

Abstract
This article is a short comprehensive review describing in vitro polyester synthesis catalyzed by a hydrolysis enzyme of lipase, most of which has been developed for these two decades. Polyesters are prepared by repeated ester bond-formation reactions; they include two major modes, ring-opening polymerization (ROP) of cyclic monomers such as cyclic esters (lactones) and condensation polymerization via the reaction between a carboxylic acid or its ester group and an alcohol group. Polyester synthesis is, therefore, a reaction in reverse way of in vivo lipase catalysis of ester bond-cleavage with hydrolysis. The lipase-catalyzed polymerizations show very high chemo-, regio-, and enantio-selectivities and involve various advantageous characteristics. Lipase is robust and compatible with other chemical catalysts, which allows novel chemo-enzymatic processes. New syntheses of a variety of functional polyesters and a plausible reaction mechanism of lipase catalysis are mentioned. The polymerization characteristics are of green nature currently demanded for sustainable society, and hence, desirable for conducting ‘green polymer chemistry’.
Keywords: polyester synthesis, lipase catalyst, lactone, ring-opening polymerization, condensation polymerization, green polymer chemistry
Introduction
Following the three major natural biomacromolecules, nucleic acids (DNA and RNA), proteins (polypeptides), and polysaccharides, polyesters are in fourth place.1) Practically, polyesters are very important materials, which are widely used like poly(ethylene terephthalate) (PET) as an aromatic polyester, poly(butylene succinate) (PBS), poly(ɛ-caprolactone) (PCL), and poly(lactic acid) (PLA) as aliphatic polyesters (Scheme 1). In industry, the former two are produced via condensation polymerization (polycondensation), and the latter two via ring-opening polymerization. Polyesters are also currently employed as biomaterials for medical purposes such as surgical sutures, screws, and reinforcing plates as well as controlled release drug carriers, since they are bio-compatible and non-toxic.
Scheme 1.

Typical examples of practically used four polyesters.
Historically, synthesis of aliphatic polyesters was first achieved via condensation polymerization between a dicarboxylic acid and a glycol in the 1930s by Carothers soon after his invention of nylon-66. Mainly due to the lower melting point and difficulty in reaching higher molecular weight, their commercialization had not been long realized. However, an aromatic polyester of PET was successfully industrialized and has been extensively utilized as an excellent polymer material for this half a century. Aliphatic polyesters found their wide utilities and recently their commercial production has been developing. In particular, an increasing environmental concern of our society demands scientists to explore the materials from renewable resources for reducing the use of fossil resources as well as the processes using non-toxic substances for catalyst in the production. This movement is in the context of “green chemistry”.2)
A class of aliphatic polyesters called poly(hydroxyalkanoate)s (PHAs) were produced via fermentation (in vivo) in the 1980s, which attracted much attention as biodegradable polymers. In contrast to the in vivo synthesis in living cells, in vitro synthesis of polyesters was first noted in the mid 1980s via a condensation reaction by using lipase (a hydrolysis enzyme) as catalyst to give oligomers.3) In vitro polyester synthesis catalyzed by enzymes, however, started actually in 1993, when lipase-catalyzed ring-opening polymerization was found by two independent groups.4)–6) Following the studies using enzymes as polymerization catalyst (“enzymatic polymerization”) to produce polysaccharides, polyesters, poly(aromatic)s, and so forth,7)–15) the present review describes recent progress of lipase-catalyzed polyester synthesis developed mainly by our group and also by other groups, with emphasizing the aspects of green polymer chemistry.10)–12),14),15)
Reaction modes of polyester synthesis
Polyester synthesis is classified into two major polymerization modes; (A) ring-opening polymerization in two sub-divisions (1) and (2), and (B) condensation polymerization (polycondensation) between a carboxyl group and an alcohol group in two subdivisions (3) and (4) (Scheme 2). All polymerizations are catalyzed by lipase, a hydrolysis enzyme.
Scheme 2.

Two major modes of reaction for enzymatic polyester synthesis.
Catalyst enzymes and their functions
Lipases (triacylglycerol acylhydrolase, EC 3.1.1.3) catalyze the hydrolysis of fatty acid glycerol esters in vivo with bond-cleavage (Scheme 3). As already studied, a hydrolase enzyme induced polymerizations of various sugar fluoride and sugar oxazoline monomers to yield natural and unnatural polysaccharides.1),9)–15) A hydrolysis enzyme of lipase was found to catalyze similarly the polymerization reactions to give polyesters in vitro with bond-formation.9)–16)
Scheme 3.

Lipase is an enzyme to catalyze the hydrolysis of glycerol tri-esters in vivo.
Scheme 4 shows a model expression of the difference in catalysis function of lipase in vivo hydrolysis (a) and in vitro polymerization to produce a polyester (b), that is, lipase catalyses the reaction in a reverse direction in vitro. It is to be emphasized that in both in vivo and in vitro reactions a common transition-state (or intermediate) is involved to induce the reactions, which is generally accepted as a new concept of “transition-state analogue substrate” (TSAS) proposed by us.1),9)–15) In the lipase-catalyzed reactions, a common intermediate corresponds to an acyl-enzyme intermediate discussed in Fig. 2, Fig. 4 and Scheme 11.
Scheme 4.

A model expression of lipase-catalyzed reactions: (a) In vivo hydrolysis and (b) In vitro polymerization of monomers to produce a polyester molecule.
Fig. 2.

Illustration of the mechanism of lipase-catalyzed hydrolysis (Nu-H = H2O) and transesterification (Nu-H = R”O-H).
Fig. 4.

A general illustrative mechanism of lipase-catalyzed ROP of lactones.
Scheme 11.

Main elemental reactions in lipase-catalyzed ROP of lactones.
Actually, a variety of polyester synthesis reactions have been developed in these two decades.9)–17) Typical examples of lipases so far employed for poly-ester synthesis are shown in Table 1 together with their abbreviations.
Table 1.
Origin of lipases used for in vitro polyester synthesis and their abbreviations
| lipase origin | abbreviation |
|---|---|
| Candida cylindracea | lipase CC |
| Pseudomonas fluorescens | lipase PF |
| porcine pancreas lipase | PPL |
| Aspergillus niger | lipase A |
| Candida rugosa | lipase CR |
| Penicillium roqueforti | lipase PR |
| Pseudomonas cepacia | lipase PC |
| Rhizopus japonicus | lipase RJ |
| Rhizomucor meihei | lipase RM |
| Mucor meihei | lipase MM |
| Candida antarctica | lipase CA |
| Candida antarctica lipase B | CALB (Novozym 435)a |
| Yarrowia lipolytica | lipase YL |
CALB immobilized on an acrylic resin is commercially called as Novozym 435.
In accord with developments of X-ray crystallographic analysis technique as well as of isolation and crystallization techniques of enzymes, three-dimensional structures of enzymes have become available since the 1990s. Such structure determination of CALB was conducted in 1994,18) the 3D structure of which is shown in Fig. 1.19) CALB is constituted of 317 amino acid residues having a formula weight of 33,273. The active center has a catalytic triad, serine (Ser105)-histidine (His224)-aspartic acid (Asp187), containing a large hydrophobic pocket above the Ser-His-Asp triad and a medium-sized pocket below it. It is considered that in the catalysis pathway the acyl moiety of the substrate lies in the large subsite, while the leaving group/nucleophile moiety lies in the medium pocket. The catalytic triad, Ser-His-Asp, is common to serine hydrolases like lipases and esterases.
Fig. 1.
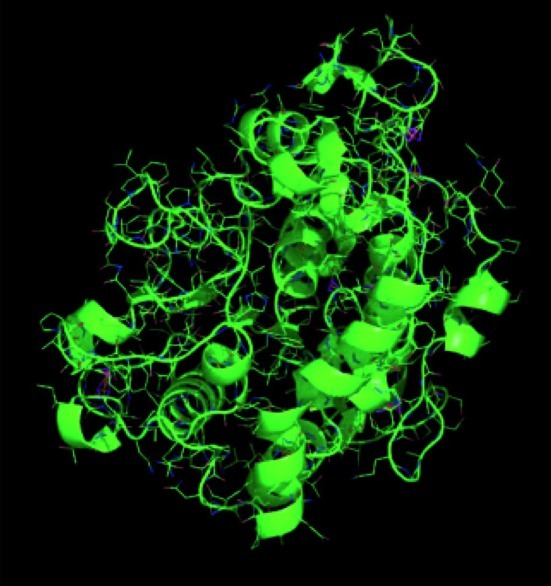
3D structure of CALB. Reproduced with permission from ref. 19), Wiley-VCH Verlag GmbH & Co.
A generally accepted catalytic mechanism involving the triad is illustrated in Fig. 2, where an ester RC(=O)-OR’ (substrate) is hydrolyzed when a nucleophile Nu-H is water (HO-H) and transesterified when Nu-H is an alcohol (R”O-H). The catalyst site is –CH2OH of Ser residue. During the reaction, the imidazole group of His residue acts as a general base catalyst to pull the proton from –CH2OH and increases the nucleophilicity of the oxygen to attack the carbonyl carbon of the substrate. At the same time, the carboxylate group of Asp residue helps the imidazole group to pull the proton, and the acyl-enzyme intermediate is formed with liberating R’O-H (acylating step). Then, in the deacylating step, also the imidazole and carboxylate groups act like a general acid base mode to facilitate the production of RC(=O)-Nu.
Ring-opening polymerization
Cyclic monomers
In the ring-opening polymerization (ROP) in Scheme 2, a number of cyclic monomers have been polymerized by enzyme catalysts. Typical examples of these monomers are shown as two classes of cyclic ester (lactone) monomers and other cyclic monomers (Fig. 3).
Fig. 3.

Typical examples of cyclic monomers for enzyme-catalyzed ring-opening polymerizations (ROP).
Ring-opening polymerization of cyclic ester (lactone) monomers
Discovery of lipase catalyst
There was a breakthrough in ring-opening polymerization (ROP) of lactones. In 1993 two research groups discovered independently that lipase enzymes induce ROP of ɛ-caprolactone (ɛ-CL, 7-membered) (Scheme 5a) and δ-valerolactone (δ-VL, 6-membered).4)–6) ROP of ɛ-CL by lipase PF in bulk at 75 °C for 10 days gave poly(ɛ-CL) in 92% yields, having Mn (number-average molecular weight) of 7,700 with Mw/Mn (molecular weight distribution) = 2.4. Similarly, δ-VL yielded at 60 °C poly(δ-VL) having Mn 1,900 with Mw/Mn = 3.0.4) Other lipases like lipase CC and PPL were also active for ROP of the monomers. These polyesters possessed the terminal structure of a carboxylic acid group at one end and a hydroxyl group at the other, indicating that the ROP was initiated by water molecule and terminated also by water present in the reaction system.
Scheme 5.

Lipase-catalyzed ROP of ɛ-CL (a) and ring-opening copolymerization of ɛ-CL (b).
Enzymatic ring-opening copolymerization by lipase PF catalyst between ɛ-CL and δ-VL was achieved in bulk at 60 °C for 10 days, giving rise to a copolyester of Mn value 3,700 with Mw/Mn = 2.9, with a random copolyester structure (Scheme 5b, m = 4). Copolymerization of ɛ-CL with other lactones like 15-pentadecanolactone (PDL) (Scheme 5b, m = 14) and d-lactide was also achieved.5)
These studies opened a new door for enzymatic polyester synthesis. Since then, ring-opening polymerization and copolymerization of various lactones (cyclic esters) of different ring size, unsubstituted and substituted, and also other related cyclic monomers, have been extensively studied.9)–17) Scheme 6 shows unsubstituted 4- to 17-membered lactones so far polymerized by lipase catalyst.20)–24) All these monomers showed a good or excellent ROP reactivity catalyzed by lipase in various solvents.
Scheme 6.

Lipase-catalyzed ROP of unsubstituted lactones with different ring size.
Scope of ring-opening polymerization of various ring-sized lactones
General features of ring-opening polymerization (ROP) of lactones are mentioned from smaller ring-sized lactones to larger ones.
A 4-membered lactone (β-propiolactone, β-PL) and substituted β-PLs were polymerized with lipase catalyst in bulk in 1996 by four groups, yielding linear polymers with molecular weight up to 2 × 104,25)–28) and also cyclic oligomers.26)
A 6-membered lactone (δ-VL) was first polymerized by lipase PF catalyst.4) α-Methyl-δ-VL afforded the polyester (Mn 8,400; Mw/Mn = 2.0) in 93% yields at 45 °C for 24 h by using lipase CA catalyst.29) Copolymerization of δ-VL with PDL (a 16-membered lactone) was conducted by lipase PF to yield a copolymer with Mn of 1,900.20)
1,4-Dioxan-2-one (DO) was polymerized with lipase catalyst. Metal free polyDO (Scheme 7a) is a desirable biocompatible polymer having good flexibility and tensile strength. Lipase CA (CALB) catalyzed the ring-opening copolymerization of DO with PDL at 70 °C for 26 h to give a copolyester poly(DO-co-PDL) with high Mw (>30,000).30)
Scheme 7.

Lipase-catalyzed ROP of DO (a) and enantio-selective ROP of lactide (LA) (b).
Lactide (LA), also a 6-membered lactone, is currently an important starting monomer for the production of poly(lactic acid) (PLA) as green plastics, which is produced commercially via Sn(II)-catalyzed ROP of LA. Lipase PS-catalyzed ROP of lactide was reported in 1997, where the ROP was carried out in bulk at a temperature between 80 and 130 °C to produce PLA with Mw up to 1.26 × 105. DLLA gave the higher molecular weight in comparison with LL-LA (LLA) and DD-LA (DLA) monomers.31) Noticeably, a recent paper has revealed that Novozym 435-catalyzed ROP of DLA was enantioselectively induced to produce polyDLA of Mn value 3,300 (Mw/Mn = 1.2) in 33% yields, whereas that of LLA did not take place. The polymerization employed the catalyst amount of 12.5% for DLA in toluene for 3 days at a lower reaction temperature of 70 °C (Scheme 7b).32)
A 7-membered lactone of ɛ-CL is the most extensively studied among various lactone monomers (m = 5 in Scheme 6), after the findings of lipase catalyst for ROP.4)–6),20)–23),29),33)–39) ROP of ɛ-CL was examined with addition of nucleophiles like butanol and water; both acted as an initiator. It was found that lipase CA is the most effective for the polymerization of ɛ-CL; the catalyst amount was reduced to 1%, which is compared with the reported systems of 20 to 50% of lipase. The reaction time could be reduced from 10 days to less than 10 h in bulk at 60 °C. The high catalytic activity was observed also for 12- and 13-membered lactones (UDL and DDL, respectively, Scheme 6). The molecular weight of poly(ɛ-CL) of 25,000 was readily reached in toluene at 70 °C.40)
ROP of ɛ-CL was carried out using Novozym 435 as catalyst at 60 °C in bulk or in an organic solvent. In bulk, cyclic dimer of ɛ-CL and linear poly(ɛ-CL) were formed in 2% and in 98%, respectively. Similar results were noted in isooctane. In acetonitrile, however, the reaction gave a major portion of cyclic oligomers in total 70% (17% of dilactone and 53% of cyclic oligomers) and a minor portion of linear poly(ɛ-CL) in 30%. Macrocycles up to 23 monomer units were produced in 1,4-dioxane (Scheme 8).41)
Scheme 8.

Lipase-catalyzed ROP of ɛ-CL produced linear polymers and cyclic oligomers depending on the reaction solvent.
α-Methyl-ɛ-CL produced the corresponding polyester with Mn 11,400 (Mw/Mn = 1.9) in 74% yields in bulk at 60 °C for 24 h with using lipase CA catalyst.29) ROP reactivity of methyl substituted ɛ-CL monomers was examined with lipase catalysis, and it was found that ω-methyl-ɛ-CL shows the least polymerizability among unsubstituted, α-and γ-substituted ɛ-CL monomers.39)
A 8-membered lactone (HL) was polymerized by Novozym 435 catalyst at 45 °C to give polyHL with Mn 23,600 (Mw/Mn = 2.8) having Tm 65.2 °C.24)
A 9-membered lactone (8-octanolide, OL) was polymerized by lipase catalyst, producing the polymer with molecular weight of 1.6 × 104 at 75 °C.42)
A 10-membered lactone (NL) and a 11-membered lactone (DL) were polymerized by Novozym 435 to produce polyNL having Mn 16,000 (Mw/Mn = 2.1, Tm 70.1 °C) and polyDL having Mn 20,000 (Mw/Mn = 3.2, Tm 77.0 °C), respectively.24)
Larger ring-sized lactones (called ‘macrolides’), 11-undecanolide (UDL, 12-membered), 12-dodecano-lide (DDL, 13-membered), 15-pentadecanolide (PDL, 16-membered), and 16-hexadecanolide (HDL, 17-membered) in Scheme 6, were enzymatically polymerized.20)–23),43)–47) At 75 °C in bulk, polyUDL of Mn = 23,000 (Mw/Mn = 2.6) was obtained quantitatively by lipase PF and the same polyester Mn = 25,000 (Mw/Mn = 2.2) in 95% yields by lipase CC. These results suggested a much higher ring-opening polymerizability of the macrolides than ɛ-CL.43)
A 12-membered lactone, 2-oxo-12-crown-4-ether (OC) showed a high ROP reactivity by Novozym 435 to give polyOC, a poly(ester-ether) (Mn = 3,500) having Tg value −40 °C, soluble in water (Scheme 9a). Copolymerization of OC with PDL tuned the copolymer properties (Mn 4,400–12,200).48)
Scheme 9.

Lipase-catalyzed ROP of (a) crown ether-ester monomer, (b) cyclic dimer of ɛ-CL, and (c) cyclic diester monomers.
A 14-membered monomer, cyclic dimer of ɛ-CL, was polymerized by lipase CA catalyst to give poly(ɛ-CL) reaching to Mn = 89,000 at a higher temperature (Scheme 9b).35)
A 16-membered lactone (PDL) was polymerized via ROP at a low water content in the reaction system at 70 °C, affording polyPDL reaching Mn 62,000 and Mw/Mn 1.9, respectively.46) 16- and 17-Memembered diester monomers were polymerized via lipase CA-catalyzed ROP, which produced the corresponding polyesters up to Mn = 4,100 (Mw/Mn = 2.2) in high yields (Scheme 9c).49)
Ring-opening copolymerization of PDL with four monomers, δ-VL, ɛ-CL, UDL, and DDL, by lipase PF and lipase PC at 60 °C in bulk was carried out to give copolymers with Mn ranging 1,200–6,300, being not statistically random (Scheme 10).20)
Scheme 10.

Ring-opening copolymerization of PDL with four lactones of different ring size.
A 17-membered lactone (HDL) is the largest ring-sized monomer having a simple unsubstituted structure so far studied. ROP of HDL was performed by lipase CA, lipase CC, lipase PC, lipase PF, or PPL in bulk at 45–75 °C for 5 days, giving rise to polyHDL with Mn reaching 5,800 (Mw/Mn = 2.0) in good or quantitative yields.47) ROP of Ambrettolide (Am) epoxide, also a 17-membered functional macrolide having an epoxy group at 10-position (Fig. 3), was performed by Novozym 435 catalyst.50)
A 24-membered lactone derived from natural sophorolipid was polymerized via lipase-catalyzed ROP to give a glycolipid-based polyester. The reaction proceeded in two modes depending on the reacted position of OH group with formation of mono-acylated products to oligomers and polymers.51)
Cyclic oligomers from ɛ-CL were polymerized via ROP to give poly(ɛ-CL) with Mn greater than 7.0 × 104, when the reaction was carried out at 70 °C in toluene. A cyclic dimer of ɛ-CL (14-membered, Scheme 9b) and other larger cyclic oligomers were also polymerized at a higher reaction temperature.35),52)
Reaction solvents
Recently, reaction solvents are considered very important to conduct green chemistry.2) Lipase-catalyzed ROP is normally carried out in bulk or in an organic solvent like toluene, heptane, 1,4-dioxane, and diisopropyl ether. However, as typical “green solvents”, water, super-critical carbon dioxide (scCO2) and ionic liquids have been often employed.
The first example using a water solvent is the lipase-catalyzed ROP of five lactone monomers, ɛ-CL, OL, UDL, DDL and PDL (Scheme 6). Macrolides of UDL, DDL and PDL were polymerized by lipase in water to produce the corresponding polyesters up to 89% yields. Lipase PC showed the best results in terms of polyester yields and molecular weight.22),53) DDL is hardly soluble in water; however, addition of the lipase gave a white emulsion-like solution, which allowed the ROP. In contrast, a mixture of the lipase and ɛ-CL or OL did not induce the ROP, which failed to form an emulsion-like system. The second example of the water medium system is the lipase-catalyzed ROP of a lactone in miniemulsions.54) As a typical run, a mixture of PDL monomer (10 mmol), water (10 mL, containing 1.0 wt% of a non-ionic surfactant), and hexadecane (100 mg) was vigorously stirred for 1 h at 45 °C to give miniemulsions. Then, a suspension of lipase PC (50 mg) in 5.0 g of the surfactant solution was added to the mixture, and the resulting miniemulsions consisting of PDL nanodroplets were subjected to ROP with stirring at 45 or 60 °C up to 24 h to reach a full conversion of PDL. PolyPDL nanoparticles were thus obtained, which is considered to be a direct synthesis of biodegradable polymer nanoparticles (size < 100 nm). PolyPDL showed a bimodal molecular weight distribution; the majority was that of high molecular weight (>2.0 × 105).
Supercritical carbon dioxide (scCO2) is an inexpensive, inert, nontoxic, and nonflammable green solvent, and possesses potentials for polymer production and recycling. scCO2 was used for the first time for the lipase CA-catalyzed ROP of ɛ-CL to produce poly(ɛ-CL) with molecular weight Mw ~ 1.1 × 104 in high yields and for the ring-opening copolymerization between ɛ-CL and DDL to produce poly(ɛ-CL-co-DDL) with Mw 1.3 × 104.55) A further work reported the synthesis of poly(ɛ-CL) having a higher molecular weight (Mw) reaching 7.4 × 104. The enzyme and scCO2 were repeatedly used for the polymerization.56) A chemoenzymatic method combining the enzymatic ROP and atom-transfer radical polymerization (ATRP) techniques used scCO2 as solvent for the synthesis of block copolymers containing ɛ-CL and MMA chains.57)
Ionic liquids are nonvolatile, thermally stable, and highly polar liquids, which allow to dissolve many organic, inorganic, metallo-organic compounds, and also polymeric materials. Thus, ionic liquids became popular for the synthesis and modification of polymers from the standpoint of green character.58) Lipase-catalyzed ROP of ɛ-CL was realized for the first time in an ionic liquid solvent, such as 1-butyl-3-methyl-imidazolium salts ([bmim][X−]), giving rise to poly(ɛ-CL) with Mn 4,200 (Mw/Mn = 2.7) in 97% yields at 60 °C.59) Novozym 435-catalyzed ROP of ɛ-CL in three ionic liquids, [bmim][BF4], [bmim][PF6], and [bmim][(CF3SO2)2N] at 60 °C for 24 h produced poly(ɛ-CL) of a higher Mn 7,000–9,500 (Mw/Mn ~ 2.4) in good yields.60) ROP of ɛ-CL was performed by lipase catalyst in ionic liquids such as 1-ethyl-3-methyl-imidazolium salt [emim][BF4], [bmim][BF4], n-butyl pyridinium salts [BuPy][BF4], [BuPy]]CF3CO2], and [emim][NO3]. As lipase, lipase YL, lipase CR, and PPL were employed. The poly-merization was carried out at 60 °C for 24 h to produce poly(ɛ-CL) of Mn ranged of 300–9,000, which is a telechelic polymer having OH group at one end and CO2H group at the other.61)
Reaction mechanism and monomer reactivity
An accepted reaction mechanism of lipase-catalyzed ROP of lactones is given by the principal reaction course involving an acyl-enzyme intermediate (Scheme 11).9)–11),33),43) Lipase-catalyzed hydrolysis of an ester in vivo is generally explained by the steps to proceed via a similar acyl-enzyme intermediate as shown in Fig. 2.62) Catalytic site of lipase is known to be –CH2OH group of a serine-residue in the triad Ser-His-Asp. The key step is the reaction of lactone with lipase catalyst involving an enzyme-lactone complex and ring-opening of lactone to give an acyl-enzyme intermediate (enzyme-activated monomer, EM), which corresponds to an acylation of enzyme. The initiation is nucleophilic attack of a nucleophile such as water (or another like an alcohol) at the acyl carbon of the intermediate to produce ω-hydroxycarboxylic acid (n = 1); the shortest propagating species. In the propagation, EM is nucleophilically attacked by the terminal hydroxyl group of a propagating chain end to form a one-monomer-unit elongated polymer chain. Both initiation and propagation steps correspond to a deacylation of enzyme. Thus, the polymerization proceeds via an “activated monomer mechanism”. This mechanism is different from an “active chain-end mechanism” observed normally for many other polymerizations, typically a vinyl polymerization, where a propagating chain-end is active, reacting with a monomer to give a one-more-monomer unit elongated, propagating polymer chain.
ROP reactivities of the monomers have been examined, the data of which are given in Table 2 together with dipole moment values and reaction rate values of five kinds of reaction of unsubstituted lactones with different ring size. These data show that the ring-opening reactivity is governed primarily by the ring strain, except for enzymatic polymer-ization. The dipole moment value of monomers is indicative of polarity of the molecule and is taken as a measure of their ring strain. The value of macrolides is lower than that of ɛ-CL and close to that of an acyclic fatty acid ester (butyl caproate), suggesting a very small ring strain of the macrolides. In fact, the rate constants of the macrolides in alkaline hydrolysis and anionic polymerization with NaOMe initiator are much smaller than those of ɛ-CL, although the hydrolysis took a little peculiar behaviors for medium sized lactones, paticularly NL and DL. These data generally imply that the macrolides have much lower ring strain, and hence, show less reactivity and polymerizability than ɛ-CL with an anionic catalyst.
Table 2.
Dipole moment values and reaction rate values of unsubstituted lactones with different ring size
| rate constant | relative rate of polymerization | |||||
|---|---|---|---|---|---|---|
|
|
|
|||||
| lactone (ring size) | dipole moment (C·m) | alkaline hydrolysisa (L·mol−1·s−1, × 104) | propagationb (s−1, × 103) | enzymatic polymerization | Zn-catalyzed polymerizatione | |
| Ac | Bd | |||||
| δ-VL (6) | 4.22 | 55,000 | 0.10 | 0.07 | 2,500 | |
| ɛ-CL (7) | 4.45 | 2,550 | 120 | 0.10 | 0.15 | 330 |
| HL (8) | 3.70 | 3,530 | 3.8 | |||
| OL (9) | 2.25 | 116 | 0.45 | 21 | ||
| NL (10) | 2.01 | 0.22 | 0.04 | |||
| DL (11) | 1.88 | 0.53 | 0.02 | |||
| UDL (12) | 1.86 | 3.3 | 2.2 | 0.13 | 0.06 | 0.9 |
| DDL (13) | 1.86 | 6.0 | 15 | 0.19 | 0.37 | 1.0 |
| PDL (16) | 1.86 | 6.5 | 0.74 | 1.0 | 0.9 | |
| HDL (17) | 1.0 | 1.0 | ||||
| butyl caproate | 1.75 | 8.4 | ||||
With alkaline of NaOH in 1,4-dioxane/water at 0 °C.63)
Anionic ROP by NaOMe catalyst in THF at 0 °C.64)
Lipase PF as catalyst in diisopropyl ether at 60 °C; the relative rate is given by normalizing the Vmax/Km values with respect to HDL.65)
CALB as catalyst in toluene at 45 °C; the relative rate is given by normalizing the Vmax/Km values with respect to PDL.24)
Zn(Oct)2 initiator in bulk polymerization at 100 °C; the relative rate is given by normalizing the initial rate constants with respect to HDL.66)
In 1997, the first kinetic analysis based on the Michaelis-Menten equation was performed for the lipase PF-catalyzed ROP of ɛ-CL and DDL.21) As the results, the formation of an acyl-enzyme intermediate (EM in Scheme 11) was considered to be the rate-determining of the overall reaction, because the reactivity of the intermediate must be high and hence the subsequent steps are rapid. This hypothesis was supported also by the experiments using a different amount of nucleophiles in the polymerization reaction. The Michaelis-Menten kinetics parameters, Km and Vmax values, were determined and it was concluded quantitatively that DDL is 1.9 times more reactive than ɛ-CL.21),22)
The Vmax/Km (s−1) value is a good measure for the overall rate of the enzymatic ROP. Thus, the relative rate ratio of lactone monomers with different ring size was derived by normalizing the values with respect to a specific lactone monomer (Table 2).20)–22),24),65)–68) There is a tendency that with lipase PF the larger the ring-size, the larger the polymerization rate in one order magnitude difference as seen in column A, that is, the higher polymerizability of the macrolides was explained by the higher rate in the formation of EM. As for the Michaelis-Menten constant Km [(k −1 + kcat)/k+1 mol·L −1], the values are not much different each other; they are from 0.61 to 1.1, on the other hand, the maximal reaction rate Vmax (kcat [E]0, mol·L−1·s−1) values are much more varied from 0.66 to 7.2.65) These results suggested that the reaction rate is mainly governed by the larger value of Vmax, and much less by the binding ability judged from the Km value. Namely, the reaction step from the lipase-lactone complex to form the intermediate EM is the key step in the lipase-catalyzed ROP in Scheme 11.
In contrast, the Zn-catalyzed anionic polymerizability of lactones showed the reverse direction with big difference, for example, δ-VL and ɛ-CL showed 2,500 and 330 times more reactive than the macrolides (Table 2). The polymerizability is mainly governed by the ring-strain. In the propagation reaction, the rate-determining is of SN2 type reaction; the nucleophilic attack of the propagating Zn-alkoxide species occurs at the carbonyl carbon to cause the acyl-oxgen bond scission, forming the alkoxide species again (Scheme 12).66)
Scheme 12.

Propagation reaction of anionic ROP of lactone by Zn-catalyst.
Recently, CALB (Novozym 435) is increasingly more often used. In comparison with lipase PF, catalysis of CALB was examined and found much different in behaviors. CALB exhibited a higher catalytic activity for ɛ-CL than for macrolides. Qualitatively, initial rates (×105, L·mol−1·h−1·mg−1) of ROP by CALB catalyst were obtained as 2,300 for ɛ-CL, 48 for OL, 1,600 for DDL, and 4,700 for PDL. For these four monomers, the corresponding values of lipase PF were 1.3, 1.8, 2.5, and 8.5, and those of lipase PC were 0.42, 2.2, 4.8, and 11.40),42)
Catalysis of CALB for ROP of nine lactones was investigated according to Michaelis-Menten kinetics.24) Values of Km are in a narrow range between 0.09 and 0.73 mol·L−1, suggesting close affinities of the lipase for all lactones. On the other hand, values of Vmax varied between 0.07 and 6.10 mol · L−1·h−1 and did not show a trend. These are partially similar to the results observed above for lipase PF catalysis,22),23),65) indicating that the ROP reactivity with CALB catalyst is likely operated also by the process from the lipase-lactone complex to form EM in Scheme 11. The relative rate of ROP of nine monomers was derived from Vmax/Km values (column B, Table 2). The most reactive monomer HL (8-membered) showed almost 200 times more reactive than the least reactive DL (11-membered). The relative rate values look very complicated; there is no monotonous change depending on the ring size. Medium sized lactones showed rather smaller reactivity. These results cannot be related to variations in physical properties such as the dipole moment of lactones only. Simple explanations are not available, but in addition to values of the dipole moment and the ring strain, other factors such as the transoid and cisoid structure of lactones,63) and the conformational strain and transannular interactions in medium ring lactones were taken into account for the respective ring size cases.24)
A copolymerization approach gave new insights into the ROP mechanism. CALB-catalyzed copolymerization using two ω-methyl-substituted lactones (racemic) and four achiral lactones (unsubstituted) was investigated.67) The copolymerization of β-butyro-lactone (β-BL, 4-membered) with an achiral lactone at 60 °C induced enantioselection of the racemic monomer, where (S)-β-BL was preferentially consumed to give (S)-enriched copolyester (Scheme 13, p = 1). In the propagation, there are four different elemental steps, which are given as (a), (b), (c), and (d) in Scheme 14, showing the steps where an acyl-enzyme intermediate EM (or transition-state) is attacked by a nucleophile of propagating end of hydroxy group.67),68) From the 13C NMR analysis of the four diad peaks of the product copolymer, the intensity ratio was approximately a:b:c:d = 10:6:4:0, respectively. If the rate-determining step of the lipase-catalyzed ROP is the formation of EM because of its high reactivity, the formation of diads a and c, and of diads b and d should be the same in amount; however, the results were different; they are 10:4 and 6:0, respectively. This suggests that the reaction steps of Scheme 14 involve a propagating chain end of sterically two different nucleophile structures (primary and secondary alcohols), and hence, the structure of the nucleophile greatly affects the overall polymerization rate. In particular, step (d) does not actually occur.
Scheme 13.

Ring-opening copolymerization of a racemic ω-methyl-substituted lactone with an unsubstituted lactone.
Scheme 14.

Four modes of the propagation step in the copolymerization of β-BL with an unsubstituted lactone; steps (a) and (d) are homo-propagations, while steps (b) and (c) are cross-propagations.
The CALB-catalyzed copolymerization of racemic δ-CL (4-methyl-VL, 6-membered) with DDL proceeded in a similar way; however, the product copolymers were all enriched in (R)-isomer of δ-CL, in a reverse enantioselection to β-BL (Scheme 13, p = 3). The peaks intensity corresponding to a, b, c, and d of the δ-CL reaction was in about 10:3:3:0.5, which is a similar trend to the above copolymerization of β-BL with DDL.67) Similar views can be made again as to the lipase-catalyzed ROP mechanism.68)
These observations suggest that the rate-determining step is not always the formation of the acyl-enzyme intermediate (EM) in Scheme 11. It seems more general to consider a mechanism of the lipase-catalyzed ROP, in which the formation of EM (acylation of lipase) and/or the subsequent reaction of EM with OH group of propagating ends (deacylation of lipase) are operative depending on the monomer structure. In particular, the deacylation step becomes more important when the propagating alcohol end is of sterically bulky nucleophile like a secondary alcohol, and then this step determines the overall rate of reaction. Enantioselection is therefore induced possibly at both acylation and deacylation steps; these situations are indicated in more details in Fig. 4.68)
The above consideration was further supported by the lipase PF-catalyzed copolymerization, giving the structure of propagating chain-end with both a primary alcohol like a case between unsubstituted lactones, PDL and ɛ-CL. Four diad sequence ratios of the product copolyester were roughly in the following values: PDL – PDL = 10, PDL – ɛ-CL = 2.4, ɛ-CL – PDL = 9.6, ɛ-CL – ɛ-CL = 2.7, where the italics denote the monomer of EM. The homo-polymerizability ratio of PDL to ɛ-CL is 10/2.7 = 3.7, which can be compared with the value of 7.4 obtained from each homo-polymerization in Table 2. The competitive reaction rate ratio of PDL and ɛ-CL chain-ends toward EM of PDL is 10/9.6 = 1.04, which is almost the same, and that of PDL and ɛ-CL chain-ends toward EM of ɛ-CL is 2.4/2.7 = 0.89, which is also very close. These results imply that the EM mainly governs the overall reactivity of the monomer, regardless of the kind of the propagating chain-end so far as its structure is similar (a primary alcohol in all cases), i.e., the ROP mechanism involving the formation of the EM as a rate-determining step is plausible.20),21) A similar case was reported in the lipase PF-catalyzed copolymerizations between δ-VL and ɛ-CL (unsubstituted 6- and 7-membered, respectively) to give a random co-polyester, where reaction rates of homo-propagation and cross-propagation are close each other for these monomers.5)
A mechanism suggesting the importance of the deacylating step was previously described, from the results of lipase CA-catalyzed ROP of 4-alkyl-substituted ɛ-CL where methyl, ethyl, or n-propyl group was employed as an alkyl group.69) The results implied that the deacylation step is more likely to be the rate-determining for the polymerization; the reaction of nucleophile alcohols having different structures (with bulky or less bulky alkyl) with EM caused the polymerization rate difference and the shift of enantioselection. However, in the hydrolysis, the rate-determining step is most likely the formation of the acyl-enzyme intermediate EM, because water is a very small nucleophile and its reaction with EM must be fast while a propagating chain end alcohol is bigger.
Besides the main reaction course in Scheme 11 and Fig. 4, a transesterification often takes place under severe reaction conditions during the lipase-catalyzed ROP of lactones. For example, when ROP of a macrolide (DDL or PDL) was carried out in the presence of an aliphatic polyester (poly(ɛ-CL) or poly(1,4-butylene adipate)), a polyester copolymer from the cyclic monomer and the polyester was produced (Scheme 15a).70),71) Also, lipase CA- or lipase PF-catalyzed intermolecular transesterification between two different linear polyesters took place to give a polyester copolymer composed from the two polyester repeating units (Scheme 15b).70),72)
Scheme 15.

Transesterifications during the lipase-catalyzed reaction: (a) Between a lactone monomer and a linear polyester of poly(ɛ-CL), and (b) Between two linear polyesters.
End-functionalized polyesters
Typical end-functionalized polymers, macromonomers and telechelics, are fundamentally and practically important in polymer chemistry. These syntheses require a precise control of the polymer terminal structure. Lipase catalysis provides a novel method for a single-step synthesis of end-functionalized polyesters via a relatively simple reaction. Moreover, lipase-catalyzed synthesis gives product polymers free from any metals, that broadens the application scope of the products in particular for biomedical areas.
Initiator method
A mechanism of lipase catalysis involves a nucleophile like water and an alcohol to initiate the ROP of lactones. In fact, lipase CA-catalyzed ROP of ɛ-CL or DDL at 60 °C in bulk in the presence of a functional alcohol produced end-functionalized polyesters (“initiator method”).73) Functional alcohols include 2-hydroxyethyl methacrylate (HEMA), 5-hexen-1-ol and 5-hexyn-1-ol for the synthesis of methacryl-, ω-alkenyl- and alkynyl-type polyester macromonomers having Mn 1,000–3,100. The functionality reached 100% in most reaction runs (Scheme 16a). These methacryl-type poly-ester macromonomer can be radically polymerized to lead to polymers of a comb structure. As macro-initiators, poly(ethylene glycol) and poly(ɛ-CL)-diol, initiated the ROP of ɛ-CL or 1,5-dioxepane-2-one (DXO) to give a triblock polyester with a hydroxyl group at both ends (Scheme 16b).74)
Scheme 16.

Initiator method afforded various macromonomers and telechelics.
To synthesize highly end-functionalized polymers, the initiation process of ROP was examined under various reaction conditions. Among initiating nucleophiles, water, an alcohol, an amine, and a thiol, water was most reactive and hence a good ‘initiator’ in the Novozym 435-catalyzed ROP of ɛ-CL. For high functionality, the enzyme and the reaction system were to be well dried to avoid the initiation by water.75) Thiol end-functionalization was achieved by CALB catalyst to induce the ROP of ɛ-CL with 2-mercaptoethanol initiator. The fraction of thiol functionalized-ends of poly(ɛ-CL) was 70% with Mn 3,100 (Scheme 16a).76)
A new approach to a biodegradable polyester system was performed by the lipase-catalyzed ROP of ɛ-CL and 1,4-dioxan-2-one (DO) monomers initiated from an alcohol attached on the gold surface. The polyester system can be used as a biocompatible/biodegradable polymer for coating materials in biomedical area such as passive or active coatings of stents. This method would be beneficial in the applications where the minimization of harmful species is critical.77)
A macroinitiator of a linear or a four arm star-shaped polyglycidol was used for synthesis of densely grafted poly(glycidol-graft-ɛ-CL) and loosely grafted poly[(glycidol-graft-ɛ-CL)-co-glycidol] copolymers via lipase-catalyzed ROP of ɛ-CL or Sn-catalyzed chemical process using ɛ-CL monomer. Architecture, microstructures, molecular weight, and chemical compositions of the copolymers were examined and compared. In comparison with linear poly(ɛ-CL), the latter copolymer showed a change of degradation mechanism and much enhanced degradability, probably due to high concentration of hydroxy groups at the polyglycidol backbone.78)
The initiator method is useful for a single-step synthesis of end-functionalized polyesters as well as other polymers, which can be prepared via ROP of monomers induced by a functional (macromolecular) alcohol (or another nucleophile) system.
Terminator method
A single-step, convenient production of end-functionalized polyesters was developed by lipase-catalyzed ROP of DDL in the presence of vinyl esters (vide infra).79) The vinyl ester acted as terminator by reacting with a hydroxyl group during the polymerization (“terminator method”). In use of vinyl (meth)acrylate as terminator, the (meth)acryloyl group was quantitatively introduced at the polymer terminal at 60 °C in bulk to give a (meth)acryl-type polyester macromonomer with Mn 2,000–4,000 and functionality > 0.95. The polymerization in the presence of vinyl 10-undecanoate produced the ω-alkenyl-type macromonomer (Scheme 17a). This system was applied to the synthesis of telechelics having a carboxylic acid group at both ends by using divinyl sebacate in the reaction mixture at 60 °C in bulk; Mn of the telechelics was 2,900 with functionality of 1.95 (Scheme 17b).80)
Scheme 17.

Terminator method produced designed macro-monomers and telechelics.
Lipase-catalyzed one-pot synthesis of diepoxide-functionalized polyester telechelics was achieved from PDL, divinyl adipate, and the epoxide species glycidol via a combined ROP and condensation polymerization (Scheme 17c).81) The DP value (m + n = 4, 6, and 10) was controlled by the feed ratio of the starting monomers. The telechelics were UV homo-polymerized and also UV co-polymerized with 20 wt% of a cycloaliphatic diepoxide to produce crosslinked films, whose properties were studied with functions of crosslinking degree and crystallinity of the PDL segment.
The terminator method involves a lipase-catalyzed single-step acylation of an alcohol end-group with a vinyl ester, the process of which can be applied basically for other polymer alcohols to give an end-functionalized polymer.
Chemoselective and regioselective polymerizations
Lipase catalyst chemoselectively induced the ROP of 2-methylene-4-oxa-12-dodecanolide, a cyclic derivative of methyl methacrylate, yielding a polyester having the reactive exo-methylene group in the main chain (Scheme 18). This type of polymer is hardly obtained by using a conventional chemical initiator. The chemoselective polymerization of α-methylene-macrolides having various groups in the ring, e.g., aromatic, ether or amine group, was enzymatically, anionically, and radically carried out. Lipase catalyst induced ROP of lactones selectively to afford polyesters, whereas anionic and radical initiators induced the vinyl polymerization. The polyester was readily crosslinked radically at 60 °C in toluene in 15 min via reaction through the reactive methylene group to produce a crosslinked polymer.82),83)
Scheme 18.

Chemoselective ROP of an α-methylene-macrolide.
Chemoselective ROP of Ambrettolide (Am) epoxide, a 17-membered macrolide having an epoxy group at 10-position (Fig. 3), was performed by Novozym 435 catalyst to give the polyester with Mn value of 9,700 (Mw/Mn = 1.9). The epoxide group remained unaffected during the polymerization.50) Related functional macrolides, Globalide (Gl) and Am, are presently used in fragrance industry. Gl is a 16-membered lactone having the double bond at 11 or 12 position, and Am is a 17-membered lactone with the double bond at 10 position. They can be compared with simple macrolides of PDL and HDL, respectively. Novozym 435-catalyzed ROP of Gl and Am was performed in toluene at 60 °C to give polyGl and polyAm, having Mn both around 2.4 × 104. Both polyesters had melting points 46–55 °C, which are compared with those of saturated polyesters, polyPDL and polyHDL, both around 95 °C. PolyGl and polyAm are nontoxic, hydrolytically and enzymatically nonde-gradable biomaterials.84)
Regioselective, lipase-catalyzed graft-polymerization of ɛ-CL and β-butyrolactone (β-BL) onto chitin and chitosan was accomplished in one-pot in bulk at 70 °C. The ROP of the cyclic monomers was initiated regioselectively from 6-OH group in chitin and from 6-OH and NH groups in chitosan to produce chitin-graft-polyester and chitosan-graft-polyester, respectively. In the products, both the stem polymer cellulose and the graft polyester are biodegradable.85)
Enantioselective polymerization
Two studies on enantioselctive ROP at early stage are cited. Lipase PF induced an enantioselective ROP of α-methyl-β-propiolactone (4-membered) in toluene to produce an optically active (S)-enriched (up to 75% (S)) polymer with Mn values from 2,000 to 2,900 and [α]25D + 12.2 ° to +19.0°.27) The enantioselective ROP of 3-methyl-4-oxa-6-hexanolide (MOHEL) was catalyzed by lipase PC in bulk at 60 °C (Scheme 19).22) The apparent initial reaction rate of (S)-isomer was seven times larger than that of (R)-isomer, indicating that the enantioselective polymerization of MOHEL effectively occurred.
Scheme 19.

Enantioselective ROP of MOHEL.
In the lipase CA-catalyzed copolymerization of β-butyrolactone (β-BL) with DDL, (S)-β-BL was preferentially reacted to give the (S)-enriched optically active copolymer with ee of β-BL unit = 69% (Scheme 13, p = 1, m = 11). δ-Caprolactone (6-membered) was also enantioselectively copolymerized with achiral lactones to give the (R)-enriched optically active polyesters reaching 76% ee, in which the enantioselectivity became opposite (Scheme 13, p = 3, m = 11).67),68)
Lipase CA-catalyzed ROP of ω-methylated six different ring-sized lactones was studied for elucidation of the relationships between enantioselectivity and lactone structure (Scheme 20).86) The rate of polymerization was much affected by the ring size as suggested by kcat values and the enantioselectivity was switched from (S)-selective for small (4-, 6-, and 7-membered) lactones to (R)-selective for large (8-, 9-, and 13-membered) lactones. From the kcat values (s−1) [for 4-, 6-, 7-, 8-, 9-, and 13-membered: (S)-enantiomers; 45.7, 7.9, 49.3, 0.01, nd, and nd, respectively; (R)-enantiomers; nd, 7.6, 8.5, 204.4, 10.3, and 23.3, respectively, where nd meaning ‘not detected’], the enantiomeric ratio E was very large, in particular, for the large lactones. The lactone takes transoid and cisoid conformations; virtually small lactones for cisoid and large lactones for transoid. ROP of the small cisoid lactones was (S)-selective (3-MePL and 6-MeCL) or aselective (5-MeVL). ROP of the larger transoid lactones was (R)-selective with very high enantioselectivity. For the intermediate ring sizes, 7-MeHL and 8-MeOL, the significant amount of cisoid conformers present did not affect the enantioselectivity. The interpretation of the enantioselectivity and lactone structure relationship is attractive, but it seems actually not simple, particularly for understanding the intermediate ring-sized case.86) A selectivity switch was also observed in the copolymerization from (S)-selective for 3-MePL (4-membered) to (R)-selective for 5-MeVL (6-membered) (Scheme 13).67)
Scheme 20.

Enantioselectivity switching in ROP of ω-methyl-substituted lactones catalyzed by lipase CA.
Interestingly, Novozym 435 did not induce the ROP of ll-lactide but induced the ROP of dd-lactide (DLA) enantioselectively to produce poly-DLA of Mn value 3,300 in toluene at a lower reaction temperature of 70 °C for 3 days (Scheme 7).32)
A new method of ‘iterative tandem catalysis’ (ITC) was proposed, by which optically active oligoesters were obtained via ROP of 6-methyl-ɛ-CL (6-MeCL).87),88) ITC means a polymerization in which the chain growth is effectuated by a combination of two different catalytic processes that are both compatible and complementary. By combining the lipase CA-catalyzed enantioselective ROP of 6-MeCL and the Ru-catalyzed racemization of a propagating secondary alcohol in one-pot (chemoenzymatic process), optically pure oligoesters were produced, which was developed based on the ‘dynamic kinetic resolution’ (DKR) method (Scheme 21). First, lipase CA catalyzes the ring-opening of an (S) monomer enantioselectively to give the benzyl alcohol adduct, but the (S) alcohol is less favored to react with the monomer via ring-opening, and hence, the Ru-catalyzed racemization takes place to give a racemic alcohol. Then, an (R) alcohol selectively reacts with the monomer to facilitate one monomer-unit elongated. This reaction cycle repeats and ended up with the production of (R) oligoesters from the racemic monomer. The cycle could be repeated up to five times, being up to five monomer units of the product oligomers. Since the ring-opening of racemic 6-MeCL by lipase CA catalyst took place only for the (S)-isomer, (R)-6-MeCL virtually remained unreacted. In principle, this method provides with a flexible tool for producing chiral macromolecules from racemic or prochiral monomers.
Scheme 21.

Lipase-catalyzed enantioselective ROP of 6-methyl-ɛ-CL monomer to produce (R)-oligoesters based on dynamic kinetic resolution (DKR) through Ru-catalysis.
Chemoenzymatic polymerization
Enzyme is a “green” natural catalyst, in contrast to a “chemical” metal catalyst, which is sometimes toxic and not renewable. More importantly, catalysis of these two classes is quite different in function and mechanism, but can be mutually compatible each other to allow the catalysis concurrently in the same reaction system, if the combination of two classes of catalyst is appropriate. With utilizing these advantages of enzymes, a chemoenzymatic method has been developed for the synthesis of new polymeric materials in one-pot, in particular, various block copolymers, which are difficult to prepare via conventional method.
A chemoenzymatic method was newly developed by combination of an enzymatic polymerization and a chemical polymerization; enzyme (lipase)-catalyzed ring-opening polymerization (eROP) of lactones and atom transfer radical polymerization (ATRP).89) The method allows a versatile synthesis of block copolymers consisting from a polyester chain and a vinyl polymer chain by using a designed bi-functional initiator, whose reaction routes are generally shown in Scheme 22.89),90) For example, in route A an initiator having OH group for eROP and Br atom for ATRP is to be used. Lipase CA (Novozym 435) catalyzed the ROP of ɛ-CL at 60 °C in toluene to give poly(ɛ-CL). Then, the poly(ɛ-CL) having Br was used for the Cu-catalyzed radical polymerization of styrene (St) at 85 °C in 1,4-dioxane to give poly(ɛ-CL-block-St) in a high yield, in which the ɛ-CL chain exhibited Mn 5.8 × 103 and the St chain 1.5 × 104.89) These data indicate that the two catalyst systems are robust under the reaction conditions and can tolerate each other. Block copolymer synthesis via routes B and C was also possible.
Scheme 22.

Chemoenzymatic method combining an enzymatic process (eROP) and a radical process (ATRP) for the synthesis of a block copolymer.
A spontaneous single-step chemoenzymatic synthesis of block copolymers was facilitated in supercritical carbon dioxide (scCO2) via route B in Scheme 22.57) For example, the reaction was carried out in scCO2; at 35 °C, 1 500 psi (10.3 MPa) using MMA, ɛ-CL, Novozym 435, CuCl, and 2,2′-bipyridine. With reaction time of 20 h, a block copolymer, poly(ɛ-CL-block-MMA), of Mn = 41,000 (Mw/Mn = 2.11) was obtained in 60% yields. PolyMMA block part showed Mn = 10,000 (Mw/Mn = 1.02), indicating a living radical polymerization of MMA.
A chemoenzymatic process combining eROP and ATRP was employed for synthesizing heterografted molecular bottle brushes (HMBB) based on poly(glycidol-graft-ɛ-CL) core polymers. The resulting HMBB possessed following Mn values (reaction time): 55,000 (75 min) and 83,000 (120 min) for MMA grafts; 60,000 (120 min) and 100,000 (210 min) for n-BuMA grafts.91)
With utilizing the chemoenzymatic method for synthesis of multifunctional poly(meth)acrylates, functional monomers of (meth)acrylates were derived from Novozym 435-catalyzed transacylation of methyl methacrylate (MMA) and methyl acrylate (MA) with various functional alcohols. These monomers thus prepared were radically polymerized by AIBN initiator to give poly(meth)acrylates via cascade reactions. The obtained polymers possessed hydrophilic, hydrophobic, as well as cationic natures, and can be used for various surface coatings.92)
Ring-opening polymerization of other cyclic monomers
In addition to lactone monomers to lead to a variety of polyesters, other types of cyclic monomers (Fig. 3) have been polymerized by lipase catalyst. ROP of these several cyclic monomers is mentioned here in relation to the synthesis of polyesters and related polymers for showing extended lipase catalysis.
Polyester synthesis
It was found in 1993 that a cyclic acid anhydride underwent the ring-opening addition-condensation polymerization with a glycol by lipase PF catalyst, giving rise to a polyester having Mn~2,000 (Mw/Mn = 1.4) in a good yield. During the reaction, the ring-opening as well as the dehydration occurred (Scheme 23a).93) Various cyclic anhydrides, succinic, glutaric, and diglycolic anhydrides, were polymerized by lipase CA catalyst with α,ω-alkylene glycols in toluene at 60 °C to give the polyesters with Mn reaching 1.0 × 104. This ring-opening addition-condensation polymerization involving dehydration proceeded also in water and scCO2.94)
Scheme 23.

Synthesis of polyesters via ROP of succinic anhydride (a), and l-lacOCA (b).
ROP of a new cyclic monomer of an O-carboxylic anhydride derived from lactic acid (lacOCA) has been achieved with catalysis of lipase (Scheme 23b).95) The polymerization proceeded within a few hours at 80 °C in toluene with liberating carbon dioxide and gave poly(l-lactic acid) (PLLA) in high yields having high molecular weight, Mn up to 38,400 with low polydispersity Mw/Mn < 1.4. Slight preference in reactivity for l-lacOCA over d-lacOCA was observed.
Synthesis of polycarbonate, polyamide, and poly(ester-amide)
In 1997, polycarbonate synthesis from 1,3-dioxan-2-one (trimethylene carbonate, TMC) was first reported (Scheme 24a); lipase CA efficiently catalyzed ROP of TMC at 70 °C to give the product having Mn higher than 10,000.96) At a higher temperature of 100 °C, the ROP with PPL catalyst afforded a polycarbonate of higher Mw~ 1.6 × 105. Cyclic dicarbonates, cyclobis(hexamethylene carbonate) (18-membered) and cyclobis(diethylene glycol carbonate) (16-membered), were homo-polymerized via lipase CA-catalyzed ROP to give corresponding polycarbonates with Mn up to 1.3 × 104 (Mw/Mn = 2.1). Their copolymerizations with ɛ-CL and DDL produced poly(carbonate-co-ester)s having Mn 1.9 × 104 (Mw/Mn = 1.8) in high yields.97) It is to be noted that no carbon dioxide was liberated during the polymerization, in contrast to a chemical anionic polymerization which often involves the liberation of carbon dioxide to produce a non-carbonate unit.
Scheme 24.

Lipase-catalyzed ROP to produce polycarbonates, (a) and (b), poly(β-alanine) (c), and poly(ester-amide) (d).
A new 7-membered cyclic carbonate monomer having a ketal group was derived from naturally occurring l-tartaric acid. Lipase-catalyzed ROP of the monomer was induced at 80 °C in bulk, to afford the polycarbonate with Mn 15,500 with Mw/Mn = 1.7 (Scheme 24b). Deprotection of the ketal group resulted in optically pure polycarbonate (Mn 1.0 × 104, Mw/Mn = 2.0, [α]D20 = +56 °) having hydroxy functional groups. The polycarbonate is considered to have potentials for various biomedical applications.98) A degradable polycarbonate copolymer for pH-dependent controlled drug release with micelle formation was synthesized via a chemoenzymatic route. A tri-block copolymer of ABA type was designed, where A block is poly(trimethylene carbonate) (PTMC) (Scheme 24a) and B block is poly(PEG-co-cyclic acetal) (PECA). PECA is an α,ω-glycol synthesized chemically, and lipase CA-catalyzed polymerization of TMC was induced from the glycol OH groups to give the triblock copolymer, which was shown to form micelles and to be a biomaterial for drug carrier.99)
A 4-membered cyclic amide (β-lactam) was polymerized via Novozyme 435-catalyzed ROP to afford poly(β-alanine) with a degree of polymerization 8, having a linear structure (Scheme 24c).100)
A poly(ester-alt-amide) copolymer was prepared by the lipase-catalyzed ROP of a six-membered cyclic depsipeptide, 3(S)-isopropylmorpholin-2,5-dione, in bulk at 100 and 130 °C, ending up with the polymer having Mn up to 3.0 × 104 (Scheme 24d).101) ROP of a large cyclic ester-urethane oligomer was achieved by lipase CA to yield a poly(ester-urethane) with Mw reaching 101,000.52)
Synthesis of polyphosphate and polythioester
It is notable that lipase catalyzes the ROP of cyclic compounds containing atoms like phosphorus and sulfur that are larger than oxygen or nitrogen (Fig. 3).
Polyphosphates are useful materials for various applications such as flame retardation, plasticizing, and biomedical utilizations. Lipase-catalyzed ROP of a cyclic phosphate was achieved in bulk with PPL catalyst (Scheme 25a). The product polyphosphate has Mn value up to 5,800.102)
Scheme 25.

Lipase-catalyzed synthesis of polyphosphate (a) and polythioester (b).
A new class of bio-polyesters having a thioester linkage S–C(=O)– was synthesized. A cyclic monomer of 1,6-hexanedithiolsebacate (18-membered ring) was polymerized via ROP catalyzed by lipase CA in bulk in the presence of molecular sieves to give a polythioester (Scheme 25b). The highest Mw of 1.2 × 105 was obtained at 120 °C for 48 h.103)
Condensation polymerization
Condensation reaction to form an ester includes four reaction modes, an esterification (dehydration) and three kinds of transesterifications (Scheme 26). The lipase-catalyzed polyester synthesis via condensation polymerization (polycondensation) utilizes the reactions of all four modes, although acidolysis mode (c) is least involved.
Scheme 26.

Four condensation reaction modes for the formation of esters.
Condensation polymerization of oxyacids or their esters
Dehydration (Scheme 26a) is the simplest condensation polymerization mode. The first paper appeared in 1985, reporting a lipase-catalyzed condensation polymerization of an oxyacid monomer, 10-hydroxydecanoic acid. The degree of polymerization (DP) value of the product was small (≥5). A poly(ethylene glycol)-modified esterase also induced the oligomerization of glycolic acid, the smallest oxyacid (R = H, Scheme 27).104) A lipase-catalyzed polymerization of lactic acid gave a low molecular weight poly(lactic acid) (PLA) under various reaction conditions (R = CH3, Scheme 27).105)
Scheme 27.
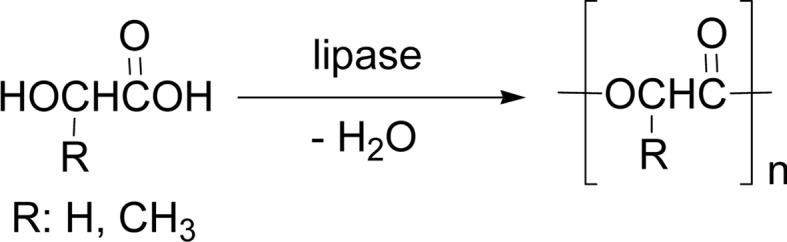
Lipase-catalyzed synthesis of oligo(glycolic acid) (R = H) and PLA (R = CH3).
Polyesters of a higher molecular weight were enzymatically produced from hydrophobic oxyacids. In the lipase CA-catalyzed polymerization of 16-hydroxyhexadecanoic acid, 12-hydroxydodecanoic acid, or 10-hydroxydecanoic acid under vacuum at a higher temperature (90 °C) in bulk for 24 h, the degree of polymerization (DP) value was beyond 100, whereas the polyester with a lower molecular weight was formed from 6-hydroxyhexanoic acid.106) Copolymerization of 12-hydroxydodecanoic acid (12HD) with methyl 12-hydroxystearate (12HS) (both from seed oils) was catalyzed with Novozym 435 in toluene in the presence of molecular sieves at 90 °C to give poly(12HD-co-12HS) having a high Mw~1.0 × 105 with showing elasticity and biodegradability.107)
For the polyester synthesis via transesterification of (b), (c), and (d) in Scheme 26, activation of carboxylic acid groups is normally needed.
A regioselective polymerization of isopropyl aleuriteate was achieved by Novozym 435 catalyst, where the only primary alcohol was involved in the reaction at 90 °C in toluene (Scheme 28). The polymer of Mn 5,600 was obtained in 43% yields. Copolymerization of isopropyl aleuriteate with ɛ-CL gave a random copolymer having Mn up to 10,600 in ~70% yields.50)
Scheme 28.

Regioselective polyester synthesis from isopropyl aleuriteate.
Transesterification polymerization of racemic AB type monomers having a secondary hydroxy group and a methyl ester group led to chiral polyesters by iterative tandem catalysis (ITC, see also Scheme 21). The concurrent actions of an enantioselective acylation catalyst (Novozym 435) and a racemization catalyst (Ru(Shvo)) brought about a high conversion of the racemic monomers to enantio-enriched polyesters having Mn of several thousands in high yields. AB type monomers used were typically methyl 6-hydroxyheptanoate, methyl 7-hydroxyoctanoate, methyl 8-hydroxynonanoate, and methyl 13-hydroxytetradecanoate.108)
Condensation polymerization of carboxylic acids or their esters with alcohols
Dehydration (esterification) polymerization is again the simplest condensation mode (Scheme 29).
Scheme 29.

Dehydration condensation polymerization between a dicarboxylic acid and a glycol.
The first paper on the lipase-catalyzed polymerization appeared in 1984, reporting a lipase A-catalyzed dehydration polymerization between a free dicarboxylic acid and a diol, which produced oligoesters.3) For example, dehydration polymerization of adipic acid and 1,4-butanediol in diisopropyl ether gave a polyester with degree of polymerization (DP) of 20 (p = q = 4, Scheme 29).109) It is notable that lipase is active in ionic liquids and a dehydration polymerization gave effectively the product polyester.59) In a solvent-free system, lipase CA efficiently catalyzed the condensation polymerization of dicarboxylic acids and glycols under mild reaction conditions at 60 °C. The polyester with molecular weight higher than 1 × 104 was readily obtained under reduced pressure.110)
A dehydration reaction is generally conducted in non-aqueous media. Since the product water of the dehydration is in equilibrium with starting materials, the solvent water disfavors the dehydration to proceed in an aqueous medium due to the “law of mass action”. Nevertheless, lipase catalysis enabled a dehydration condensation polymerization of a dicarboxylic acid and a glycol in water at 45 °C, to afford a polyester in good yields. Lipases CA and other lipases were active for the dehydration polymerization of sebacic acid and 1,8-octanediol (Scheme 30).111),112) In the polymerization of an α,ω-dicarboxylic acid and a glycol, the polymerization behavior was greatly affected by the methylene chain length of the monomers. The polyester was obtained in good yields from 1,10-decanediol, whereas no polymer formation was observed from 1,6-hexanediol, suggesting that combination of the monomers with appropriate hydrophobicity is needed for the polymer production. This finding of “dehydration in water” is a new aspect in organic chemistry and much attracted organic chemists.
Scheme 30.

Dehydration polymerization in water to produce a polyester with lipase catalyst.
Furthermore, the reaction of a linear polyanhydride like poly(azelaic anhydride) (x = 7 in Scheme 31) and a glycol like 1,8-octanediol was induced with lipase CA catalyst involving a dehydration to give a polyester with molecular weight of several thousands.113)
Scheme 31.

Lipase-catalyzed polyester synthesis from a linear polyanhydride via dehydration.
Condensation polymerization via transesterification normally needs activation of carboxylic acid groups. The activation is conducted ordinarily by esterification of the acid group. In the beginning, alkyl or haloalkyl esters (Scheme 32) and then vinyl esters (Scheme 34) have been often used.
Scheme 32.

Lipase-catalyzed condensation polymerization of alkyl and haloalkyl esters.
Scheme 34.

Transesterification of a (halo)alkyl ester with an alcohol is reversible (a), whereas that of a vinyl ester becomes irreversible (b).
Condensation polymerization of bis(2,2,2-tri-chloroethyl) glutarate and 1,4-butanediol proceeded with PPL catalyst at room temperature in diethyl ether to produce the polyesters with molecular weight of 8.2 × 103 (Scheme 32).114) Lipase (Novozym 435)-catalyzed synthesis of poly(butylene succinate) (PBS) via condensation polymerization was achieved using a monophasic reaction mixture of dimethyl succinate and 1,4-butanediol in bulk and in solution. Diphenyl ether was a preferred solvent to give a higher molecular weight PBS; at 60–90 °C after 24 h, Mn values of PBS were from 2,000 to 8,000. The reaction at 95 °C after 21 h gave PBS with Mn value of 38,000.115)
Dynamic kinetic resolution (DKR, see also Scheme 21) method was used to synthesize an optically active polyester from a racemic monomer via condensation polymerization. A mixture of stereo-isomers of a secondary diol, α,α′-dimethyl-1,4-benze-nedimethanol, was polymerized with dimethyl adipate (Scheme 33).116) Due to the enantioselectivity of lipase CA, only the hydroxy groups at the (R) center are preferentially reacted to form the ester bond with liberation of methanol. The reactivity ratio was estimated as (R)/(S) = ~1 × 106. The DKR polymerization was carried out for 4 days; during the reaction the molecular weight increased to 3,000–4,000 and the optical rotation of the reaction mixture increased from −0.6 ° to 128 °.
Scheme 33.

Enantioselective polyester synthesis using dynamic kinetic resolution (DKR) method.
In an ionic liquid like 1-butyl-3-methy-imidazolium tetrafluoroborate ([bmim][BF4]), a similar polycondensation between diethyl adipate or diethyl sebacate and 1,4-butanediol under reduced pressure at 60 °C after 72 h gave the polyester having Mn~1,500 in 97% yields. Since the ionic liquid is non-volatile, ethanol was removed under vacuum during the reaction.59) Lipase CA-catalyzed polymerization of di-methyl adipate or dimethyl sebacate with 1,4-buta-nediol was also performed in an ionic liquid such as [bmim][BF4], [bmim][PF6], and [bmim][(CF3SO2)2N] at 70 °C for 24 h to give a higher molecular weight polyester with Mn reaching several thousands. Using ionic liquids as solvent involves the wide range of tunablity of solvent hydrophilicity and monomer solubility for the reaction.60)
Although transesterification of a (halo)alkyl ester is reversible (Scheme 34a), an irreversible process was developed by using a vinyl ester for the lipase-catalyzed acylation, where the product of vinyl alcohol tautomerizes to acetaldehyde (Scheme 34b). The reaction of an alcohol with a vinyl ester proceeds much faster than with an alkyl ester or a haloalkyl ester to form the desired product in higher yields, as seen above in Scheme 17.
In 1994, a divinyl ester was employed for the first time; the lipase PF-catalyzed condensation polymerization of divinyl adipate and 1,4-butanediol was performed at 45 °C in diisopropyl ether for 48 h to afford a polyester with Mn of 6.7 × 103 (Scheme 35), whereas the use of adipic acid and diethyl adipate did not produce the polymeric materials under the similar reaction conditions. As diol, ethylene glycol, 1,6-hexenediol, and 1,10-decanediol were also reacted to give the corresponding polyester with molecular weight of several thousands.117) The same polymerization of divinyl adipate and 1,4-butanediol with lipase PC catalyst produced the polyester with Mn of 2.1 × 104.118) By varying the molar ratio of the divinyl ester and the glycol, telechelics having end-structures of the glycol, the dicarboxylic acids, or the acid-alcohol can be obtained.
Scheme 35.

Divinyl adipate showed higher reactivity toward a glycol for polyester synthesis.
Supercritical carbon dioxide (scCO2) was shown to be a good solvent for the lipase-catalyzed polymerization of divinyl adipate and 1,4-butanediol. Quantitative consumption of both monomers was readily achieved to give the polyester with Mn of 3.9 × 103.55)
Aromatic polyesters were produced efficiently by the lipase CA-catalyzed polymerization of aromatic diacid divinyl esters. Divinyl esters of isophthalic acid, terephthalic acid, and p-phenylene diacetic acid were polymerized with various glycols to give polyesters containing aromatic groups in the main chain with Mn of 7,200 in heptane at 60 °C for 48 h.119)
Regioselective condensation polymerization of polyols was achieved with using divinyl esters. The reaction of divinyl sebacate and glycerol with lipase CA catalyst produced water-soluble polyesters with Mw up to 2,700 in bulk at 60 °C. The chloroform-soluble part with Mw of 19,000 was isolated in 63%, which indicated the regioselectivity of primary OH/secondary OH ratio of 74/26. At a lower temperature of 45 °C, however, the regioselectivity was perfectly controlled to give a linear polymer consisting exclusively of 1,3-glyceride unit.120) The lipase CA catalysis gave a reduced sugar-containing polyester regio-selectively from divinyl sebacate and sorbitol, in which sorbitol was exclusively acylated at the primary alcohol of 1- and 6-positions in acetonitrile at 60 °C for 72 h (Scheme 36). Mannitol and meso-erythritol were also regioselectively polymerized with divinyl sebacate.121)
Scheme 36.

Lipase-catalyzed regioselective polymerization of sorbitol to produce a polyester.
Terpolymerization of divinyl esters, glycols, and lactones produced ester terpolymers with Mn higher than 1 × 104 (Scheme 37). Lipases showed high catalytic activity for the terpolymerization involving both condensation polymerization and ROP simultaneously in one-pot to produce ester terpolymers, without involving homo-polymer formation.122) A similar terpolymerization was performed using three kinds of monomers, ω-pentadecalactone, diethyl succinate, and 1,4-butanediol, by CALB catalyst desirably at 95 °C via a two-stage vacuum technique. The polymerization produced a terpolyester reaching Mw 77,000 with Mw/Mn~1.7 × 4.0123) These results accord with the frequent occurrence of (intermolecular) transesterifications during the polymerization (Scheme 15).72)
Scheme 37.

Terpolymerization proceeds in both condensation polymerization and ROP, with involving transesterifications, to produce a terpolyester.
Crosslinkable polyesters were prepared by the lipase-catalyzed condensation polymerization of divinyl sebacate with glycerol in the presence of an unsaturated higher fatty acid (i) such as linoleic acid and linolenic acid obtained from renewable plant oils (route A, Scheme 38). Product polyester (iii) is biodegradable and contains an unsaturated fatty acid moiety in the side chain. Curing of (iii) induced by oxidation with cobalt naphthenate catalyst or thermal treatment gave a crosslinked transparent film. Biodegradability of the film obtained was verified by biochemical oxygen demand (BOD) measurement.124),125) Furthermore, epoxide-containing polyesters were enzymatically synthesized via two routes, A and B, using unsaturated fatty acids. In route A, (iii) was enzymatically epoxidized to give (iv), and in route B the lipase-catalyzed epoxidization of the fatty acid (i) was first conducted to give (ii) and lipase-catalyzed condensation polymerization of (ii) was performed to produce (iv). Curing of (iv) proceeded thermally, yielding transparent polymeric films with high gloss surface. Pencil scratch hardness of film from (iv) was higher than that from (iii). Both films showed good biodegradability.126)
Scheme 38.

Crosslinkable polyesters were derived from renewable plant oils with lipase catalysis.
In addition to olefinic and epoxy reactive groups, a mercapto group can also be a crosslinkable group. Direct lipase CA-catalyzed condensation polymerization of 1,6-hexanediol and dimethyl 2-mercaptosuccinate at 70 °C in bulk gave an aliphatic polyester having free pendant mercapto groups with Mw = 14,000 in good yields. The polyester was readily crosslinked by the air-oxidation via the disulfide linkage formation.127)
Green polymer chemistry and future perspectives
Polyester synthesis using enzymes (mainly lipase) as catalyst has been briefly reviewed. Compared with conventional chemical processes, in vitro lipase-catalyzed polyester synthesis involves several characteristics as follows.
-
Lipase-catalyzed reactions:
Reactions can be achieved under mild conditions; normally at a lower temperature, at around normally pH, under ordinary pressure, etc.
Reactions are highly selective in all respects, in enantio-, regio-, and chemo-selectivities, and hence, they are clean with producing no- or minimal by-products. These reactions can hardly be achieved via conventional methods.
Reactions can be carried out not only in an organic solvent, but also in a green solvent, like water, supercritical carbon dioxide, ionic liquids, or in other green sovents.
-
Lipase enzymes:
Lipase is a non-toxic, renewable biocatalyst, which is free from a (toxic) metal. This nature is desirable to prepare polymers for biomedical or pharmaceutical applications.
Lipase is relatively cheap compared with other available enzymes. Recently, catalytic activity of lipase is remarkably enhanced, and a higher molecular weight can be easily reached. An immobilized lipase can be readily separated, recovered, and recycled for repeated use.
Lipase is robust enough to be used in combination with other chemical catalysts, allowing a new chemoenzymatic process.
-
Reaction substrates and products:
Renewable biobased materials (substrates) can be often used as starting raw materials. Environmentally benign reagents like water, oxygen from air, hydrogen peroxide, and carbon dioxide can be employed.
Product polyesters are almost all biodegradable. Functionalized polyesters are often highly value-added products applicable for medical usages.
In contrast to the above advantageous characteristics, lipase catalyst possesses also limitations and problems such as the cost and availability of enzymes, less varieties of reaction substrates and conditions, the reaction rate for the catalyst amount, etc. Nevertheless, owing to the above advantages, lipase catalysis has potentials to contribute to the mitigation of environmental problems for maintaining green sustainable society as green polymer chemistry (GPC) first noted by us.10)–12),14),15) Some of the lipase-catalyzed polyester synthesis already provided with good examples of GPC from the viewpoint of clean-process, energy savings, natural resource problems, carbon dioxide emission, etc. In spite of practical applications of enzyme catalysts including lipase to food industry and chemicals production in various ways, lipase catalyst has scarcely been utilized for the polymer synthesis so far on an industrial production scale.
Another possible way of conducting GPC is a lipase-catalyzed recycling of polyesters.17) Industrial examples of chemical recycling are few but known like an alcoholysis method of poly(ethylene terephthalate) (PET) and poly(butylene terephthalate). Thus, a new method of chemical recycling of polymers using lipase catalysis was proposed.35),52),128) The principle lies in that the ROP system of lactones by lipase catalysis is reversible between linear polymers and cyclic oligomers, which can be controlled by changing the reaction conditions.
We are faced to the global environmental problems of earth warming, fossil resource shortage, and the world population increase, etc. Hereafter, we are not allowed to use the fossil resources as before. In the polymer materials, the starting raw materials are to be shifted gradually from the petroleum-based to the renewable-based.
Nature is our teacher. We believe it is definitely the right direction to conduct GPC with utilizing natural catalysts of enzymes. A hard barrier for the practical utilization of enzymatic polymer synthesis on the industrial scale may be the cost of enzymes. This problem should be overcome and new highly active but cheaper enzymes are to be created. The above mentioned environmental problems are too big to be argued here; various ways can also be conceivable for the problems. Yet, it is still greatly hoped that conducting GPC of enzyme (lipase)-catalyzed polymer synthesis will contribute to the future society, even in small portions at the beginning.
Acknowledgements
The author is deeply grateful to Professor Hitosi Nozaki, m.j.a., for the encouragement to contribute the present article. He appreciates all the coworkers appearing in the references for their enthusiastic collaborations. He also acknowledges financial supports, in particular, from a Grant-in-Aid for the Specially Promoted Research, the Ministry of Education, Culture, Sports, Science, and Technology, Japan, and two NEDO projects on Technology for Novel High Functional Materials and for Precision Polymerization.
Profile
Shiro Kobayashi was born in 1941. He studied polymer chemistry and organic chemistry in the graduate course of Kyoto University and received Ph.D. degree in 1969. He stayed at Case Western Reserve University as a postdoctoral fellow for two years and investigated cationic reaction mechanisms. Then, he joined Kyoto University and restarted to study polymer synthesis. He stayed at Mainz University as a Humboldt fellow in 1976. Following the lectureship in Kyoto University, he was appointed as a full professor of Tohoku University in 1986 and started research on enzymatic polymerization. He moved to Kyoto University, Department of Materials Chemistry in 1997 and officially retired in 2005 to become a professor emeritus. Since then, he is a distinguished professor at Kyoto Institute of Technology. His research interests include polymer synthesis chemistry, organic reactions, materials chemistry, in particular, enzymatic polymerization, which involves an opportunity of green polymer chemistry and enabled the first chemical synthesis of various natural and unnatural polysaccharides, functionalized polyesters, and polymers from phenolic compounds. His pioneering work on enzymatic polymerization is now well known worldwide. He received the Award of the Chemical Society of Japan for Young Chemists (1976), the Award of the Society of Polymer Science Japan (1987), the Distinguished Invention Award (1993), the Cellulose Society of Japan Award (1996), the Humboldt Research Award (1999), the Chemical Society of Japan Award (2001), the John Stauffer Distinguished Lecture Award (2002), the SPSJ Award for Outstanding Achievement in Polymer Science and Technology (2004), the Medal with Purple Ribbon (2007), and others. He is a foreign member of the Northrhine Westfalian Academy of Science since 1999. He currently serves as a member of (executive) advisory board and editorial (advisory) board for fourteen international journals.

References
- 1).Kobayashi S. (2007) New developments of polysaccharide synthesis via enzymatic polymerization. Proc. Jpn. Acad., Ser. B 83, 215–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2).Anastas P. T., Warner J. C. (1998) Green Chemistry: Theory and Practice. Oxford University Press, Oxford [Google Scholar]
- 3).Okumura S., Iwai M., Tominaga Y. (1984) Synthesis of ester oligomer by Aspergilliusniger lipase. Agri. Biol. Chem. 48, 2805–2808 [Google Scholar]
- 4).Uyama H, Kobayashi S. (1993) Enzymatic ring-opening polymerization of lactones catalyzed by lipase. Chem. Lett., 1149–1150 [Google Scholar]
- 5).Uyama H., Takeya K., Kobayashi S. (1993) Synthesis of polyesters by enzymatic ring-opening copolymerization using lipase catalyst. Proc. Jpn. Acad., Ser. B 69, 203–207 [Google Scholar]
- 6).Knani D., Gutman A. L., Kohn D. H. (1993) Enzymatic polyesterification in organic media. Enzyme-catalyzed synthesis of linear polyesters. I. Condensation polymerization of linear hydroxyesters. II. Ring-opening polymerization of ɛ-caprolactone. J. Polym. Sci. Part A: Polym. Chem. 31, 1221–1232 [Google Scholar]
- 7).Kobayashi S., Shoda S., Uyama H. (1995) Enzymatic polymerization and oligomerization. Adv. Polym. Sci. 121, 1–30 [Google Scholar]
- 8).Kobayashi S., Shoda S, Uyama H. (1997) Enzymatic catalysis. InCatalysis in Precision Polymerization (ed. Kobayashi S.). John Wiley & Sons, Chichester, England, pp. 417–441 [Google Scholar]
- 9).Kobayashi S. (1999) Enzymatic polymerization: A new method of polymer synthesis. J. Polym. Sci. Part A: Polym. Chem. 37, 3041–3056 [Google Scholar]
- 10).Kobayashi S., Uyama H., Ohmae M. (2001) Accounts: Enzymatic polymerization for precision polymer synthesis. Bull. Chem. Soc. Jpn. 74, 613–635 [Google Scholar]
- 11).Kobayashi S., Uyama H., Kimura S. (2001) Enzymatic polymerization. Chem. Rev. 101, 3793–3818 [DOI] [PubMed] [Google Scholar]
- 12).Kobayashi S, Uyama H. (2003) Enzymatic polymerization. InEncyclopedia of Polymer Science and Technology (ed. Kroschwitz J. I.). Wiley, New York, pp. 328–364 [Google Scholar]
- 13).Kobayashi S., Ritter H, Kaplan D. (2006) Adv. Polym. Sci. Special issue: Enzyme-catalyzed synthesis of polymers. Vol. 194, Springer, Berlin [Google Scholar]
- 14).Kobayashi S, Ohmae M. (2007) Polymer synthesis and modification by enzymatic catalysis InMacromolecular Engineering: Precise Synthesis, Materials Properties, Applications (eds. Matyjaszewski K., Gnanou Y., Leibler L.). Wiley-VCH, Weinheim, pp. 400–477 [Google Scholar]
- 15).Kobayashi S., Makino A. (2009) Enzymatic polymer synthesis: An opportunity for green polymer chemistry. Chem. Rev. 109, 5288–5353 [DOI] [PubMed] [Google Scholar]
- 16).Gross R. A., Kumar A., Kalra B. (2001) Polymer synthesis by in vitro enzyme catalysis. Chem. Rev. 101, 2097–2124 [DOI] [PubMed] [Google Scholar]
- 17).Matsumura S. (2002) Enzyme-catalyzed synthesis and chemical recycling of polyesters. Macromol. Biosci. 2, 105–126 [Google Scholar]
- 18).Uppenberg J., Hansen M. T., Patkar S., Jones T. A. (1994) Sequence, crystal-structure determination and refinement of 2 crystal forms of lipase B from Candida antarctica. Structure 2, 293–308 [DOI] [PubMed] [Google Scholar]
- 19).Gotor-Fernandez V., Busto E., Gotor V. (2006) Candida antarctica lipase B: An ideal biocatalyst for the preparation of nitrogenated organic compounds. Adv. Synth. Catal. 348, 797–812 [Google Scholar]
- 20).Uyama H., Kikuchi H., Takeya K., Kobayashi S. (1996) Lipase-catalyzed ring-opening polymerization and copolymerization of 15-pentadecanolide. Acta Polym. 47, 357–360 [Google Scholar]
- 21).Uyama H., Namekawa S., Kobayash S. (1997) Mechanistic studies on the lipase-catalyzed ring-opening polymerization of lactones. Polym. J. 29, 299–301 [DOI] [PubMed] [Google Scholar]
- 22).Kobayashi S., Uyama H., Namekawa S. (1998) In vitro biosynthesis of polyesters with isolated enzymes in aqueous systems and organic solvents. Polym. Degrad. Stabil. 59, 195–201 [Google Scholar]
- 23).Kobayashi S., Uyama H. (1999) Precision enzymatic polymerization to polyesters with lipase catalysts. Macromol. Symp. 144, 237–246 [Google Scholar]
- 24).van der Mee L., Helmich F., de Bruijn R., Vekemans J., Palmans A. R. A., Meijer E. W. (2006) Investigation of lipase-catalyzed ring-opening polymerizations of lactones with various ring sizes: Kinetic evaluation. Macromolecules 39, 5021–5027 [Google Scholar]
- 25).Matsumura S., Beppu H., Nakamura K., Osanai S, Toshima K. (1996) Preparation of poly(β-malic acid) by enzymatic ring-opening polymerization of benzyl β-malolactonate. Chem. Lett., 795–796 [Google Scholar]
- 26).Namekawa S., Uyama H., Kobayashi S. (1996) Lipase-catalyzed ring-opening polymerization and copolymerization of β-propiolactone. Polym. J. 28, 730–731 [Google Scholar]
- 27).Svirkin Y. Y., Xu J., Gross R. A., Kaplan D. L., Swift G. (1996) Enzyme-catalyzed stereoelective ring-opening polymerization of α-methyl-β-propiolactone. Macromolecules 29, 4591–4597 [Google Scholar]
- 28).Nobes G. A. R., Kazlauskas R. J., Marchessault R. H. (1996) Lipase-catalyzed ring-opening polymerization of lactones: A novel route to poly(hydroxyalkanoate)s. Macromolecules 29, 4829–4833 [Google Scholar]
- 29).Küllmer K., Kikuchi H., Uyama H., Kobayashi S. (1998) Lipase-catalyzed ring-opening polymerization of α-methyl-δ-valerolactone and α-methylɛ-caprolactone. Macromol. Rapid Commun. 19, 127–130 [Google Scholar]
- 30).Jiang Z. Z., Azim H., Gross R. A., Focarete M. L., Scandola M. (2007) Lipase-catalyzed copolymerization of ω-pentadecalactone with p-dioxanone and characterization of copolymer thermal and crystalline properties. Biomacromolecules 8, 2262–2269 [DOI] [PubMed] [Google Scholar]
- 31).Matsumura S., Mabuchi K., Toshima K. (1997) Lipase-catalyzed ring-opening polymerization of lactide. Macromol. Rapid Commun. 18, 477–482 [Google Scholar]
- 32).Hans M., Keul H., Moeller M. (2009) Ring-opening polymerization of dd-lactide catalyzed by Novozyme 435. Macromol. Biosci. 9, 239–247 [DOI] [PubMed] [Google Scholar]
- 33).MacDonald R. T., Pulapura S. K., Svirkin Y. Y., Gross R. A., Kaplan D. L., Akkara J., et al. (1995) Enzyme-catalyzed ɛ-caprolactone ring-opening polymerization. Macromolecules 28, 73–78 [Google Scholar]
- 34).Matsumura S., Ebata H., Toshima K. (2000) A new strategy for sustainable polymer recycling using an enzyme: Poly(ɛ-caprolactone). Macromol. Rapid Commun. 21, 860–863 [Google Scholar]
- 35).Ebata H., Toshima K., Matsumura S. (2000) Lipase-catalyzed transformation of poly(ɛ-caprolactone) into cyclic dicaprolactone. Biomacromolecules 1, 511–514 [DOI] [PubMed] [Google Scholar]
- 36).Mei Y., Kumar A., Gross R. A. (2002) Probing water-temperature relationships for lipase-catalyzed lactone ring-opening polymerizations. Macromolecules 35, 5444–5448 [Google Scholar]
- 37).Kumar A., Gross R. A. (2000) Candida antartica lipase B catalyzed polycaprolactone synthesis: Effects of organic media and temperature. Biomacromolecules 1, 133–138 [DOI] [PubMed] [Google Scholar]
- 38).Kobayashi S., Takeya K., Suda S., Uyama H. (1998) Lipase-catalyzed ring-opening polymerization of medium-size lactones to polyesters. Macro-mol. Chem. Phys. 199, 1729–1736 [Google Scholar]
- 39).Kikuchi H., Uyama H., Kobayashi S. (2002) Lipase-catalyzed ring-opening polymerization of substituted lactones. Polym. J. 34, 835–840 [Google Scholar]
- 40).Uyama H., Suda S., Kikuchi H, Kobayashi S. (1997) Extremely efficient catalysis of immobilized lipase in ring-opening polymerization of lactones. Chem. Lett., 1109–1110 [Google Scholar]
- 41).Cordova A., Iversen T., Martinelle M. (1998) Lipase-catalysed formation of macrocycles by ring-opening polymerisation of ɛ-caprolactone. Polymer 39, 6519–6524 [Google Scholar]
- 42).Kobayashi S., Uyama H., Namekawa S., Hayakawa H. (1998) Enzymatic ring-opening polymerization and copolymerization of 8-octanolide by lipase catalyst. Macromolecules 31, 5655–5659 [Google Scholar]
- 43).Uyama H., Takeya K., Kobayashi S. (1995) Enzymatic ring-opening polymerization of lactones to polyesters by lipase catalyst. Unusually high reactivity of macrolides. Bull. Chem. Soc. Jpn. 68, 56–61 [Google Scholar]
- 44).Uyama H., Takeya K., Hoshi N., Kobayashi S. (1995) Lipase-catalyzed ring-opening polymerization of 12-dodecanolide. Macromolecules 28, 7046–7050 [Google Scholar]
- 45).Uyama H., Kikuchi H., Takeya K., Hoshi N, Kobayashi S. (1996) Immobilized lipase showing high catalytic activity toward enzymatic ring-opening polymerization of macrolides. Chem. Lett., 107–108 [Google Scholar]
- 46).Bisht K. S., Henderson L. A., Gross R. A., Kaplan D. L., Swift G. (1997) Enzyme-catalyzed ring-opening polymerization of ω-pentadecalactone. Macromolecules 30, 2705–2711 [Google Scholar]
- 47).Namekawa S., Uyama H., Kobayashi S. (1998) Lipase-catalyzed ring-opening polymerization of 16-hexadecanolide. Proc. Jpn. Acad., Ser. B 74, 65–68 [Google Scholar]
- 48).van der Mee L., Antens A., van de Kruijs B., Palmans A. R. A., Meijer E. W. (2006) Oxo-crown-ethers as comonomers for tuning polyester properties. J. Polym. Sci. Part A: Polym. Chem. 44, 2166–2176 [Google Scholar]
- 49).Mueller S., Uyama H, Kobayashi S. (1999) Lipase-catalyzed ring-opening polymerization of cyclic diesters. Chem. Lett., 1317–1318 [Google Scholar]
- 50).Veld M. A. J., Palmans A. R. A., Meijer E. W. (2007) Selective polymerization of functional monomers with Novozym 435. J. Polym. Sci. Part A: Polym. Chem. 45, 5968–5978 [Google Scholar]
- 51).Hut Y. M., Ju L. K. (2003) Lipase-mediated de-acetylation and oligomerization of lactonic sophorolipids. Biotechnol. Prog. 19, 303–311 [DOI] [PubMed] [Google Scholar]
- 52).Soeda Y., Toshima K., Matsumura S. (2005) Synthesis and chemical recycling of novel poly (ester-urethane)s using an enzyme. Macromol. Bio-sci. 5, 277–288 [DOI] [PubMed] [Google Scholar]
- 53).Namekawa S., Uyama H., Kobayashi S. (1998) Lipase-catalyzed ring-opening polymerization of lactones in water. Polym. J. 30, 269–271 [DOI] [PubMed] [Google Scholar]
- 54).Taden A., Antonietti M., Landfester K. (2003) Enzymatic polymerization towards biodegradable polyester nanoparticles. Macromol. Rapid Commun. 24, 512–516 [Google Scholar]
- 55).Takamoto T., Uyama H., Kobayashi S. (2001) Lipase-catalyzed synthesis of aliphatic polyesters in supercritical carbon dioxide. e-Polymers 4, 1–6 [Google Scholar]
- 56).Loeker F. C., Duxbury C. J., Kumar R., Gao W., Gross R. A., Howdle S. M. (2004) Enzyme-catalyzed ring-opening polymerization of ɛ-caprolactone in supercritical carbon dioxide. Macromolecules 37, 2450–2453 [Google Scholar]
- 57).Duxbury C. J., Wang W. X., de Geus M., Heise A., Howdle S. M. (2005) Can block copolymers be synthesized by a single-step chemoenzymatic route in supercritical carbon dioxide? J. Am. Chem. Soc. 127, 2384–2385 [DOI] [PubMed] [Google Scholar]
- 58).Kubisa P. (2005) Ionic liquids in the synthesis and modification of polymers. J. Polym. Sci. Part A: Polym. Chem. 43, 4675–4683 [Google Scholar]
- 59).Uyama H., Takamoto T., Kobayashi S. (2002) Enzymatic synthesis of polyesters in ionic liquids. Polym. J. 34, 94–96 [Google Scholar]
- 60).Marcilla R., de Geus M., Mecerreyes D., Duxbury C. J., Koning C. E., Heise A. (2006) Enzymatic polyester synthesis in ionic liquids. Eur. Polym. J. 42, 1215–1221 [Google Scholar]
- 61).Barrera-Rivera K. A., Marcos-Fernandez A., Vera-Graziano R., Martinez-Richa A. (2009) Enzymatic ring-opening polymerization of ɛ-caprolactone by Yarrowia lipolytica lipase in ionic liquids. J. Polym. Sci. Part A: Polym. Chem. 47, 5792–5805 [Google Scholar]
- 62).Anderson E. M., Karin M., Kirk O. (1998) One biocatalyst – Many applications: The use of Candida antarctica B-lipase in organic synthesis. Biocatal. Biotransform. 16, 181–204 [Google Scholar]
- 63).Huisgen R., Ott H. (1959) Die konfiguration der carbonestergruppe und die sondereigenschaften der lactone. Tetrahedron 6, 253–267 [Google Scholar]
- 64).Nomura R., Ueno A., Endo T. (1994) Anionic ring-opening polymerization of macrocyclic esters. Macromolecules 27, 620–621 [Google Scholar]
- 65).Namekawa S., Suda S., Uyama H., Kobayashi S. (1999) Lipase-catalyzed ring-opening polymerization of lactones to polyesters and its mechanistic aspects. Int. J. Biol. Macromol. 25, 145–151 [DOI] [PubMed] [Google Scholar]
- 66).Duda A., Kowalski A., Penczek S., Uyama H., Kobayashi S. (2002) Kinetics of the ring-opening polymerization of 6-, 7-, 9-, 12-, 13-, 16-, and 17-membered lactones. Comparison of chemical and enzymatic polymerizations. Macromolecules 35, 4266–4270 [Google Scholar]
- 67).Kikuchi H., Uyama H., Kobayashi S. (2000) Lipase-catalyzed enantioselective copolymerization of substituted lactones to optically active poly-esters. Macromolecules 33, 8971–8975 [Google Scholar]
- 68).Kobayashi S. (2006) Enzymatic ring-opening polymerization of lactones by lipase catalyst: Mechanistic aspects. Macromol. Symp. 240, 178–185 [Google Scholar]
- 69).Peeters J. W., van Leeuwen O., Palmans A. R. A., Meijer E. W. (2005) Lipase-catalyzed ring-opening polymerizations of 4-substituted ɛ-caprolactones: Mechanistic considerations. Macromolecules 38, 5587–5592 [Google Scholar]
- 70).Namekawa S., Uyama H., Kobayashi S. (2001) Lipase-catalyzed ring-opening polymerization of lactones in the presence of aliphatic polyesters to ester copolymers. Macromol. Chem. Phys. 202, 801–806 [Google Scholar]
- 71).Kumar A., Gross R. A. (2000) Candida antarctica lipase B-catalyzed transesterification: New synthetic routes to copolyesters. J. Am. Chem. Soc. 122, 11767–11770 [Google Scholar]
- 72).Takamoto T., Kerep P., Uyama H., Kobayashi S. (2001) Lipase-catalyzed transesterification of polyesters to ester copolymers. Macromol. Bio-sci. 1, 223–227 [Google Scholar]
- 73).Uyama H., Suda S., Kobayashi S. (1998) Enzymatic synthesis of terminal-functionalized polyesters by initiator method. Acta Polym. 49, 700–703 [Google Scholar]
- 74).Srivastava R. K., Albertsson A. C. (2006) Enzyme-catalyzed ring-opening polymerization of seven-membered ring lactones leading to terminal-functionalized and triblock polyesters. Macromolecules 39, 46–54 [Google Scholar]
- 75).de Geus M., Peters R., Koning C. E., Heise A. (2008) Insights into the initiation process of enzymatic ring-opening polymerization from monofunctional alcohols using liquid chromatography under critical conditions. Biomacromolecules 9, 752–757 [DOI] [PubMed] [Google Scholar]
- 76).Hedfors C., Ostmark E., Malmstrom E., Hult K, Martinelle M. (2005) Thiol end-functionalization of poly(ɛ-caprolactone), catalyzed by Candida antarctica lipase B. Macromolecules 38, 647–649 [Google Scholar]
- 77).Yoon K. R., Lee K. B., Chi Y. S., Yun W. S., Joo S. W., Choi I. S. (2003) Surface-initiated, enzymatic polymerization of biodegradable polyesters. Adv. Mater. 15, 2063–2066 [Google Scholar]
- 78).Hans M., Keul H., Moeller M. (2008) Poly(ether-ester) conjugates with enhanced degradation. Bio-macromolecules 9, 2954–2962 [DOI] [PubMed] [Google Scholar]
- 79).Uyama H., Kikuchi H, Kobayashi S. (1995) One-shot synthesis of polyester macromonomer by enzymatic ring-opening polymerization of lactone in the presence of vinyl ester. Chem. Lett., 1047–1048 [Google Scholar]
- 80).Uyama H., Kikuchi H., Kobayashi S. (1997) Single-step acylation of polyester terminals by enzymatic ring-opening polymerization of 12-dodeca-nolide in the presence of acyclic vinyl esters. Bull. Chem. Soc. Jpn. 70, 1691–1695 [Google Scholar]
- 81).Eriksson M., Fogelstrom L., Hult K., Malmstrom E., Johansson M., Trey S., Martinelle M. (2009) Enzymatic one-pot route to telechelic poly-pentadecalactone epoxide: synthesis, UV curing, and characterization. Biomacromolecules 10, 3108–3113 [DOI] [PubMed] [Google Scholar]
- 82).Uyama H., Kobayashi S., Morita M., Habaue S., Okamoto Y. (2001) Chemoselective ring-opening polymerization of a lactone having exomethylene group with lipase catalysis. Macromolecules 34, 6554–6556 [Google Scholar]
- 83).Habaue S., Asai M., Morita M., Okamoto Y., Uyama H., Kobayashi S. (2003) Chemo-specific ring-opening polymerization of α-methyle-nemacrolides. Polymer 44, 5195–5200 [Google Scholar]
- 84).van der Meulen I., de Geus M., Antheunis H., Deumens R., Joosten E. A. J., Koning C. E., Heise A. (2008) Polymers from functional macro-lactones as potential biomaterials: Enzymatic ring opening polymerization, biodegradation, and bio-compatibility. Biomacromolecules 9, 3404–3410 [DOI] [PubMed] [Google Scholar]
- 85).Fujioka M., Okada H., Kusaka Y., Nishiyama S., Noguchi H., Ishii S., Yoshida Y. (2004) Enzymatic synthesis of chitin- and chitosan-graft-aliphatic polyesters. Macromol. Rapid Commun. 25, 1776–1780 [Google Scholar]
- 86).van Buijtenen J., van As B. A. C., Verbruggen M., Roumen L., Vekemans J., Pieterse K., Hilbers P. A. J., Hulshof L. A., Palmans A. R. A., Meijer E. W. (2007) Switching from S- to R-selectivity in the Candida antarctica lipase B-catalyzed ring-opening of ω-methylated lactones: Tuning polymerizations by ring size. J. Am. Chem. Soc. 129, 7393–7398 [DOI] [PubMed] [Google Scholar]
- 87).van As B. A. C., van Buijtenen J., Heise A., Broxterman Q. B., Verzijl G. K. M., Palmans A. R. A., Meijer E. W. (2005) Chiral oligomers by iterative tandem catalysis. J. Am. Chem. Soc. 127, 9964–9965 [DOI] [PubMed] [Google Scholar]
- 88).van Buijtenen J., van As B. A. C., Meuldijk J., Palmans A. R. A., Vekemans J., Hulshof L. A., Meijer E. W. (2006) Chiral polymers by iterative tandem catalysis. Chem. Commun., 3169–3171 [DOI] [PubMed] [Google Scholar]
- 89).Meyer U., Palmans A. R. A., Loontjens T., Heise A. (2002) Enzymatic ring-opening polymerization and atom transfer radical polymerization from a bifunctional initiator. Macromolecules 35, 2873–2875 [Google Scholar]
- 90).van As B. A. C., Thomassen P., Kalra B., Gross R. A., Meijer E. W., Palmans A. R. A., Heise A. (2004) One-pot chemoenzymatic cascade poly-merization under kinetic resolution conditions. Macromolecules 37, 8973–8977 [Google Scholar]
- 91).Hans M., Keul H., Heise A., Moeller M. (2007) Chemoenzymatic approach toward heterografted molecular bottle brushes. Macromolecules 40, 8872–8880 [Google Scholar]
- 92).Popescu D., Keul H., Moeller M. (2009) Highly functional poly(meth)acrylates via cascade reaction. Macromol. Chem. Phys. 210, 123–139 [Google Scholar]
- 93).Kobayashi S., Uyama H. (1993) Enzymatic polymerization of cyclic acid anhydrides and glycols by a lipase catalyst. Makromol. Chem. Rapid Commun. 14, 841–844 [Google Scholar]
- 94).Uyama H., Wada S., Fukui T., Kobayashi S. (2003) Lipase-catalyzed synthesis of polyesters from anhydride derivatives involving dehydration. Biochem. Eng. J. 16, 145–152 [Google Scholar]
- 95).Bonduelle C., Martin-Vaca B., Bourissou D. (2009) Lipase-catalyzed ring-opening polymerization of the O-carboxylic anhydride derived from lactic acid. Biomacromolecules 10, 3069–3073 [DOI] [PubMed] [Google Scholar]
- 96).Kobayashi S., Kikuchi H., Uyama H. (1997) Lipase-catalyzed ring-opening polymerization of 1,3-dioxan-2-one. Macromol. Rapid Commun. 18, 575–579 [Google Scholar]
- 97).Namekawa S., Uyama H., Kobayashi S., Kricheldorf H. R. (2000) Lipase-catalyzed ring-opening polymerization and copolymerization of cyclic dicarbonates. Macromol. Chem. Phys. 201, 261–264 [Google Scholar]
- 98).Wu R., Al-Azemi T. F., Bisht K. S. (2008) Functionalized polycarbonate derived from tartaric acid: Enzymatic ring-opening polymerization of a seven-membered cyclic carbonate. Biomacromolecules 9, 2921–2928 [DOI] [PubMed] [Google Scholar]
- 99).Kaihara S., Fisher J. P., Matsumura S. (2009) Chemo-enzymatic synthesis of degradable PTMC-b-PECA-b-PTMC triblock copolymers and their micelle formation for pH-dependent controlled release. Macromol. Biosci. 9, 613–621 [DOI] [PubMed] [Google Scholar]
- 100).Schwab L. W., Kroon R., Schouten A. J., Loos K. (2008) Enzyme-catalyzed ring-opening polymerization of unsubstituted β-lactam. Macromol. Rapid Commun. 29, 794–797 [Google Scholar]
- 101).Feng Y. K., Klee D., Keul H., Höcker H. (2000) Lipase-catalyzed ring-opening polymerization of morpholine-2,5-dione derivatives: A novel route to the synthesis of poly(ester amide)s. Macromol. Chem. Phys. 201, 2670–2675 [Google Scholar]
- 102).He F., Zhuo R. X., Liu L. J., Jin D. B., Feng J., Wang X. L. (2001) Immobilized lipase on porous silica beads: preparation and application for enzymatic ring-opening polymerization of cyclic phosphate. React. Funct. Polym. 47, 153–158 [Google Scholar]
- 103).Kato M., Toshima K., Matsumura S. (2007) Enzymatic synthesis of polythioester by the ring-opening polymerization of cyclic thioester. Biomacromolecules 8, 3590–3596 [DOI] [PubMed] [Google Scholar]
- 104).Ajima A., Yoshimoto T., Takahashi K., Tamaura Y., Saito Y., Inada Y. (1985) Polymerization of 10-hydroxydecanoic acid in benzene with poly-ethylene glycol-modified lipase. Biotechnol. Lett. 7, 303–306 [Google Scholar]
- 105).Kiran K. R., Divakar S. (2003) Lipase-catalysed polymerization of lactic acid and its film forming properties. World J. Microbiol. Biotechnol. 19, 859–865 [Google Scholar]
- 106).Mahapatro A., Kumar A, Gross R. A. (2004) Mild, solvent-free ω-hydroxy acid polycondensations catalyzed by Candida antarctica Lipase B. Biomacromolecules 5, 62–68 [DOI] [PubMed] [Google Scholar]
- 107).Ebata H., Toshima K., Matsumura S. (2008) Lipase-catalyzed synthesis and properties of poly[(12-hydroxydodecanoate)-co-(12-hydroxystearate)] directed towards novel green and sustainable elastomers. Macromol. Biosci. 8, 38–45 [DOI] [PubMed] [Google Scholar]
- 108).Kanca U., Van Buijtenen J., Van As B. A. C., Korevaar P. A., Vekemans J., Palmans A. R. A., Meijer E. W. (2008) Iterative tandem catalysis of racemic AB monomers. J. Polym. Sci. Part A: Polym. Chem. 46, 2721–2733 [Google Scholar]
- 109).Binns F., Roberts S. M., Taylor A., Williams C. F. (1993) Enzymatic polymerization of an un-activated diol diacid system. J. Chem. Soc. Perkin Trans. 1, 899–904 [Google Scholar]
- 110).Uyama H., Inada K, Kobayashi S. (1998) Enzymatic polymerization of dicarboxylic acid and glycol to polyester in solvent-free system. Chem. Lett., 1285–1286 [Google Scholar]
- 111).Kobayashi S., Uyama H., Suda S, Namekawa S. (1997) Dehydration polymerization in aqueous medium catalyzed by lipase. Chem. Lett., 105 [Google Scholar]
- 112).Suda S., Uyama H., Kobayashi S. (1999) Dehydration polycondensation in water for synthesis of polyesters by lipase catalyst. Proc. Jpn. Acad., Ser. B 75, 201–206 [Google Scholar]
- 113).Uyama H., Wada S, Kobayashi S. (1999) Synthesis of aliphatic polyester by insertion of glycols into polyanhydrides using lipase catalyst. Chem. Lett., 893–894 [Google Scholar]
- 114).Wallace J. S., Morrow C. J. (1989) Biocatalytic synthesis of polymers. 2. Preparation of [AA-BB]X polyesters by porcine pancreatic lipase catalyzed transesterification in anhydrous, low polarity organic solvents. J. Polym. Sci. Part A: Polym. Chem. 27, 3271–3284 [Google Scholar]
- 115).Azim H., Dekhterman A., Jiang Z. Z., Gross R. A. (2006) Candida antarctica Lipase B-catalyzed synthesis of poly(butylene succinate): Shorter chain building blocks also work. Biomacromolecules 7, 3093–3097 [DOI] [PubMed] [Google Scholar]
- 116).Hilker I., Rabani G., Verzijl G. K. M., Palmans A. R. A., Heise A. (2006) Chiral polyesters by dynamic kinetic resolution polymerization. Angew. Chem. Int. Ed. 45, 2130–2132 [DOI] [PubMed] [Google Scholar]
- 117).Uyama H, Kobayashi S. (1994) Lipase-catalyzed polymerization of divinyl adipate with glycols to polyesters. Chem. Lett., 1687–1690 [Google Scholar]
- 118).Uyama H., Yaguchi S., Kobayashi S. (1999) Lipase-catalyzed polycondensation of dicarboxylic acid-divinyl esters and glycols to aliphatic poly-esters. J. Polym. Sci. Part A: Polym. Chem. 37, 2737–2745 [Google Scholar]
- 119).Uyama H., Yaguchi S., Kobayashi S. (1999) Enzymatic synthesis of aromatic polyesters by lipase-catalyzed polymerization of dicarboxylic acid divinyl esters and glycols. Polym. J. 31, 380–383 [Google Scholar]
- 120).Uyama H., Inada K., Kobayashi S. (2001) Regioselectivity control in lipase-catalyzed polymerization of divinyl sebacate and triols. Macromol. Biosci. 1, 40–44 [Google Scholar]
- 121).Uyama H., Klegraf E., Wada S, Kobayashi S. (2000) Regioselective polymerization of sorbitol and divinyl sebacate using lipase catalyst. Chem. Lett., 800–801 [Google Scholar]
- 122).Namekawa S., Uyama H., Kobayashi S. (2000) Enzymatic synthesis of polyesters from lactones, di-carboxylic acid divinyl esters, and glycols through combination of ring-opening polymerization and polycondensation. Biomacromolecules 1, 335–338 [DOI] [PubMed] [Google Scholar]
- 123).Jiang Z. Z. (2008) Lipase-catalyzed synthesis of aliphatic polyesters via copolymerization of lactone, dialkyl diester, and diol. Biomacromolecules 9, 3246–3251 [DOI] [PubMed] [Google Scholar]
- 124).Tsujimoto T., Uyama H., Kobayashi S. (2001) Enzymatic synthesis of cross-linkable polyesters from renewable resources. Biomacromolecules 2, 29–31 [DOI] [PubMed] [Google Scholar]
- 125).Tsujimoto T., Uyama H., Kobayashi S. (2002) Enzymatic synthesis and curing of biodegradable crosslinkable polyesters. Macromol. Biosci. 2, 329–335 [Google Scholar]
- 126).Uyama H., Kuwabara M., Tsujimoto T., Kobayashi S. (2003) Enzymatic synthesis and curing of biodegradable epoxide-containing polyesters from renewable resources. Biomacromolecules 4, 211–215 [DOI] [PubMed] [Google Scholar]
- 127).Kato M., Toshima K., Matsumura S. (2009) Direct enzymatic synthesis of a polyester with free pendant mercapto groups. Biomacromolecules 10, 366–373 [DOI] [PubMed] [Google Scholar]
- 128).Kobayashi S., Uyama H., Takamoto T. (2000) Lipase-catalyzed degradation of polyesters in organic solvents, a new methodology of polymer recycling using enzyme as catalyst. Biomacromolecules 1, 3–5 [DOI] [PubMed] [Google Scholar]