

Abstract
In this work, we introduce a novel network synthesis model that can generate families of evolutionarily related synthetic protein–protein interaction (PPI) networks. Given an ancestral network, the proposed model generates the network family according to a hypothetical phylogenetic tree, where the descendant networks are obtained through duplication and divergence of their ancestors, followed by network growth using network evolution models. We demonstrate that this network synthesis model can effectively create synthetic networks whose internal and cross-network properties closely resemble those of real PPI networks. The proposed model can serve as an effective framework for generating comprehensive benchmark datasets that can be used for reliable performance assessment of comparative network analysis algorithms. Using this model, we constructed a large-scale network alignment benchmark, called NAPAbench, and evaluated the performance of several representative network alignment algorithms. Our analysis clearly shows the relative performance of the leading network algorithms, with their respective advantages and disadvantages. The algorithm and source code of the network synthesis model and the network alignment benchmark NAPAbench are publicly available at http://www.ece.tamu.edu/bjyoon/NAPAbench/.
Introduction
Protein-protein interactions (PPIs) lie at the core of a wide range of biological processes in cells, including transcriptional, signaling, and metabolic processes [1]. Recent technological advances have enabled the high-throughput measurement of these interactions in various species [2]–[4], and a variety of computational methods have been developed for in-silico prediction of protein interactions [5]–[8]. Availability of large-scale protein interaction data, typically represented as networks of interacting proteins, has opened up new ways for the systematic study of biological networks. Especially, cross-species comparison of genome-scale PPI networks can provide important insights into the structure and organization of biological networks, as well as important similarities and variations across different species [9]. In recent years, a large number of computational methods have been developed for comparative analysis of biological networks, where their main focus has been on the identification of functional modules that are conserved in the networks of multiple species [10]–[39]. These methods can be broadly divided into two categories, namely, network querying and network alignment. Network querying aims to identify subnetwork regions in the network of a target species that are similar to a small subnetwork of another species, used as query [32]–[39]. For example, this could be used for querying a known functional pathway in a well-studied species to identify putative homologous pathways in different species, thereby allowing knowledge transfer across species. Network alignment can be viewed as a generalization of network querying, and it aims to predict the best mapping between a set of networks, based on the similarity of the constituent molecules and their interaction patterns [10]–[31]. Network alignment methods may be used to analyze the cross-species variations of biological networks, to predict conserved functional modules, or to infer the function of unannotated proteins.
Research in comparative network analysis is still at an early stage, but many existing studies have demonstrated its potential as an effective tool for gaining important insights into biological systems, that would be otherwise difficult to obtain.
Unfortunately, further advance in comparative network analysis research is critically impeded by the lack of a gold standard for evaluating network alignment algorithms. Currently, there is no comprehensive and reliable benchmark dataset that can be used for validating these algorithms [12]. For this reason, it is common practice to assess the performance of network alignment algorithms in indirect ways, for instance, based on the functional coherence of the aligned nodes in the predicted network alignment or simply through anecdotal examples. Functional annotations based on Gene Ontology (GO) [40] or KEGG orthology (KO) [41] are often employed for this purpose. However, these annotations are mainly curated based on the sequence similarity between molecules, hence they may fail to effectively capture the actual functional coherence between the molecules [28], [42]. Considering that network alignment aims to incorporate molecular interaction data with sequence data to make predictions that are biologically more relevant, evaluating network alignment algorithms based on annotations that are strongly influenced by sequence similarity is certainly less than ideal. Besides, currently available protein interaction databases, such as BioGRID [43], MIPS [44], DIP [45], IntAct [46], MINT [47], and Human Protein Reference Database (HPRD) [48], include the protein interaction networks for only a few species, where the interaction data are very incomplete even for meta-databases – such as PINA [49] and APID [50] – that have been constructed by integrating multiple databases. For example, BioGRID v. 3.1.82 (November 2011), which is one of the most comprehensive among the existing PPI databases, contains the PPI networks of just 25 organisms, where the networks of 7 organisms – A. thaliana, C. elegans, D. melanogaster, H. sapiens, M. musculus, S. cerevisiae, and S. pombe – include more than few hundred interactions. It is widely suspected that a significant number of interactions in the current PPI networks may be spurious, while many true interactions may be still missing. As discussed in [51], based on the analysis of synthetic networks, incomplete knowledge poses a major challenge for interactome-level comparison between different species.
Considering the incompleteness of the current PPI networks, as well as the difficulty of accurately assessing the functional correspondence between proteins, a network synthesis model that can generate families of protein interaction networks with biologically realistic properties may provide a practical and effective alternative. Recently, Ali and Dean [51] have performed a simulation-based study, where a pair of evolutionary related synthetic networks were analyzed to investigate the source of low level of interaction conservation in network alignment results. Erten et al. [52] also proposed a simulation scheme for generating a set of networks with known phylogeny, where the driving motivation was to evaluate the accuracy of their network-based phylogeny reconstruction algorithm. These studies [51], [52] serve as interesting showcases of the important role of synthetic network models. However, these models have also a number of practical limitations. For example, the model presented in [51] cannot be used to synthesize a network family with an arbitrary phylogeny. Furthermore, both models in [51] and [52] do not explicitly represent the functional correspondence between individual proteins across different networks, which is indispensable for evaluating the accuracy of network alignment algorithms.
In this paper, we present a general network synthesis model that can effectively address these issues. Following a pre-specified phylogenetic tree, the model can generate a family of evolutionarily related protein interaction networks, whose properties closely mimic those of real networks – in terms of both the internal properties of the individual networks as well as the comparative properties across networks – as will be shown in our analysis. By internal network properties, we refer to the local characteristics (such as the node degree and the clustering coefficient) and their distributions over each network, which are important in understanding the overall topology. On the other hand, by comparative or cross-network properties, we refer to the properties that can be estimated through network comparison (e.g., sequence similarity between proteins that belong to different networks) and reflect the similarity (or the lack thereof) between networks, which arise from their evolutionary relationship. To demonstrate the utility of the network synthesis model, we created a comprehensive network alignment benchmark based on the proposed model and carried out an extensive performance analysis of select state-of-the-art network alignment algorithms.
Methods
Network Growth Models
In this section, we briefly review existing network growth models that aim to computationally simulate the evolutionary growth of a single biological network. Recently, there has been significant interest in developing network growth models [53]–[70] that can capture the characteristics of real biological networks, including PPI networks. As pointed out in [71], PPI networks do not follow the Erdös-Rényi's model for random graphs. Instead, the structure of biological networks appears to be governed by a scale-free degree distribution, which is also the case for social networks. The scale-free model suggests that the probability that a given node will have a degree (i.e., number of edges) of 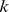 follows a power-law
follows a power-law 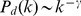 , for some degree exponent
, for some degree exponent  . In general, a scale-free network possesses a few highly connected nodes (often referred as hubs), while the rest of the nodes have only a relatively small number of connections. This trend is generally observed in many PPI networks, which can be explained at a molecular level, at least in part, by the different degrees of protein binding specificity – i.e., the number of binding surfaces or binding partners – required by the cell for carrying out various biological functions [42]. Preferential attachment (PA) growth model
[56] is one of the network evolution models that can generate such a distribution. In the PA model, the network is grown by iteratively adding a new node to the network and adding random connections to existing nodes. The probability of adding an edge to a given node is proportional to its degree, hence the model prefers to connect the new node to nodes that have many interacting partners. The PA model can also capture another important property of PPI networks called the “small-world effect”, which means that any node in the network can be typically reached from other nodes within a few links. Despite its effectiveness in modeling the scale-free degree distribution in PPI networks as well as their small-world property, the PA mechanism fails to capture other important properties, such as the graphlet distribution in real networks and their structural modularity [53], [65], [72], [73].
. In general, a scale-free network possesses a few highly connected nodes (often referred as hubs), while the rest of the nodes have only a relatively small number of connections. This trend is generally observed in many PPI networks, which can be explained at a molecular level, at least in part, by the different degrees of protein binding specificity – i.e., the number of binding surfaces or binding partners – required by the cell for carrying out various biological functions [42]. Preferential attachment (PA) growth model
[56] is one of the network evolution models that can generate such a distribution. In the PA model, the network is grown by iteratively adding a new node to the network and adding random connections to existing nodes. The probability of adding an edge to a given node is proportional to its degree, hence the model prefers to connect the new node to nodes that have many interacting partners. The PA model can also capture another important property of PPI networks called the “small-world effect”, which means that any node in the network can be typically reached from other nodes within a few links. Despite its effectiveness in modeling the scale-free degree distribution in PPI networks as well as their small-world property, the PA mechanism fails to capture other important properties, such as the graphlet distribution in real networks and their structural modularity [53], [65], [72], [73].
Inspired by the gene duplication model used to explain genome evolution [74], several duplication-based techniques have been proposed to simulate network evolution [53]–[55], [57]–[63], [66], [67], [69]. Basically, the gene duplication models assumes that the primary source of protein diversity is the repetitive duplication of existing genes followed by mutation of the duplicated genes leading to functional divergence [74]. A recent analysis of protein interaction networks [75] showed that gene duplication may play important roles in increasing the organismal complexity. The duplication-divergence model can generate networks that retain many of the generic characteristics of biological networks, such as the power-law degree distribution [76], hence it can provide an alternative framework for modeling PPI networks. The duplication-mutation-complementation (DMC) model [53] and the duplication with random mutation (DMR) model [54], [55] are two examples of duplication-divergence based network growth models that have been investigated in depth. Given a seed network, the DMC model [53] grows it by iterating the following steps:
Add a new node
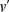 to the network by duplicating a randomly chosen node
to the network by duplicating a randomly chosen node  in the current network. Connect
in the current network. Connect 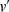 to all neighbors
to all neighbors 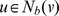 of the node
of the node  .
.For every neighbor
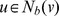 , randomly pick either edge
, randomly pick either edge  or
or  , and randomly remove the edge with probability
, and randomly remove the edge with probability 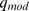 .
.Add a new edge between
 and
and 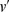 with probability
with probability 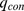 .
.
It was shown that the above DMC model can capture various biological features of PPI networks [72], [77], including their hierarchical modularity. The DMR model is another well-studied network growth model based on the duplication-divergence principle [54], [55], where the network is obtained by repetitively applying the following steps:
As in the DMC model, add a new node
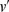 to the network by duplicating a randomly chosen node
to the network by duplicating a randomly chosen node  in the current network. Connect
in the current network. Connect 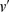 to all neighbors
to all neighbors 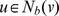 of the node
of the node  .
.Randomly remove the edges between
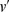 and
and  with probability
with probability 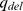 .
.Introduce random edges between
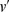 and other nodes in the network (that are not connected to the original node
and other nodes in the network (that are not connected to the original node  ) with probability
) with probability 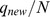 , where
, where 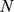 is the size of the current network.
is the size of the current network.
As shown in [73], [78], the DMR model can generate networks that resemble real PPI networks in various aspects, such as the 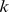 -hop reachability (i.e, the number of distinct nodes that can be reached from a given node via a path of
-hop reachability (i.e, the number of distinct nodes that can be reached from a given node via a path of 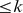 edges), the graphlet distribution, as well as the betweenness, closeness, and degree distributions.
edges), the graphlet distribution, as well as the betweenness, closeness, and degree distributions.
Another notable network growth model that is not based on the duplication-divergence principle is the crystal growth (CG) model, recently proposed by Kim and Marcotte [65]. The CG model takes a highly module-oriented approach, which tries to emulate the physical process of growing protein crystals in solution. Kim and Marcotte [65] showed that the CG model can better explain many features of real PPI networks, including their network topology, their characteristic age distribution, and the spatial distribution of the subunits of different ages within protein complexes, hinting at a plausible physical mechanism of network evolution. Specifically, the capability to accurately capture age-dependent interaction patterns in PPI networks is an important advantage of the CG model, as this is one major drawback of existing models (e.g., duplication-based techniques). The CG model grows a seed network by iteratively adding new nodes as follows:
Define modules (i.e., dense local network regions) in the current network using Newman's algorithm [79]. Let
 be the number of modules in the network.
be the number of modules in the network.Introduce a new node
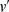 to the network. Either define the node
to the network. Either define the node 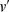 as a new module by itself (with probability
as a new module by itself (with probability 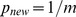 ) or add it to one of the existing modules (with probability
) or add it to one of the existing modules (with probability 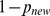 ).
).If
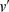 is defined as a new module, add
is defined as a new module, add  random connections to other nodes in the network according to the anti-preferential attachment (AP) rule. (Note that, according to the AP rule, nodes prefer to add edges to low-degree nodes.)
random connections to other nodes in the network according to the anti-preferential attachment (AP) rule. (Note that, according to the AP rule, nodes prefer to add edges to low-degree nodes.)Otherwise, randomly select one of the
 modules in the network and choose an anchor node
modules in the network and choose an anchor node  in the selected module, based on the AP rule. Add
in the selected module, based on the AP rule. Add  connections between
connections between 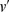 and the randomly selected neighbors of
and the randomly selected neighbors of  . Repeat this step if
. Repeat this step if  has less than
has less than  neighbors.
neighbors.
In addition to these three network growth models, there are also other randomized network generation schemes based on different approaches. For example, the scheme proposed in [70] does not generate a random network by growing a small seed network. Instead, this algorithm, which is developed based on Tailored random graphs, initiates from another random graph with the same dimensionality and the same degree sequence (i.e., the sequence of node degrees of the desired network) as the final network. Then it iteratively rewires the network (e.g., by edge swapping) to reach the desired degree distribution and joint degree statistics for connected nodes. However, this method is not well-suited for modeling network families, as it requires a predefined degree sequence (which may not be available in practice). Furthermore, as this scheme does not follow a growth model, it cannot effectively simulate evolutionarily related networks.
In the current work, we adopt and compare the three network growth models discussed above–i.e., DMC, DMR, and CG–to generate families of synthetic PPI networks. Note that the variables 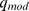 ,
, 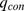 ,
, 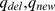 and
and  are user defined parameters for DMC, DMR, and CG schemes. Incorporation of other network evolution models is straightforward.
are user defined parameters for DMC, DMR, and CG schemes. Incorporation of other network evolution models is straightforward.
Characteristics of Protein Interaction Networks
To develop a biologically realistic model for generating families of synthetic protein interaction networks, we first study the characteristics of real PPI networks of five organisms: C. elegans, D. melanogaster, H. sapiens, M. musculus, and S. cerevisiae. We present the analysis results for D. melanogaster, H. sapiens, and S. cerevisiae, which have the largest PPI networks among the five organisms, while the rest can be found in the supplementary data. The protein interaction data for these organisms have been obtained from IsoBase [80], a recently developed database of functionally related protein orthologs. IsoBase consists of the PPI networks of these five species, along with the homology scores between all pairs of proteins across different species, measured in terms of BLAST bit-value similarity of the protein sequences. The PPI networks in the IsoBase have been constructed by integrating the data in three different public databases: DIP [45], BioGRID [43], and HPRD [48]. Table 1 summarizes the statistics of IsoBase, which currently contains 48,120 proteins and 114,897 protein-protein interactions. From this table, we can also observe the incompleteness of the current PPI networks, evidenced by the large number of isolated proteins (i.e., proteins without known interactions). Furthermore, it also shows that only a small portion of the included proteins have known functional annotations according to the KEGG orthology. In the following, we investigate several important features that can be observed in these PPI networks.
Table 1. Statistics of the IsoBase database.
| Species | C. elegans | D. melanogaster | H. sapiens | M. musculus | S. cerevisiae |
| # Proteins | 19,756 | 14,098 | 22,369 | 24,855 | 6,659 |
| # Interactions | 5,853 | 26,726 | 43,757 | 452 | 38109 |
| # Connected proteins | 2,745 | 6,700 | 8,966 | 218 | 4,928 |
| Average Degree | 3.19 | 5.89 | 8.09 | 1.56 | 13.36 |
| # Proteins with KO | 2,102 | 3,366 | 4,195 | 3,805 | 1,605 |
| # Connected proteins with KO | 628 | 1,912 | 2,740 | 71 | 1,470 |
| # Unique KO's | 1,510 | 1,979 | 3,486 | 3,073 | 1,212 |
For each organism, the following numbers are shown: number of proteins in the network, number of interactions, number of connected proteins (those with interactions), average degree, number of proteins with KO annotations, number of connected proteins with KO annotations, and number of unique KO annotations in the network.
Intra-network properties of individual PPI networks
Two important network properties that we can typically observe in a real PPI network is the scale-free property and the modularity. The scale-free property manifests itself in the degree distribution 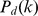 , defined as the probability that a given node in the network will have
, defined as the probability that a given node in the network will have 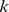 connections to other nodes, that follows a power-law distribution:
connections to other nodes, that follows a power-law distribution: 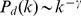 for some
for some  . One measure that can be used to evaluate the modularity of a network is the clustering coefficient function
. One measure that can be used to evaluate the modularity of a network is the clustering coefficient function 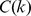 . We define the clustering coefficient of a node
. We define the clustering coefficient of a node  of degree
of degree 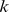 as
as 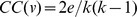 , where
, where  is the number of connections among the neighbors of
is the number of connections among the neighbors of  . The clustering function
. The clustering function 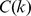 is defined as the average clustering coefficient of all nodes with
is defined as the average clustering coefficient of all nodes with 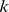 neighbors, and it is expected to scale down with
neighbors, and it is expected to scale down with 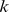 in a modular network. Figures 1(A)–1(F) and Figures S1(A)–S1(D) show the degree distribution
in a modular network. Figures 1(A)–1(F) and Figures S1(A)–S1(D) show the degree distribution 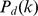 and the clustering coefficient function
and the clustering coefficient function 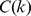 for the five organisms. These figures show that the degree distribution of each organism clearly follows a power-law distribution
for the five organisms. These figures show that the degree distribution of each organism clearly follows a power-law distribution 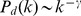 , where
, where  ranges between 1.8 and 2.3. We can also see that the clustering coefficient
ranges between 1.8 and 2.3. We can also see that the clustering coefficient 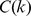 quickly scales down with
quickly scales down with 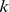 for all organisms, indicating the hierarchical modularity present in the PPI networks [71], [81].
for all organisms, indicating the hierarchical modularity present in the PPI networks [71], [81].
Figure 1. Network properties of various organisms.

(A), (B), and (C) show the degree distributions, and (D), (E), (F) show the clustering coefficient profiles.
Cross-network properties between different PPI networks
In order to devise a practical model for synthesizing a family of related networks, instead of a single network, it is important to investigate the cross-network properties that can be observed when comparing the PPI networks of different organisms. As discussed earlier, two aspects that are important in the comparative analysis of PPI networks are the structural similarity of the networks and the molecular similarity between the proteins that belong to different networks. The molecular similarity between proteins and their potential orthology is typically assessed based on their sequence similarity using a sequence alignment algorithm, such as BLAST [82] or FASTA [83]. Two questions of practical interest are: (i) how many potential orthologs would exist in different networks, for a specific protein in a given network, and (ii) how the protein similarity scores are distributed when comparing a network pair.
Distribution of potential orthologs
Let 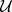 be the set of nodes (i.e., protein) in a PPI network
be the set of nodes (i.e., protein) in a PPI network 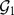 and
and 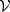 be the set of nodes in
be the set of nodes in 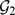 . For a given node
. For a given node 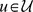 in the network
in the network 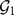 , how many potential orthologs exist in the network
, how many potential orthologs exist in the network 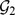 ? By potential orthologs, we refer to pairs of proteins (in different PPI networks) that are candidates for being true orthologs according to their sequence similarity. Sequence similarity is often used as practical evidence for predicting protein orthology, and we assume that nodes with relatively high sequence similarity are more likely to be orthologous. Thus, we estimate the number of potential orthologs of each node
? By potential orthologs, we refer to pairs of proteins (in different PPI networks) that are candidates for being true orthologs according to their sequence similarity. Sequence similarity is often used as practical evidence for predicting protein orthology, and we assume that nodes with relatively high sequence similarity are more likely to be orthologous. Thus, we estimate the number of potential orthologs of each node  as
as
which is the number of nodes 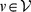 in the network
in the network 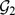 whose similarity score
whose similarity score 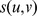 exceeds some threshold
exceeds some threshold 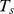 . In practice, we may use a sequence alignment score, such as the BLAST bit score, to estimate
. In practice, we may use a sequence alignment score, such as the BLAST bit score, to estimate 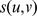 . For any integer
. For any integer 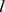 , we define
, we define 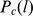 as the fraction of nodes
as the fraction of nodes 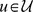 with
with 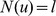 . This relative frequency
. This relative frequency 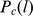 can provide useful insights regarding the presence of potential orthologs across different networks. Figures 2(A)–2(F) and Figures S2(A)–S2(N) show
can provide useful insights regarding the presence of potential orthologs across different networks. Figures 2(A)–2(F) and Figures S2(A)–S2(N) show 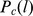 across all pairs of the five organisms in IsoBase, where a threshold of
across all pairs of the five organisms in IsoBase, where a threshold of 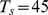 was used in all experiments. As shown in these figures, potential orthologs are generally sparse across networks. The results in Figure 2 and Figure S2 clearly reveal that the distribution
was used in all experiments. As shown in these figures, potential orthologs are generally sparse across networks. The results in Figure 2 and Figure S2 clearly reveal that the distribution 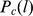 closely follows a power-law distribution
closely follows a power-law distribution 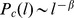 with an exponent
with an exponent 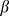 that ranges between 1.4 and 2.1. For example, let us consider the number of proteins in the D. melanogaster network that are potentially orthologous to proteins in the S. cerevisiae network. Among the 6,659 proteins in the S. cerevisiae network, 3,369 proteins do not have any potential orthologs in D. melanogaster whose sequence similarity score exceeds the threshold
that ranges between 1.4 and 2.1. For example, let us consider the number of proteins in the D. melanogaster network that are potentially orthologous to proteins in the S. cerevisiae network. Among the 6,659 proteins in the S. cerevisiae network, 3,369 proteins do not have any potential orthologs in D. melanogaster whose sequence similarity score exceeds the threshold 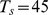 . Among the rest, 1,707 proteins have no more than two potential orthologs in the D. melanogaster PPI network, 578 proteins have
. Among the rest, 1,707 proteins have no more than two potential orthologs in the D. melanogaster PPI network, 578 proteins have 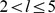 potential orthologs, 291 proteins have
potential orthologs, 291 proteins have 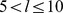 potential orthologs, 246 proteins have
potential orthologs, 246 proteins have 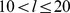 potential orthologs, 295 proteins have
potential orthologs, 295 proteins have  potential orthologs, 130 proteins have
potential orthologs, 130 proteins have 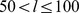 potential orthologs, and only 43 proteins have more than 100 potential orthologs. The general trend does not significantly change for choosing a different threshold
potential orthologs, and only 43 proteins have more than 100 potential orthologs. The general trend does not significantly change for choosing a different threshold 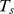 . For example, even when we raise the threshold to
. For example, even when we raise the threshold to 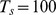 , the number of proteins in S. cerevisiae with more than 50 potential orthologs in D. melanogaster would just decrease to 33. The results are similar for other network pairs, which show that there are typically only a few nodes in a PPI network with a relatively large number of potential orthologs, while most nodes only have a small number of potential orthologs, if any, in other organisms. This observation reveals an important challenge in network alignment, namely, strong reliance on sequence similarity can lead to predictions that are biologically insignificant and misleading, and effective incorporation of interaction data is crucial to minimize this risk.
, the number of proteins in S. cerevisiae with more than 50 potential orthologs in D. melanogaster would just decrease to 33. The results are similar for other network pairs, which show that there are typically only a few nodes in a PPI network with a relatively large number of potential orthologs, while most nodes only have a small number of potential orthologs, if any, in other organisms. This observation reveals an important challenge in network alignment, namely, strong reliance on sequence similarity can lead to predictions that are biologically insignificant and misleading, and effective incorporation of interaction data is crucial to minimize this risk.
Figure 2. Cross-species network properties for different pairs of organisms.

(A)–(F) show how the number of potential orthologs (i.e., nodes with high sequence similarity) are distributed between a given pair of networks. 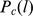 is the fraction of nodes with
is the fraction of nodes with 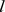 potential orthologs in the other network. (G)–(I) illustrate the sequence similarity (BLAST bit score) distribution for orthologous and non-orthologous node pairs.
potential orthologs in the other network. (G)–(I) illustrate the sequence similarity (BLAST bit score) distribution for orthologous and non-orthologous node pairs.
Distribution of sequence similarity scores
Now, let us consider the distribution of the similarity score between nodes in different networks. As before, let 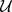 be the set of nodes in a PPI network
be the set of nodes in a PPI network 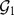 and let
and let 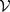 be the set of nodes in a different PPI network
be the set of nodes in a different PPI network 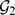 . We define the set of orthologous proteins in the two networks as
. We define the set of orthologous proteins in the two networks as
and the set of non-orthologous proteins as
where  (in network
(in network 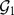 ) and
) and  (in
(in 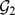 ) are regarded as orthologs if they belong to the same KEGG ortholog group, thus share the same functional annotation. We define
) are regarded as orthologs if they belong to the same KEGG ortholog group, thus share the same functional annotation. We define 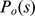 as the distribution of the similarity score
as the distribution of the similarity score 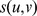 for orthologous nodes
for orthologous nodes 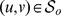 . Similarly, we define
. Similarly, we define 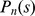 as the score distribution for non-orthologous node pairs
as the score distribution for non-orthologous node pairs 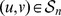 . These distributions are shown in Figures 2(G)–2(I) and Figures S3(A)–S3(G) across all pairs of the considered organisms. These results show that the score distribution can be closely approximated by the Gamma distribution
. These distributions are shown in Figures 2(G)–2(I) and Figures S3(A)–S3(G) across all pairs of the considered organisms. These results show that the score distribution can be closely approximated by the Gamma distribution 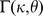 , whose probability density function
, whose probability density function 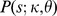 is defined as follows
is defined as follows
| (1) |
for some shape parameter 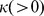 and scale parameter
and scale parameter 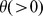 . These figures also show that there is a substantial overlap between
. These figures also show that there is a substantial overlap between 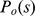 and
and 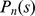 , the similarity score distribution for orthologs and that for non-orhologs, which again reveals the the importance of incorporating interaction data into comparative networks analysis. This observation also confirms the results in previous studies [28], [42], [67], which showed that proteins that are conserved at the sequence level may fail to have conserved functionalities at the network level.
, the similarity score distribution for orthologs and that for non-orhologs, which again reveals the the importance of incorporating interaction data into comparative networks analysis. This observation also confirms the results in previous studies [28], [42], [67], which showed that proteins that are conserved at the sequence level may fail to have conserved functionalities at the network level.
Proposed Network Synthesis Model
Following the previous discussions, in this section, we propose a novel network synthesis model that can generate a family of evolutionarily related protein-protein interaction networks. Suppose we want to generate a family of  synthetic PPI networks
synthetic PPI networks 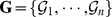 . Each network
. Each network 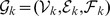 consists of set
consists of set 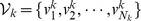 of
of 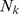 nodes; a set
nodes; a set 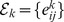 of
of 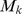 edges, where
edges, where 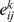 denotes the edge between node
denotes the edge between node 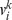 and
and 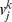 ; and a set
; and a set 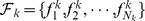 , which maps each node
, which maps each node 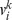 to a functional group
to a functional group 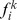 in
in 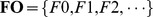 , the set of all functional orthology (FO) annotations. A node
, the set of all functional orthology (FO) annotations. A node 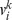 with
with 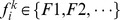 is regarded as an annotated protein with a known function
is regarded as an annotated protein with a known function 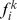 , while it is regarded as an unannotated protein if
, while it is regarded as an unannotated protein if 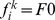 . We define
. We define 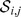 as a
as a 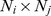 similarity score matrix that contains the sequence similarity score between all pairs of proteins for the networks
similarity score matrix that contains the sequence similarity score between all pairs of proteins for the networks 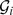 and
and 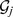 . The set
. The set 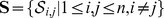 consists of the scoring matrices for all pairs of networks.
consists of the scoring matrices for all pairs of networks.
To generate the  networks, we first specify the hypothetic phylogenetic tree
networks, we first specify the hypothetic phylogenetic tree 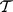 that describes the evolutionary relationship among the networks. The tree
that describes the evolutionary relationship among the networks. The tree 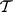 , which is assumed to be a binary tree, will have exactly
, which is assumed to be a binary tree, will have exactly  leaf nodes, in addition to a number of internal nodes, which correspond to the
leaf nodes, in addition to a number of internal nodes, which correspond to the  networks to be generated by the model. The basic idea of the proposed method is to follow the phylogenetic tree
networks to be generated by the model. The basic idea of the proposed method is to follow the phylogenetic tree 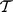 to create a set of related networks through repetitive network duplication, mutation, and network extension, starting from a single hypothetical ancestral network
to create a set of related networks through repetitive network duplication, mutation, and network extension, starting from a single hypothetical ancestral network 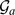 . In order to create a biologically realistic ancestral network
. In order to create a biologically realistic ancestral network 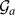 , we begin by generating a small seed network and iteratively extend it using one of the network growth models – DMC, DMR, and CG models – described earlier. As discussed in [73], choosing the right seed network is crucial to capture the key topological features of real PPI networks. For the duplication-based models (i.e., DMC and DMR), we use a seed network that is similar to the one presented in [73], which was shown to accurately characterize the attributes of the S. cerevisiae PPI network. This seed network of size 50 includes two cliques (complete subgraphs), one with 10 nodes and the other with 7 nodes. Nodes in each of these two cliques are randomly connected to a few nodes in the other clique. The other 33 nodes are randomly connected to one of the 17 clique nodes. The nodes in the first and the second cliques are assigned to distinct functional groups
, we begin by generating a small seed network and iteratively extend it using one of the network growth models – DMC, DMR, and CG models – described earlier. As discussed in [73], choosing the right seed network is crucial to capture the key topological features of real PPI networks. For the duplication-based models (i.e., DMC and DMR), we use a seed network that is similar to the one presented in [73], which was shown to accurately characterize the attributes of the S. cerevisiae PPI network. This seed network of size 50 includes two cliques (complete subgraphs), one with 10 nodes and the other with 7 nodes. Nodes in each of these two cliques are randomly connected to a few nodes in the other clique. The other 33 nodes are randomly connected to one of the 17 clique nodes. The nodes in the first and the second cliques are assigned to distinct functional groups 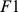 and
and 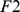 , respectively. Each of the remaining 33 nodes is assigned to a different functional group, from
, respectively. Each of the remaining 33 nodes is assigned to a different functional group, from 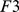 to
to 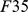 . For the CG model, we use a seed graph of size 4 as in [65]. The initial seed network is grown into the ancestral network
. For the CG model, we use a seed graph of size 4 as in [65]. The initial seed network is grown into the ancestral network 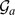 of size
of size 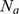 by employing one of the network extension models. While growing the network, every new node is assigned to a new functional group of its own.
by employing one of the network extension models. While growing the network, every new node is assigned to a new functional group of its own.
Once the ancestral PPI network 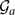 is created, we traverse the phylogenetic tree
is created, we traverse the phylogenetic tree 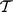 to generate descendant networks that are evolutionarily related to
to generate descendant networks that are evolutionarily related to 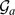 . Figure 3 illustrates an example of a phylogenetic tree
. Figure 3 illustrates an example of a phylogenetic tree 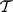 for five hypothetical species, which correspond to the five leaf nodes
for five hypothetical species, which correspond to the five leaf nodes 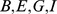 , and
, and 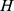 . The tree also includes three internal nodes
. The tree also includes three internal nodes  ,
, 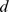 and
and 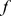 , and the root node
, and the root node  . Since the phylogenetic tree is assumed to be binary, each internal node (including the root node) branches off to two child nodes. For each child node, we create a network by duplicating the parent network and evolving it into a larger network. For example, according to the tree in Figure 3, we generate two networks
. Since the phylogenetic tree is assumed to be binary, each internal node (including the root node) branches off to two child nodes. For each child node, we create a network by duplicating the parent network and evolving it into a larger network. For example, according to the tree in Figure 3, we generate two networks 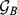 (for the leaf node
(for the leaf node 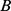 ) and
) and 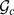 (for the internal node
(for the internal node  ) based on the ancestral network
) based on the ancestral network 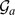 that corresponds to the root node
that corresponds to the root node  , which is the parent of
, which is the parent of 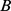 and
and  . We will traverse the tree
. We will traverse the tree 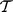 through a breadth-first search [84] and repeat this bifurcation process until all
through a breadth-first search [84] and repeat this bifurcation process until all  networks are generated. It is straightforward to see that this will require
networks are generated. It is straightforward to see that this will require  bifurcations, in total.
bifurcations, in total.
Figure 3. The phylogenetic tree of five hypothetical organisms.

The bifurcation step is carried out as follows. Suppose 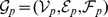 is the network that corresponds to the current internal node. We denote
is the network that corresponds to the current internal node. We denote 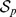 as the set of scoring matrices that contain the similarity scores between proteins in
as the set of scoring matrices that contain the similarity scores between proteins in 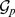 and those in the networks for other nodes in
and those in the networks for other nodes in 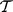 that have been previously visited. We generate the networks
that have been previously visited. We generate the networks 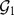 and
and 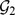 for the two child nodes by duplicating the parent network:
for the two child nodes by duplicating the parent network: 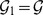 and
and 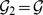 . Both networks inherit the functional annotations of their parent
. Both networks inherit the functional annotations of their parent 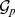 and the set
and the set 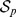 of scoring matrices. For every pair of nodes
of scoring matrices. For every pair of nodes  in
in 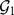 and
and  in
in 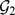 , we randomly assign their similarity score according to a Gamma distribution as follows:
, we randomly assign their similarity score according to a Gamma distribution as follows:
| (2) |
where 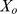 and
and 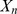 are random numbers sampled according to
are random numbers sampled according to 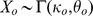 and
and 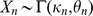 . Note that the similarity score
. Note that the similarity score 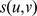 takes a different distribution, depending on whether or not
takes a different distribution, depending on whether or not  and
and  have the same functional annotation:
have the same functional annotation: 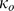 and
and 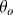 are the shape and scale parameters of the Gamma distribution for orthologs (with identical FO annotations);
are the shape and scale parameters of the Gamma distribution for orthologs (with identical FO annotations); 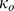 and
and 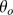 are the parameters for non-orthologs (with different FO annotations).
are the parameters for non-orthologs (with different FO annotations). 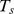 is used to simulate the thresholding effect of sequence similarity scores. As we have seen in our analysis of real PPI networks, potential orthologs across different networks are generally sparse. In the proposed model, we enforce the number of potential orthologs to follow a power-law distribution
is used to simulate the thresholding effect of sequence similarity scores. As we have seen in our analysis of real PPI networks, potential orthologs across different networks are generally sparse. In the proposed model, we enforce the number of potential orthologs to follow a power-law distribution 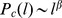 , as in real PPI networks.
, as in real PPI networks.
To diverge the child networks 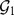 and
and 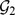 from the parent network
from the parent network 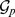 , we independently apply a network growth algorithm (DMC, DMR, or CG) to each of these networks. In this step, the number of new nodes added to each child network may be specified according to the evolutionary distance between the corresponding hypothetical species in the tree
, we independently apply a network growth algorithm (DMC, DMR, or CG) to each of these networks. In this step, the number of new nodes added to each child network may be specified according to the evolutionary distance between the corresponding hypothetical species in the tree 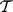 . For instance, in Figure 3, the number of additional nodes (referred as the “length” of a given branch) are shown along the branches. In this example, if the ancestral network has
. For instance, in Figure 3, the number of additional nodes (referred as the “length” of a given branch) are shown along the branches. In this example, if the ancestral network has 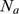 nodes, the PPI network
nodes, the PPI network 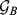 for node
for node 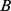 will have
will have 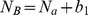 nodes and the PPI network
nodes and the PPI network 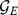 for node
for node 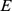 will have
will have 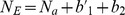 nodes. Consider a new node
nodes. Consider a new node 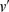 that was either (i) obtained by duplicating an existing node
that was either (i) obtained by duplicating an existing node  (when using either the DMC or the DMR model) or (ii) a new node whose anchor node was chosen to be
(when using either the DMC or the DMR model) or (ii) a new node whose anchor node was chosen to be  (when using the CG model). We transfer the functional annotation and the similarity scores from an existing node
(when using the CG model). We transfer the functional annotation and the similarity scores from an existing node  to a new node
to a new node 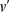 as follows:
as follows:
With probability
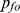 , assign
, assign 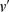 to the same functional group as
to the same functional group as  by setting
by setting 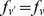 . With probability
. With probability 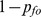 , set
, set 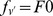 , which implies that
, which implies that 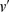 takes a new unknown function.
takes a new unknown function.- For every protein
 in the networks that correspond to previously visited nodes in
in the networks that correspond to previously visited nodes in 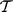 , assign the similarity score between
, assign the similarity score between  and
and 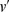 as:
as:
where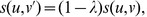
(3) 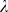 is a random scaling factor with a uniform distribution over
is a random scaling factor with a uniform distribution over 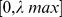 . The upper bound
. The upper bound 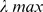 (
(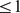 ) specifies the extent of the sequence-level divergence between
) specifies the extent of the sequence-level divergence between  and
and 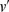 .
.
In this way, we can model the functional inheritance and the sequence similarity between the duplicated nodes, where a duplicated node may have a different function from the original node. Finally, when using the CG model, a new node 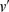 that forms a new functional module by itself, hence not anchored to any of the existing nodes, will be assigned a new unannotated function (i.e.,
that forms a new functional module by itself, hence not anchored to any of the existing nodes, will be assigned a new unannotated function (i.e., 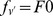 ).
).
Results and Discussion
Attributes of Synthetic Networks
To validate the proposed network synthesis model, we generated synthetic PPI networks according to the model and analyzed the individual and cross-species characteristics of the synthesized networks. We first generated an ancestral network 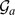 of size
of size 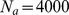 . A simple binary tree with two leaves was used to evolve
. A simple binary tree with two leaves was used to evolve 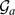 into two networks
into two networks 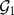 and
and 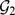 , respectively with 5,000 nodes and 7,000 nodes. For network extension, we applied all three network growth models – DMC, DMR, and CG – discussed in this paper. For DMC, we used
, respectively with 5,000 nodes and 7,000 nodes. For network extension, we applied all three network growth models – DMC, DMR, and CG – discussed in this paper. For DMC, we used 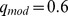 and
and 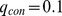 as in [65]. For DMR, we set the parameters to
as in [65]. For DMR, we set the parameters to 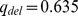 and
and 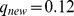 as in [73]. We used
as in [73]. We used 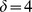 for CG as in [65]. The scaling and shape parameters of the Gamma distributions in (2) were set to
for CG as in [65]. The scaling and shape parameters of the Gamma distributions in (2) were set to 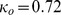 ,
, 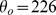 ,
, 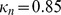 ,
, 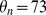 , and the exponent
, and the exponent 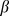 in the distribution
in the distribution 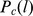 was set to
was set to 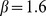 , such that the cross-network properties between
, such that the cross-network properties between 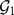 and
and 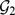 resemble those between the D. melanogaster PPI network and the S. cerevisiae PPI network. The parameters
resemble those between the D. melanogaster PPI network and the S. cerevisiae PPI network. The parameters 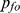 and
and 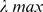 that control the functional inheritance and sequence similarity between orthologous nodes were set to
that control the functional inheritance and sequence similarity between orthologous nodes were set to 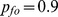 and
and 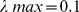 , so that protein function and sequence similarity is conserved at the
, so that protein function and sequence similarity is conserved at the 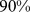 level. Although it is practically difficult to accurately determine these two parameters in real networks, the analysis in [85] shows this rate of functional conservation for duplicated genes.
level. Although it is practically difficult to accurately determine these two parameters in real networks, the analysis in [85] shows this rate of functional conservation for duplicated genes.
In the case of CG algorithm, we made a slight modification in the first step of the algorithm as follows. In the original algorithm proposed in [65], when adding a new node, the modules of the current network are recomputed at each iteration. To speed up the CG algorithm, we instead redefine the modules every 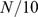 steps, where
steps, where 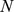 is the size of the current network. In other words, in the early iterations, we redefine modules in every iteration, while as the network grows larger, we apply the module redefinition step only occasionally and use these modules over multiple iterations. Simulation results show that the CG method can still accurately capture the generic features of real PPI networks with this modification. We leave the module redefinition frequency as a control parameter that can be freely adjusted.
is the size of the current network. In other words, in the early iterations, we redefine modules in every iteration, while as the network grows larger, we apply the module redefinition step only occasionally and use these modules over multiple iterations. Simulation results show that the CG method can still accurately capture the generic features of real PPI networks with this modification. We leave the module redefinition frequency as a control parameter that can be freely adjusted.
The properties of the synthetic PPI network are shown in Figure 4, Figure 5, and Figure 6, for using DMC, DMR, and CG, respectively. As can be seen in these figures, all three schemes can accurately model the scale-free degree distribution. However, it appears that the hierarchical modularity can be better captured by using either DMC or CG, rather than DMR. Regarding the cross-network properties, these results also clearly show that the proposed network synthesis model can effectively capture the attributes of real PPI networks. For example, this can be immediately seen by comparing the network properties of 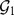 and
and 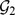 in Figures 4(E) and 4(F) (when using DMC) with those of the D. melanogaster and the S. cerevisiae PPI networks shown in Figures 2(B) and 2(H). Similar observations can be made from Figures 5(E) and 5(F) (for DMR) as well as Figures 6(E) and 6(F) (for CG).
in Figures 4(E) and 4(F) (when using DMC) with those of the D. melanogaster and the S. cerevisiae PPI networks shown in Figures 2(B) and 2(H). Similar observations can be made from Figures 5(E) and 5(F) (for DMR) as well as Figures 6(E) and 6(F) (for CG).
Figure 4. Properties of the networks generated using the DMC model.

(A)–(B) Degree distribution. (C)–(D) Clustering coefficient profile. (E) Distribution of the number of potential orthologs. (F) Sequence similarity distribution for orthologous nodes and the distribution for non-orthologous nodes. (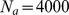 ,
, 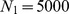 ,
, 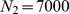 ,
, 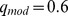 , and
, and 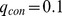 ).
).
Figure 5. Properties of the networks generated using the DMR model.

(A)–(B) Degree distribution. (C)–(D) Clustering coefficient profile. (E) Distribution of the number of potential orthologs. (F) Sequence similarity distribution for orthologous nodes and the distribution for non-orthologous nodes. (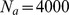 ,
, 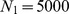 ,
, 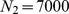 ,
, 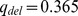 , and
, and 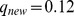 ).
).
Figure 6. Properties of the networks generated using the CG model.

(A)–(B) Degree distribution. (C)–(D) Clustering coefficient profile. (E) Distribution of the number of potential orthologs. (F) Sequence similarity distribution for orthologous nodes and the distribution for non-orthologous nodes. (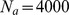 ,
, 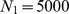 ,
, 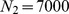 , and
, and 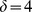 ).
).
Construction of Network Alignment Benchmark
The network synthesis model presented in this paper provides an effective framework for generating network families with diverse characteristics. Such network sets may be used to assess the performance of various alignment techniques to identify their respective strengths and weaknesses under different conditions and problems settings. Furthermore, the proposed network synthesis model may be potentially used to expose previously unknown biases that a network alignment technique may have towards specific types of networks, thereby leading to better alignment techniques.
To demonstrate the utility of the proposed network generation scheme, we used it to create synthetic benchmark datasets that can be used for evaluating and comparing the performance of various network alignment algorithms. We call the proposed Network Alignment Performance Assessment benchmark as NAPAbench. In total, we generated three suites of datasets. The first suite (referred as the pairwise alignment dataset) contains three pairs of networks, where the respective network pairs were generated using DMC, DMR, and CG, respectively. Each pair consists of a network 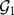 with
with 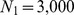 nodes and another network
nodes and another network 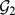 with
with 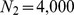 nodes, both evolved from an ancestral network
nodes, both evolved from an ancestral network 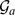 with
with 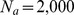 nodes, following a binary tree with two leaves. The second suite (referred as the 5-way alignment dataset) contains three network families, each with five networks generated using DMC, DMR, or CG. To generate the network family, we first created an ancestral network
nodes, following a binary tree with two leaves. The second suite (referred as the 5-way alignment dataset) contains three network families, each with five networks generated using DMC, DMR, or CG. To generate the network family, we first created an ancestral network 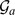 with
with 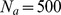 nodes. The phylogenetic tree
nodes. The phylogenetic tree 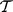 in Figure 3 was used to evolve
in Figure 3 was used to evolve 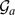 into five networks –
into five networks – 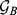 ,
, 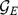 ,
, 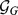 ,
, 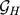 , and
, and 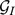 – which correspond to the five leaf nodes. For every branch, we set its length to 500. Thus, the size of the five networks were
– which correspond to the five leaf nodes. For every branch, we set its length to 500. Thus, the size of the five networks were 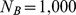 ,
, 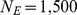 ,
, 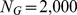 ,
, 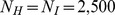 . This dataset simulates a family of PPI networks that correspond to distantly related species. Finally, the third suite (referred as the 8-way alignment dataset) also consists of three network families, each with eight networks generated by one of the three network extension models. The eight networks were obtained by evolving an ancestral network
. This dataset simulates a family of PPI networks that correspond to distantly related species. Finally, the third suite (referred as the 8-way alignment dataset) also consists of three network families, each with eight networks generated by one of the three network extension models. The eight networks were obtained by evolving an ancestral network 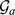 of size
of size 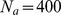 according to a full binary tree with eight leaf nodes. The branch length was set to 200 for all branches, which gave rise to eight equally sized networks, each with 1,000 nodes. This 8-way alignment dataset tries to simulate a network family of closely-related species. All the datasets in NAPAbench are publicly available at http://www.ece.tamu.edu/bjyoon/NAPAbench/.
according to a full binary tree with eight leaf nodes. The branch length was set to 200 for all branches, which gave rise to eight equally sized networks, each with 1,000 nodes. This 8-way alignment dataset tries to simulate a network family of closely-related species. All the datasets in NAPAbench are publicly available at http://www.ece.tamu.edu/bjyoon/NAPAbench/.
Performance Analysis of Network Alignment Algorithms
The created benchmark datasets, NAPAbench, can be used for reliable and comprehensive performance evaluation of existing network alignments. In this work, we used this synthetic benchmark to assess the performance of five well-known multiple network alignment algorithms: IsoRank [13], IsoRankN [12], NetworkBLAST-M [15], Græ mlin 2.0 [11], and MI-GRAAL [25]. IsoRank [13] uses spectral graph theory to evaluate the overall similarity between nodes that belong to different networks. This pairwise alignment score is computed for every node pair across all pairs of networks, which is then used to build the multiple network alignment according to a greedy approach. IsoRankN [12] further extends the idea in IsoRank by employing a spectral clustering scheme based on the pairwise node alignment scores. NetworkBLAST-M [15] computes the network alignment by first constructing a layered alignment graph based on the potential orthologous nodes, and then greedily searching for highly conserved local regions in the alignment graph. Græ mlin 2.0 [11] takes a progressive approach to construct a global alignment of multiple networks, where it repeatedly performs pairwise network alignments according to a given phylogenetic tree that describes the relationship among the networks. The alignment is predicted by maximizing an objective function based on parameters that are learned from a set of known alignments. Finally, MI-GRAAL [25] is a recently proposed pairwise network alignment scheme that can integrate any number and type of similarity measures between network nodes, such as sequence similarity, structural similarity, and topological similarity.
Recall that the node similarity score in the proposed model tries to mimic the BLAST bit scores. Since NetworkBLAST-M and MI-GRAAL employ the BLAST E-values, instead of the BLAST bit scores, we should transform the bit scores into the corresponding E-values for these two algorithms. As discussed in [86], [87], the simulated bit score (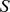 ) is related to the E-value (
) is related to the E-value (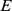 ) as
) as 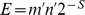 , where
, where 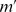 is the length of the BLAST query and
is the length of the BLAST query and 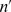 is the length of the target sequence. Here, we transform our simulated bit scores to E-values using
is the length of the target sequence. Here, we transform our simulated bit scores to E-values using 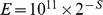 (assuming, for instance, the case when we BLAST a protein sequence with 500 residues in a database that contains a total of 200,000,000 residues). In this paper, we used the restricted-order version of NetworkBLAST-M as the running time of the relaxed-order version increases exponentially with respect to the number of networks to be aligned. As Græ mlin needs to learn the parameters of its scoring function in advance, we generated a training set that consists of five networks (with
(assuming, for instance, the case when we BLAST a protein sequence with 500 residues in a database that contains a total of 200,000,000 residues). In this paper, we used the restricted-order version of NetworkBLAST-M as the running time of the relaxed-order version increases exponentially with respect to the number of networks to be aligned. As Græ mlin needs to learn the parameters of its scoring function in advance, we generated a training set that consists of five networks (with 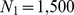 ,
, 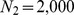 ,
, 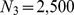 ,
, 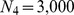 , and
, and 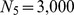 nodes, respectively), using the proposed scheme with the DMC model by following the tree shown in Figure 3. MI-GRAAL can integrate different kinds of similarity measures into the search process. Here, we adopt the graphlet degree signature distance and the E-values (measuring the sequence similarity) for MI-GRAAL alignment algorithm. For IsoRank and IsoRankN, the parameter
nodes, respectively), using the proposed scheme with the DMC model by following the tree shown in Figure 3. MI-GRAAL can integrate different kinds of similarity measures into the search process. Here, we adopt the graphlet degree signature distance and the E-values (measuring the sequence similarity) for MI-GRAAL alignment algorithm. For IsoRank and IsoRankN, the parameter  , which determines the balance between sequence similarity and topological similarity, was set to 0.6.
, which determines the balance between sequence similarity and topological similarity, was set to 0.6.
The accuracy of each network alignment algorithm was assessed using four measures – specificity, correct nodes, mean normalized entropy, and coverage – which had been previously used in [11] and [12]. We refer the set of aligned nodes (i.e., potential orthologs) as the equivalence class. Each equivalence class may include an arbitrary number of nodes from each species. To compute the accuracy measures, we first removed the unannotated nodes from the alignment (i.e, nodes with the annotation  ) and then removed equivalence classes containing only a single node. A given equivalence class is viewed as being correct if all the included nodes belong to the same FO group. The four measures are defined as follows:
) and then removed equivalence classes containing only a single node. A given equivalence class is viewed as being correct if all the included nodes belong to the same FO group. The four measures are defined as follows:
Specificity (SPE): The relative number of correctly predicted equivalence classes.
Correct Nodes (CN): The total number of nodes (i.e., proteins) that are assigned to the correct equivalence class. This measure reflects the sensitivity of the prediction [11].
- Mean normalized entropy (MNE): The mean normalized entropy of the predicted equivalence classes can provide an effective measure of the consistency of the predicted network alignment. The normalized entropy of a given equivalence class
 is computed as:
is computed as:
where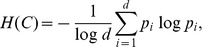
(4) 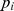 is the fraction of proteins in
is the fraction of proteins in  with the FO annotation
with the FO annotation 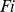 , and
, and 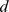 is the number of different FO groups. Thus, a cluster that consists of nodes with higher functional consistency will have lower entropy.
is the number of different FO groups. Thus, a cluster that consists of nodes with higher functional consistency will have lower entropy. Coverage: For any integer
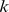 , the total number of equivalence classes that contain nodes from
, the total number of equivalence classes that contain nodes from 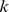 species. We report this measure only for multiple network alignment experiments (and not for pairwise alignments).
species. We report this measure only for multiple network alignment experiments (and not for pairwise alignments).
NetworkBLAST-M reports only the local alignment of the input networks, while the other four algorithms yield the global alignment of the given networks. For a fair comparison between these algorithms, we first convert the local alignment predicted by NetworkBLAST-M into a global network alignment by merging all local node correspondences. For example, if nodes  and
and 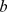 are aligned in one local alignment while
are aligned in one local alignment while  and
and  are aligned in another local alignment, we assume that
are aligned in another local alignment, we assume that  ,
, 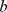 , and
, and  belong to the same equivalence class.
belong to the same equivalence class.
The SPE, CN, and MNE of the five algorithms are summarized in Table 2, Table 3, and Table 4, for the pairwise alignment dataset, 5-way alignment dataset, and the 8-way alignment dataset, respectively. Figure 7 and Figure 8 shows the coverage of different algorithms for the 5-way and 8-way dataset, respectively.
Table 2. Performance of different alignment algorithms on the pairwise alignment dataset of NAPAbench.
| DMC | DMR | CG | |||||||
| SPE | CN | MNE | SPE | CN | MNE | SPE | CN | MNE | |
| IsoRank | 77.53 | 3883 | 24.29 | 77.77 | 3914 | 23.92 | 77.22 | 3986 | 24.47 |
| IsoRankN | 82.69 | 3836 | 14.13 | 83.55 | 3915 | 13.40 | 83.16 | 3868 | 13.34 |
| NetworkBLAST-M | 96.34 | 3354 | 5.33 | 96.60 | 3005 | 4.28 | 95.86 | 4646 | 4.44 |
| Græ mlin | 77.37 | 2137 | 15.70 | 81.03 | 2322 | 13.33 | 90.72 | 2549 | 7.96 |
| MI-GRAAL | 66.13 | 3612 | 35.27 | 69.97 | 3852 | 31.59 | 79.48 | 4385 | 22.76 |
Performance comparison based on the pairwise alignment of two networks of size 3,000 and 4,000. The performance of each method is assessed using the following metrics: specificity(SP), number of correct nodes (CN), and mean normalized entropy (MNE). In each column, best performance is shown in bold.
Table 3. Performance Comparison on the 5-way network alignment dataset of NAPAbench.
| DMC | DMR | CG | |||||||
| SPE | CN | MNE | SPE | CN | MNE | SPE | CN | MNE | |
| IsoRankN | 80.91 | 5538 | 10.27 | 79.58 | 5496 | 11.14 | 82.68 | 5689 | 9.72 |
| NetworkBLAST-M | 62.18 | 1774 | 12.72 | 67.66 | 1591 | 10.62 | 69.90 | 3225 | 9.31 |
| Græ mlin | 51.07 | 3028 | 16.32 | 50.88 | 3100 | 16.94 | 62.89 | 4451 | 13.19 |
| IsoRankN (only 5-species) | 69.67 | 1859 | 9.67 | 68.07 | 1610 | 10.26 | 73.83 | 2223 | 7.99 |
| Græ mlin (only 5-species) | 35.90 | 1575 | 19.50 | 36.60 | 1581 | 20.29 | 54.44 | 2394 | 14.17 |
Performance comparison based on the 5-way alignment of five networks of size 1500, 2000, 2500, 3000 and 3000. The last two rows are obtained by considering only equivalence classes that contain at least one node from every species. The performance of each method is assessed using the following metrics: specificity(SP), number of correct nodes (CN), and mean normalized entropy (MNE). In each metrics, best performance is shown in bold.
Table 4. Performance Comparison on 8-way network alignment dataset of NAPAbench.
| DMC | DMR | CG | |||||||
| SPE | CN | MNE | SPE | CN | MNE | SPE | CN | MNE | |
| IsoRankN | 64.50 | 4069 | 13.62 | 62.52 | 3938 | 14.58 | 61.18 | 3890 | 14.58 |
| NetworkBLAST-M | 54.06 | 1166 | 13.97 | 63.72 | 1203 | 10.65 | 63.66 | 2236 | 10.84 |
| Græ mlin | 58.67 | 2315 | 16.51 | 51.34 | 1939 | 19.38 | 49.29 | 2729 | 17.24 |
| IsoRankN (only 8-species) | 56.74 | 1987 | 10.06 | 54.36 | 1797 | 10.81 | 54.30 | 2172 | 10.33 |
| Græ mlin (only 8-species) | 13.08 | 345 | 29.83 | 9.87 | 291 | 31.63 | 25.66 | 802 | 20.78 |
Performance comparison based on the 8-way alignment of eight networks of equal size 1,000. The last two rows are obtained by considering only equivalence classes that contain at least one node from every species. The performance of each method is assessed using the following metrics: specificity(SP), number of correct nodes (CN), and mean normalized entropy (MNE). In each column, best performance is shown in bold.
Figure 7. Number of equivalence classes in the 5-way alignment experiment that contain nodes from .

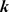 species (
species (
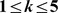 ).
).
Figure 8. Number of equivalence classes in the 8-way alignment experiment that contain nodes from .

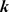 species (
species (
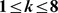 ).
).
For pairwise network alignments, NetworkBLAST-M boasts significantly higher specificity and consistency (reflected in lower MNE) compared to other algorithms. IsoRank, IsoRankN, and MI-GRAAL yielded the highest number of correctly aligned nodes (i.e., CN) for networks generated using the DMC/DMR growth models, implying high sensitivity. For the networks created using the CG model, which yield highly modular networks, NetworkBLAST-M showed highest sensitivity, closely followed by MI-GRAAL.
For the 5-way and 8-way alignment experiments, we can clearly observe the degradation in sensitivity of NetworkBLAST-M, as shown in Table 3 and Table 4. This may be due to the fact that NetworkBLAST-M aims to predict equivalence classes that are conserved across all the compared species, as illustrated in Figure 7 and Figure 8. In these experiments, Græ mlin showed moderate performance, where the sensitivity was higher than NetworkBLAST-M, but the specificity and the consistency were lower. The multiple network alignment experiments based on the 5-way and the 8-way benchmark datasets in NAPAbench show that IsoRankN can yield the most accurate network alignment results, in terms of specificity, sensitivity, and consistency. This observation is in agreement with the performance assessment in [12], based on five real biological networks.
To compared the performance of different algorithms in predicting equivalence classes conserved across all networks, we also estimated the accuracy of IsoRankN and Græ mlin only for such classes. These results are shown in the last two rows of Table 3 and Table 4. We can see that IsoRankN still outperforms NetworkBLAST-M in most cases for 5-way alignment. In the 8-way network alignment, IsoRankN appears to outperform NetworkBLAST-M for networks generated using the DMC growth model. However, NetworkBLAST-M is more sensitive on networks obtained using the DMR model, and it is also more sensitive and more specific for networks generated using the CG model. These results also show that Græ mlin is outperformed by the other two algorithms in this case, which implies that it may not be effective in predicting orthologous nodes that are conserved across all species.
Figure 7 shows the number of equivalence classes (i.e., the coverage) that are predicted in the 5-way alignment dataset by the respective algorithms. In each case, the total number of equivalence classes is split into the number of classes that consist of nodes from 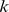 different networks (
different networks (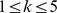 ). As shown in this figure, all three algorithms predicted similar number of equivalence classes that contain nodes from all
). As shown in this figure, all three algorithms predicted similar number of equivalence classes that contain nodes from all 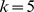 networks. However, we can see that IsoRankN predicts a significantly larger number of equivalence classes with
networks. However, we can see that IsoRankN predicts a significantly larger number of equivalence classes with 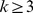 compared to the other algorithms. Considering that the 5-way alignment dataset consists of networks with varying size, equivalence classes that contain nodes from
compared to the other algorithms. Considering that the 5-way alignment dataset consists of networks with varying size, equivalence classes that contain nodes from 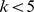 networks are fairly common, hence the ability of identifying such equivalence classes is certainly an important advantage of IsoRankN. Figure 8 shows coverage of different algorithms on the 8-way dataset. The trends are similar as in the 5-way alignment, and we can see that IsoRankN results in greater coverage for equivalence classes spanning
networks are fairly common, hence the ability of identifying such equivalence classes is certainly an important advantage of IsoRankN. Figure 8 shows coverage of different algorithms on the 8-way dataset. The trends are similar as in the 5-way alignment, and we can see that IsoRankN results in greater coverage for equivalence classes spanning 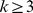 networks. Another interesting observation is that Græ mlin predicts a large number of equivalence classes that contain only nodes from
networks. Another interesting observation is that Græ mlin predicts a large number of equivalence classes that contain only nodes from 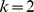 networks.
networks.
Next, we investigate the effect of sequence similarity on the performance of the various network alignment algorithms. To this aim, we add a bias term 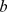 to the similarity score distribution of potential orthologs in (2), such that the score is randomly sampled as
to the similarity score distribution of potential orthologs in (2), such that the score is randomly sampled as 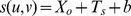 , where
, where 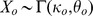 . Increasing the bias
. Increasing the bias 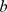 will further separate the similarity score distributions of orthologous and non-orthologous nodes. As a result, the larger
will further separate the similarity score distributions of orthologous and non-orthologous nodes. As a result, the larger 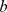 is, the easier it becomes to align the networks (and to predict the potential orthologs across networks) based on sequence similarity alone, without utilizing the topological similarity between networks. For this experiment, we generated two networks with 1,000 nodes from an ancestral network of size
is, the easier it becomes to align the networks (and to predict the potential orthologs across networks) based on sequence similarity alone, without utilizing the topological similarity between networks. For this experiment, we generated two networks with 1,000 nodes from an ancestral network of size 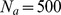 . Figure 9 shows how specificity (SPE) and CN (which reflects sensitivity), change for varying values of
. Figure 9 shows how specificity (SPE) and CN (which reflects sensitivity), change for varying values of 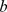 between 0 and 250. As can be seen in this figure, as the separation between the score distributions of orthologs and non-orthologs increases, both the specificity and the sensitivity are improved for IsoRank, IsoRankN, and Græ mlin. On the other hand, NetworkBLAST-M and MI-GRAAL display a constant level of accuracy that does not depend on the amount of separation. This implies that the first three alignment algorithms rely on the similarity between nodes relatively strongly when predicting the network alignment, while NetworkBLAST-M and MI-GRAAL use the similarity score mainly to predict potential orthology and do not rely too much on the extent of the similarity. In these experiments, Græ mlin appears to most strongly rely on the node similarity among the compared algorithms. In fact, Græ mlin achieves the highest specificity and sensitivity when there is a large separation between the score distributions (e.g.,
between 0 and 250. As can be seen in this figure, as the separation between the score distributions of orthologs and non-orthologs increases, both the specificity and the sensitivity are improved for IsoRank, IsoRankN, and Græ mlin. On the other hand, NetworkBLAST-M and MI-GRAAL display a constant level of accuracy that does not depend on the amount of separation. This implies that the first three alignment algorithms rely on the similarity between nodes relatively strongly when predicting the network alignment, while NetworkBLAST-M and MI-GRAAL use the similarity score mainly to predict potential orthology and do not rely too much on the extent of the similarity. In these experiments, Græ mlin appears to most strongly rely on the node similarity among the compared algorithms. In fact, Græ mlin achieves the highest specificity and sensitivity when there is a large separation between the score distributions (e.g., 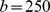 ), while resulting in the lowest sensitivity when the separation is small (e.g.,
), while resulting in the lowest sensitivity when the separation is small (e.g., 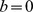 ).
).
Figure 9. The specificity (SPE) and the CN (which reflects the sensitivity) of different alignment algorithms for varying level of separation between the similarity score distribution for orthologs and the score distribution for non-orthologs.

Increasing the bias 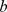 increases the separation between the two score distributions, hence increase the discriminative power of the node similarity score for predicting potential orthologs.
increases the separation between the two score distributions, hence increase the discriminative power of the node similarity score for predicting potential orthologs.
Table 5 compares the computational complexity of the five algorithms, in terms of the total CPU time needed to align the networks in the respective datasets. All experiments have been performed on a desktop computer with a 2.2GHz Intel Core2Duo CPU and 4GB memory. It should be noted that Græ mlin requires a training stage for estimating the parameters used by the algorithm, which took more than a day in our experiments. The CPU time shown in Table 5 reveals that Græ mlin (without considering the training stage) and NetworkBLAST-M are the fastest among the five algorithms, while IsoRankN and MI-GRAAL are computationally more complex than these two algorithms.
Table 5. Total CPU time (min) for aligning the networks.
| DMC | DMR | CG | |||||||
| pairwise | 5-way | 8-way | pairwise | 5-way | 8-way | pairwise | 5-way | 8-way | |
| IsoRank | 2.5 | N/A | N/A | 2.5 | N/A | N/A | 5 | N/A | N/A |
| IsoRankN | 25 | 65 | 60 | 20 | 65 | 57 | 56 | 170 | 150 |
| NetworkBLAST-M | 0.5 | 10 | 6 | 0.5 | 10 | 6 | 0.5 | 10 | 6 |
| Græmlin | 0.3 | 5.5 | 7 | 0.2 | 3.5 | 7.5 | 0.5 | 5 | 10 |
| MI-GRAAL | 45 | N/A | N/A | 45 | N/A | N/A | 45 | N/A | N/A |
Discussion
Absence of a comprehensive and reliable network alignment benchmark has been a critical obstacle that has been hindering research progress in comparative network analysis. In this work, we addressed this problem by proposing a novel network synthesis model that can generate network families with biologically realistic properties. The proposed model allows us to effectively generate families of evolutionarily related networks, where the network family may contain any number of networks with arbitrary phylogenetic relationships. We demonstrated that the internal as well as the cross-network properties of the synthesized networks closely resemble those of real protein-protein networks. Based on the proposed model, we synthesized a number of network benchmark datasets and evaluated the performance of several representative network alignment algorithms. These experiments allow us to clearly delineate the advantages and disadvantages of the respective algorithms in contrast to other algorithms. As demonstrated throughout this paper, the proposed network synthesis model provides an effective framework for generating large-scale network benchmarks, which can be used to reliably assess the performance of current and future network alignment algorithms under various conditions and problem settings.
Supporting Information
Network properties of different organisms. (A), (B) show the degree distributions, and (C), (D) show the clustering coefficient profiles.
(EPS)
Cross-species network properties for different pairs of organisms. (A)–(N) show how the number of potential orthologs are distributed between a given pair of networks.
(EPS)
Cross-species network properties for different pairs of organisms. (A)–(G) illustrate the sequence similarity distribution for orthologous and non-orthologous node pairs.
(EPS)
Funding Statement
This work was supported in part by the National Science Foundation through NSF Award CCF-1149544. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Zhang A (2009) Protein Interaction Networks: Computational Analysis, 1st ed. New York, NY: Cambridge University Press.
- 2. Uetz P, Giot L, Cagney G, Mansfield TA, Judson RS, et al. (2000) A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature 403: 623–627. [DOI] [PubMed] [Google Scholar]
- 3. Ho Y, Gruhler A, Heilbut A, Bader GD, Moore L, et al. (2002) Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 415: 180–183. [DOI] [PubMed] [Google Scholar]
- 4. Ge H (2000) UPA, a universal protein array system for quantitative detection of protein-protein, protein-DNA, protein-RNA and protein-ligand interactions. Nucleic Acids Res 28: e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Enright AJ, Iliopoulos I, Kyrpides NC, Ouzounis CA (1999) Protein interaction maps for complete genomes based on gene fusion events. Nature 402: 86–90. [DOI] [PubMed] [Google Scholar]
- 6. Pellegrini M, Marcotte EM, Thompson MJ, Eisenberg D, Yeates TO (1999) Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proc Natl Acad Sci USA 96: 4285–4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Deng M, Mehta S, Sun F, Chen T (2002) Inferring domain-domain interactions from proteinprotein interactions. Genome Res 12: 1540–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Matthews LR, Vaglio P, Reboul J, Ge H, Davis BP, et al. (2001) Identification of potential interaction networks using sequence-based searches for conserved protein-protein interactions or “interologs”. Genome Res 11: 2120–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sharan R, Ideker T (2006) Modeling cellular machinery through biological network comparison. Nat Biotechnol 24: 427–433. [DOI] [PubMed] [Google Scholar]
- 10. Flannick J, Novak A, Srinivasan B, McAdams H, Batzoglou S (2006) Græmlin: general and robust alignment of multiple large interaction networks. Genome Res 16: 1169–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Flannick J, Novak A, Do CB, Srinivasan BS, Batzoglou S (2009) Automatic parameter learning for multiple local network alignment. J Comput Biol 16: 1001–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liao CS, Lu K, Baym M, Singh R, Berger B (2009) IsoRankN: spectral methods for global alignment of multiple protein networks. Bioinformatics 25: i253–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Singh R, Xu J, Berger B (2008) Global alignment of multiple protein interaction networks with application to functional orthology detection. Proc Natl Acad Sci USA 105: 12763–12768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chindelevitch L, Liao CS, Berger B (2010) Local optimization for global alignment of protein interaction networks. Pac Symp Biocomput 123–132. [DOI] [PubMed] [Google Scholar]
- 15. Kalaev M, Smoot M, Ideker T, Sharan R (2008) NetworkBLAST: comparative analysis of protein networks. Bioinformatics 24: 594–596. [DOI] [PubMed] [Google Scholar]
- 16. Sharan R, Suthram S, Kelley RM, Kuhn T, McCuine S, et al. (2005) Conserved patterns of protein interaction in multiple species. Proc Natl Acad Sci USA 102: 1974–1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Koyuturk M, Kim Y, Topkara U, Subramaniam S, Szpankowski W, et al. (2006) Pairwise alignment of protein interaction networks. J Comput Biol 13: 182–199. [DOI] [PubMed] [Google Scholar]
- 18. Guo X, Hartemink AJ (2009) Domain-oriented edge-based alignment of protein interaction networks. Bioinformatics 25: i240–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dutkowski J, Tiuryn J (2007) Identification of functional modules from conserved ancestral protein-protein interactions. Bioinformatics 23: i149–158. [DOI] [PubMed] [Google Scholar]
- 20. Berg J, Lassig M (2006) Cross-species analysis of biological networks by Bayesian alignment. Proc Natl Acad Sci USA 103: 10967–10972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zaslavskiy M, Bach F, Vert JP (2009) Global alignment of protein-protein interaction networks by graph matching methods. Bioinformatics 25: i259–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Denilou YP, Boyer F, Viari A, Sagot MF (2009) Multiple alignment of biological networks: A flexible approach. In: Kucherov G, Ukkonen E, editors. Combinatorial Pattern Matching, volume 5577 of Lecture Notes in Computer Science. Heidelberg: Springer. pp. 263–273.
- 23. Klau GW (2009) A new graph-based method for pairwise global network alignment. BMC Bioinformatics 10 Suppl 1: S59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bradde S, Braunstein A, Mahmoudi H, Tria F, Weigt M, et al. (2010) Aligning graphs and finding substructures by a cavity approach. Europhysics Letters (epl) 89. [Google Scholar]
- 25. Kuchaiev O, Przulj N (2011) Integrative network alignment reveals large regions of global network similarity in yeast and human. Bioinformatics 27: 1390–1396. [DOI] [PubMed] [Google Scholar]
- 26. Li Z, Zhang S, Wang Y, Zhang X, Chen L (2007) Alignment of molecular networks by integer quadratic programming. Bioinformatics 23: 1631–1639. [DOI] [PubMed] [Google Scholar]
- 27. Ali W, Deane CM (2009) Functionally guided alignment of protein interaction networks for module detection. Bioinformatics 25: 3166–3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bandyopadhyay S, Sharan R, Ideker T (2006) Systematic identification of functional orthologs based on protein network comparison. Genome Res 16: 428–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bayati M, Gerritsen M, Gleich D, Saberi A, Wang Y (2009) Algorithms for large, sparse network alignment problems. In: Ninth IEEE International Conference on Data Mining (ICDM), 2009. pp. 705–710.
- 30. Qian X, Yoon BJ (2009) Effective identification of conserved pathways in biological networks using hidden Markov models. PLoS ONE 4: e8070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ay F, Kellis M, Kahveci T (2011) SubMAP: aligning metabolic pathways with subnetwork mappings. J Comput Biol 18: 219–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kelley BP, Yuan B, Lewitter F, Sharan R, Stockwell BR, et al. (2004) PathBLAST: a tool for alignment of protein interaction networks. Nucleic Acids Res 32: W83–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pinter R, Rokhlenko O, Yeger-Lotem E, Ziv-Ukelson M (2005) Alignment of metabolic pathways. Bioinformatics 21: 3401–3408. [DOI] [PubMed] [Google Scholar]
- 34. Shlomi T, Segal D, Ruppin E, Sharan R (2006) QPath: a method for querying pathways in a proteinprotein interaction network. BMC Bioinformatics 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dost B, Shlomi T, Gupta N, Ruppin E, Bafna V, et al. (2008) QNet: A tool for querying protein interaction networks. J Comput Biol 15: 913–925. [DOI] [PubMed] [Google Scholar]
- 36. Bruckner S, Huffner F, Karp RM, Shamir R, Sharan R (2010) Topology-free querying of protein interaction networks. J Comput Biol 17: 237–252. [DOI] [PubMed] [Google Scholar]
- 37. Qian X, Sze SH, Yoon BJ (2009) Querying pathways in protein interaction networks based on hidden Markov models. Journal of Computational Biology 16: 145–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sahraeian SME, Yoon BJ (2011) Fast network querying algorithm for searching large-scale biological networks. In: 2011 IEEE International Conference on Acoustics, Speech, and Signal Processing (ICASSP). pp. 6008–6011.
- 39. Sahraeian SME, Yoon BJ (2012) RESQUE: Network reduction using semi-Markov random walk scores for efficient querying of biological networks. Bioinformatics doi: 10.1093/bioinformatics/bts341. [DOI] [PubMed] [Google Scholar]
- 40. Ashburner M, Ball C, Blake J, Botstein D, Butler H, et al. (2000) Gene Ontology: Tool for the unification of biology. the gene ontology consortium. Nat Genet 25: 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kanehisa M, Goto S (2000) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28: 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Beltrao P, Serrano L (2007) Specificity and evolvability in eukaryotic protein interaction networks. PLoS Comput Biol 3: e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stark C, Breitkreutz BJ, Chatr-Aryamontri A, Boucher L, Oughtred R, et al. (2011) The BioGRID Interaction Database: 2011 update. Nucleic Acids Res 39: 698–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mewes HW, Frishman D, Mayer KF, Munsterkotter M, Noubibou O, et al. (2006) MIPS: analysis and annotation of proteins from whole genomes in 2005. Nucleic Acids Res 34: D169–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Salwinski L, Miller CS, Smith AJ, Pettit FK, Bowie JU, et al. (2004) The Database of Interacting Proteins: 2004 update. Nucleic Acids Res 32: D449–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kerrien S, Aranda B, Breuza L, Bridge A, Broackes-Carter F, et al. (2012) The IntAct molecular interaction database in 2012. Nucleic Acids Res 40: D841–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Licata L, Briganti L, Peluso D, Perfetto L, Iannuccelli M, et al. (2012) MINT, the molecular interaction database: 2012 update. Nucleic Acids Res 40: D857–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Keshava Prasad TS, Goel R, Kandasamy K, Keerthikumar S, Kumar S, et al. (2009) Human Protein Reference Database–2009 update. Nucleic Acids Res 37: D767–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cowley MJ, Pinese M, Kassahn KS, Waddell N, Pearson JV, et al. (2012) PINA v2.0: mining interactome modules. Nucleic Acids Res 40: D862–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Prieto C, De Las Rivas J (2006) APID: Agile Protein Interaction DataAnalyzer. Nucleic Acids Res 34: 298–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ali W, Deane CM (2010) Evolutionary analysis reveals low coverage as the major challenge for protein interaction network alignment. Mol Biosyst 6: 2296–2304. [DOI] [PubMed] [Google Scholar]
- 52. Erten S, Li X, Bebek G, Li J, Koyuturk M (2009) Phylogenetic analysis of modularity in protein interaction networks. BMC Bioinformatics 10: 333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Vazquez A, Flammini A, Maritan A, Vespignani A (2003) Modeling of Protein Interaction Networks. Complexus 1: 38–44. [Google Scholar]
- 54. Sole RV, Pastor-Satorras R, Smith E, Kepler TB (2002) A model of large-scale proteome evolution. Advances in Complex Systems (ACS) 5: 43–54. [Google Scholar]
- 55. Pastor-Satorras R, Smith E, Sole RV (2003) Evolving protein interaction networks through gene duplication. J Theor Biol 222: 199–210. [DOI] [PubMed] [Google Scholar]
- 56. Barabasi AL, Albert R (1999) Emergence of scaling in random networks. Science 286: 509–512. [DOI] [PubMed] [Google Scholar]
- 57. Qian J, Luscombe NM, Gerstein M (2001) Protein family and fold occurrence in genomes: powerlaw behaviour and evolutionary model. J Mol Biol 313: 673–681. [DOI] [PubMed] [Google Scholar]
- 58. Bhan A, Galas DJ, Dewey TG (2002) A duplication growth model of gene expression networks. Bioinformatics 18: 1486–1493. [DOI] [PubMed] [Google Scholar]
- 59. Rzhetsky A, Gomez SM (2001) Birth of scale-free molecular networks and the number of distinct DNA and protein domains per genome. Bioinformatics 17: 988–996. [DOI] [PubMed] [Google Scholar]
- 60. i Goh K, Kahng B, Kim D (2005) Evolution of the protein interaction network of budding yeast: Role of the protein family compatibility constraint. J Korean Phys Soc 46: 551–555. [Google Scholar]
- 61.Bebek G, Berenbrink P, Cooper C, Friedetzky T, Nadeau JH, et al.. (2007) Improved duplication models for proteome network evolution. In: Proceedings of the 2005 joint annual satellite conference on systems biology and regulatory genomics (RECOMB' 05). Berlin: Springer-Verlag. pp. 119–137.
- 62. Przulj N, Kuchaiev O, Stevanović A, Hayes W (2010) Geometric evolutionary dynamics of protein interaction networks. Pac Symp Biocomput 178–189. [DOI] [PubMed] [Google Scholar]
- 63. Berg J, Lassig M, Wagner A (2004) Structure and evolution of protein interaction networks: a statistical model for link dynamics and gene duplications. BMC Evol Biol 4: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Guillaume JL, Latapy M (2006) Bipartite graphs as models of complex networks. Physica A: Statistical and Theoretical Physics 371: 795–813. [Google Scholar]
- 65. Kim WK, Marcotte EM (2008) Age-dependent evolution of the yeast protein interaction network suggests a limited role of gene duplication and divergence. PLoS Comput Biol 4: e1000232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ispolatov I, Krapivsky PL, Yuryev A (2005) Duplication-divergence model of protein interaction network. Phys Rev E Stat Nonlin Soft Matter Phys 71: 061911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Evlampiev K, Isambert H (2008) Conservation and topology of protein interaction networks under duplication-divergence evolution. Proc Natl Acad Sci USA 105: 9863–9868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Levy ED, Pereira-Leal JB (2008) Evolution and dynamics of protein interactions and networks. Current Opinion in Structural Biology 18: 349–357. [DOI] [PubMed] [Google Scholar]
- 69. Ratmann O, Jrgensen O, Hinkley T, Stumpf M, Richardson S, et al. (2007) Using likelihood-free inference to compare evolutionary dynamics of the protein networks of H. pylori and P. falciparum. PLoS Comput Biol 3: e230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Coolen ACC, Fraternali F, Annibale A, Fernandes L, Kleinjung J (2011) Modelling Biological Networks via Tailored Random Graphs, JohnWiley and Sons, Ltd. pp. 309–329.
- 71. Barabasi AL, Oltvai ZN (2004) Network biology: understanding the cell's functional organization. Nat Rev Genet 5: 101–113. [DOI] [PubMed] [Google Scholar]
- 72. Middendorf M, Ziv E, Wiggins CH (2005) Inferring network mechanisms: the Drosophila melanogaster protein interaction network. Proc Natl Acad Sci USA 102: 3192–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hormozdiari F, Berenbrink P, Przulj N, Sahinalp SC (2007) Not all scale-free networks are born equal: the role of the seed graph in PPI network evolution. PLoS Comput Biol 3: e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ohno S (1970) Evolution by gene duplication, 1st ed. Heidelberg: Springer-Verlag. 160 p.
- 75. D'Antonio M, Ciccarelli FD (2011) Modification of Gene Duplicability during the Evolution of Protein Interaction Network. PLoS Comput Biol 7: e1002029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wagner A (2003) How the global structure of protein interaction networks evolves. Proc Biol Sci 270: 457–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Colak R, Hormozdiari F, Moser F, Schonhuth A, Holman J, et al. (2009) Dense graphlet statistics of protein interaction and random networks. Pac Symp Biocomput 178–189. [DOI] [PubMed] [Google Scholar]
- 79. Newman ME (2004) Fast algorithm for detecting community structure in networks. Phys Rev E Stat Nonlin Soft Matter Phys 69: 066133. [DOI] [PubMed] [Google Scholar]
- 80. Park D, Singh R, Baym M, Liao CS, Berger B (2011) IsoBase: a database of functionally related proteins across PPI networks. Nucleic Acids Res 39: 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Vazquez A, Dobrin R, Sergi D, Eckmann JP, Oltvai ZN, et al. (2004) The topological relationship between the large-scale attributes and local interaction patterns of complex networks. Proc Natl Acad Sci USA 101: 17940–17945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215: 403–410. [DOI] [PubMed] [Google Scholar]
- 83. Lipman DJ, Pearson WR (1985) Rapid and sensitive protein similarity searches. Science 227: 1435–1441. [DOI] [PubMed] [Google Scholar]
- 84.Knuth DE (1997) The Art of Computer Programming, 3rd ed, vol. 1. Boston, MA: Addison-Wesley Professional. 672 p.
- 85. Baudot A, Jacq B, Brun C (2004) A scale of functional divergence for yeast duplicated genes revealed from analysis of the protein-protein interaction network. Genome Biol 5: R76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Altschul SF, Gish W (1996) Local alignment statistics. Meth Enzymol 266: 460–480. [DOI] [PubMed] [Google Scholar]
- 87. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, et al. (1997) Gapped BLAST and PSIBLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Network properties of different organisms. (A), (B) show the degree distributions, and (C), (D) show the clustering coefficient profiles.
(EPS)
Cross-species network properties for different pairs of organisms. (A)–(N) show how the number of potential orthologs are distributed between a given pair of networks.
(EPS)
Cross-species network properties for different pairs of organisms. (A)–(G) illustrate the sequence similarity distribution for orthologous and non-orthologous node pairs.
(EPS)