terminus. (B) Superpositions of the structures determined by automated (blue) and manual (yellow) methods in the trigonal (Left) and tetragonal (Right) crystal forms. (C) Cross section showing the Ala-16-Leu-12-Leu-12 layer with the van der Waals surfaces of the core amino acids filling the space in the core of the trimer. (D) Close packing of the Leu-12-Ala-16-Ala-16 layer. (E) In contrast, a 165-Å3 cavity exists in the Ala-16 layer of the parallel trimer stabilized by benzene (29).
For the 2.7-Å resolution data from the P43 crystals, elves found 16 heavy atom sites and stopped after producing the electron density map, because the arp/warp algorithm was not expected to succeed at this resolution. The electron density map showed four antiparallel trimers (48 kDa) in the au displaying the same core packing arrangements seen in the two high-resolution structures. Crystal contacts were similar to the other tetragonal form. Because of the reduced resolution, this structure was not analyzed in more detail.
Program Timing. The automatic analysis of the 816 diffraction images recorded from the P43212 crystals into a refined molecular model was completed in 9.5 h on eight 2-GHz Athlon Linux (AMD, Sunnyvale, CA) computers (Table 2). Data processing took 39 min, and the electron density map (Fig. 2) was produced in <5 h. Using 270 frames, a clear electron density map was produced with two-wavelength MAD phases in <1 h. Because the crystal symmetry was not specified by the input parameters, elves used eight computer processors to simultaneously perform the phasing calculations in eight possible tetragonal space groups to avoid misassignment of the symmetry. For the trigonal crystal form, 1,023 x-ray diffraction images at four wavelengths were converted to the refined model in 165 h on four 450-MHz Pentium processors (Intel, Santa Clara, CA).
Table 2. Progress of the crystal structure of the GCN4 Asn-16–Ala variant determined by elves running on an eight-processor, 2-GHz Athlon, Linux computer.
| elf | Task | Timing | Results |
|---|---|---|---|
| wedger | Indexing | 38 sec | P4 symmetry |
| processer | Data integration (mosflm) | 39 min | 816 frames, 5 λs |
| scaler | Scale wedges (scala) | 3.5 hr | Rsym = 0.066-0.075 |
| phaser | Locate Ses (shelx) | 3 min | 3 sites |
| phaser | Refine Ses (mlphare) | 13 min | 〈m〉 = 0.507 |
| phaser | Solvent flatten (dm) | 15 min | 〈m〉 = 0.708 |
| phaser | Calculate electron density map (CCP4) | <1 min | |
| phaser | Se search and phasing in seven alternate space groups | 23 min | P43212 symmetry |
| processer | Build and refine model (arp/warp) | 4 hr | R/Rfree = 0.20/0.31 |
| Total time | 9.5 hr |
Helix Flip in the Mutant Leucine Zipper. All previous crystal structures of GCN4 variants have revealed coiled coils of two, three, or four parallel helices (26-30). In the presence of benzene, the Asn-16-Ala mutant crystallized as a parallel trimer with benzene in the core (29). Correspondingly, in the absence of benzene, we expected the structure to form a parallel dimer or trimer. To the contrary, the new crystal structures reveal that the Asn-16-Ala variant forms a trimeric coiled coil with antiparallel chains (Fig. 3). The observation of the antiparallel, trimeric structure in three different crystal forms supports the conclusion that crystal-packing forces are not responsible for the reversal in helix direction.
Although each chain is chemically identical, the two-up-one-down arrangement makes the helices structurally distinct. The Cα rms deviation (rmsd) between individual helices ranged from 0.19 to 0.50 Å, indicating the limits of structural adaptation. Characteristic of antiparallel coiled coils (33-36), residues occupying both core hydrophobic positions (called a and d) of the seven-residue sequence repeat occur together in each layer of the Asn-16-Ala structure (Fig. 3). Layers containing two a positions and one d position (a-a-d) alternate with layers containing two d positions and one a position (d-d-a). Ala-16 occurs in adjacent layers containing Ala-16-Ala-16-Leu-12 and Leu-12-Leu-12-Ala-16 (Figs. 2 and 3).
Discussion
Determinants of Structural Uniqueness. Formation of a unique protein structure requires a free-energy gap between the native fold and all alternate conformational ensembles. Even closely related structures must be destabilized. How is such a free energy gap created? The structural polymorphism of GCN4 leucine zipper variants provides unique information about the basis for specificity, because similar or identical sequences adopt different structures. The Asn-16-Ala variant illustrates two mechanisms of destabilizing alternate folds. As anticipated (24), burial of the WT Asn-16 in the core layers containing Leu-12 destabilizes antiparallel arrangements relative to the WT, parallel dimer (37-39). These findings reveal that the WT core asparagine strongly influences not only the number of helices, but also the parallel direction of helices in the GCN4 leucine zipper.
In addition, the Asn-16-Ala mutation favors the antiparallel helical trimer over the parallel trimer observed in the presence of benzene (29). The benzene molecule occupies a large cavity (165 Å3) formed by the three Ala-16 residues grouped in the central layer of the parallel trimer. In contrast, no cavities large enough to accommodate water occur in the layers containing Ala-16 in the antiparallel trimer. Because parallel trimers form when Asn-16 is replaced by larger hydrophobic residues such as valine (26), avoidance of the core cavity provides the driving force for the observed antiparallel arrangement of Asn-16-Ala chains. Based on the energetic penalty of 5.0 kcal/mol measured for a 150-Å3 cavity in phage T4 lysozyme (40), the central cavity can be estimated to destabilize the parallel Asn-16-Ala trimer by ≈5.5 kcal/mol. Thus, in addition to strong forces such as steric overlap and burial of polar residues (26-28, 36-38), structural relaxations that reduce core cavities significantly influence the energy gap between alternate protein folds.
Accelerating Crystallography. The use of elves to determine the GCN4 Asn-16-Ala trimer structures demonstrates that high-resolution crystal structures with well-ordered metals can be determined automatically. The Cα rmsd of residues 1-30 in the structures determined by elves and manual methods was 0.09 Å for the tetragonal form and 0.16 Å for the trigonal form. By comparison, the rmsd for Cα atoms in the finished models from the trigonal and tetragonal crystal forms was 0.35 Å. The helical and superhelical parameters for the structures determined by automated and manual methods also coincided closely (Table 3). These results indicate that the program runs managed by elves produced models that accurately revealed the protein structure.
Table 3. Helical and superhelical parameters of the Asn-16—Ala GCN4 leucine zipper trimers.
| Crystal form
|
||||
|---|---|---|---|---|
| P3121
|
P43212
|
|||
| Structural methods | Auto | Hand | Auto | Hand |
| rmsd from ideal superhelix, Å | 0.71 | 0.69 | 0.79 | 0.80 |
| Supercoil radius, R0, Å | 6.1 | 6.1 | 6.0 | 6.0 |
| Residues per supercoil turn | 99 | 100 | 98 | 99 |
| Supercoil pitch, Å | 144 | 146 | 143 | 144 |
| Radius of curvature, Å | 92 | 95 | 92 | 93 |
| Superhelix crossing angle, χ | 29.9° | 29.3° | 29.6° | 29.5° |
| Position a orientation angle, ϕ | 68.5° | 67.9° | 67.8° | 67.8° |
| Residues per α-helix turn, n | 3.62 | 3.62 | 3.63 | 3.63 |
| Rise/residue, d, Å | 1.50 | 1.51 | 1.50 | 1.50 |
| α-helix radius (Cα), R1, Å | 2.24 | 2.24 | 2.22 | 2.22 |
| Pairwise helix-crossing angle, ω0 | 25.9° | 25.3° | 25.6° | 25.4° |
| Pairwise interhelix distance, D, Å | 10.6 | 10.5 | 10.5 | 10.4 |
The limitations of elves are those inherent in the underlying algorithms. The GCN4 Asn-16-Ala structures contain 12 or 48 kDa in the au. Reconstruction experiments indicate that the previously solved structures of MIA (24 kDa/au, 14 Se sites, ref. 20) and DnaG (38 kDa/au, 9 Se sites, ref. 17) can be determined automatically (data not shown). elves also automatically produced interpretable electron density maps of TRAF2/CD40 (64 kDa/au, 6 Hg sites, ref. 14) and PknB (66 kDa/au, 11 Se sites, ref. 23) at 2.4- and 3.0-Å resolution, respectively. These results indicate that diverse structures can be solved automatically by using elves. Currently, the most stringent limitations are imposed by the model-building step. Success of the arp/warp program used to build the initial model requires an accurately phased electron density map calculated at ≈2.3-Å resolution (4). As additional methods are developed, they can be incorporated into the elves system as long as they are driven by scripts. In this sense, elves provides a general, flexible, computational framework that reduces the time and training required to determine macromolecular crystal structures.
In our experience, elves is incapable of overcoming problems arising from poor data or inadequate phasing signal. Problems such as radiation damage, weak heavy atom signals, twinning, poor heavy atom models, low resolution, or crystal disorder that hinder crystallographic projects are not overcome by automation. Instead, the elves system accelerates standard procedures that are often sufficient to either determine a structure or indicate rapidly what problems must be overcome. Trivial errors (such as typos) are avoided, and the systematic exploration of parameters (e.g., the crystal symmetry, the number of heavy atoms, the hand of the heavy atom solution, and the solvent content) affords a comprehensive analysis of the data. Automated analysis of a single-wavelength anomalous diffraction data set, for example, can demonstrate the utility of a given derivative crystal while more comprehensive data collection is in progress.
An important feature of elves is the CUI that interprets English-language commands and communicates with users. For the structures of the Asn-16-Ala mutant leucine zipper, the CUI was used only to interpret the initial command at the highest level of elves. For semiautomated operation (14-23), the CUI affords a simple means of interpreting any input and implementing a defined computational strategy. The hierarchical structure of elves (Fig. 1) allows experienced users to select particular automation features of the system and rapidly implement novel computational strategies. This flexibility increases throughput even on difficult crystallographic problems that require more experience and manual input to solve (14-23). The combination of automated analysis demonstrated here and the semiautomated operation driven by the elves CUI decreases the time and training required to perform the computational steps of x-ray crystallography. This capability can increase the efficiency of protein crystallography beamlines at synchrotron sources and facilitate projects in structural genomics and structural biology.
Acknowledgments
We thank the many scientists who tested elves during development. D. King carried out peptide synthesis and mass spectrometry. X-ray data were collected at the Stanford Synchrotron Radiation Laboratory, which is operated by the Department of Energy and supported by the National Institutes of Health. Calculations used Beam Line 8.3.1 at the Advanced Light Source, which was funded by the National Science Foundation, the University of California, and Henry Wheeler. This work was supported by a University of California Campus-Laboratory Collaboration Grant and a grant from the National Institutes of Health. elves is distributed by the University of California (http://ucxray.berkeley.edu/~jamesh/elves).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: au, asymmetric unit; CUI, conversational user interface; MAD, multiwavelength anomalous diffraction; rmsd, rms deviation.
Data deposition: The coordinates and structure factors have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID codes 1RB1, 1RB4, 1RB5, and 1RB6).
References
- 1.Hendrickson, W. A. (1991) Science 254, 51-58. [DOI] [PubMed] [Google Scholar]
- 2.Terwilliger, T. C. & Berendzen, J. (1999) Acta Crystallogr. D 55, 849-861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Terwilliger, T. C. (2000) Acta Crystallogr. D 56, 965-972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morris, R. J., Perrakis, A. & Lamzin, V. S. (2002) Acta Crystallogr. D 58, 968-975. [DOI] [PubMed] [Google Scholar]
- 5.Collaborative Crystallography Project 4 (1994) Acta Crystallogr. D 50, 760-763.15299374 [Google Scholar]
- 6.Leslie, A. G. W. (1991) in Crystallographic Computing 5: From Chemistry to Biology, eds. Moras, D., Podjarny, A. D. & Thiery, J. C. (Oxford Univ. Press, Oxford), pp. 27-38.
- 7.Kabsch, W. (1988) J. Appl. Crystallogr. 21, 916-924. [Google Scholar]
- 8.Sheldrick, G. M. (1990) Acta Crystallogr. A 46, 467-473. [Google Scholar]
- 9.Otwinowski, Z. (1991) in Proceedings of the CCP4 Study Weekend, eds. Wolf, W., Evans, P. R. & Leslie, A. G. W. (Science and Engineering Research Council, Daresbury Laboratory, Warrington, U.K.), pp. 80-86.
- 10.Cowtan, K. (1994) Joint CCP4 ESF-EACBM Newlett. Protein Crystallogr. 31, 34-38. [Google Scholar]
- 11.Jones, T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard, M. (1991) Acta Crystallogr. A 47, 110-119. [DOI] [PubMed] [Google Scholar]
- 12.Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997) Acta Crystallogr. D 53, 240-255. [DOI] [PubMed] [Google Scholar]
- 13.Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., et al. (1998) Acta Crystallogr. D 54, 905-921. [DOI] [PubMed] [Google Scholar]
- 14.McWhirter, S. M., Pullen, S. S., Holton, J. M., Crute, J. J., Kehry, M. R. & Alber, T. (1999) Proc. Natl. Acad. Sci. USA 96, 8408-8413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harris, S. F. & Botchan, M. R. (1999) Science 284, 1673-1677. [DOI] [PubMed] [Google Scholar]
- 16.Endrizzi, J. A., Beernink, P. T., Alber, T. & Schachman, H. K. (2000) Proc. Natl. Acad. Sci. USA 97, 5077-5082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keck, J. L., Roche, D. D., Lynch, A. S. & Berger, J. M. (2000) Science 287, 2482-2486. [DOI] [PubMed] [Google Scholar]
- 18.Jacobson, R. H., Ladurner, A. G., King, D. S. & Tjian, R. (2000) Science 288, 1422-1425. [DOI] [PubMed] [Google Scholar]
- 19.Rose, R. B., Endrizzi, J. A., Cronk, J. D., Holton, J. & Alber, T. (2000) Biochemistry 39, 15062-15070. [DOI] [PubMed] [Google Scholar]
- 20.Lougheed, J. C., Holton, J. M., Alber, T., Bazan, J. F. & Handel, T. M. (2001) Proc. Natl. Acad. Sci. USA 98, 5515-5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Erzberger, J. P., Pirruccello, M. M. & Berger, J. M. (2002) EMBO J. 21, 4763-4773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mosavi, L. K., Minor, D. L., Jr., & Peng, Z. Y. (2002) Proc. Natl. Acad. Sci. USA 99, 16029-16034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Young, T. A., Delagoutte, B., Endrizzi, J. A. & Alber, T. (2003) Nat. Struct. Biol. 10, 168-174. [DOI] [PubMed] [Google Scholar]
- 24.O'Shea, E. K., Klemm, J. D., Kim, P. S. & Alber, T. (1991) Science 254, 539-544. [DOI] [PubMed] [Google Scholar]
- 25.Wolf, E., Kim, P. S. & Berger, B. (1997) Protein Sci. 6, 1179-1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harbury, P. B., Zhang, T., Kim, P. S. & Alber, T. (1993) Science 262, 1401-1407. [DOI] [PubMed] [Google Scholar]
- 27.Gonzalez, L., Jr., Brown, R. A., Richardson, D. & Alber, T. (1996) Nat. Struct. Biol. 3, 1002-1010. [DOI] [PubMed] [Google Scholar]
- 28.Gonzalez, L., Jr., Woolfson, D. N. & Alber, T. (1996) Nat. Struct. Biol. 3, 1011-1018. [DOI] [PubMed] [Google Scholar]
- 29.Gonzalez, L., Jr., Plecs, J. J. & Alber, T. (1996) Nat. Struct. Biol. 3, 510-515. [DOI] [PubMed] [Google Scholar]
- 30.Harbury, P. B., Kim, P. S. & Alber, T. (1994) Nature 371, 80-83. [DOI] [PubMed] [Google Scholar]
- 31.Kleywegt, G. J. & Jones, T. A. (1994) Acta Crystallogr. D 50, 178-185. [DOI] [PubMed] [Google Scholar]
- 32.Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993) J. Appl. Crystallogr. 26, 283-291. [Google Scholar]
- 33.Lovejoy, B., Choe, S., Cascio, D., McRorie, D. K., DeGrado, W. F. & Eisenberg, D. (1993) Science 259, 1288-1293. [DOI] [PubMed] [Google Scholar]
- 34.Yan, Y., Winograd, E., Viel, A., Cronin, T., Harrison, S. C. & Branton, D. (1993) Science 262, 2027-2030. [DOI] [PubMed] [Google Scholar]
- 35.Biou, V., Yaremchuk, A., Tukalo, M. & Cusack, S. (1994) Science 263, 1404-1410. [DOI] [PubMed] [Google Scholar]
- 36.Walshaw, J. & Woolfson, D. N. (2001) J. Mol. Biol. 307, 1427-1450. [DOI] [PubMed] [Google Scholar]
- 37.Lumb, K. J. & Kim, P. S. (1995) Biochemistry 34, 8642-8648. [DOI] [PubMed] [Google Scholar]
- 38.Hendsch, Z. S., Jonsson, T., Sauer, R. T. & Tidor, B. (1996) Biochemistry 35, 7621-7625. [DOI] [PubMed] [Google Scholar]
- 39.Akey, D. L., Malashkevich, V. N. & Kim, P. S. (2001) Biochemistry 40, 6352-6360. [DOI] [PubMed] [Google Scholar]
- 40.Eriksson, A. E., Baase, W. A., Zhang, X. J., Heinz, D. W., Blaber, M., Baldwin, E. P. & Matthews, B. W. (1992) Science 255, 178-183. [DOI] [PubMed] [Google Scholar]