

Abstract
Analyses of humoral responses against different infectious agents are critical for infectious disease diagnostics, understanding pathogenic mechanisms, and the development and monitoring of vaccines. While ELISAs are often used to measure antibody responses to one or several targets, new antibody-profiling technologies, such as protein microarrays, can now evaluate antibody responses to hundreds, or even thousands, of recombinant antigens at one time. These large-scale studies have uncovered new antigenic targets, provided new insights into vaccine research and yielded an overview of immunoreactivity against almost the entire proteome of certain pathogens. However, solid-phase antigen arrays also have drawbacks that limit the type of information obtained, including suboptimal detection of conformational epitopes, high backgrounds due to impure antigens and a narrow dynamic range of detection. We have developed a solution-phase antibody-profiling technology, luciferase immunoprecipitation systems (LIPS), which harnesses light-emitting recombinant antigen fusion proteins to quantitatively measure patient antibody titers. Owing to the highly linear light output of the luciferase reporter, some antibodies can be detected without serum dilution in a dynamic range of detection often spanning seven orders of magnitude. When LIPS is applied iteratively with multiple target antigens, a high-definition antibody profile is obtained. Here, we discuss the application of these different antibody-profiling technologies and their associated limitations with particular emphasis on protein microarrays. We also describe LIPS in detail and discuss several clinically relevant uses of the technology. Together, these new technologies offer new tools for understanding humoral responses to known and emerging infectious agents.
Keywords: antibody-profiling technologies, antigen array, luciferase immunoprecipitation systems, vaccine targets
Detection of antibodies against infectious agents is an important part of basic and clinical research. For clinical diagnosis, the detection of antibody responses to some pathogens provides a sensitive assay for current, as well as past, infections. Antibody responses are also used to evaluate specific clinical symptoms associated with some pathogens. In vaccine research, antibody titers to specific proteins of an infectious agent often correlate strongly with the extent of protection afforded by the vaccine. Identifying protective B-cell responses against antigens, particularly against conformational epitopes that provide broad-spectrum protection against a given infection, are also critical for vaccine development and monitoring. The identification of antigens that induce the most effective immune response and which are the most diagnostically useful can be a daunting task. Improved approaches to increase both the spectrum of antigens tested and the quality of humoral responses detected has tremendous potential for many of these types of studies.
Although routine detection of antibodies is generally performed one antigen at a time by ELISA, there is increasing interest in studying antibody responses to whole proteomes for some infectious agents. While the availability of full genomic DNA sequences to many infectious agents has provided a framework to systematically identify antigenic targets, significant challenges remain to study antibody responses to large numbers of recombinant proteins. One approach involves using protein arrays to analyze the complete proteome of an infectious agent to obtain a better understanding of immunodominant antigens, and efficiently identifying antigens useful for both diagnosis and vaccination. Although these solid-phase array studies are useful for defining new antigen targets and revealing global insight into humoral responses, more sensitive, specific and robust immunoassays are also needed to convert these target antigens into useful diagnostic tests, and to monitor infection and vaccine development. While a comprehensive review of all immunoassays is beyond the scope of this paper, the goal of this article is to describe the more recently developed technologies to measure antibody titers to panels of antigens (Table 1). With these new technologies, investigators are now able to generate more extensive information than previously possible.
Table 1. Advantages and limitations of common antibody-profiling technologies.
| Assay | Advantages | Limitations |
|---|---|---|
|
In vivo-produced antigen arrays |
Evaluate antibody responses to 100–900 recombinant antigens at one timeAntigen identification in an unbiased fashionUseful for monitoring and understanding responses to vaccinesSimilar diagnostic performance to ELISA |
Labor intensive because requires antigen purificationContains variable amounts of contaminantsPoor recombinant coating efficiencyHigh background due to cross-reactivity to Escherichia coli or yeast proteinsLow dynamic range of detectionPoor detection of conformational epitopes |
| IVVT antigen arrays |
Evaluate antibody responses to hundreds to thousands of recombinant antigens at one timeAntigen identification in an unbiased fashionHigh-throughput antigen productionSimilar diagnostic performance to ELISA |
Cross-reactivity with E. coli proteinsVariations in the amount of immobilized proteinPoor detection of conformational epitopesLow dynamic range of detection |
| Luminex® |
Detects multiple antibodies in a given sampleRequires little serum |
Labor-intensive antigen purification and coupling to beadsPossible false-positives and high level of nonspecific backgroundLow dynamic range of detection |
| RBA |
Detection of many conformational epitopesHigh sensitivity and specificityModerate dynamic range of detection |
Requires radioactivityRequires large amount of seraCannot profile multiple antigens simultaneously |
| LIPS | Rapid recombinant antigen productionDetection of many conformational epitopesHigh sensitivity and specificityHigh dynamic range of detection Inexpensive | Cannot profile multiple antigens simultaneouslyMay still require testing multiple antigen constructs for detecting conformational epitopes for some cell-surface proteins |
IVVT: In vitro transcription/translation; LIPS: Luciferase immunoprecipitation systems; RBA: Radiobinding assay.
Antibody profiling using protein arrays & other technologies
Immunoassays such as western blotting and ELISA have been used extensively to detect antibody responses against defined recombinant antigens from infectious agents. In most of these studies, one or a very small number of target recombinant antigens have been used for detecting humoral responses. In recent years, newer high-throughput technologies have enabled large-scale analysis of protein antigens. In this section, we describe a variety of antibody-profiling studies that employ technologies that use large numbers of recombinant protein antigens (e.g., 80–2000) to investigate humoral responses against infectious agents. As described later, these technologies markedly enhance antigen discovery, provide new diagnostics and tools for vaccine monitoring, and yield new insights into global host humoral responses to pathogens.
Protein arrays based on production of antigens in Escherichia coli or yeast
Large numbers of Escherichia coli or yeast recombinant proteins were initially employed in the first solid-phase protein arrays. The large-scale production of recombinant proteins requires significant time and effort, and the production process itself can introduce various problems that later influence the performance of these proteins in immunoassays. For these studies, large numbers of protein-coding sequences are cloned in appropriate expression vectors and then expressed in bacteria or yeast. These recombinant proteins are then isolated from lysate using affinity tags such as glutathione-S-transferase (GST), which can be purified on glutathione-affinity supports. Owing to the fact that recombinant protein extracts contain variable amounts of E. coli or other contaminants after purification, the serum samples to be tested must be preincubated with bacterial or yeast lysates to block nonspecific binding. Additional blocking agents are typically needed to decrease the background and increase the signal-to-noise ratio. Owing to the toxicity of the expressed proteins and insolubility issues, recombinant proteins generated in bacteria or yeast often show variable expression and low yields. This can result in marked differences in antigen coating efficiency, which can yield false negatives and/or miss detecting some antigens.
Protein arrays are highly useful for identifying antigen targets in an unbiased fashion from complex proteomes. In one study, large-scale pathogen antibody profiling involved generating 882 recombinant E. coli-produced proteins from Treponema pallidum, the causative agent of syphilis [1]. These different proteins were produced with a GST tag and directly immobilized onto glutathione-coated microtiter plates. From ELISA testing, approximately 100 T. pallidumproteins were targets of antibodies from T. pallidum-infected rabbits. In a follow-up study using sera from syphilis-infected patients, 900 different T. pallidum antigens were arrayed on microtiter plates and used for antibody profiling [2]. A total of 34 additional proteins were found to be antigenic in infected patients and these antibody responses varied between primary, secondary and latent syphilis infection. One of the identified proteins, TP0136, was further studied and later found to be a protective antigen in experimentally infected rabbits [3].
Antibody array profiling technologies can provide new information for monitoring and understanding responses to vaccines. For example, antibody responses in a simian model for human HIV infection were evaluated using a combination of 430 distinct recombinant proteins and chemically synthesized short peptides from selected proteins [4]. Poly-L-lysine-coated microscope slides were used to spot these different recombinant and peptide antigens. Serum samples from macaques vaccinated with three different HIV multiprotein vaccines, as well as after SIV viral challenge, were analyzed. Following incubation with sera, a Cy3-conjugated secondary antibody was used to detect primary antibody binding by fluorescent scanning. The B-cell responses generated from these array data distinguished vaccinated macaques from virus-challenged macaques [4]. Immunoreactivity against several peptide epitopes also predicted survival. Interestingly, analysis of B-cell responses to these three different multiprotein vaccines revealed a convergence of immunodominant antibody responses to several linear epitopes in the envelope protein. The large-scale analysis afforded by protein arrays in this study enabled the identification of these epitopes, which would not have been possible by ELISA or western blotting.
A protein array using recombinant antigens produced in yeast was also used for diagnosis of SARS [5]. In these studies, 82 open reading frames (ORFs) from the entire proteome of the SARS coronavirus and several other proteins from related coronaviruses were amplified by PCR and cloned into a yeast expression vector that produces the viral proteins as C-terminal GST fusions. Following purification of these antigens by GST-affinity chromatography, the recombinant fusion proteins were spotted on nitrocellulose-coated slides. From probing these arrays, serum antibodies from SARS-infected patients were distinguished from healthy controls, demonstrating the diagnostic potential of this approach. Side-by-side comparison of the microarray with a diagnostic ELISA for SARS showed that both assay formats yielded similar results.
These studies demonstrate that arrays employing in vitro-produced recombinant proteins can be used to discover new antigens, identify vaccine targets, monitor vaccine outcomes and for diagnostics. While very large numbers of immunodominant antigens and epitopes can be identified from these screens, such solid-phase arrays exhibit similar analytical sensitivity to ELISA, and likely miss many conformational epitopes. These protein arrays, similar to ELISAs, also suffer from a limited dynamic range (3-log dynamic range at best). One of the major impediments for using this approach is the significant labor and resources for producing the large numbers of purified recombinant antigens produced from the in vivo bacterial and yeast expression systems.
In vitro transcribed/translated antigen arrays
As an alternative to in vivo production of recombinant proteins, antigen arrays can also be produced using in vitro transcription/translation (IVTT) [6]. Felgner’s group has developed a relatively simple and scalable PCR-based recombination approach to generate the necessary antigen expression vector constructs that are compatible for IVTT expression. The constructs are used to produce recombinant proteins via cell-free E. coliIVTT reactions and the unpurified, recombinant proteins are then printed on nitrocellulose arrays. Since the recombinant proteins produced in this cell-free system are generated from E. coli components that can also bind the array matrix, the sera must first be blocked with bacterial lysates before incubation with the arrays. Following array incubation with the sera, fluorescently labeled secondary antibodies are used to detect primary antibody binding. These large-scale screening experiments have identified a large number of antigenic proteins for a variety of human infectious agents, including vaccinia virus [6–8], Francisella tularensis[9,10], Chlamydia trachomatis[11], Plasmodium falciparum[12,13], Coxiella burnetii[14] and Burkholderia pseudomallei[15].
Davies et al. was the first to demonstrate the feasibility of the IVTT large-scale antigen array to identify new antigenic targets in an unbiased fashion [6]. In this study, 165 vaccinia virus antigens were tested with sera from naturally infected animals and humans. Over 25 new antigenic targets were identified. One of the antigens, the H3L envelope protein, was later identified as a major target of neutralizing antibodies associated with smallpox vaccination [7]. Additional testing of sera from vaccinia infection, smallpox vaccinia vaccination (Dryvax®, the licensed smallpox vaccine) and archival convalescent smallpox sera revealed similar antibody profiles [8]. While a variety of antigenic envelope and core proteins were identified as potential targets for vaccine monitoring, analysis of the antibody signals between positive and negative antigens revealed a relatively narrow dynamic range of fluorescent intensity showing 1–250-fold titer differences. Nevertheless, this approach is very useful for identifying the most immunodominant antigens within this viral proteome in an unbiased fashion.
Immunodominant antigens have also been identified as potential targets of a vaccine for malaria. Here, 250 P.falciparum proteins were generated by IVTT and printed on microarray slides to study antibody responses to both natural and experimental malarial infection [12]. From these antibody-profiling experiments, 72 highly reactive antigens that might represent malaria vaccine targets and provide new information on immune correlates of vaccine protection were identified.
While wheat germ rather than E. coli IVTT has also been used to study malaria vaccine antigen candidates [16], one major bottleneck for generating recombinant proteins is the need to clone coding sequences for ORFs into an expression vector plasmid for use with IVTT. An even faster, less labor-intensive approach called transcriptionally active PCR (TAP) can also be used, in which the priming sites for the transcription machinery for IVTT are directly incorporated into the PCR primers. In one study, TAP fragments for producing recombinant P. falciparum antigens for microarray spotting were as good as the standard plasmid-cloning approach [13]. Thus, TAP extends the versatility of this IVTT array technology to rapidly generate large numbers of recombinant proteins for array printing.
Other studies have also successfully employed IVTT antigen arrays and demonstrated the ability of this approach in detecting subtle differences in humoral responses after candidate vaccine and natural exposure. Molina et al. used antigen arrays for studying C. trachomatis, using mice inoculated with live or UV-inactivated Chlamydia. From studying 225 arrayed proteins, seven immunodominant antigens, which could be developed further for vaccine targets, were identified [11]. Immunoglobulin subtype-specific IgG1 and IgG2 antibody profiles were also different between live and UV-inactivated Chlamydia, thereby elucidating humoral response differences between these two experimental vaccine strategies. This array approach distinguished live versus UV-inactivated Chlamydia, suggesting that this array technology could be used further for monitoring subtle responses to vaccines.
Another major finding from large-scale array antibody profiling has been the global insights into the antigenicity of proteins with different subcellular localization. In the case of the intracellular bacteria, F. tularensis, 50% of the surface or membrane-associated proteins were antigenic compared with only 22% of the entire proteome [9]. These global antibody-profiling studies highlight the relative antigenicity of different types of proteins and identify new antigens that could be exploited for diagnostic purposes [10]. In another study, 1300 potential antigens from ORFs from Borrelia burgdorfi, the causative agent of Lyme disease, were studied by IVTT antigen array in sera from patients with early and late disease [17]. Overall, 15% of the ORFs were found to be antigenic. Comparison of the human antigenic profile with that of deer mice, the natural host reservoir, showed similar antibody profiles, suggesting a common set of immunogens for these different hosts.
Similar large-scale screens of 1205 B. pseudomallei proteins have identified disease-specific antigens, as well as antigens that cross-react with healthy individuals, which probably reflects shared antigenicity with other bacterial proteins [15]. In the largest protein antigen screen to date, 2000 ORFs from C. burnetii, the agent responsible for Q fever, were generated for array analysis [14]. Approximately 50 of these proteins were identified as antigenic and some may form the basis of a simple serodiagnostic immunoassay needed for this pathogen [14]. The overall information and insight from these large-scale antibody-profiling studies are unique to these arrays and provide multiple areas for further follow-up, including diagnostics, identifying vaccine targets and understanding host responses to the global protein repertoire of these pathogenic organisms.
While large amounts of information can be obtained from antigen array studies, there are several limitations. For example, some of the observed differences in antibody immunoreactivity may be due to variations in the amount of protein immobilized rather than actual serum antibody titer differences. One approach directed at improving the IVTT antigen arrays has been the development of nucleic acid programmable protein arrays (NAPPAs) [18]. In this format, recombinant proteins are also generated by IVTT, but the antigens are immobilized in situ by means of fused epitope tags. For example, C-terminal GST-tagged proteins can be captured directly on NAPPA by immobilized glutathione [19]. This approach eliminates the need to express and purify proteins separately, since they are immobilized during the IVTT reaction. This strategy eliminates potential stability issues and provides greater purity of the target antigens. If necessary, the presence of the GST moiety also allows the array to be probed with anti-GST antibodies to estimate the amounts of each protein immobilized to the array. In one NAPPA study, 262 antigens from Pseudomonas aeruginosawere produced and profiled with sera from cystic fibrosis patients and patients with acute infections [19]. From this study, 12 out of 262 outer membrane proteins of P. aeruginosawere determined to be highly antigenic targets. Immunoreactivity to other antigens on the array was also detected in the sera from healthy individuals and probably reflected antibodies directed at homologous proteins from other bacteria. In this regard, a 2760-protein array from Yersinia pestis also detected antibody cross-reactivity in rabbits immunized with several different Gram-negative bacteria [20]. Together, these findings suggest that many antigenic epitopes are shared among similar proteins from different Gram-negative bacteria and are consistent with known protein sequences for many of these bacteria. However, it is not clear if the detection of antibodies to more conformational epitopes with other immunoassay formats might better distinguish the responses to proteins from different bacteria.
In conclusion, protein arrays are extremely powerful tools for antigen discovery, providing antibody response data on thousands of proteins, which is not possible using other technologies. However, these antigen arrays are probably less practical for diagnostics because they are slow solid-phase assays that require blocking of serum and probably underestimate the breadth and strength of the humoral responses (i.e., antibody titers showing 1–50-fold differences). This limited dynamic range also may constrict the relative differences in immunoreactivity to certain antigens.
Luminex® microsphere immunoassays
In addition to protein arrays, the Luminex® microsphere immunoassay technology (Luminex Corp., TX, USA) can also be used to detect and measure multiple antibodies (≤100) in a given sample [21,22]. In these studies, solid-phase beads with different-colored fluorophores are used to immobilize different antigens. Following incubation with sera, a fluorescent secondary antibody is used. When the sample passes through the detector, one laser excites the fluorochrome in the bead, which exhibits a unique signal, and the other laser excites the fluorescent molecule (e.g., phycoerythrin) bound to the second antibody. Thus, antigen identity (bead fluorescence) and antibody titer (secondary antibody fluorescence) are obtained simultaneously. Approximately 100 different microspheres are available, potentially allowing multiple antigens to be detected.
Several studies have used multiplexed Luminex assays for detecting antibodies to different pathogens, including papillomavirus [23,24], Bordetella pertussis[25], Clostridium tetani[22], Corynebacterium diphtheria[22] and Haemophilus influenzae type b [22]. In most of these studies, the detection of multiple different pathogens is the primary goal. One intrinsic problem of Luminex is that sufficient quantities of purified antigens are needed, which must be covalently coupled to the beads. As an alternative, IVVT proteins have been used with epitope and anti-epitope tags in Luminex for immobilization and concentration [26]. In these studies, antibodies to several different Epstein–Barr virus (EBV) proteins were detected using the Luminex-based assay. However, these anti-EBV antibody signals were, at best, only fivefold above the background vector control [26]. Thus, the level of detection of anti-EBV antibody responses had a very narrow range of detection, limiting the utility of this technology for obtaining a detailed antigenic profile to understand the full range of antibody responses to this pathogen.
Recently, Waterboer et al. documented another intrinsic problem with the Luminex technology for serological assays [27]. Some human sera were found to directly bind to the carboxylated microspheres, causing false positives and a very high level of nonspecific background. A more recent study showed that an alternative microsphere (SeroMAP™; Luminex Corp., TX, USA) with improved blocking protocols reduced the nonspecific binding problem [28]. Finally, the need to prepare antigen-specific capturing beads, along with the cost and expertise needed to run these flow cytometry machines, have thus far limited this technology mainly to profiling antibodies to different infectious agents in a single multiplex Luminex assay run.
In addition to studying antibodies to infectious agents, the detection of autoantibodies to self-proteins is extremely important for predicting, diagnosing and monitoring autoimmune diseases, and probably other diseases such as cancer. Solution-phase immunoassays, such as the radiobinding assay (RBA), often show the highest sensitivity and specificity for the detection of autoantibodies in autoimmune diseases [29]. This is because RBAs can detect many more conformational epitopes than solid-phase immunoassays and are the assay of choice for detecting both low- and high-affinity antibodies to autoantigens, resulting in high sensitivity and specificity. The RBA uses radiolabeled antigen, generated typically by IVTT, in a solution-phase immunoprecipitation assay [29]. For example, RBA for a variety human pancreatic β-cell targets, including GAD65 and IA-2, are used to predict, diagnose and monitor autoantibodies in Type 1 diabetes (T1D). Of note, RBA methods for detecting autoantibodies in T1D have show high sensitivity and specificity, and to date there have been no protein arrays showing this high degree of diagnostic performance. Furthermore, RBAs also show much larger dynamic ranges in titers than solid-phase immunoassays, which makes them more useful for accurately evaluating antibody titers [30]. Unfortunately, the application of RBA has received little attention for monitoring antibodies to infectious agents mainly because it requires radioactive protein labeling. As discussed in the next section, a powerful alternative, nonradioactive solution-phase assay is luciferase immunoprecipitation systems (LIPS).
LIPS antibody profiling
One such technology to quantitatively measure antibodies is LIPS [31]. If the template is available, LIPS is a nonradioactive solution-phase immunoassay that can rapidly generate high-quality antibody titer data for most protein antigens. The most time-consuming steps are cloning and generating the appropriate plasmid expression vector containing the luciferase–antigen fusion. LIPS assays are relatively simple immunoprecipitation assays involving only a few steps (Figure 1). Since there are no available reviews on LIPS, here we summarize some of the important technical considerations for understanding and achieving optimal LIPS performance. In addition, we discuss applications of LIPS directed at humoral response profiling of infectious agents for diagnosis, partial and whole-proteome analysis, disease stratification and vaccine monitoring.
Figure 1. Luciferase immunoprecipitation systems.
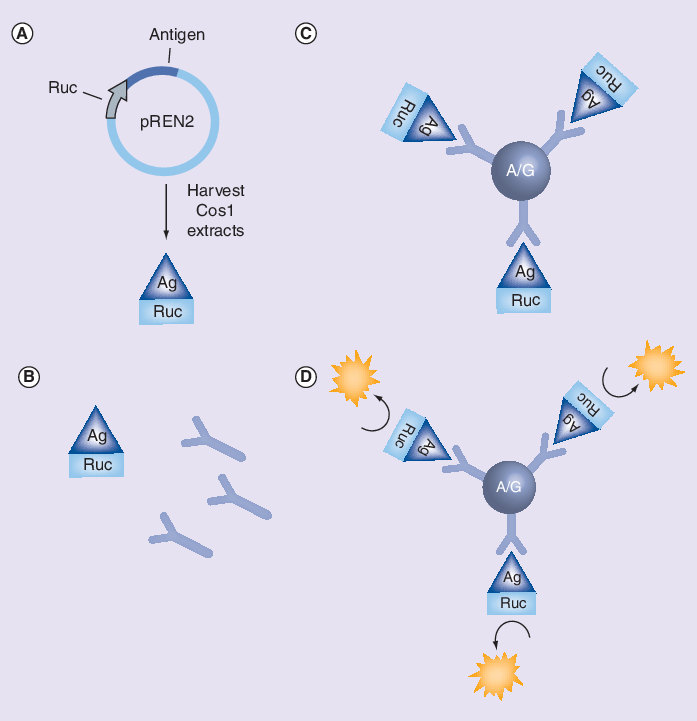
(A) DNA encoding antigens of interest are fused to Renilla luciferase (Ruc) using a mammalian plasmid expression vector and expressed in Cos-1 cells. (B) Unpurified, crude Ruc-antigen extracts are then obtained and incubated with sera. (C) The sera–Ruc-antigen mixture is transferred to a filter plate with protein A/G beads to capture Ruc-antigen complexes. (D) After washing, to remove nonspecific binding, coelenterazine substrate is added and light units are measured with a plate luminometer.
Ag: Antigen; Ruc: Renilla luciferase.
LIPS technical overview
Luciferase immunoprecipitation systems harnesses light-emitting recombinant antigen-fusion proteins to quantitatively measure patient antibody titers. Renillaluciferase (Ruc), derived from the sea pansy, is used for generating antigen fusions. Ruc is an ideal reporter owing to its small size (i.e., molecular weight: 30 kDa, approximately the same size as green fluorescent protein), wide linear detection range (100–107 light units [LUs]) and a complete lack of antigenicity with human and other animal sera [32]. Other luciferases, including the 60-kDa firefly luciferase, have also been used in LIPS. Antigen fusions to the C-terminal of Ruc are made by cloning into the pREN2 vector [32], while the pREN3S vector generates N-terminal fusions [33]. Since better yields of luciferase-tagged proteins are often observed in pREN2-expressed antigens in comparison to those expressed in pREN3S, the pREN3S vector is usually reserved for cell surface receptors and associated proteins [33], as well as other secreted proteins [Burbelo PD, Unpublished Data]. For some secreted proteins, removal of the signal peptide and cloning of the antigen as a C-terminal fusion in pREN2 is sufficient to allow proper conformational folding and exposure of the antigenic epitopes of the target. If removal of the signal sequence and producing these antigens as C-terminal fusions result in poor antigenicity, redesigning the target as an N-terminal fusion protein in the pREN3S vector should be tested as well. For example, antibodies to detect conformational and neutralizing antibodies within the EBV gp350 extracellular protein work better when the protein is cloned into pREN3S rather than in pREN2 [33]. Once Ruc-antigen mammalian expression plasmids are generated, these constructs are transiently transfected in Cos1 or other cells (e.g., HEK 293) to produce the Ruc antigens. After 36 h of transfection, the cells are scraped in cold lysis buffer containing glycerol, cleared by centrifugation and used directly in the LIPS assay. Alternatively, these extracts can be stored frozen at -80°C where they typically remain stable for at least 6 months.
Without purification, the Cos-1 cell lysates containing the directly tagged Ruc antigens are used in LIPS to evaluate antibodies. In contrast to protein arrays, the high-throughput LIPS format detects antibodies in many different serum samples against one target in any given assay. However, because of the ease with which Ruc antigens are generated, the LIPS format is easily scalable to screen hundreds of serum samples against multiple antigens. For these studies, a master microtiter plate of serum from a particular patient cohort is first made. The master plate is then used for dispensing diluted serum samples to individual testing plates, which allows reiterative profiling of the sera [31]. Antibody profiles can be generated against 45 or more antigens, in duplicate, using 100 µl of serum per patient. Since the Ruc reporter is linear over an 8-log dynamic range, in most cases there is no need to dilute the sera further for accurately quantifying antibodies. The first step of LIPS involves incubating the serum containing the antibodies and Ruc-antigen lysate together, at room temperature, for 1 h [31]. Although rarely necessary, increased antibody binding can also be achieved by incubation at 4°C. After incubation with sera, the mixture is then transferred to microtiter filter plates containing protein A/G beads for an additional hour to capture both the free immunoglobulins and Ruc-antigen–antibody complexes. For capturing antibodies, high-binding-capacity protein A/G beads (>20 mg of immunoglobulin binding per milliliter of beads) in a microtiter filter plate are used [31] and preferred over magnetic protein A/G beads because of lower cost and higher binding efficiency. While protein A/G beads efficiently immunoprecipitate IgG antibodies, they poorly detect IgA and IgM antibodies. Other immunoglobulin subtypes can also be examined using LIPS, but require specialized affinity reagents. For example, LIPS detected anti-IgG4 antibodies in sera from filarial-infected patients by coupling an anti-IgG4 secondary antibody to beads [34,35]. Further development to determine other isotype-specific antibodies and anti-IgM, IgA and IgE may be useful.
After incubation of the antigen–antibody complex with protein A/G beads, the filter plate is then extensively rinsed with wash buffer to remove unbound Ruc-tagged antigens [31]. Although manual washing and suctioning using a vacuum manifold can be performed, washing is more easily accomplished with the aid of a robotic workstation or a plate washer with vacuum filtration [31]. After filtration, the filter plate is loaded into a plate luminometer equipped with a substrate injector. Using LIPS, highly quantitative antibody titer values, reported as LUs, can be assigned to clinical and experimental serum samples. From these LIPS tests, a low LU reflects the presence of few or no antibodies, while an elevated LU reflects high antibody titers. Unlike most other immunoassays, the LIPS titer values are often compared using geometric mean titers (GMTs) because of the wide dynamic range and overdispersed nature of the LIPS data, which are typically represented on a log10 scale. Cut-offs can be determined by using the mean plus 3 or 5 standard deviations (SD) above the control group. In contrast to solid-phase assays, the diagnostic performance using either the mean plus 3 or 5 SD as cut-offs are generally identical because of the much greater analytical sensitivity of LIPS. Finally, compared with the solid-phase assays and even the solution-phase RBA, LIPS shows a greater dynamic range, reflecting a higher signal-to-noise ratio [36].
Single-antigen LIPS tests yield highly robust antibody titers for diagnosis
Owing to the low background and high signals, LIPS tests are ideal for developing diagnostics using single antigens. Furthermore, the large dynamic range of antibody titers within the seropositive range has additional clinical utilities for accurately assessing different stages of infection, substratifying disease, and for monitoring vaccines and treatment. To illustrate the high diagnostic performance of LIPS, we describe several examples of single-antigen LIPS tests that are extremely robust and feature very high diagnostic sensitivity and specificity.
One example is where LIPS was used for antibody profiling and diagnosis of herpes simplex virus (HSV)-1 and HSV-2 infection. The gold standard of detection of antibodies for diagnosis of HSV-1 and HSV-2 is western blotting. LIPS testing of a single Ruc-antigen fusion, gG-2 from HSV-2, generated highly robust antibody titers in all the HSV-2 infected samples, which were over 1000-fold higher than HSV-1 or negative samples [37]. For example, the geometric mean of seronegative samples was 2317 LU versus 2,001,600 LU in the HSV-2-positive samples. More importantly, the anti-gG2 LIPS assay correlated perfectly with western blot analysis with 100% sensitivity and 100% specificity [37]. By contrast, the anti-gG2 ELISA showed 100% sensitivity and 93% specificity. Highly quantitative antibody titers to several other HSV-1 and HSV-2 envelope and structural proteins were also detected, which were used to further study natural infection and for vaccine monitoring [38].
Another example of the high diagnostic performance of a single-antigen LIPS test is that for Loa loa, a filarial infection occurring mainly in Africa and Asia [34]. A LIPS test measuring anti-LlSXP-1 antibodies readily differentiated L. loa-infected from uninfected patients and demonstrated markedly improved sensitivity (100%) and specificity (100%) compared with an existing LlSXP-1 IgG4-based ELISA (67% sensitivity and 99% specificity). No significant cross-immunoreactivity of anti-LlSXP-1 antibodies were observed with other filarial infections [34]. Measuring anti-IgG4-specific antibodies to LlSXP-1 showed a significant correlation with the anti-IgG results, but showed no advantage over measuring the total IgG response alone. These results demonstrate the high diagnostic performance that can be achieved with LIPS using a single antigen.
Serological tests for B. burgdorfi infection, the spirocyte responsible for Lyme disease, are needed to diagnose and monitor antibiotic treatment for Lyme disease. LIPS screening of a panel of B. burgdorfi antigens revealed high antibody titers to a number of antigens in these different infected patients, but also showed marked patient heterogeneity [Burbelo PD, Issa AT, Ching KH, Cohen JI, Iadarola MJ, Marques A, Unpublished Data]. However, to achieve the highest sensitivity, a synthetic gene synthesis approach was used. This small synthetic protein, designated as VOVO, consisted of repeated antigenic VlsE-OspC-VlsE-OspC immunodominant peptide. The VOVO LIPS test showed 98% sensitivity and 100% specificity [Burbelo PD, Issa AT, Ching KH, Cohen JI, Iadarola MJ, Marques A, Unpublished Data]. Similarly, the established C6 ELISA showed only 98% sensitivity and 98% specificity. Unlike the ELISA, the VOVO LIPS test displayed a wide dynamic range of antibody detection, spanning over 10,000-fold without serum dilution. These findings suggest that LIPS screening using VOVO and other B. burgdorfi antigens offers an efficient and quantitative approach for the evaluation of antibody responses in Lyme disease. Moreover, this synthetic recombinant approach using LIPS may be highly useful for diagnosis and studying antibody responses to other pathogen proteins.
In summary, LIPS represents a universal format to develop diagnostic tests for many infectious agents. An appealing aspect of LIPS is the substantial gap between the highest titer value for the negative sera and the lowest titer value for the positive sera for many of the antigens tested, making it possible to predict the infection status of a given serum without previously determining cut-off values from a training set of samples [39,40]. LIPS also does not require significant assay optimization to obtain high-quality antibody titer data. Owing to these characteristics, it is likely that many novel antigens identified from array technologies, particularly in the area of diagnosis and monitoring vaccines, could be further evaluated using the LIPS technology.
LIPS profiling of panels of pathogen antigens
While there is an increasing interest to understand antibody responses to whole proteomes of different infectious agents, most studies use solid-phase antigen arrays, which underestimate the breadth and strength of these antibody responses. Using LIPS, a larger dynamic range of antibody titers and detection of many more conformational than other immunoassays provides a high-definition antibody profile. For example, LIPS was used to generate a high-definition antibody profile against the entire proteome of HIV from HIV-infected patients [39]. A total of 17 different HIV proteins were tested and included all of the processed HIV proteins. In these chronically HIV-infected patients, robust antibody titers were detected to at least one of the 17 HIV proteins from at least one infected patient. In most cases, multiple antigen immunoreactivity was detected. Many of the antibody titers detected by LIPS against these antigens were 1000-fold higher in the HIV-infected patients compared with the uninfected subjects. The most antigenic HIV protein was gp41, which showed similar antibody titers in all HIV patients tested. As might be expected, antibodies to different protein fragments (e.g., MA, p24 and NC) of the processed GAG protein showed similar high titers in all patients tested. In addition, antibodies were also detected to several of the small nonstructural proteins, including TAT, Rev, Vpr and Vpu in some of the patients. Owing to the large dynamic range of titer values typically obtained by LIPS to HIV and other infectious agents, a ‘flame’ heatmap is typically used to graphically visualize antibody profiles to multiple antigens from different infected and control samples simultaneously. In this heatmap, antibody titers ranging from 100 to 107 LUs are graphically presented using a color palette ranging from green to red, reflecting no/low and high antibody titers, respectively [39–41]. From analysis of antibody responses and heatmap data, there is often marked heterogeneity in patient immunoreactivity to these large panels of antigens. Further studies are underway to use these full HIV proteome responses to substratify infected patients based on clinical symptoms, length of infection and other parameters. It is likely that these antibody profiles against all the antigens of HIV and other pathogens could yield significant insight into patient complications, prognosis, monitoring and response to therapy. Along these lines, recent LIPS profiling of HCV–HIV-coinfected patients revealed anti-HCV antibody responses as biomarkers for response to therapy [42]. Finally, the relative simplicity of the LIPS technology makes it practical for small laboratories to generate antibody response profiles against infectious agents with relatively small proteomes (i.e., <100 proteins).
Moreover, LIPS can be used to screen for immunodominant antigens for diagnostic testing. Since there is a need for sensitive and specific testing to identify human herpes virus (HHV)-8/Kaposi sarcoma (KS)-associated virus-infected individuals, especially among potential blood donors, LIPS was used to identify diagnostically useful antigens [43]. A panel of 14 different HHV-8 fusion proteins, including a variety of latent and lytic antigens, were tested with serum from patients with HHV-8-associated KS. From LIPS testing, most of the HHV-8 proteins showed weak or nonexistent antibody titers with the KS sera. However, the latent HHV-8 protein, v-cyclin, was highly antigenic in approximately 80% of the KS samples. The antibody titers in the KS samples were 100–1000-fold higher than the uninfected controls [43]. These results contrast with a report using a number of bacterially produced GST fusions in western blotting, which were unable to detect immunoreactivity to v-cyclin [44]. The detection of diagnostically useful antibodies to v-cyclin by LIPS, but not with a bacterial recombinant protein in western blotting, supports the notion that the LIPS antigens may detect many more conformational epitopes, in part because the recombinant proteins are tested in solution under mild conditions. Particularly useful for diagnosis of HHV-8 infection was the finding that some of the positive anti-v-cyclin antibody responses were detected in sera that were determined to be negative by ELISA and/or LIPS for antibodies to other HHV-8 antigens [43]. Together, these studies demonstrate that LIPS is an effective yet simple and rapid method to screen proteins from the proteomes of infectious agents to identify diagnostically useful antigens.
High diagnostic performance using antigen mixtures by LIPS
While ELISA employing single antigens can be useful for the diagnosis of infection, very often detecting antibodies to one antigen is not sufficient for high diagnostic sensitivity because of HLA differences among subjects, different disease stages and other variables. To circumvent this problem, multiple ELISAs detecting different antibodies to different antigens are run in parallel followed by data analysis. For example, the optimal serological diagnosis of HHV-8 infection by ELISA involves analyzing the results from two or three antigens [45,46]. Efforts to simplify testing in an ELISA format by coating multiple antigens to the microtiter plates are usually unsuccessful because the antigen coating efficiency is quite variable, resulting in poor diagnostic performance. Alternatively, multiepitope hybrid recombinant molecules have been employed, whereby multiple antigens or immunodominant regions are fused together in a single protein [47–49]. While these hybrid antigens can be highly useful, time and effort is required to design them and optimize their performance.
As an alternative approach to running multiple individual assays, a mixture of multiple Ruc antigens can be tested simultaneously by LIPS. This is because the protein A/G beads capture a subset of all the immunoglobulins present in the serum, which allows multiple Ruc antigens to be immunoprecipitated simultaneously on the protein A/G beads. The analysis of the data is also simplified in the LIPS mixture format. For example, a mixture of four antigens containing K8.1, LANA, ORF65 and v-cyclin showed 100% sensitivity and 100% specificity for detecting HHV-8 infection in KS patients [43]. LIPS antigen mixture tests have also been highly successful for diagnosing other infections. A two-antigen LIPS mixture for Strongyloides stercoralis diagnosis [35], a four-antigen LIPS mixture for onchocerciasis diagnosis [41] and a four-antigen LIPS mixture for cytomegalovirus diagnosis [40] showed higher diagnostic performance than ELISA-based tests. Since this mixture approach is simple and based on using the most informative single-antigen tests, it will probably be a very useful general approach for the diagnosis of many other infections and disease states.
Antibody stratification of different diseases caused by the same infectious agent
Given the large range of antibody titers detected by LIPS, this technology is ideal for exploring antibody profiling to stratify different clinical features caused by infectious agents. In one LIPS study, antibodies against two latent and two lytic antigens were examined in three different diseases caused by the HHV-8 viruses: KS and two other lymphoproliferation disorders, multicentric Castleman’s disease (MCD) and primary effusion lymphoma (PEL) [50]. One of the most obvious differences was that antibody titers against the early lytic HHV-8 antigen, K8.1, were markedly higher in the PEL and MCD patients compared with the KS patients [50]. In contrast to the anti-K8.1 antibody profile, markedly higher antibodies to v-cyclin, a latent HHV-8 gene, were found in the KS and PEL patients compared with the MCD patients. Since elevated anti-latent antibodies are a common feature found of KS patients compared with MCD, the sum of the anti-v-cyclin and anti-LANA antibodies was the most useful approach to optimally segregate KS from MCD+/KS+ and MCD+/KS-. This approach, using a set cut-off value, discriminated KS from MCD+/KS+ with 93% sensitivity and 83% specificity. These results demonstrate that LIPS provides unique inventories of clinically relevant antibody responses that likely would be missed with other technologies. In another study, LIPS was used to profile antibodies against human T-cell lymphotropic virus type 1 (HTLV-1) infection. Since a small subset (5%) of HTLV-1-infected individuals develop HTLV-1-associated adult T-cell leukemia (ATLL) or HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP), antibody profiles were examined to see if these different HTLV-1 disease states could be distinguished [51]. Analysis of the antibody titer data from seven different HTLV-1 proteins revealed that the anti-GAG antibody titers were the most informative for distinguishing HTLV-infected subjects from uninfected controls, and showed 100% sensitivity and 100% specificity. However, anti-GAG antibody titers were similar between the HTLV-1 asymptomatic, ATLL and HAM/TSP subgroups [51]. By contrast, anti-Env antibody titers were over fourfold higher in HAM/TSP compared with both asymptomatic HTLV-1 (p < 0.0001) and ATLL patients (p < 0.0005). Anti-Env antibodies in asymptomatic carriers (r = 0.76) correlated well with the proviral load. Antibodies to another protein, TAX also were higher in HAM/TSP compared with the asymptomatic controls. Overall, these studies indicate that anti-HTLV-1 antibody responses detected by LIPS are useful for diagnosis and suggest that elevated anti-Env antibodies are a common feature found in HAM/TSP patients [51]. This HTLV-1 study and the HHV-8 study highlight the fact that LIPS can be employed to sensitively distinguish between different disease states caused by these and possibly other infectious agents.
Rapid testing by quick LIPS
There is a need to develop rapid and personalized serum-based diagnostic tests to detect autoimmune, cancer and infectious diseases. These types of point-of-care diagnostics could be extremely valuable in the clinical setting. Several rapid antibody-based tests have already been developed, but these tests mainly yield qualitative seronegative or seropositive status. For example, lateral flow immunoassays are used for the diagnosis of several infectious agents such as HIV and HCV, producing a qualitative result (i.e., positive or negative), but do not yield any informative data on antibody titers for clinicians.
Owing to the rapid kinetics of solution-phase assays, LIPS is ideal for development of quantitative point-of-care testing. To this end, LIPS was used in a modified rapid format called quick LIPS (QLIPS) to diagnose both infection and autoimmunity. In these QLIPS tests, only 25 min of total processing time per 94 serum samples are needed, which includes a 5-min set-up, two 5-min incubation steps, and 10 min of washing and reading of the plate with a luminometer. Even faster determinations are possible with equipment for rapid liquid handling. Nevertheless, two different QLIPS tests have shown promise for the diagnosis of infection by L. loa[34] and onchocerciasis [41]. In both these studies, QLIPS was as informative as the normal LIPS format and markedly better than ELISA. In addition, we have compared QLIPS with LIPS for detecting autoantibodies for the diagnosis of Sjögren’s syndrome, an autoimmune disease characterized by inflammation of the salivary and lacrimal glands and high levels of autoantibodies to the proteins Ro52, Ro60 and La. QLIPS was able to rapidly and sensitively detect antibodies to Ro52 that were often 1000-fold higher in the Sjögren’s syndrome patients than controls [52]. Future improvements in instrumentation are needed to move the QLIPS testing to a handheld device for office and field testing.
Vaccine research using LIPS
Almost all vaccines elicit a robust antibody response that correlates with the extent of protection afforded by the vaccine. Measurements of antibody responses are typically made by ELISA and virus neutralization assays. Most neutralizing antibodies recognize conformational epitopes, which are poorly detected by ELISA and solid-phase assays. By contrast, the solution-phase LIPS assay readily detects conformational epitopes.
Current assays to study antibody-mediated neutralization of EBV typically require 6 weeks to perform and are very labor intensive. Sashihara et al. used LIPS to measure antibody responses to two EBV surface glycoproteins, gp350 and gp42 [33]. Gp350 and gp42 are of particular interest and are ideal candidate vaccine targets because they are involved in EBV attachment and fusion to B lymphocytes, respectively. Interestingly, the LIPS anti-gp350 antibody titers correlated strongly (r = 0.86) with neutralizing activity measured by the standard 6-week transformation-based assay. Of note, previous ELISA studies have not reported antibodies that correlate with neutralizing activity [53]. For the first time, anti-gp42 antibodies were also detected, but the LIPS antibody titers only partially correlated with neutralizing activity. These results demonstrate the potential of this simple and rapid LIPS assay to monitor antibodies to possible conformational epitopes, missed by ELISA, for assessing neutralizing activity.
In addition to yielding insight into conformation-specific antibodies, the highly quantative LIPS assay allows the detection of subtle antibody titer differences elicited by different vaccine strategies. LIPS was used to monitor antibodies to test the effectiveness of HSV-2 attenuated and single-subunit vaccines [38]. In a guinea pig model of HSV, animals were vaccinated with an attenuated HSV-2 virus (dl5–29) as well as with a single recombinant subunit vaccine for the surface glycoprotein gD2 administered with two different adjuvants. With the dl5–29 vaccine, LIPS detected the induction of antibodies to three different HSV-2 surface proteins but not to ICP-8, which is deleted in the dl5–29 vaccine. In addition, statistically significant differences between anti-gD2 antibody titers were detected in animals receiving gD2 with complete and incomplete Freund’s adjuvant (CFA/IFA) compared with those receiving gD2 (alum/MPL). However, only animals receiving the gD2 (alum/MPL) had antibody titers detected by LIPS that correlated with HSV-2-neutralizing activity. These results demonstrate the intricate vaccine antibody response that can be teased out using the highly quantative antibody profiling by LIPS.
Luciferase immunoprecipitation systems was also used to monitor antibody responses to a major influenza vaccine antigen target to develop a therapeutic vaccine for H5N1, avian influenza [54]. In this study, a dose-escalation clinical trial using inactivated H5N1 vaccine was administered to healthy volunteers. Two different processed forms of the surface hemagglutinin (HA), HA-1 and HA-2, were tested by LIPS. Interestingly, high levels of anti-HA-2 were detected in many individuals before vaccination (day 0). This is possibly cross-reactivity with other influenzas as HA-2 is more conserved across influenza species. By contrast, the anti-HA-1 antibody titers were generally absent at day 0 and dramatically increased by 10,000-fold in most individuals following vaccination. Interestingly, several subject who showed robust antibody increases in the longitudinal vaccine series, showed no immune response by microneutralization assays. While these are not promising results for the vaccine study, future work with additional N-terminal fusions may hold greater promise for correct folding and the detection of surrogate neutralizing antibodies, and could be easily tested by LIPS. Lastly, these results also highlight that even with LIPS, the correct folding of any given cell surface target may not be optimal and may require testing multiple different constructs to obtain the protein in its native conformation.
Expert commentary
A variety of immunoassay formats are now available to profile antibodies to large panels of antigens. Solid-phase array technologies are increasingly being used to understand antibody responses to whole proteomes and for development of new candidates for vaccines. The use of IVTT is gaining particular attention because, rather than using recombinant proteins made in vivo from bacteria or yeast, IVTT provides a relatively rapid and simple approach for generating the needed recombinant proteins for protein arrays. Using protein arrays, it is now possible to profile antibody responses to all the proteins of a given pathogen (often >1000 proteins) at one time. Future protein arrays will allow the simultaneous study of antibodies to different pathogens.
In addition to the expansion of available protein arrays, the LIPS technology will also continue to grow. Owing to the high diagnostic performance of LIPS using one or a few immunodominant antigens in the mixture format, it may be possible to develop LIPS tests to all known human pathogens from bacteria, fungi, protozoa, filarial nematodes and viruses. The application of large-scale screening of pathogen antibodies in patients with undiagnosed diseases may yield insight into their clinical conditions. In addition, this comprehensive resource could also be developed for point-of-care testing, which could greatly enhance the diagnosis of a wide variety of infectious agents and the ability to monitor vaccine strength. More sophisticated cloning strategies could greatly enhance many LIPS studies. For example, recombination approaches and the use of the TAP system with LIPS could greatly enhance the production of any needed antigen targets. Since LIPS is not as high-throughput as protein arrays, identifying new antigens by protein arrays and studying their antigenicity further by LIPS may be an efficient way to develop antibody-based tests for diagnosis of infections and monitoring responses to vaccines.
Lastly, antibody inventories generated by these technologies may also be integrated with other types of high-throughput information, including T-cell responses, and genomic and proteomic data for systems level analysis. Although not discussed here, additional autoantibody profiling against human proteins may also be studied in parallel, which could provide further information with regard to studying adverse reactions due to vaccination.
Five-year view
It is expected that in the next 5 years, antibody profiles generated by protein arrays, LIPS and other technologies will continue to provide important information for diagnosis, antigen discovery, vaccine monitoring and understanding infectious disease pathogenesis. Although our overall knowledge of humoral responses to full proteomes of infectious agents is still limited, these technologies will provide a useful resource for years to come. The ability of these technologies to perform large-scale antibody screens will also help accelerate the study of newly discovered pathogens. Since the detection of host humoral responses to infectious agents can provide indirect evidence for in vivo expression of the pathogen, the ability to rapidly and comprehensively generate multiple recombinant proteins from a given pathogen should be very useful for epidemiological studies, confirming the relationship between exposure to the infection and disease pathogenesis. Antibody profiling will also allow exploration of various human disease states, in which exposure to a pathogen can contribute to or trigger a subsequent pathological disorder. Finally, it is likely that additional modifications of existing approaches or the development of completely new systems may further accelerate the quantity, quality and utility of antibody response profiles to infectious agents.
Key issues
• Using antigen arrays, it is now possible to screen 1000 or more proteins to identify immunodominant antigens.
• Protein array studies highlight the relative antigenicity of different types of proteins and identify candidate antigens that can be exploited for diagnostic and vaccine purposes.
• Analysis of antibody responses to large-scale protein arrays can provide global insights into proteome-wide antigenicity.
• As an alternative to ELISAs and protein arrays, luciferase immunoprecipitation systems (LIPS) represents an easy format to quantitatively measure antibodies, including conformational epitopes often missed by solid-phase formats.
• Compared with ELISA, LIPS often provides higher sensitivity and specificity for diagnosis of many infectious agents, often over a wider dynamic range.
• Identifying new antigens by protein arrays and studying their antigenicity further by LIPS may be an efficient way to develop antibody-based diagnostics for infections and monitoring responses to vaccines.
Acknowledgements
This work is dedicated to the memory of our colleague, Dr Thomas L Mattson, who was instrumental in developing the LIPS technology and who showed extraordinary kindness and friendship.
Financial & competing interests disclosure
This work was supported by the intramural research program of the National Institute of Dental and Craniofacial Research, NIH. Two of the authors, Peter D Burbelo and Michael J Iadarola, have multiple patent applications submitted using the LIPS technology. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as: • of interest •• of considerable interest
- 1.McKevitt M, Brinkman MB, McLoughlin M et al. Genome scale identification of Treponema pallidum antigens. Infect. Immun. 73(7), 4445–4450 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brinkman MB, McKevitt M, McLoughlin M et al. Reactivity of antibodies from syphilis patients to a protein array representing the Treponema pallidum proteome. J. Clin. Microbiol. 44(3), 888–891 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brinkman MB, McGill MA, Pettersson J et al. A novel Treponema pallidum antigen, TP0136, is an outer membrane protein that binds human fibronectin. Infect. Immun. 76(5), 1848–1857 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neuman de Vegvar HE, Amara RR, Steinman L, Utz PJ, Robinson HL, Robinson WH. Microarray profiling of antibody responses against simian–human immunodeficiency virus: postchallenge convergence of reactivities independent of host histocompatibility type and vaccine regimen. J. Virol. 77(20), 11125–11138 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhu H, Hu S, Jona G et al. Severe acute respiratory syndrome diagnostics using a coronavirus protein microarray. Proc. Natl Acad. Sci. USA 103(11), 4011–4016 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davies DH, Liang X, Hernandez JE et al. Profiling the humoral immune response to infection by using proteome microarrays: high-throughput vaccine and diagnostic antigen discovery. Proc. Natl Acad. Sci. USA 102(3), 547–552 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Important first publication describing the generation of templates for in vitro transcription/translation arrays.
- 7.Davies DH, McCausland MM, Valdez C et al. Vaccinia virus H3L envelope protein is a major target of neutralizing antibodies in humans and elicits protection against lethal challenge in mice. J. Virol. 79(18), 11724–11733 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davies DH, Molina DM, Wrammert J et al. Proteome-wide analysis of the serological response to vaccinia and smallpox. Proteomics 7(10), 1678–1686 (2007). [DOI] [PubMed] [Google Scholar]; • Important publication describing the use of in vitro transcription/translation arrays for identifying immunodominant antigens for the smallpox vaccine.
- 9.Eyles JE, Unal B, Hartley MG et al. Immunodominant Francisella tularensisantigens identified using proteome microarray. Proteomics 7(13), 2172–2183 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Sundaresh S, Randall A, Unal B et al. From protein microarrays to diagnostic antigen discovery: a study of the pathogen Francisella tularensis Bioinformatics 23(13), i508–i518 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Molina DM, Pal S, Kayala MA et al. Identification of immunodominant antigens of Chlamydia trachomatis using proteome microarrays. Vaccine 28(17), 3014–3024 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doolan DL, Mu Y, Unal B et al. Profiling humoral immune responses to P. falciparum infection with protein microarrays. Proteomics 8(22), 4680–4694 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Regis DP, Dobano C, Quinones-Olson P et al. Transcriptionally active PCR for antigen identification and vaccine development: in vitro genome-wide screening and in vivo immunogenicity. Mol. Biochem. Parasitol. 158(1), 32–45 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beare PA, Chen C, Bouman T et al. Candidate antigens for Q fever serodiagnosis revealed by immunoscreening of a Coxiella burnetiiprotein microarray. Clin. Vaccine Immunol. 15(12), 1771–1779 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Felgner PL, Kayala MA, Vigil A et al. A Burkholderia pseudomallei protein microarray reveals serodiagnostic and cross-reactive antigens. Proc. Natl Acad. Sci. USA 106(32), 13499–13504 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsuboi T, Takeo S, Iriko H et al. Wheat germ cell-free system-based production of malaria proteins for discovery of novel vaccine candidates. Infect. Immun. 76(4), 1702–1708 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barbour AG, Jasinskas A, Kayala MA et al. A genome-wide proteome array reveals a limited set of immunogens in natural infections of humans and white-footed mice with Borrelia burgdorferi Infect. Immun. 76(8), 3374–3389 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ramachandran N, Hainsworth E, Bhullar B et al. Self-assembling protein microarrays. Science 305(5680), 86–90 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Montor WR, Huang J, Hu Y et al. Genome-wide study of Pseudomonas aeruginosa outer membrane protein immunogenicity using self-assembling protein microarrays. Infect. Immun. 77(11), 4877–4886 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Keasey SL, Schmid KE, Lee MS et al. Extensive antibody cross-reactivity among infectious Gram-negative bacteria revealed by proteome microarray analysis. Mol. Cell. Proteomics 8(5), 924–935 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pickering JW, Martins TB, Greer RW et al. A multiplexed fluorescent microsphere immunoassay for antibodies to pneumococcal capsular polysaccharides. Am. J. Clin. Pathol. 117(4), 589–596 (2002). [DOI] [PubMed] [Google Scholar]
- 22.Pickering JW, Martins TB, Schroder MC, Hill HR. Comparison of a multiplex flow cytometric assay with enzyme-linked immunosorbent assay for auantitation of antibodies to tetanus, diphtheria, and Haemophilusinfluenzae type b. Clin. Diagn. Lab. Immunol. 9(4), 872–876 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dias D, Van Doren J, Schlottmann S et al. Optimization and validation of a multiplexed luminex assay to quantify antibodies to neutralizing epitopes on human papillomaviruses 6, 11, 16, and 18. Clin. Diagn. Lab. Immunol. 12(8), 959–969 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Opalka D, Lachman CE, MacMullen SA et al. Simultaneous quantitation of antibodies to neutralizing epitopes on virus-like particles for human papillomavirus types 6, 11, 16, and 18 by a multiplexed luminex assay. Clin. Diagn. Lab. Immunol. 10(1), 108–115 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prince HE, Lape-Nixon M, Matud J. Evaluation of a tetraplex microsphere assay for Bordetella pertussis antibodies. Clin. Vaccine Immunol. 13(2), 266–270 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wong J, Sibani S, Lokko NN, LaBaer J, Anderson KS. Rapid detection of antibodies in sera using multiplexed self-assembling bead arrays. J. Immunol. Methods 350(1–2), 171–182 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Waterboer T, Sehr P, Pawlita M. Suppression of non-specific binding in serological Luminex assays. J. Immunol. Methods 309(1–2), 200–204 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Pickering JW, Larson MT, Martins TB, Copple SS, Hill HR. Elimination of false-positive results in a luminex assay for pneumococcal antibodies. Clin. Vaccine Immunol. 17(1), 185–189 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu E, Eisenbarth GS. Accepting clocks that tell time poorly: fluid-phase versus standard ELISA autoantibody assays. Clin. Immunol. 125(2), 120–126 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li M, Yu L, Tiberti C et al. A report on the International Transglutaminase Autoantibody Workshop for Celiac Disease. Am. J. Gastroenterol. 104(1), 154–163 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burbelo PD, Ching KH, Klimavicz CM, Iadarola MJ. Antibody profiling by luciferase immunoprecipitation systems (LIPS). J. Vis. Exp. 7(32), 1549 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Provides detailed protocol and video for performing luciferase immunoprecipitation systems (LIPS).
- 32.Burbelo PD, Goldman R, Mattson TL. A simplified immunoprecipitation method for quantitatively measuring antibody responses in clinical sera samples by using mammalian-produced Renilla luciferase-antigen fusion proteins. BMC Biotechnol. 5, 22 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Important publication describing the use of Renilla luciferase-antigen fusion for measuring antibodies by LIPS.
- 33.Sashihara J, Burbelo PD, Savoldo B, Pierson TC, Cohen JI. Human antibody titers to Epstein–Barr virus (EBV) gp350 correlate with neutralization of infectivity better than antibody titers to EBV gp42 using a rapid flow cytometry-based EBV neutralization assay. Virology 391(2), 249–256 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; • Important publication describing the detection of antibodies by LIPS as a surrogate measurement of neutralizing antibodies for Epstein–Barr virus infection.
- 34.Burbelo PD, Ramanathan R, Klion AD, Iadarola MJ, Nutman TB. Rapid, novel, specific, high-throughput assay for diagnosis of Loa loa infection. J. Clin. Microbiol. 46(7), 2298–2304 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramanathan R, Burbelo PD, Groot S, Iadarola MJ, Neva FA, Nutman TB. A luciferase immunoprecipitation systems assay enhances the sensitivity and specificity of diagnosis of Strongyloides stercoralis infection. J. Infect. Dis. 198(3), 444–451 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burbelo PD, Hirai H, Issa AT et al. Comparison of radioimmunoprecipitation with luciferase immunoprecipitation for autoantibodies to GAD65 and IA-2b. Diabetes Care 33(4), 754–756 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burbelo PD, Hoshino Y, Leahy H et al. Serological diagnosis of human herpes simplex virus type 1 and 2 infections by luciferase immunoprecipitation system assay. Clin. Vaccine Immunol. 16(3), 366–371 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoshino Y, Pesnicak L, Dowdell KC et al. Protection from herpes simplex virus (HSV)-2 infection with replication-defective HSV-2 or glycoprotein D2 vaccines in HSV-1-seropositive and HSV-1-seronegative guinea pigs. J. Infect. Dis. 200(7), 1088–1095 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burbelo PD, Ching KH, Mattson TL, Light JS, Bishop LR, Kovacs JA. Rapid antibody quantification and generation of whole proteome antibody response profiles using LIPS (luciferase immunoprecipitation systems). Biochem. Biophys. Res. Commun. 352(4), 889–895 (2007). [DOI] [PubMed] [Google Scholar]
- 40.Burbelo PD, Issa AT, Ching KH et al. Highly quantitative serological detection of anti-cytomegalovirus (CMV) antibodies. Virol. J. 6, 45 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Burbelo PD, Leahy HP, Iadarola MJ, Nutman TB. A four-antigen mixture for rapid assessment of Onchocerca volvulus infection. PLoS Negl. Trop. Dis. 3(5), e438 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Burbelo PD, Kovacs JA, Ching KH et al. Proteome-wide anti-HCV and anti-HIV antibody profiling for predicting and monitoring response to HCV treatment in HIV co-infected patients. J. Infect. Dis. (2010) (In Press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burbelo PD, Leahy HP, Groot S et al. Four-antigen mixture containing v-cyclin for serological screening of human herpesvirus 8 infection. Clin. Vaccine Immunol. 16(5), 621–627 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Katano H, Iwasaki T, Baba N et al. Identification of antigenic proteins encoded by human herpesvirus 8 and seroprevalence in the general population and among patients with and without Kaposi’s sarcoma. J. Virol. 74(8), 3478–3485 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jenkins FJ, Hayes RB, Jackson A et al. Human herpesvirus 8 seroprevalence among prostate cancer case patients and control subjects. J. Infect. Dis. 196(2), 208–211 (2007). [DOI] [PubMed] [Google Scholar]
- 46.Laney AS, Peters JS, Manzi SM, Kingsley LA, Chang Y, Moore PS. Use of a multiantigen detection algorithm for diagnosis of Kaposi’s sarcoma-associated herpesvirus infection. J. Clin. Microbiol. 44(10), 3734–3741 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Andrews JA, Bligh WJ, Chiodini PL, Bradley JE, Nde PN, Lucius R. The role of a recombinant hybrid protein based ELISA for the serodiagnosis of Onchocerca volvulus J. Clin. Pathol. 61(3), 347–351 (2008). [DOI] [PubMed] [Google Scholar]
- 48.Houghton RL, Benson DR, Reynolds LD et al. A multi-epitope synthetic peptide and recombinant protein for the detection of antibodies to Trypanosoma cruzi in radioimmunoprecipitation-confirmed and consensus-positive sera. J. Infect. Dis. 179(5), 1226–1234 (1999). [DOI] [PubMed] [Google Scholar]
- 49.Nde PN, Pogonka T, Bradley JE, Titanji VP, Lucius R. Sensitive and specific serodiagnosis of onchocerciasis with recombinant hybrid proteins. Am. J. Trop. Med. Hyg. 66(5), 566–571 (2002). [DOI] [PubMed] [Google Scholar]
- 50.Burbelo PD. Distinct profiles of antibodies to Kaposi’s sarcoma-associated herpesvirus antigens in patients with kaposi sarcoma, multicentric Castleman’s disease, and primary effusion lymphoma. J. Infect. Dis. 198(3), 444–451 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Burbelo PD, Meoli E, Leahy HP et al. Anti-HTLV antibody profiling reveals an antibody signature for HTLV-I-associated myelopathy/tropical spastic paraparesis (HAM/TSP). Retrovirology 5, 96 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Burbelo PD, Ching KH, Issa AT et al. Rapid serological detection of autoantibodies associated with Sjogren’s syndrome. J. Transl. Med. 7, 83 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Randle BJ, Epstein MA. A highly sensitive enzyme-linked immunosorbent assay to quantitate antibodies to Epstein–Barr virus membrane antigen gp340. J. Virol. Methods 9(3), 201–208 (1984). [DOI] [PubMed] [Google Scholar]
- 54.Beigel JH, Voell J, Huang CY, Burbelo PD, Lane HC. Safety and immunogenicity of multiple and higher doses of an inactivated influenza A/H5N1 vaccine. J. Infect. Dis. 200(4), 501–509 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]