

Abstract
Bacteria-gut epithelial interplay and the mucosal immune response are the most critical issues in determining the fate of bacterial infection and the severity of diseases. Shigella species (abbreviated here as Shigella), the causative agent of bacillary dysentery (shigellosis), are highly adapted human pathogens that are capable of invading and colonizing the intestinal epithelium, which results in severe inflammatory colitis. Shigella secrete a large and diverse number (more then 50) of effectors via the type III secretion system (TTSS) during infection, some of which are delivered into the surrounding bacterial space and some others into the host cell cytoplasm and nucleus. The delivered effectors mimic and usurp the host cellular functions, and modulate host cell signaling and immune response, thus playing pivotal roles in promoting bacterial infection and circumventing host defense systems. This article overviews the pathogenic characteristics of Shigella, and highlights current topics related to the bacterial infectious stratagem executed by the TTSS-secreted effectors. Though bacterial stratagems and the molecular mechanisms of infection vary greatly among pathogens, the current studies of Shigella provide a paradigm shift in bacterial pathogenesis.
Keywords: infection, pathogenicity, innate immunity, epithelial cells, Shigella, shigellosis
1. Introduction
The lumen of the intestine is connected to the external environments, and the intestinal epithelium is exposed to dietary and environmental antigens and commensal bacteria. Many bacterial pathogens exploit the intestinal epithelium as an infectious foothold, and some of them exploit it as the port of entry to gain access to deeper tissues. Thereby, the intestinal epithelium is equipped with multiple layers of innate defense systems and acts as a barrier against microbial invaders. This barrier is composed of four major elements: the commensal microbiota, integrity of epithelium, rapid epithelial turnover, and mucosal immune system.1),2) The commensal microbiota in the lumen can compete with foreign bacteria to grow and interfere with their colonization over the mucosal surface, and can also contribute to the balance between immune tolerance and immune activation at the mucosa. The integrity of the epithelial monolayer, sustained by the tight cell-to-cell adherence, becomes a physical and biological barrier against microbial invaders. In addition, the epithelial surface, which is covered by a thick mucin layer, can also prevent microbes from directly accessing the epithelial-cell surface. The rapid turnover of the intestinal epithelium, which is sustained by creating progenitors at the base of crypts and by ejecting the migrated cells from the villus tips, contributes to limiting bacterial colonization. Finally, the mucosal immune system, which consists of innate and acquired immune systems, plays paramount roles as a biological defense against microbial infection.
Despite the numerous host defenses, gastrointestinal pathogens such as Shigella, Salmonella, Yersinia, enteropathogenic Escherichia coli (EPEC), and enterohemorrhagic E. coli (EHEC) are capable of circumventing the intestinal barrier functions and can rather efficiently colonize over and within the epithelium. Indeed, it has been shown that although the molecular mechanisms and the stratagems vary among pathogens, they are equipped with highly evolved infectious systems. This article will thus address how bacterial pathogens, such as Shigella, exploit normal host cell functions, evade innate immune systems, and circumvent the epithelial barrier functions as a model of versatile mucosal pathogens.
2. Shigella are a member of E. coli discovered by Shiga
Shigella was discovered by Kiyoshi Shiga (Fig. 1) during an outbreak in Japan in 1897, who investigated 36 shigellosis patients and identified the bacilli as the etiology agent in 1898.3) Now his identified bacilli are known to be a strain belonging to S. dysenteriae type 1. To credit him for his discovery, the 1930 Edition of Bergey’s Manual of Determinative Bacteriology formally renamed the genus as ‘Shigella’.4) Since his discovery, many researchers have conducted numerous studies aimed at understanding the pathogenicity of shigellosis and bacterial pathogenesis, and also developing a safer shigellosis vaccine. Earlier studies indicated that Shigella are highly adapted human pathogens that are able to cause severe inflammatory colitis, and shigellosis is one of the most easily communicable enteric diseases.5) Clinical and epidemiological studies of shigellosis revealed that as few as 10–100 bacteria are capable of leading to shigellosis, thus permitting easy spread of the diseases by person-to-person contact as well as by the drinking of contaminated water. In tropical countries, and even in developed countries under low sanitary conditions, shigellosis thus becomes frequently endemic and one of the major killers of young children less than 5 years even today.6)
Fig. 1.

Kiyoshi Shiga (1871–1957). Photo provided courtesy of the Shibasaburo Kitasato Memorial Museum at the Kitasato Institute (Tokyo).
Shigella comprises four species; S. dysenteriae, S. flexneri, S. boydii and S. sonnei, and the taxonomic classification implicates that they are a member of E. coli; however, the group of bacteria causing shigellosis is still idiomatically termed Shigella. Note that among Shigella species, S. dysenteriae type I alone produces the Shiga toxin. Shigellosis, albeit to less extent, is also caused by enteroinvasive E. coli (EIEC), a member of enteric pathogenic E. coli. Indeed, Shigella and EIEC strains share a large (210–230 kb) plasmid on which the major virulence-associated proteins, such as effectors (see below), and the proteins’ transport machinery, called the type III secretion system (TTSS), are encoded. Unlike other pathogenic E. coli, Shigella and EIEC have neither flagella nor adhesins required for traversing the mucin layer covering the epithelium and accessing the epithelial-cell surface, instead they are equipped with a highly evolved invasive and intracellular survival stratagem.7)
3. Shigella invasion of enterocytes is a long process
The stratagem of Shigella for reaching their destination, the colonic enterocytes, seemingly is programmed by a subset of genes encoded on the large plasmid. As will be described later, the processes from the entry of M cells to the colonization of epithelial cells and subsequent bacterial intercellular spreading are executed through a series of interactions between the virulence-associated proteins, delivered via the TTSS, and their target host cell factors. When Shigella are ingested via the fecal-oral route, the bacteria move directly down to the colon, where Shigella preferentially enter the M cells overlying the solitary lymphoid nodules, and the bacteria endocytosed by the apical membrane of M cells are transported to the endosomal compartment, sorted, and finally exocytosed at the basal membrane.1),2),8) The macrophages and dendritic cells residing within the M cell pocket receive the exocytosed bacteria and antigens from the M cells, digest them, and present the immunogenic information to activate the downstream immune system. However, Shigella invade the resident macrophages by themselves, where they are at once surrounded by phagocytic membranes, but can disrupt the membranes, disseminate into the cytoplasm, and multiply therein (Fig. 2). As will be described later, the bacterial multiplication within the macrophages results in the induction of a strong inflammatory response, and then macrophage cell death. Following their release from killed macrophages, Shigella invade the surrounding enterocytes from the basolateral surface of the polarized epithelial cells by inducing macropinocytosis. Shigella are entrapped by the membrane vacuoles within epithelial cells, but they can rupture the vacuole membranes and disseminate into the epithelial-cell cytoplasm, where Shigella multiply and spread within, as well as, into the adjacent epithelial cells (Fig. 2). By continuing cell-to-cell spreading, they are able to expand the replicative niches among enterocytes (Fig. 2).1),2),7),8)
Fig. 2.

A model for Shigella infection of intestinal epithelium. See the text for details.
4. Host innate immune response to bacterial infection
Bacterial invasion of the host cell cytoplasm is recognized by various innate immune systems, thus leading to a strong inflammatory response, and occasionally cell death. Macrophages express various pattern-recognition receptors (PRRs) that recognize pathogen-associated molecular patterns (PAMPs) derived from intracellular invading bacteria. Within the macrophage-cell cytoplasm, invading bacteria release the lipopolysaccharide (LPS), peptidoglycan (PGN), flagellin, nucleic acids, toxins, and some virulence-associated proteins (called effectors hereafter) secreted via the TTSS, which are recognized by the cytoplasmic PRRs, such as NOD (nucleotide binding oligomerization domain)-like receptors (NLRs).1),2),8) Upon detection of various PAMPs (and also bacterial effectors and toxins) and dangerous host signals, NLRs form high molecular complexes, called inflammasomes, and recruit and activate caspase-1, which in turn proteolytically processes pro-IL-1β and IL-18.2) For example, upon invasion of macrophages by Shigella, NLRC4 (IPAF)-inflammasome executes the activation of caspase-1, thus inducing the production and secretion of IL-1β and IL-18, and resulting in macrophage cell death specialized pyroptosis.9),10) Pyroptosis is a class of cell death distinctive from the silent apoptotic cell death, which is occasionally associated with a high inflammatory condition and dependent on caspase-1 activation.11) The pyroptosis cell death has been best studied in the context of Shigella infection of macrophages through our study.10) The activation of IPAF-inflammasome by Shigella is thus the major cause of inflammation and macrophage cell death at the initial stage of infection.8)–10)
Infection of the intestinal epithelium by Shigella also plays a major role in inducing inflammation, since the intestinal epithelium also expresses a wide range of PRRs over and inside the cells, thus acting as sensing cells toward cytoplasmic intruding microbes as well as becoming a major cause for induction of inflammation during bacterial infection. Upon multiplication of Shigella within epithelial cells, the PGN released from bacteria is recognized by NOD1, a prominent NLR expressed from epithelial cells, which in turn stimulates the NOD1-RICK pathway. The NOD1-RICK pathway in turn mediates the activation of downstream NF-κB and mitogen-activated protein kinases pathways, thus leading to the production and secretion of a tremendous amount of proinflammatory chemokine and cytokines, and anti-microbial peptides (Fig 3).1),2) A recent study indicated that NOD1 is recruited to the bacterial entry site by moving from the cytoplasm to the plasma membrane, thus facilitating NOD1 to sensitize PGN.12) In addition, NLRX1, which is a newly identified cytoplasmic NLR family protein that is localized in the mitochondria via its N-terminal mitochondrial targeting sequence, can also sense intruding Shigella and stimulate the production of reactive oxygen species.13) Another study indicated that Shigella induce mitochondrial dysfunction, resulting in caspase-independent necrotic cell death through a new pathway depending on Bnip3 and cyclophilin D, two key regulators of mitochondrial permeability transition and cell death during oxidative cell stress in epithelial cells.14) Therefore, bacterial multiplication within intestinal epithelial cells that is recognized by various innate immune systems can lead to an inflammatory response and necrotic epithelial-cell death.
Fig. 3.

A model for Shigella-induced inflammatory response in epithelial cells and the bacterial down-regulation of the host inflammatory signals. During the multiplication of Shigella in epithelial cells, the peptidoglycan (PGN), including lipopolysaccharide (LPS), released from bacteria, can be recognized by the NOD1, which activates the downstream RICK and MAPK-mediated signal pathways, thus leading to the production of inflammatory chemokines, cytokines, and anti-microbial peptides. Intracellular Shigella delivers a subset of effectors, including IpaHs, OspF, and OspG via the TTSS, into the host cell cytoplasm and nucleus, enabling the effectors to target the host signaling pathways or factors involved in the inflammatory responses and dampen the inflammatory responses.
5. Shigella invasion of intestinal epithelium
Many bacterial pathogens are capable of invading various host cells. The bacterial capability is often called ‘invasiveness’, and such pathogens are termed ‘invasive bacteria’.7) The aim of entering host cells varies among pathogens; bacteria such as Shigella and Listeria monocytogenes enter the host-cell cytoplasm to gain a place to multiply, Salmonella invade epithelial cells to further disseminate to internal tissues, uropathogenic E. coli invade bladder epithelial cells to evade the immune surveillance system, and Mycobacterium tuberculosis and Legionella pneumophila enter macrophages to survive and multiply by becoming sequestered in phagosomes.
Based on the various mechanisms that bacteria use to enter host cells, they are categorized into two major classes; those in which a microbial ligand interacts with a host target receptor, called a zipper-like mechanism, and those in which entry is mediated by the delivery of effectors via TTSS into the host cells, called a trigger mechanism.7) The delivered effectors can trigger the formation of membrane ruffling and macropinocytosis represented by Shigella and Salmonella. Despite their different aims, invasive bacteria can variously remodel the host cell surface, for example, by stimulating Rho GTPases, protein tyrosine phosphorylation, or microtubule dynamics in mammalian cells.1),2) Thus, the ability to invade host cells is a prominent feature of bacterial pathogenesis.
5.1. Shigella invasion of epithelium
As mentioned above, Shigella possess a highly evolved invasive system; bacteria deliver more than 50 effectors via the TTSS through infection of the intestine, and some of which are delivered upon bacterial invasion. When the bacterium comes into contact with epithelial cells, the TTSS can be activated and secrete the effectors around the bacterial space and into the host cells, where the secreted effectors target host proteins residing over or within the cells to stimulate cellular signaling pathways involved in actin polymerization. Shigella can provoke the formation of large membrane ruffles that protrude from the bacterial entry site, by which bacteria are entrapped and endocytosed into the cell cytoplasm (Fig. 4).1),2) For example, IpaB and IpaC effectors are secreted in the surrounding bacterial space via the TTSS of Shigella, and the IpaB effector interacts with CD44 and β1-integrin and the IpaC effector interacts with β1-integrin. Since CD44 and β1-integrin are expressed from the basolateral surface of polarized enterocytes, the interactions of bacterial effectors with these host receptors contribute to the basolateral entry and stimulation of the out-side-in signals involved in inducing actin polymerization. In addition, some of the secreted IpaC, which can integrate in the epithelial-cell plasma membrane, stimulate the recruitment of the cellular Src and contribute to actin polymerization via the activation of the Arp2/3 complex. The IpgD effector injected via the TTSS into the host-cell cytoplasm, exhibits phosphatidylinositol (4, 5) bisphosphate phosphatase activity, contributing to local actin polymerization. We found that the IpgB1 effector, delivered from extracellular Shigella into the epithelial cytoplasm, plays a central role in Shigella invasion, since IpgB1 can activate the Rac1-WAVE-Arp2/3 pathway and induction of actin polymerization (Fig. 4).15),16) Further characterization of IpgB1 has indicated that it belongs to the Map/IpgB/Sif family distributing among EPEC, Shigella, and Salmonella, and acts as a GEF (GTP exchange factor) for Rac1.17) Thus, the series of studies imply that synergistic activities arising from the interplay between bacterial effectors and target host proteins, orchestrated by Rho-GTPases, play key roles in processing Shigella internalization into epithelial cells (Fig. 4).
Fig. 4.

A model of Shigella invasive mechanism for epithelial cells. Upon contact of Shigella to epithelial cells, the bacterium delivers several effectors (red circles) via the TTSS around the bacterial surface and into the host-cell cytoplasm. The bacterial effectors interact with the host target molecules to stimulate several signal transduction pathways capable of activating the Rac1-WAVE-Arp2/3 pathway, and induce local actin polymerization and protrude the membrane ruffles. See the text for details.
5.2. Shigella cell-to-cell spreading
In 1968, Ogawa and his coworkers reported for the first time that intracellular Shigella are motile and the bacterial movement is highly dynamic; where bacterial movement and speed depend on the cellular location and the stage of bacterial growth.18) They also demonstrated that the bacterial movement is almost completely abolished when HeLa cells are treated with tetracycline, suggesting that de novo synthesized bacterial protein(s) are involved.18) Twenty years after Ogawa’s discovery, our group defined for the first time one of the Shigella outer membrane proteins, VirG, as the protein playing the central role in the bacterial intra-and intercellular spreading (Fig. 5).19) Our and other groups characterized VirG as the protein able to mediate actin polymerization with the aid of several host proteins.20),21) At the same period, extensive studies were also conducted with other pathogens and revealed that some invasive microbes such as Listeria monocytogenes, Rickettsia, Mycobacterium marinum, Burkholderia pseudomallei, and some viruses, such as Vaccinia, are also capable of inducing actin polymerization and moving within as well as into adjacent cells.22),23) The bacterial intracellular movement is thus called ‘actin-based motility’. Intriguingly, to gain a propulsive force within the host cells, the pathogens share a universal activity to induce local actin polymerization by exploiting the host Arp2/3 complex, which directly executes actin polymerization within mammalian cells.22),23) In the case of Shigella, bacterium accumulates the VirG protein at one pole of the bacterium during multiplication, and the VirG protein recruits N-WASP (neural Wiskott-Aldrich syndrome protein), a member of the WASP family.24),25) The N-WASP is subsequently activated by interacting with Cdc42 and Toca-1, where N-WASP acts as the adapter as well as the stimulator to interact with and activate the Arp2/3 complex (Fig. 5).25),26) By virtue of some additional host proteins such as monomeric actin and profilin, the activated Arp2/3 complex can direct actin polymerization at one pole of the bacterium, thus allowing the bacterium to be pushed forward within the cytoplasm (Fig. 5). Finally, motile bacterium impinges on the host plasma membrane, causing the membrane to protrude (Fig. 2). The tips of these bacteria-containing protrusions are endocytosed by neighboring epithelial cells, leaving the bacterium transiently contained within double host plasma membrane-bound vacuoles (Fig. 2). The bacterium then disrupts the protrusion vacuoles, thereby disseminating into the new cytoplasm and multiply again (Figs. 2 and 5).
Fig. 5.
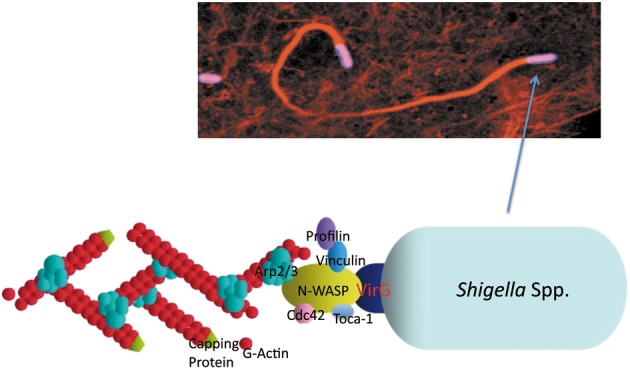
The actin-dependent Shigella motility. (a) A confocal immunofluorescence image of the actin tail from one pole of the moving bacterium. (b) The machinery required for bacterial motility consists of VirG (a bacterial outer-membrane protein), N-WASP, Arp2/3 complex, Profilin, and Toca-1, and accumulates at one pole of the bacterium to polymerize actin.
In addition to the bacterial activity to induce actin polymerization, Shigella require the activity to destroy the microtubules (MTs) during movement, since the MT networks become obstacles for motile bacterium. Indeed, the bacterial movement within the cytoplasm is severely hindered by areas rich with MT network structures.27) Nevertheless, motile Shigella can destroy the surrounding MTs; a process executed by the VirA effector secreted via the TTSS to the surrounding bacterial surface.28) Conversely, smooth bacterial movement within host cells and dissemination into the neighboring cells are almost completely abolished when the virA gene is deleted from Shigella. Consequently, the virA mutants are incapable of cell-to-cell spreading within an infected epithelial monolayer and become attenuated when they are inoculated into mice lung via the nasal route.28) The series of our studies thus defined the two distinctive intracellular bacterial activities as the essential mechanisms for promoting bacterial cell-to-cell spreading.
6. Bacterial circumvention of host innate immune system
6.1. Modulation of host inflammatory response by Shigella
Bacterial ability to circumvent the host innate and acquired immune system is the most critical pathogenic feature of pathogenic bacteria to survive inside host tissues. As has been described in many current excellent reviews,1),2),8) enteric bacterial pathogens need various factors for circumvention of the host innate immune system, those of which include the LPS, capsule, outer membrane proteins, effectors, and toxins. In the case of Shigella, as mentioned above, bacterial multiplication within macrophages and epithelial cells induce a strong inflammatory response (Fig. 3), and many numbers of effectors are needed to modulate an inflammatory response. Shigella deliver a set of IpaH effectors, the OspG and OspF effectors, during multiplication within epithelial cells, where the delivered effectors target various host proteins involved in modulating the inflammatory signals such as those upstream and downstream of NF-κB, and dampen the host inflammatory response (Fig. 3). For example, one of the IpaH effectors, IpaH9.8, is delivered into the host cell cytoplasm as well as the nucleus, in which the cytoplasmic IpaH9.8 targets the NEMO (IKKγ), a component of the IKK complex, which interferes with the NF-κB activation, while the nuclear translocated IpaH9.8 targets U2AF35, a mRNA splicing factor, which dampens the U2AF35-dependent splicing reaction (Fig. 3).1),2),29),30) A recent study has indicted that the IpaH cognate proteins are also produced by Salmonella, and Pseudomonas species, which act as E3 ubiquitin ligases in host cells, and that the E3 liagse activity is encoded by the highly conserved C-terminal region of each protein,31) though the host target molecules for each of the IpaH family proteins remains largely unclear. We have found that IpaH9.8 elicits aberrant ubiquitination of NEMO, by which the NEMO undergoes proteasomal degradation, thus suppressing the NF-κB activation.30) The biological significance of the IpaH9.8 activity can be demonstrated by using a mice lung infection model, a conventional assay system for evaluating Shigella activity to induce inflammation; infection with the ipaH9.8-deleting mutant or the E3 ligase-deficient mutant caused a more severe inflammatory response and a greater production of proinflammatory cytokine than that of infection with wild-type Shigella. Most important, the colonization rate of the ipaH9.8-deleting mutant or the E3 ligase-deficient mutant in the infected lung tissues greatly reduced to less than one-thirtieth of the wild-type level. These studies imply that bacterial ability to modulate the host inflammatory response is essential for surviving and promoting colonization within host tissues.
6.2. Autophagy recognizes Shigella but they can escape
Autophagy is a bulky degradation system via lysosomal fusion, through which eukaryotic cells apprehend and destroy damaged organelles, misfolded protein aggresomes, and cytoplasmic intruders.32) Autophagy targets undesirable cytoplasmic contents and encloses it by using a double-layered isolation membrane, and the entrapped materials are delivered to an autophagosomal compartment and finally degraded after autophagosome fusion with lysosomes. Autophagy is thus pivotal for eliminating or limiting the growth of intracellular invading bacteria.32),33) For example, Group A Streptococcus (GAS) can invade epithelial cells but is finally targeted and destroyed by autophagy. Rickettsia is sequestered in autophagosome-like double-membranes, in which bacterial replication is limited and eventually degraded. Mycobacterium tuberculosis that survives in the phagosomes within the macrophages, can also be targeted by autophagy at an early stage of infection as long as the host innate immune response is intact (Fig. 6).32) Although there are a number of controversial reports, some intra-cellular pathogens, such as Legionella pneumophila, Coxiella burnetti, and Porphyromonas gingivalis are enclosed by vacuoles within the macrophages that they can modify to resist fusion with lysosomes, allowing them to survive and multiply unless autophagy is activated (Fig. 6).32)
Fig. 6.

Autophagy affects the fate of bacterial colonization within host cells. Shigella, L. monocytogenes, and Burkholderia pseudomallei are capable of escaping from autophagy by exploiting the activities of IcsB, ActA, and BopA, respectively. Group A Streptoccus (GAS) internalized into the host cytoplasm is apprehended by autophagosomes and undergoes lysosomal degradation. M. tuberculosis, L. pneumophila, Brucella abortus, and Coxiella brunetti are sequestered by autophagosome-like membranes and undergo lysosomal degradation, if they cannot modulate the autophagy activity in macrophages.
Among the cytoplasmic invading pathogens, Shigella, Burkholderia pseudomallei, and L. monocytogenes are exceptional in their ability to evade autophagic recognition (Fig. 6). In the case of Shigella, IcsB, one of the effectors secreted via the TTSS by intracellular Shigella, plays a crucial role in evading autophagic recognition (Fig. 7).33),34) An icsB-deficient mutant is still invasive for epithelial cells,35) but it cannot eventually multiply within the cells by enclosing autophagic membranes.34) Intriguingly, a recent study has indicated that the vacuolar membrane remnants, ruptured by Shigella, contribute to triggering the autophagy activation by recruiting the autophagy markers LC3 and adaptor p62.36) We previously showed that the VirG protein, which mediates actin-based Shigella motility, can be targeted by autophagy through its binding to Atg5, an essential autophagy-associated protein involving the elongating isolation membrane, a seed for autophagosome formation. In In vitro binding assays, both IcsB and Atg5 exhibit the ability to interact with VirG, and IcsB and Atg5 share the same interacting region on VirG.34) However, IcsB can competitively bind to VirG, compared to that of Atg5, thus acting as an anti-Atg5 binding protein, and camouflaging the target VirG protein from autophagic recognition.34) Although the mechanism of the Atg5-VirG interplay-mediated autophagy remains to be elucidated, it is clear that Shigella invasion itself stimulates autophagy, but the pathogen can execute a sophisticated system for evading autophagy during multiplication within epithelial cells.
Fig. 7.

A model of Shigella evasion of autophagy (upper panel), and the strategy used by intracellular Shigella to escape from autophagy (lower panel). Shigella are capable of multiplying within the cytoplasm of epithelial cells and moving into adjacent epithelial cells. However, Shigella lacking the icsB gene, which encodes the IcsB effector, and acts as an anti-Atg5 binding factor for VirG, succumbs to autophagy and undergo lysosomal degradation. The upper right panel shows the VirG protein at one pole of bacterium, which is required for the actin-based bacterial motility.
L. monocytogenes adopt a distinctive system for evading autophagy to that of Shigella. We recently uncovered that the L. monocytogenes ActA protein plays a central role in evading autophagic recognition (Fig. 8). The ActA proteins are expressed on the bacterial surface, which also accumulates at one pole of the bacterium and mediates actin polymerization by directly linking to the host proteins such as the Arp2/3 complex and VASP/Ena. Several assays demonstrate that the ability of ActA to recruit the Arp2/3 complex or VASP/Ena, which is required for inducing actin polymerization and bacterial motility, is also essential for disguising the bacteria with the host protein, since the bacterial surface is decorated with the host proteins, the Arp2/3 complex or VASP/Ena, the surface becomes camouflaged from autophagic recognition (Fig. 8).37) Our findings corroborate that Shigella and L. monocytogenes exploit distinctive systems for evading autophagy.
Fig. 8.

A model of L. monocytogenes escaping from autophagy recognition. L. monocytogenes disrupts the membrane enclosing bacterium by secreting LLO (Listeriolysin O), the pore-forming toxin, and disseminates into the host cell cytoplasm. During multiplication within the cytoplasm, L. monocytogenes expresses ActA over the bacterial surface, which recruits the Arp2/3 complex, Ena/VASP, and actin, thus disguising the bacterium against autophagic recognition, and allowing the bacterium to mediate actin polymerization and move within the host cells as well as into the neighboring cells. However, L. monocytogenes lacking ActA or expressing ActA mutants deficient in recruiting any of the host proteins become target for autophagic clearance, because the bacterium is ubiquitinated, which is followed by binding with p62 and LC3, thus allowing the bacterium to be apprehended by autophagosomes.37)
7. Prolonging of epithelial-cell life span by bacteria
Self-renewal of tissues like skin, stomach, and intestine, are the essential innate activity for preserving tissue homeostasis, which is supported by providing the regenerative potential via supplying progenitors. The intestinal epithelium is formed by a tight-sealed monolayer but exists in a highly dynamic state by renewing every 4–5 days; the epithelial cells created by the stem cells at the bottom of crypts migrate up to the villus tips, undergoing proliferation, differentiation, maturation, and apoptosis, and then shed into the lumen.38) This renewal is also called epithelial turnover, which is crucial for limiting bacterial colonization. Nevertheless, many bacterial pathogens are able to colonize intestinal mucosa, implying that they have some maneuver to circumvent the innate defense system.
7.1. Shigella slow down rapid turnover of epithelium
We have recently discovered that some bacterial pathogens such as Shigella and Helicobactor pylori possess the activity to dampen rapid turnover of epithelial cells, which contribute to the promotion of bacterial colonization.39),40) Shigella have the ability to slow down the cell cycle progression of epithelial progenitors, by which they can prolong colonization within the intestinal epithelial cells.39) This bacterial activity can be executed by IpaB, one of the effectors, secreted via the TTSS from intracellular Shigella into intestinal progenitor cells at the crypts. A rabbit ileal loop assay infected with Shigella demonstrated that bacteria allow direct access to the intestinal crypts at the middle stage of infection, where bacteria invade the non-polarized progenitor cells.39) At this stage, while few PCNA (proliferation cell nuclear antigen, representing growing progenitor cells)-positive cells are detected in the crypts after wild-type Shigella infection, abundant progenitor cells are detected after infection with the ipaB mutant (Fig. 9). An in vitro study also indicated that IpaB secreted from intracellular Shigella into epithelial cells causes cell cycle arrest by targeting Mad2L2, an anaphase-promoting complex (APC) inhibitor. The APC is a multi-subunit complex possessing E3 ligase activity for the degradation of mitotic Cyclin A and Cyclin B1 during mitosis and G1 phases, allowing mitotic progression.39) Cyclin B1 ubiquitination assays showed that APC undergoes unscheduled activation in response to IpaB interaction with Mad2L2. Synchronized HeLa cells infected with Shigella fail to accumulate APC substrates, such as Cyclin B1, Cdc20, and Plk1, causing cell cycle arrest at the G2/M phase in an IpaB/Mad2L2-dependent manner. The IpaB/Mad2L2-dependent cell cycle arrest by Shigella infection can be visualized in the intestinal crypt progenitors of rabbit ileal loops, and the IpaB-Mad2L2-mediated arrest contributes to the efficient colonization of the host cells (Fig. 9).39) Recent studies have indicated that a growing family of bacterial small compounds, toxins, and effectors are capable of modulating the mammalian cell cycle, called ‘cyclomodulins’, though the target host cells for each cyclomodulin remains largely speculative.41) Accordingly, our study indicates that Shigella IpaB, which targets the Mad2L2 associated with APC, can act as a cyclomodulin in promoting colonization of the intestinal epithelium.
Fig. 9.
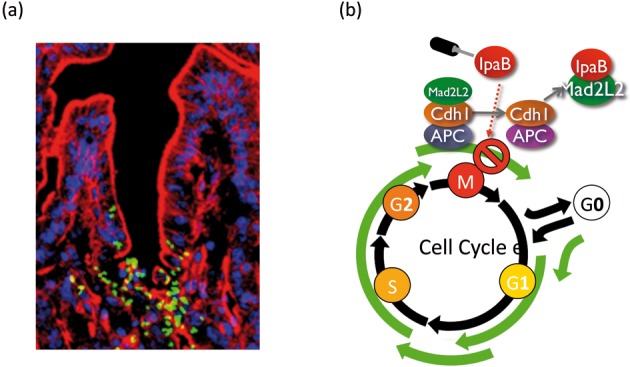
(a) Shigella can directly access the cryptic progenitor cells in rabbit intestine. Shown is a rabbit intestinal tissue 12 h after inoculation with 2 × 108 GFP-Shigella (green) stained with rhodamine-phalloidin (red) and TO-PRO3 (blue). (b) A model for Shigella strategy to cause cell cycle arrest via the interaction of the IpaB effector with Mad2L2. Once inside the epithelial progenitors, invaded bacterium delivers IpaB via TTSS, which interferes with the binding of Mad2L2 to Cdh1, leading to unscheduled activation of APC and subsequent Cyclin B1 degradation.39)
Although the mechanism is different from that of Shigella, we have recently reported that Helicobacter pylori can dampen gastric epithelial renewal by interfering with gastric pit cell apoptosis by delivering the CagA protein, a major virulence factor,40) via the type IV secretion system.42) Importantly, this bacterial activity was also pivotal to the promotion of H. pylori colonization of the gastric epithelium.40) Although the bacterial tactics to slow down epithelial-cell turnover are distinctive to each pathogen, the desperate bacterial effort to dampen rapid epithelial-cell turnover displayed by Shigella and H. pylori represent an intriguing adaptation of pathogens that are specialized to colonize the human gastrointestinal epithelium.
7.2. Shigella antagonize epithelial-cell exfoliation
As a countermeasure against bacterial colonization, infected epithelial cells undergo rapid exfoliation from the epithelium followed by the rapid sealing of neighboring cells, required for maintaining the epithelial integrity. Despite the host defense reaction, Shigella and many other gastroenteric pathogens are capable of colonizing over and within the host cells. We recently found that Shigella are capable of antagonizing the detachment of their infected foothold during multiplication within epithelial cells.43) This bacterial activity can be executed by the OspE effector during multiplication within epithelial cells, which is important to sustain Shigella cell-to-cell spreading among enterocytes, thus promoting bacterial colonization of the intestinal epithelium. In vitro studies supported the premise; OspE delivered from intracellular Shigella accumulates at the focal adhesions (FAs), reinforcing the host cells adhesion to the basement membrane by interacting with integrin-linked kinase (ILK) (Fig. 10).43) ILK is a unique intracellular Ser/Thr kinase that links the cell-adhesion receptors, integrin, and growth factors to the actin cytoskeleton and to a range of signaling pathways.44),45) The interaction between OspE and ILK increases formation of FAs, and surface levels of β1-integrin, and also suppresses phosphorylation of FAK and paxillin, thus resulting in dampening the rapid turnover of FAs in cell motility and promoting cell adhesion to the extracellular matrix (Fig. 10). The impact of OspE-ILK interplay can also be visualized when polarized epithelial cell monolayers or guinea pig colons are infected with the ospE mutant. Of importance, ospE cognate genes also distribute among many other enteric bacterial pathogens such as EPEC, EHEC, Citrobacter rodentium, and Salmonella,43) and the OspE activities are interchangeable with Shigella OspE. Therefore, the hitherto unknown bacterial activity, driven from the interplay between OspE and ILK to counteract infection-induced cell detachment, represents the pivotal bacterial activity required for promoting colonization of the intestine (Fig. 11).
Fig. 10.
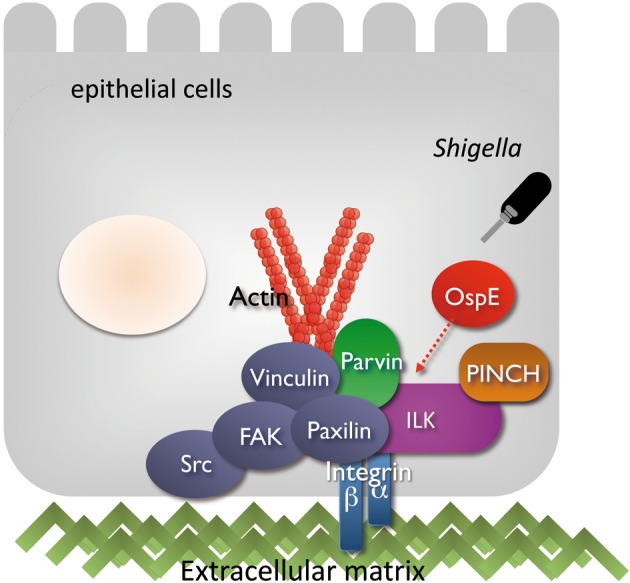
A model of Shigella OspE targeting epithelial-cell ILK for the reinforcement of focal adhesion. Shigella deliver OspE via TTSS during multiplication within epithelial cells. This effector protein, OspE, reinforces epithelial adherence to the basement membrane by interacting with ILK. The interaction between OspE and ILK increases formation of focal adhesions and surface levels of β1-integrin, while suppressing phosphorylation of FAK and paxillin, thus dampening rapid turnover of focal adhesions, reducing cell motility and promoting cell adhesion to extracellular matrix.43)
Fig. 11.

A model of Shigella strategy to antagonize the detachment of infected epithelial cells by delivering the OspE effectors. See the text for details.
Although bacterial strategies vary greatly, similar mechanisms that prevent cell detachment were also identified in UPEC, Neisseria gonorrhoeae, Neisseria meningitides, Haemophilus influenzae, and Moraxella catarrhalis.46),47) Therefore, the bacterial strategy to reinforce host cell adherence to extracellular matrices may be a universal countermeasure against host cell detachment that occurs in response to bacterial infection of epithelial cells. The findings in our recent studies thus add new information that expands the complexity of our knowledge on how bacteria employ strategies to preserve their replicative niches during infection.
8. Conclusion
Bacteria-host interplay and the host immune response are the most critical aspects in determining the fate of infection, severity of diseases, and convalescent outcome. As illustrated in the present and current excellent reviews, in-depth studies of bacterial pathogenesis have provided a new paradigm shift of bacteria-host interplay and also many insights into understanding highly evolved bacterial infectious systems. As exemplified in this article, the recent development of a variety of areas related to innate immunity, cell biology, protein structure biology, bioinformatics, and animal model development enable us to manipulate both the determinants of host defense and bacterial virulence, thus providing us the opportunity to uncover new infectious aspects. Furthermore, by taking a multidisciplinary approach and taking advantage of the new technologies we are now able to dissect almost any class of virulence-associated factors and evaluate the impact of each bacterial and host cellular factor on the pathogenesis and the outcome of infectious diseases. Clearly, the molecular and cellular details about bacterial pathogenesis will also provide us lots of clues and ideas for the development of a safer, well-attenuated vaccine48)–50) and new animal models.51)
9. Acknowledgements
The author sincerely thanks Prof. Tamio Yamakawa, the Editor-in-Chief for the Proceedings of the Japan Academy Series B, for the invitation to the journal. The author thanks Drs. Hitomi Mimuro, Michinaga Ogawa, Minsoo Kim, Masato Suzuki and Yuko Yoshikawa for preparing figures. The author also thanks the members of my laboratory for their contributions, as well as, the contributions of my collaborators to the studies highlighted in this review. This work was supported by a Grant-in-Aid for Scientific Research (S) (20229006); a Grant-in-Aid for Exploratory Research (20659067); a Grant-in-Aid for Scientific Research on Priority Areas (18073003); the Strategic Cooperation to Control Emerging and Reemerging Infections funded by the Special Coordination Funds for Promoting Science and Technology; a contract research fund for the Program of Founding Research Centers for Emerging and Re-emerging Infectious Diseases from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT); and the Core Research for Evolutional Science and Technology (CREST) from the Japan Science and Technology Agency (JST).
Profile
Chihiro Sasakawa was born in 1948. He graduated from the Faculty of Science and Postgraduate School of Pharmaceutical Science at Chiba University, entered the Postgraduate School of Medicine, University of Tokyo, and received his Ph.D. in 1978 from University of Tokyo. In 1978, he was promoted to research associate at the Institute of Medical Science, University of Tokyo. After three years of postdoctoral working in Department of Microbiology and Immunology, Washington University School of Medicine, he started to study bacterial pathogenesis in 1983 at the Institute of Medical Science, University of Tokyo, and was promoted to associated professor in 1986 and professor in 1995. He was the Head of Department of Microbiology and Immunology at the Institute of Medical Science from 1999 to 2005, and appointed also professor of Research Institute for Microbial Diseases, Osaka University from 1999 to 2001. At present he is a professor of Department of Microbiology and Immunology, and International Research Center for Infectious Diseases, at the Institute of Medical Science, University of Tokyo. Over the past 30 years he has been studying molecular and cellular interaction between bacterial pathogens and host. His current research interests are mechanisms of bacterial countermeasure against host innate immune defense systems and the outcome of infectious diseases. He is currently a member of editorial or advisory boards of Nature Reviews Microbiology, Cell Host Microbe, Trends in Microbiology and several other international journals. He was awarded the Noguchi Hideyo Memorial Prize in 1998 and the Takeda Medical Prize in 2006

References
- 1).Ogawa M., Handa Y., Ashida H., Suzuki M., Sasakawa C. (2008) The versatility of Shigella effectors. Nat. Rev. Microbiol. 6, 11–16 [DOI] [PubMed] [Google Scholar]
- 2).Ashida H., Ogawa M., Mimuro H., Sasakawa C. (2009) Shigella infection of intestinal epithelium and circumvention of the host innate defense system. Curr. Top. Microbiol. Immunol. 337, 231– 255 [DOI] [PubMed] [Google Scholar]
- 3).Shiga K. (1898) Ueber den errenger der dysenterie in Japan. Zent. Bakteriol. Microbiol. Hyg. 23, 599–600 [Google Scholar]
- 4).Trofa A.F., Ueno-Olsen H., Oiwa R., Yoshikawa M. (1999) Dr. Kiyoshi Shiga: discovery of the dysentery bacillus. Clin. Infect. Dis. 29, 1303–1306 [DOI] [PubMed] [Google Scholar]
- 5).DuPont H.L., Levine M.M., Hornick R.B., Formal S.B. (1989) Inoculum size in shigellosis and implications for expected mode of transmission. J. Infect. Dis. 159, 1126–1128 [DOI] [PubMed] [Google Scholar]
- 6).Keusch G.T. (2001) Shigella. Mol. Med. Microbiol. (ed. Sussman M.) 2, Academic Press, London, pp. 1279–1290 [Google Scholar]
- 7).Sasakawa C. (2004) Shigella invasion. Adv. Mol. Cell. Microbiol. (ed. Lamont R.J.). Cambridge University Press, Cambridge, U.K., pp. 25–58 [Google Scholar]
- 8).Sansonetti P.J., Di Santo J.P. (2007) Debugging how bacteria manipulate the immune response. Immunity 26, 149–161 [DOI] [PubMed] [Google Scholar]
- 9).Suzuki T., Nakanishi K., Tsutsui H., Iwai H., Akira S., Inohara N., et al. (2005) A novel Caspase-1/Toll-like receptor 4-independent pathway of cell death induced by cytosolic Shigella in infected macrophages. J. Biol. Chem. 280, 14042–14050 [DOI] [PubMed] [Google Scholar]
- 10).Suzuki T., Franchi L., Toma C., Ashida H., Ogawa M., Yoshikawa Y., et al. (2007) Differential regulation of caspase-1 activation, pyroptosis and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLos. Pathog. 3, e111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11).Bergsbaken T., Fink S.L., Cookson B.T. (2009) Pyroptosis: host cell death and inflammation. Nat. Rev. Microbiol. 7, 99–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12).Kufer T.A., Kremmer E., Adam A.C., Philpott D.J., Sansonetti P.J. (2007) The pattern-recognition molecule Nod1 is localized at the plasma membrane at sites of bacterial interaction. Cell. Microbiol. 10, 477–486 [DOI] [PubMed] [Google Scholar]
- 13).Tattoli I., Carneiro L.A., J3 ehanno M., Magalhaes J.G., Shu Y., Philpott D.J., et al. (2008) NLRX1 is a mitochondrial NOD-like receptor that amplifies NF-κB and JNK pathways by inducing reactive oxygen species production. EMBO Rep. 9, 293– 300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14).Carneiro L.A., Travassos L.H., Soares F., Tattoli I., Magalhaes J.G., Bozza M.T., et al. (2009) Shigella induces mitochondrial dysfunction and cell death in nonmyleoid cells. Cell. Host. Microbe. 5, 123–136 [DOI] [PubMed] [Google Scholar]
- 15).Ohya K., Handa Y., Ogawa M., Suzuki M., Sasakawa C. (2005) IpgB1 is a novel Shigella effector protein involved in bacterial invasion of host cells: its activity to promote membrane ruflng via Rac1 and Cdc42 activation. J. Biol. Chem. 280, 24022–24034 [DOI] [PubMed] [Google Scholar]
- 16).Handa Y., Suzuki M., Ohya K., Iwai H., Ishijima N., Koleske A.J., et al. (2007) Shigella IpgB1 promotes bacterial entry through the ELMO-Dock180 machinery. Nat. Cell. Biol. 9, 121–128 [DOI] [PubMed] [Google Scholar]
- 17).Huang Z., Sutton S.E., Wallenfang A.J., Orchard R.C., Wu X., Feng Y., et al. (2009) Structure insights into host GTPase isoform selection by a family of bacterial GEF mimics. Nat. Strct. Mol. Biol. 16, 853–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18).Ogawa H., Nakamura A., Nakaya R. (1968) Cinemicroscopic study of tissue cell cultures infected with Shigella flexneri. Jpn. J. Med. Sci. Biol. 21, 259–273 [DOI] [PubMed] [Google Scholar]
- 19).Makino S., Sasakawa C., Kamata K., Kurata T., Yoshikawa M. (1986) A genetic determinant required for continuous reinfection of adjacent cells on large plasmid in S. flexneri 2a. Cell 46, 551–555 [DOI] [PubMed] [Google Scholar]
- 20).Lett M.C., Sasakawa C., Okada N., Sakai T., Makino S., Yamada M., et al. (1989) virG, a plasmid-coded virulence gene of Shigella flexneri: identification of the virG protein and determination of the complete coding sequence. J. Bacteriol. 171, 353–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21).Bernardini M.L., Mounier J., d’Hauteville H., Coquis-Rondon M., Sansonetti P.J. (1989) Identification of icsA, a plasmid locus of Shigella flexneri that governs bacterial intra- and inter-cellular spread through interaction with F-actin. Proc. Natl. Acad. Sci. USA 86, 3867–3871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22).Cossart P., Sansonetti P.J. (2004) Bacterial invasion: the paradigms of enteroinvasive pathogens. Science 303, 242–248 [DOI] [PubMed] [Google Scholar]
- 23).Gouin E., Welch M.D., Cossart P. (2005) Actin-based motility of intracellular pathogens. Curr. Opin. Microbiol. 8, 35–45 [DOI] [PubMed] [Google Scholar]
- 24).Suzuki T., Miki H., Takenawa T., Sasakawa C. (1998) Neural Wiskott-Aldrich syndrome protein is implicated in the actin-based motility of Shigella flexneri. EMBO J. 17, 2767–2776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25).Suzuki T., Mimuro H., Miki H., Takenawa T., Sasaki T., Nakanishi H., et al. (2000) Rho family GTPase Cdc42 is essential for the actin-based motility of Shigella in mammalian cells. J. Exp. Med. 191, 1905–1920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26).Leung Y., Ally S., Goldberg M.B. (2008) Bacterial actin assembly requires Toca-1 to relieve N-WASP autoinhibition. Cell Host Microbe 3, 39–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27).Yoshida S., Katayama E., Kuwae A., Mimuro H., Suzuki T., Sasakawa C. (2002) Shigella deliver an effector protein to trigger host microtubule destabilization, which promotes Rac1 activity and efficient bacterial internalization. EMBO J. 21, 2923–2935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28).Yoshida S., Handa Y., Suzuki T., Ogawa M., Suzuki M., Tamai A., et al. (2006) Microtuble-severing activity of Shigella is pivotal for intercellular spreading. Science 314, 985–989 [DOI] [PubMed] [Google Scholar]
- 29).Ashida H., Toyotome T., Nagai T., Sasakawa C. (2007) Shigella chromosomal IpaH proteins are secreted via the type III secretion system and act as effectors. Mol. Microbiol. 63, 680–693 [DOI] [PubMed] [Google Scholar]
- 30).Ashida H., Kim M., Schmidt-Supprian M., Ma A., Ogawa M., Sasakawa C. (2010) A bacterial E3 ubiquitin ligase IpaH9.8 effector targets NEMO/ IKKγ to dampen the host NF-κB-mediated inflammatory response. Nat. Cell Biol. 12, 66–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31).Rohde J.R., Breitkreutz A., Chenal A., Sansonetti P.J., Parsot C. (2007) Type III secretion effectors of the IpaH family are E3 ubiquitin ligase. Cell Host Microbe 1, 77–83 [DOI] [PubMed] [Google Scholar]
- 32).Deretic V., Levine B. (2009) Autophagy, immunity, and microbial adaptations. Cell Host Microbe 18, 527–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33).Ogawa M., Sasakawa C. (2006) Shigella and autophagy. Autophagy 2, 171–174 [DOI] [PubMed] [Google Scholar]
- 34).Ogawa M., Yoshimori T., Suzuki T., Sagara H., Mizushima N., Sasakawa C. (2005) Escape of intracellular Shigella from autophagy. Science 307, 727–731 [DOI] [PubMed] [Google Scholar]
- 35).Ogawa M., Suzuki T., Tatsuno I., Abe H., Sasakawa C. (2003) IcsB, secreted via the type III secretion system, is chaperoned by IpgA and required at the post-invasion stage of Shigella pathogenicity. Mol. Microbiol. 48, 913–931 [DOI] [PubMed] [Google Scholar]
- 36).Dupont N., Lacas-Gervais S., Bertout J., Paz I., Freche B., Tran V., et al. (2009) Shigella phagocytic vacuolar membrane remnants participate in the cellular response to pathogen invasion and are regulated by autophagy. Cell Host Microbe 6, 137–149 [DOI] [PubMed] [Google Scholar]
- 37).Yoshikawa Y., Ogawa M., Hain T., Yoshida M., Kim M., Mimuro H., et al. (2009) Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat. Cell Biol. 11, 1233–1240 [DOI] [PubMed] [Google Scholar]
- 38).Radtke F., Clevers H. (2005) Self-renewal and cancer of the gut: two sides of a coin. Science 307, 1904–1909 [DOI] [PubMed] [Google Scholar]
- 39).Iwai H., Kim M., Yoshikawa Y., Ashida H., Ogawa M., Fujita Y., et al. (2007) A bacterial effector targets Mad2L2, an APC inhibitor, to modulate host cell cycling. Cell 130, 611–623 [DOI] [PubMed] [Google Scholar]
- 40).Mimuro H., Suzuki T., Nagai S., Rieder G., Suzuki M., Fujita Y., et al. (2007) Helicobacter pylori dampens gut epithelial self-renewal by inhibiting apoptosis, a bacterial strategy to enhance colonization of the stomach. Cell Host Microbe 11, 250–263 [DOI] [PubMed] [Google Scholar]
- 41).Nougayrède J.P., Taieb F., De Rycke J., Oswald E. (2005) Cyclomodulins: bacterial effectors that modulate the eukaryotic cell cycle. Trends Microbiol. 13, 103–110 [DOI] [PubMed] [Google Scholar]
- 42).Tanaka J., Suzuki T., Mimuro H., Sasakawa C. (2003) Structural definition on the surface of Helicobacter pylori type IV secretion apparatus. Cell Microbiol. 5, 395–404 [DOI] [PubMed] [Google Scholar]
- 43).Kim M., Ogawa M., Fujita Y., Yoshikawa Y., Nagai T., Nagai S., et al. (2009) Bacteria hijack integrin-linked kinase to stabilize focal adhesions and block cell detachment. Nature 459, 578–582 [DOI] [PubMed] [Google Scholar]
- 44).Legate K.R., Montanez E., Kudlacek O., Fassler R. (2006) ILK, PINCH and parvin: the tIPP of integrin signalling. Nat. Rev. Mol. Cell Biol. 7, 20–31 [DOI] [PubMed] [Google Scholar]
- 45).McDonald P.C., Fielding A.B., Dedhar S. (2008) Integrin-linked kinase–essential roles in physiology and cancer biology. J. Cell Sci. 121, 3121–3132 [DOI] [PubMed] [Google Scholar]
- 46).Mulvey M.A., Schilling J.D., Martinez J.J., Hultgren S.J. (2000) Bad bugs and beleaguered bladders: interplay between uropathogenic Escherichia coli and innate host defenses. Proc. Natl. Acad. Sci. USA 97, 8829–8835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47).Muenzner P., Rohde M., Kneitz S., Hauck C.R. (2005) CEACAM engagement by human pathogens enhances cell adhesion and counteracts bacteria-induced detachment of epithelial cells. J. Cell Biol. 170, 825–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48).Kotloff K.L., Winickoff J.P., Ivanoff B., Clemens J.D., Swerdlow D.L., Sansonetti P.J., et al. (1999) Global burden of Shigella infections: implications for vaccine development and implementation of control strategies. Bulletin of the World Health Organization 77, 651–666 [PMC free article] [PubMed] [Google Scholar]
- 49).Yoshikawa M., Sasakawa C., Okada N., Takasaka N., Nakayama M., Yoshikawa Y., et al. (1995) Construction and evaluation of a virG thyA double mutant of Shigella flexneri 2a as a candidate live-attenuated oral vaccine. Vaccine 13, 1436–1440 [DOI] [PubMed] [Google Scholar]
- 50).Suzuki T., Yoshikawa Y., Ashida H., Iwai H., Toyotome T., Matsui H., Sasakawa C. (2006) High vaccine efficacy against shigellosis of recombinanat noninvasive Shigella mutant that express Yersinia invasin. J. Immunol. 177, 4709– 4717 [DOI] [PubMed] [Google Scholar]
- 51).Shim D.H., Suzuki T., Chang S.Y., Park S.M., Sansonetti P.J., Sasakawa C., Kweon M.N. (2007) New animal model of shigellosis in the guinea pig: its usefulness for protective efficacy studies. J. Immunol. 178, 2476–2482 [DOI] [PubMed] [Google Scholar]